 A. Wahid Ansari1*
A. Wahid Ansari1* Adeeba Kamarulzaman1 and Reinhold E. Schmidt2
Adeeba Kamarulzaman1 and Reinhold E. Schmidt2
- 1Faculty of Medicine, Centre of Excellence for Research in AIDS (CERiA), University of Malaya, Kuala Lumpur, Malaysia
- 2Clinic for Immunology and Rheumatology, Hannover Medical School, Hannover, Germany
Active tuberculosis remains the leading cause of death among the HIV-1 seropositive individuals. Although significant success has been achieved in bringing down the number of HIV/AIDS-related mortality and morbidity following implementation of highly active anti-retroviral therapy (HAART). Yet, co-infection of Mycobacterium tuberculosis (Mtb) has posed severe clinical and preventive challenges in our efforts to eradicate the virus from the body. Both HIV-1 and Mtb commonly infect macrophages and trigger production of host inflammatory mediators that subsequently regulate the immune response and disease pathogenesis. These inflammatory mediators can impose beneficial or detrimental effects on each pathogen and eventually on host. Among these, inflammatory C–C chemokines play a central role in HIV-1 and Mtb pathogenesis. However, their role in lung-specific mechanisms of HIV-1 and Mtb interaction are poorly understood. In this review we highlight current view on the role of C–C chemokines, more precisely CCL2, on HIV-1: Mtb interaction, potential mechanisms of action and adverse clinical consequences in a setting HIV-1/Mtb co-infection. Targeting common chemokine regulators of HIV-1/Mtb pathogenesis can be an attractive and potential anti-inflammatory intervention in HIV/AIDS-related comorbidities.
Introduction
According to United Nations Program on HIV/AIDS (1) nearly 14 million individuals are living with HIV-1/ Mycobacterium tuberculosis (Mtb) co-infection (http://www.unaids.org/documents/20101123_GlobalReport_Chap2_em.pdf), estimating around 26% of HIV/AIDS-related deaths each year (2). The host immune response elicited against these pathogens can impose beneficial or detrimental effects on each other and the host. HIV/AIDS is characterized by severe immune dysfunction associated with marked reduction in CD4+ T cell counts and high plasma HIV-1 viral load. Under such immune-deficient condition HIV-1+ individuals become susceptible to infection by opportunistic pathogens, including Mtb. Pathologically, both HIV-1 and Mtb infect alveolar macrophages in a setting of pulmonary co-infection. Seminal contributions have been made in past decades to understand the role of host derived soluble factors in HIV-1 and Mtb mono-infections (3–8), while lesser is known in HIV-1/Mtb co-infection setting. Nonetheless, clinical data has supported the production of some of the common soluble factors induced by these pathogens. For example production of pro-inflammatory mediators like IFN-γ, TNF-α, and CCL2 (MCP-1) by both HIV-1 and Mtb contribute significantly in disease control.
Chemokines are small molecular weight proteins involved in immuno-regulatory and inflammatory functions (9, 10). Based on their N-terminal cysteine residues they are categorized into: C–, C–C, C–X–C, and C–X3–C sub-families (10, 11). However, based on function additional classification has also been suggested into homeostatic or inflammatory chemokines (9). For example, homeostatic C–C chemokines such as CCL19 and CCL21 control homing of CCR7+ dendritic cells (DCs) and lymphocytes in the secondary lymphoid organs for optimal immune reactions (11). While inflammatory chemokines, CCL3 (MIP-1α), CCL3 (MIP-1β), CCL5 (RANTES), CXCL8 (IL-8), CXCL9 (MIG), CXCL10 (IP-10), and CXCL11 (I-TAC) participate in inflammation, autoimmune disorders, and malignancies (12–16). The pro-inflammatory chemokine CCL2 is linked to a number of human acute and chronic viral infections including HIV-1 (3, 17, 18). In addition to HIV-1+ individuals, a higher CCL2 levels are also detected in the broncho alveolar lavage (BAL) fluid of pulmonary TB patients (19) and pleural fluid of both HIV-1 infected and un-infected patients (20). Thus induction of CCL2 by both pathogens is an interesting aspect that needs to be addressed in a setting of HIV-1/Mtb co-infection.
The most striking feature of Mtb infection is the formation of granuloma, a highly organized cellular structure composed of macrophages, T cells, NK cells, B-cells, neutrophils, and DCs. Functionally granuloma restricts the Mtb bacilli within this specialized microenvironment. Several hypotheses are drawn to describe the mechanism by which HIV-1 increases the risk of TB reactivation. Some of the potential mechanisms include (1) persistent HIV-1 replication in the lung causes immune dysfunction (20). (2) HIV-1 induced CD4+ T cell apoptosis and subsequent granuloma disruption (21). (3) Depletion of Mtb-specific CD4+ T cells by HIV-1 increases the risk of latent TB reactivation (22, 23). Most of the previous studies pertaining to host derived soluble factors in HIV-1/Mtb co-infection highlighted the cytokines with limited information on chemokines. Since the chemokine biology itself is a large area and to discuss the relevance of each chemokine sub-families is beyond the scope of current review. Thus, we focus on most relevant C–C chemokines associated with each pathogen and in a setting of HIV-1/Mtb-co-infection.
Chemokines in HIV-1 Pathogenesis and Disease Progression
HIV-1 induced inflammatory chemokines exhibit dual function. For example, C–C chemokine CCL3, CCL4, CCL5, and C–X–C chemokine CXCL12 (SDF-1α) function to block the entry of R5 and R4 HIV-1 strains (24–26) thereby preventing HIV-1 replication. While another C–C family member, CCL2 has been suggested to support HIV-1 replication (3, 17, 18). The most compelling evidence in support of chemokines in HIV-1 pathogenesis is exhibited by the individuals homozygous for 32 bp-CCR5 deletion conferring resistance toward HIV-1 (27) and recently reported stem cell transplant study from individual with 32 bp-CCR5 deletion to HIV-1 infected patient showing a long-term HIV-1 control (28). Thus early production of above suppressive chemokines in the lymph nodes can benefit host by restricting HIV-1 dissemination (29). This unique feature of HIV-1 suppressive chemokines made the basis of developing CCR5 antagonist Maraviroc, which has now progressed to clinical practice (30).
CCL2, is a strong chemo-attractant of CCR2+ monocytes/macrophages and CD4+ T cells (9, 10). In human peripheral blood, CCL2 is mainly produced by circulating monocytes, in particular by the CD14+ CD16+ inflammatory monocyte subsets of HIV-1 patients (3, 31). Clinical data including ours showed an elevated CCL2 levels in the serum and cerebrospinal fluid (CSF) and that significantly correlates with plasma viral load of HIV-1 patients (3, 18, 32–34). Further we showed a selective up-regulation CCL2 mRNA and serum CCL2 by HIV-1 viremic than aviremic patients (3), suggesting differential CCL2 response by host depends on the status of HIV-1 replication. This observation was further strengthened in a case study where a highly viremic HIV-1+ patient who received a short-term prednisolone treatment for severe uveitis, displayed a drastic viral load reduction paralleled with declined CCL2 expression (35). Another evidence supporting the association of viremia with CCL2 production is recently reported in the CSF specimens of “Elite controllers” (EC) (36). ECs are a rare group of HIV-1+ individuals with persistently suppressed viral load (37). Strikingly, both EC and HIV-negative individuals show lower level of serum CCL2 compared to HIV-1+ individuals. Based on our own and others findings a hypothetical model on impact of CCL2 in HIV-1 pathogenesis is described elsewhere (4, 17). This explains (1) recruitment of HIV-1 permissive monocytes/macrophages and CD4+ T cells at the site of infection for new rounds of replication (feed-back loop model), (2) induction of HIV-1 co-receptor CXCR4 by CCL2 (38), (3) CCL2-mediated polarization of helper T cell (Th0) toward Th2 phenotype (39), (4) IL-4 induction CXCR4 expression on resting CD4 T cells (40), and (5) enhanced HIV-1 progeny release by CCL2 (34). Thus, a selective inhibition of CCL2 could provide an attractive anti-inflammatory intervention in HIV/AIDS.
Chemokines in M. tuberculosis Infection
It is well established now that Mtb infection can occur throughout the course of HIV-1 infection (2) and that eventually results in diagnostic and preventive challenges. The protective immunity against Mtb is mainly driven by CD4+ T cells and macrophages, supported by a network of inflammatory cytokines and chemokines. Among these IFN-γ and TNF-α are the two major cytokines conferring protective immunity against Mtb (41). TNF-α, in addition to macrophage activation also induces secretion of several C–C and C–X–C chemokines including CCL2 (42, 43). Availability of animal models including mice, guinea pig, and non-human primate (NHP) have immensely contributed to our understanding of inflammatory reactions in Mtb infection (21). For example in murine model of Mtb infection, secretion of C–X–C chemokines, CXCL3, and CXCL5 (44) lead to influx of CXCR2+ neutrophils and NK cells while CXCL13 (45) recruits follicular helper (Tfh) CXCR5+ T cell into lung to provide immune protection against TB (46). Similarly, chemokine CCL5 tends to play an important role in T cell priming by recruiting lymphocytes into the lung, thereby helping in controlling murine Mtb infection. Recent study on BAL samples of HIV-1/Mtb co-infected patients showed a significant correlation of viremia with CCL5 and its receptor CCR5 (47) suggesting persistent HIV-1 replication in the lung drives activation of local T cells as evident by high expression of CCR5 in HIV/latent Mtb co-infection.
Owing to its strong chemotactic and pro-inflammatory properties, CCL2 has been shown to participate in granuloma formation (48) and to certain extent protection against Mtb (49). While other Mtb-induced C–C chemokine CCL3, CCL4, and CCL5 has been described to inhibit bacilli growth (50). Among the C–X–C chemokine, CXCL8 (IL-8), is reported to be the most important soluble factor which got elevated and recruits monocytes and lymphocytes in the lung of TB patients (51). Looking closely on CCL2 role in Mtb infection, studies have described induction of CCL2 by both the BCG and Mtb in in vitro as well as in BCG vaccinated individuals in vivo (52). Interestingly, a high CCL2 level was found to be associated with disease severity of TB patients (52). In addition to pathogen and induced soluble factors, host genetic make-up is considered as key factor in determining the Mtb infection susceptibility and progression to active TB. Recent population genetics studies have described the association of CCL2 polymorphism and TB disease. For example individuals with CCL2-2518G allele show significant association with risk of developing active TB in Asian and Hispanic population (53).
Potential Impact of Mtb-Induced CCL2 in HIV-1 Pathogenesis
Previous studies have described the exploitation of lung and pleura-specific cellular environment by HIV-1 for replication and subsequent induction of inflammatory mediators such as CCL2 (19). In pulmonary TB infection, bacilli enter the lung via respiratory pathway which are subsequently encountered and phagocytosed by alveolar macrophages and DC (20). During this event activated macrophages secrete TNF-α to control Mtb bacilli growth (54). It is should be noted that TNF-α is also known to activate HIV-1 replication (55), that means the detrimental effects of one pathogen proving beneficial to the other. Nevertheless, Mtb has been shown to induce HIV-1 replication in acutely and chronically infected macrophages or T cells (56) as well as in alveolar macrophages and lymphocytes of HIV-1 infected individuals (57, 58). These effects are clinically displayed as high viral load in the plasma of HIV-1/Mtb co-infected (59, 60) as well as in BAL (61) of TB patients, suggesting Mtb could support HIV-1 replication by manipulating the lung microenvironment.
A hypothetical model based on available data can be is summarized (Figure 1). Firstly recruitment of HIV-1 permissive CCR2+ monocytes/macrophages and CD4+ T cells (20) by CCL2 released from Mtb-infected alveolar macrophages (55, 62) increases the risk of HIV-1 infection. Secondly, CCL2-mediated activation of HIV-LTR (long-terminal repeats) as shown in infected macrophages and CD4+ T cells (63) also results in induction of pro-inflammatory genes such as TNF-α, CCL2, and IL-6 (59, 60), that mean CCL2–CCR2 axis has dual effects on HIV-1 infected cells by inducing HIV-LTR and pro-inflammatory genes. Studies have also shown, activation of latent HIV-1 by Mtb-purified protein derivatives (PPD) in the alveolar macrophages of infected patients (64). Thus, one can argue that both CCL2 and bacilli can affect the HIV-1 replication in HIV-1/Mtb co-infection scenario (Figure 1). A number of studies have described Mtb-derived products to trans-activate HIV-LTR coupled with pro-inflammatory genes expression upon recognition by cell surface molecules (Figure 1). This event is regulated by several innate molecular signaling pathways (6) including MAPKinases, NFkB, C/EBPs, and very recently identified NFAT5 (65). A sustained and prolonged activation of signaling pathways and subsequent secretion of inflammatory mediators including CCL2 may cause chronic inflammation and that may prove fatal to HIV-1/Mtb co-infected individuals.
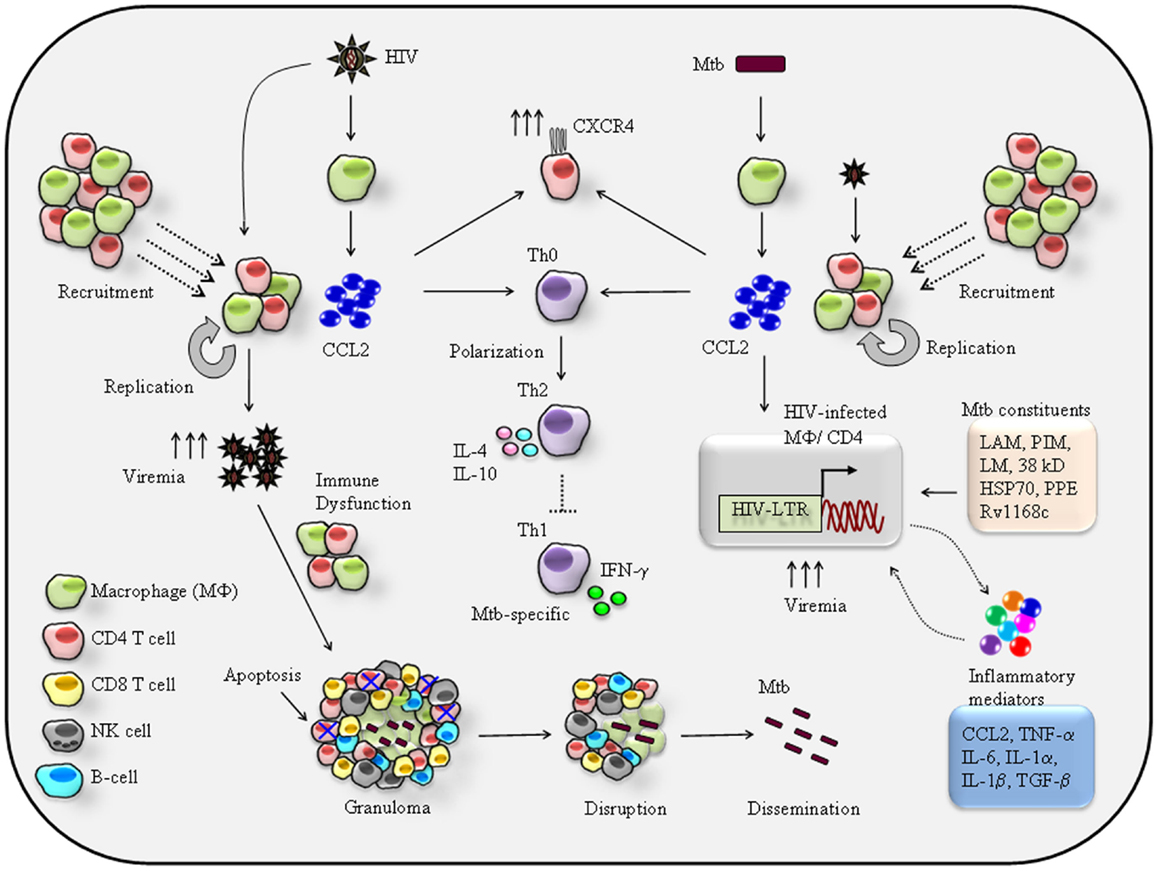
Figure 1. Proposed mechanisms of reciprocal effects of CCL2-mediated immuno-pathogenesis of HIV/Mtb co-infection. HIV-1 infection of alveolar macrophage releases CCL2 that recruits monocytes/macrophages and CD4+ T cells at the site of infection hence increase the pool of HIV-1 permissive cells for new round of replication-the feed-back loop model (4, 17) eventually persistence of a high viremia in the BAL. CCL2 can acts on resting CD4+ T cells to induce expression of HIV-1 co-receptor CXCR4, thereby rendering them susceptible to infection by X4 strains (38). CCL2 known to trigger differentiation of Th0 toward Th2 phenotype (39) via CCL2–CCR2 axis. Therefore, in the lung a high CCL2 creates a Th2 dominant environment that presumably suppresses Mtb-specific Th1 response. Persistence HIV-1 replication and high viremia in the lung impairs the macrophage and CD4+ T cell effector function against Mtb. Most importantly, the targeted apoptosis of CD4+ T cells leads to granuloma disruption leading to reactivation and dissemination of latent TB (21). On other hand secretion of CCL2 by Mtb infection may shares the similar effects like cellular recruitment, CXCR4 induction, and suppression of Mtb-specific Th1 immune response very much similar to those imposed by HIV-1. In addition to CCL2, Mtb and its cell wall constituents like lipo-arabinomannan (LAM), phosphatidylinositol (PIM), lipomannan (LM), and 19-kD Mtb protein (56), the 38-kD glycoprotein and HSP70 recognition by TLR4 (66) proline–proline-glutamic acid (PPE) protein Rv1168c (67, 68) by pattern recognition receptor (PPR) and C-type lectin receptor (69) result in secretion of pro-inflammatory cytokines and chemokines including TNF-α (5), CCL2 (54, 70, 71), IL-1α/β (5, 72), IFN-γ (56, 73), and IL-6 (74–76) that can trigger HIV-1 replication by activating HIV-LTR of the infected macrophages or CD4+ T cells eventually a high viremia. While the secreted inflammatory molecules can act in autocrine manner to activate HIV-LTR. Thus, productions of these inflammatory mediators lead to local immune reaction that eventually enhances the severity of HIV/Mtb comorbidity.
HIV-Induced CCL2 in Latent TB Reactivation and Pathogenesis
As estimated nearly one third population of world is living with Mtb infection and individuals with HIV/AIDS are at high risk of developing active TB (77). Entry of Mtb bacilli into the lung, trigger early immune response where DC captures Mtb and migrate to nearby draining lymph nodes to start Mtb-specific adaptive immune responses (78). This leads to activation, expansion, and functional maturation of Mtb-specific CD4+ T cells that home to the site of primary infection in lung and activates innate immune cells such as macrophages to release IFN-γ and TNF-α to control infection (55, 79). Some of the potential mechanisms by which HIV-1 facilitate Mtb pathogenesis are, up-regulation of Mtb receptor on macrophages (80, 81), impaired leukocyte recruitment (82), altered Th1/Th2 balance (83), and impaired TNF-α mediated macrophage apoptosis (84). In fact, the greater impact of HIV-1 in Mtb disease is the reactivation of latent TB by disruption of granuloma, a feature commonly associated with immune compromised conditions such as HIV/AIDS (85). Based on in vitro and ex vivo clinical findings some of the potential pathways by which HIV-1 induced CCL2 can contribute to TB reactivation and associated pathology (Figure 1) include (1) HIV-1 infected alveolar macrophages secreted CCL2 recruits CCR2+ leukocytes including macrophages, CD4+ T cells, and NK cells to participate in immune reactions. This gives HIV-1 opportunity to infect and replicate within these freshly recruited permissive cells resulting in high viremia as detected in the BAL fluid of HIV-1/Mtb co-infected patients (7, 21, 86, 87). (2) Persistence of high HIV-1 viremia causes macrophage dysfunction to kill Mtb (20). (3) Entry of HIV-1 into granuloma causes CD4+ T cell apoptosis, depletion, and disorganization of granuloma (85, 88, 89), resulting in Mtb dissemination associated with extra-pulmonary TB manifestations. (4) IFN-γ producing CD4+ T cells are crucial for Mtb control (21) thus, depletion of Mtb-specific CD4+ T cells by HIV-1 certainly is an important factor to increase the risk of latent TB reactivation (80, 81). (5) Given that CCL2 favors Th2 response which is evident from CCL2−/− mice that confer resistance against parasitic infection (22, 23) under this scenario, a higher CCL2 level in the BAL of HIV-1/Mtb patients (39) will generate a Th2 dominant environment that presumably suppresses Mtb-specific IFN-γ mediated Th1 immunity (Figure 1). Taken together, this hypothetical model explains the CCL2 mediated on-going lung-specific HIV-1 and Mtb interplay and a mechanistic insight how HIV-1 and Mtb reciprocate each other in a setting of HIV-1/Mtb co-infection.
IRIS Associated Complications in HIV-1/Mtb Co-Infection
Although highly active anti-retroviral therapy (HAART) dramatically declines the HIV/AIDS-associated morbidity and mortality by restoring CD4 T cell counts in HIV-1 infected individuals but an unwanted phenomena of immune reconstitution inflammatory syndrome (IRIS) hampers the successful treatment of HIV-1/Mtb co-infection (90, 91). There are two forms of TB-IRIS, the “paradoxical” which occurs in patient receiving anti-TB drugs before HAART and “unmasking” TB-IRIS in patients with initiation of HAART without prior clinical symptoms of TB (92). Despite serious clinical efforts, the pathogenic mechanism of IRIS is largely unknown. Some of the most widely accepted potential mechanisms includes (a) the magnitude of immune restoration, (b) the antigenic burden, and (c) the host genetic susceptibility. Moreover, early prediction of developing either forms of IRIS may benefit susceptible individuals by introducing preventive approaches. In this regard, some of the predictive chemokine markers such as CCL2, CXCL8, and CXCL10 have been suggested (93). Of which, the pro-inflammatory CCL2 is argued as a strong predictor of TB-IRIS in HIV patients after commencing HAART. Further studies are required to understand the mechanism and identification of a set of biomarkers to predict IRIS for improved disease management.
Conclusion and Perspectives
As a major public health issue it is critical to understand and exploit the beneficial and detrimental effects imposed by each pathogen on host survival in a situation of HIV-1/Mtb co-infection. Under this complex scenario, inflammatory mediators tend to play pivotal role in containing pathogens and disease progression. Although Mtb-specific CD4+ T cells are critical for controlling active TB at the same time they are prone to attack by HIV-1 (20). Therefore, in addition to reconstitution of CD4+ T cells by anti-retroviral and anti-TB therapy regimens, strategies should be developed to reduce CCL2 expression to contain severity of co-infection. Due to ethical limitations, a direct study on HIV-1/Mtb co-infected individuals is not feasible, animal models could prove an alternative and valuable tool for such studies. Efforts have been made in this regard to generate CD4+ T cell-deficient mouse model mimicking HIV/AIDS-associated features (81) and humanized mice (94) to study HIV-1 pathogenesis and behavior within granuloma (95). Moreover, several NHP models of HIV-1/Mtb co-infection have also been developed (96) including new cynomolgus macaque model to address SIV induced reactivation of latent TB (86, 97, 98). Further studies focusing CCL2 in these animal models will allow us to unravel the mechanism of CCL2-mediated co-infection pathogenesis and consequences of HIV-1: Mtb interactions on disease outcome. We hope this will lead to manipulate CCL2 as an anti-inflammatory intervention point in HIV/AIDS-related comorbidities.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the University Malaya Research Grant (UMRG 501-13HTM) of the Health and Translational Medicine Cluster to A. Wahid Ansari. Adeeba Kamarulzaman received funding from High Impact Research Grant (HIRGA E000001-20001) of the Ministry of Higher Education (MoHE) Malaysia.
References
1. UNAIDS. Chapter 2: Epidemic Update. UNAIDS Report on the Global AIDS Epidemic 2010. (2010). Available from http://www.unaids.org/documents/20101123_GlobalReport_Chap2_em.pdf
2. Getahun H, Gunneberg C, Granich R, Nunn P. HIV infection-associated tuberculosis: the epidemiology and the response. Clin Infect Dis (2010) 50(Suppl 3):S201–7. doi: 10.1086/651492
3. Ansari AW, Bhatnagar N, Dittrich-Breiholz O, Kracht M, Schmidt RE, Heiken H. Host chemokine (C-C motif) ligand-2 (CCL2) is differentially regulated in HIV type 1 (HIV-1)-infected individuals. Int Immunol (2006) 18:1443–51. doi:10.1093/intimm/dxl078
4. Ansari AW, Heiken H, Moenkemeyer M, Schmidt RE. Dichotomous effects of C-C chemokines in HIV-1 pathogenesis. Immunol Lett (2007) 110:1–5. doi:10.1016/j.imlet.2007.02.012
5. Collins KR, Quinones-Mateu ME, Toossi Z, Arts EJ. Impact of tuberculosis on HIV-1 replication, diversity, and disease progression. AIDS Rev (2002) 4:165–76.
6. Falvo JV, Ranjbar S, Jasenosky LD, Goldfeld AE. Arc of a vicious circle: pathways activated by Mycobacterium tuberculosis that target the HIV-1 long terminal repeat. Am J Respir Cell Mol Biol (2012) 45:1116–24. doi:10.1165/rcmb.2011-0186TR
7. Toossi Z. Virological and immunological impact of tuberculosis on human immunodeficiency virus type 1 disease. J Infect Dis (2003) 188:1146–55. doi:10.1086/378676
8. Toossi Z, Mayanja-Kizza H, Lawn SD, Hirsch CS, Lupo LD, Butera ST. Dynamic variation in the cellular origin of HIV type 1 during treatment of tuberculosis in dually infected subjects. AIDS Res Hum Retroviruses (2007) 23:93–100. doi:10.1089/aid.2006.0050
9. Moser B, Loetscher P. Lymphocyte traffic control by chemokines. Nat Immunol (2001) 2:123–8. doi:10.1038/84219
11. Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity (2000) 12:121–7. doi:10.1016/S1074-7613(00)80165-X
12. Fridlender ZG, Buchlis G, Kapoor V, Cheng G, Sun J, Singhal S, et al. CCL2 blockade augments cancer immunotherapy. Cancer Res (2010) 70:109–18. doi:10.1158/0008-5472.CAN-09-2326
13. Kusano KF, Nakamura K, Kusano H, Nishii N, Banba K, Ikeda T, et al. Significance of the level of monocyte chemoattractant protein-1 in human atherosclerosis. Circ J (2004) 68:671–6. doi:10.1253/circj.68.671
14. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475:222–5. doi:10.1038/nature10138
15. Rafei M, Berchiche YA, Birman E, Boivin MN, Young YK, Wu JH, et al. An engineered GM-CSF-CCL2 fusokine is a potent inhibitor of CCR2-driven inflammation as demonstrated in a murine model of inflammatory arthritis. J Immunol (2009) 183:1759–66. doi:10.4049/jimmunol.0900523
16. Sorensen TL, Ransohoff RM, Strieter RM, Sellebjerg F. Chemokine CCL2 and chemokine receptor CCR2 in early active multiple sclerosis. Eur J Neurol (2004) 11:445–9. doi:10.1111/j.1468-1331.2004.00796.x
17. Ansari AW, Heiken H, Meyer-Olson D, Schmidt RE. CCL2: a potential prognostic marker and target of anti-inflammatory strategy in HIV/AIDS pathogenesis. Eur J Immunol (2011) 41:3412–8. doi:10.1002/eji.201141676
18. Mengozzi M, De Filippi C, Transidico P, Biswas P, Cota M, Ghezzi S, et al. Human immunodeficiency virus replication induces monocyte chemotactic protein-1 in human macrophages and U937 promonocytic cells. Blood (1999) 93:1851–7.
19. Sadek MI, Sada E, Toossi Z, Schwander SK, Rich EA. Chemokines induced by infection of mononuclear phagocytes with mycobacteria and present in lung alveoli during active pulmonary tuberculosis. Am J Respir Cell Mol Biol (1998) 19:513–21. doi:10.1165/ajrcmb.19.3.2815
20. Toossi Z, Johnson JL, Kanost RA, Wu M, Luzze H, Peters P, et al. Increased replication of HIV-1 at sites of Mycobacterium tuberculosis infection: potential mechanisms of viral activation. J Acquir Immune Defic Syndr (2001) 28:1–8. doi:10.1097/00126334-200109010-00001
21. Diedrich CR, Flynn JL. HIV-1/Mycobacterium tuberculosis coinfection immunology: how does HIV-1 exacerbate tuberculosis? Infect Immun (2011) 79:1407–17. doi:10.1128/IAI.01126-10
22. Lewinsohn DA, Heinzel AS, Gardner JM, Zhu L, Alderson MR, Lewinsohn DM. Mycobacterium tuberculosis-specific CD8+ T cells preferentially recognize heavily infected cells. Am J Respir Crit Care Med (2003) 168:1346–52. doi:10.1164/rccm.200306-837OC
23. Lewinsohn DA, Winata E, Swarbrick GM, Tanner KE, Cook MS, Null MD, et al. Immunodominant tuberculosis CD8 antigens preferentially restricted by HLA-B. PLoS Pathog (2007) 3:1240–9. doi:10.1371/journal.ppat.0030127
24. Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, et al. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature (1996) 382:829–33. doi:10.1038/382829a0
25. Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science (1995) 270:1811–5. doi:10.1126/science.270.5243.1811
26. Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature (1996) 382:833–5. doi:10.1038/382833a0
27. Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science (1996) 273:1856–62.
28. Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med (2009) 360:692–8. doi:10.1056/NEJMoa0802905
29. Trumpfheller C, Tenner-Racz K, Racz P, Fleischer B, Frosch S. Expression of macrophage inflammatory protein (MIP)-1alpha, MIP-1beta, and RANTES genes in lymph nodes from HIV+ individuals: correlation with a Th1-type cytokine response. Clin Exp Immunol (1998) 112:92–9. doi:10.1046/j.1365-2249.1998.00555.x
30. Fatkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med (2005) 11:1170–2. doi:10.1038/nm1319
31. Ansari AW, Meyer-Olson D, Schmidt RE. Selective expansion of pro-inflammatory chemokine CCL2-loaded CD14(+)CD16(+) monocytes subset in HIV-infected therapy naive individuals. J Clin Immunol (2013) 33:302–6. doi:10.1007/s10875-012-9790-0
32. Bernasconi S, Cinque P, Peri G, Sozzani S, Crociati A, Torri W, et al. Selective elevation of monocyte chemotactic protein-1 in the cerebrospinal fluid of AIDS patients with cytomegalovirus encephalitis. J Infect Dis (1996) 174:1098–101. doi:10.1093/infdis/174.5.1098
33. Cinque P, Vago L, Mengozzi M, Torri V, Ceresa D, Vicenzi E, et al. Elevated cerebrospinal fluid levels of monocyte chemotactic protein-1 correlate with HIV-1 encephalitis and local viral replication. AIDS (1998) 12:1327–32. doi:10.1097/00002030-199811000-00014
34. Fantuzzi L, Spadaro F, Vallanti G, Canini I, Ramoni C, Vicenzi E, et al. Endogenous CCL2 (monocyte chemotactic protein-1) modulates human immunodeficiency virus type-1 replication and affects cytoskeleton organization in human monocyte-derived macrophages. Blood (2003) 102:2334–7. doi:10.1182/blood-2002-10-3275
35. Ansari AW, Schmidt RE, Heiken H. Prednisolone mediated suppression of HIV-1 viral load strongly correlates with C-C chemokine CCL2: in vivo and in vitro findings. Clin Immunol (2007) 125:1–4. doi:10.1016/j.clim.2007.07.003
36. Card CM, Keynan Y, Lajoie J, Bell CP, Dawood M, Becker M, et al. HIV controllers are distinguished by chemokine expression profile and HIV-specific T-cell proliferative potential. J Acquir Immune Defic Syndr (2012) 59:427–37. doi:10.1097/QAI.0b013e3182454fcd
37. Okulicz JF, Lambotte O. Epidemiology and clinical characteristics of elite controllers. Curr Opin HIV AIDS (2011) 6:163–8. doi:10.1097/COH.0b013e328344f35e
38. Campbell GR, Watkins JD, Singh KK, Loret EP, Spector SA. Human immunodeficiency virus type 1 subtype C Tat fails to induce intracellular calcium flux and induces reduced tumor necrosis factor production from monocytes. J Virol (2007) 81:5919–28. doi:10.1128/JVI.01938-06
39. Gu L, Tseng S, Horner RM, Tam C, Loda M, Rollins BJ. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature (2000) 404:407–11. doi:10.1038/35006097
40. Jourdan P, Abbal C, Noraz N, Hori T, Uchiyama T, Vendrell JP, et al. IL-4 induces functional cell-surface expression of CXCR4 on human T cells. J Immunol (1998) 160:4153–7.
41. Orme IM. Immunity to mycobacteria. Curr Opin Immunol (1993) 5:497–502. doi:10.1016/0952-7915(93)90029-R
42. Algood HM, Chan J, Flynn JL. Chemokines and tuberculosis. Cytokine Growth Factor Rev (2003) 14:467–77. doi:10.1016/S1359-6101(03)00054-6
43. Collins HL, Kaufmann SH. The many faces of host responses to tuberculosis. Immunology (2001) 103:1–9. doi:10.1046/j.1365-2567.2001.01236.x
44. Kang DD, Lin Y, Moreno JR, Randall TD, Khader SA. Profiling early lung immune responses in the mouse model of tuberculosis. PLoS One (2011) 6:e16161. doi:10.1371/journal.pone.0016161
45. Kahnert A, Hopken UE, Stein M, Bandermann S, Lipp M, Kaufmann SH. Mycobacterium tuberculosis triggers formation of lymphoid structure in murine lungs. J Infect Dis (2007) 195:46–54. doi:10.1086/508894
46. Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J Clin Invest (2012) 123:712–26. doi:10.1172/JCI65728
47. Kalsdorf B, Skolimowska KH, Scriba TJ, Dawson R, Dheda K, Wood K, et al. Relationship between chemokine receptor expression, chemokine levels and HIV-1 replication in the lungs of persons exposed to Mycobacterium tuberculosis. Eur J Immunol (2013) 43:540–9. doi:10.1002/eji.201242804
48. Saunders BM, Britton WJ. Life and death in the granuloma: immunopathology of tuberculosis. Immunol Cell Biol (2007) 85:103–11. doi:10.1038/sj.icb.7100027
49. Kipnis A, Basaraba RJ, Orme IM, Cooper AM. Role of chemokine ligand 2 in the protective response to early murine pulmonary tuberculosis. Immunology (2003) 109:547–51. doi:10.1046/j.1365-2567.2003.01680.x
50. Saukkonen JJ, Bazydlo B, Thomas M, Strieter RM, Keane J, Kornfeld H. Beta-chemokines are induced by Mycobacterium tuberculosis and inhibit its growth. Infect Immun (2002) 70:1684–93. doi:10.1128/IAI.70.4.1684-1693.2002
51. Moser B, Wolf M, Walz A, Loetscher P. Chemokines: multiple levels of leukocyte migration control. Trends Immunol (2004) 25:75–84. doi:10.1016/j.it.2003.12.005
52. Hasan Z, Cliff JM, Dockrell HM, Jamil B, Irfan M, Ashraf M, et al. CCL2 responses to Mycobacterium tuberculosis are associated with disease severity in tuberculosis. PLoS One (2009) 4:e8459. doi:10.1371/journal.pone.0008459
53. Feng WX, Flores-Villanueva PO, Mokrousov I, Wu XR, Xiao J, Jiao WW, et al. CCL2-2518 (A/G) polymorphisms and tuberculosis susceptibility: a meta-analysis. Int J Tuberc Lung Dis (2012) 16:150–6. doi:10.5588/ijtld.11.0205
54. Quesniaux VF, Jacobs M, Allie N, Grivennikov S, Nedospasov SA, Garcia I, et al. TNF in host resistance to tuberculosis infection. Curr Dir Autoimmun (2010) 11:157–79. doi:10.1159/000289204
55. Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol (2007) 179:2509–19.
56. Kedzierska K, Crowe SM. Cytokines and HIV-1: interactions and clinical implications. Antivir Chem Chemother (2001) 12:133–50.
57. Shattock RJ, Friedland JS, Griffin GE. Modulation of HIV transcription in and release from human monocytic cells following phagocytosis of Mycobacterium tuberculosis. Res Virol (1993) 144:7–12. doi:10.1016/S0923-2516(06)80005-1
58. Zhang Y, Nakata K, Weiden M, Rom WN. Mycobacterium tuberculosis enhances human immunodeficiency virus-1 replication by transcriptional activation at the long terminal repeat. J Clin Invest (1995) 95:2324–31. doi:10.1172/JCI117924
59. Goletti D, Weissman D, Jackson RW, Collins F, Kinter A, Fauci AS. The in vitro induction of human immunodeficiency virus (HIV) replication in purified protein derivative-positive HIV-infected persons by recall antigen response to Mycobacterium tuberculosis is the result of a balance of the effects of endogenous interleukin-2 and proinflammatory and antiinflammatory cytokines. J Infect Dis (1998) 177:1332–8.
60. Toossi Z, Nicolacakis K, Xia L, Ferrari NA, Rich EA. Activation of latent HIV-1 by Mycobacterium tuberculosis and its purified protein derivative in alveolar macrophages from HIV-infected individuals in vitro. J Acquir Immune Defic Syndr Hum Retrovirol (1997) 15:325–31. doi:10.1097/00042560-199708150-00001
61. Goletti D, Weissman D, Jackson RW, Graham NM, Vlahov D, Klein RS, et al. Effect of Mycobacterium tuberculosis on HIV replication. Role of immune activation. J Immunol (1996) 157:1271–8.
62. Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol (2009) 10:943–8. doi:10.1038/ni.1781
63. Fietta AM, Morosini M, Meloni F, Bianco AM, Pozzi E. Pharmacological analysis of signal transduction pathways required for Mycobacterium tuberculosis-induced IL-8 and MCP-1 production in human peripheral monocytes. Cytokine (2002) 19:242–9. doi:10.1006/cyto.2002.1968
64. Melgarejo E, Medina MA, Sanchez-Jimenez F, Urdiales JL. Monocyte chemoattractant protein-1: a key mediator in inflammatory processes. Int J Biochem Cell Biol (2009) 41:998–1001. doi:10.1016/j.biocel.2008.07.018
65. Ranjbar S, Jasenosky LD, Chow N, Goldfeld AE. Regulation of Mycobacterium tuberculosis-dependent HIV-1 transcription reveals a new role for NFAT5 in the toll-like receptor pathway. PLoS Pathog (2012) 8:e1002620. doi:10.1371/journal.ppat.1002620
66. Harding CV, Boom WH. Regulation of antigen presentation by Mycobacterium tuberculosis: a role for toll-like receptors. Nat Rev Microbiol (2010) 8:296–307. doi:10.1038/nrmicro2321
67. Bulut Y, Michelsen KS, Hayrapetian L, Naiki Y, Spallek R, Singh M, et al. Mycobacterium tuberculosis heat shock proteins use diverse Toll-like receptor pathways to activate pro-inflammatory signals. J Biol Chem (2005) 280:20961–7. doi:10.1074/jbc.M411379200
68. Jung SB, Yang CS, Lee JS, Shin AR, Jung SS, Son JW, et al. The mycobacterial 38-kilodalton glycolipoprotein antigen activates the mitogen-activated protein kinase pathway and release of proinflammatory cytokines through Toll-like receptors 2 and 4 in human monocytes. Infect Immun (2006) 74:2686–96. doi:10.1128/IAI.74.5.2686-2696.2006
69. Bhat KH, Chaitanya CK, Parveen N, Varman R, Ghosh S, Mukhopadhyay S. Proline-proline-glutamic acid (PPE) protein Rv1168c of Mycobacterium tuberculosis augments transcription from HIV-1 long terminal repeat promoter. J Biol Chem (2012) 287:16930–46. doi:10.1074/jbc.M111.327825
70. Keane J, Ni Cheallaigh C. Making tumor necrosis factor blockers safer: a prescriber’s consumptive challenge. J Rheumatol (2008) 35:1238–9.
71. Salgame P. Host innate and Th1 responses and the bacterial factors that control Mycobacterium tuberculosis infection. Curr Opin Immunol (2005) 17:374–80. doi:10.1016/j.coi.2005.06.006
72. Ranjbar S, Boshoff HI, Mulder A, Siddiqi N, Rubin EJ, Goldfeld AE. HIV-1 replication is differentially regulated by distinct clinical strains of Mycobacterium tuberculosis. PLoS One (2009) 4:e6116. doi:10.1371/journal.pone.0006116
73. Zhang Y, Broser M, Rom WN. Activation of the interleukin 6 gene by Mycobacterium tuberculosis or lipopolysaccharide is mediated by nuclear factors NF-IL6 and NF-kappa B. Proc Natl Acad Sci U S A (1994) 91:2225–9. doi:10.1073/pnas.91.6.2225
74. Ottenhoff TH, Verreck FA, Hoeve MA, van de Vosse E. Control of human host immunity to mycobacteria. Tuberculosis (Edinb) (2005) 85:53–64. doi:10.1016/j.tube.2004.09.011
75. Warfel AH, Thorbecke GJ, Belsito DV. Synergism between interferon-gamma and cytokines or lipopolysaccharide in the activation of the HIV-LTR in macrophages. J Leukoc Biol (1995) 57:469–76.
76. Poli G, Bressler P, Kinter A, Duh E, Timmer WC, Rabson A, et al. Interleukin 6 induces human immunodeficiency virus expression in infected monocytic cells alone and in synergy with tumor necrosis factor alpha by transcriptional and post-transcriptional mechanisms. J Exp Med (1990) 172:151–8. doi:10.1084/jem.172.1.151
77. Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO global surveillance and monitoring project. JAMA (1999) 282:677–86. doi:10.1001/jama.282.7.677
78. Selwyn PA, Sckell BM, Alcabes P, Friedland GH, Klein RS, Schoenbaum EE. High risk of active tuberculosis in HIV-infected drug users with cutaneous anergy. JAMA (1992) 268:504–9. doi:10.1001/jama.1992.03490040080029
79. Reiley WW, Calayag MD, Wittmer ST, Huntington JL, Pearl JE, Fountain JJ, et al. ESAT-6-specific CD4 T cell responses to aerosol Mycobacterium tuberculosis infection are initiated in the mediastinal lymph nodes. Proc Natl Acad Sci U S A (2008) 105:10961–6. doi:10.1073/pnas.0801496105
80. Egen JG, Rothfuchs AG, Feng CG, Horwitz MA, Sher A, Germain RN. Intravital imaging reveals limited antigen presentation and T cell effector function in mycobacterial granulomas. Immunity (2011) 34:807–19. doi:10.1016/j.immuni.2011.03.022
81. Gallegos AM, Pamer EG, Glickman MS. Delayed protection by ESAT-6-specific effector CD4+ T cells after airborne M. tuberculosis infection. J Exp Med (2008) 205:2359–68. doi:10.1084/jem.20080353
82. Rosas-Taraco AG, Arce-Mendoza AY, Caballero-Olin G, Salinas-Carmona MC. Mycobacterium tuberculosis upregulates coreceptors CCR5 and CXCR4 while HIV modulates CD14 favoring concurrent infection. AIDS Res Hum Retroviruses (2006) 22:45–51. doi:10.1089/aid.2006.22.45
83. Wahl SM, Allen JB, Gartner S, Orenstein JM, Popovic M, Chenoweth DE, et al. HIV-1 and its envelope glycoprotein down-regulate chemotactic ligand receptors and chemotactic function of peripheral blood monocytes. J Immunol (1989) 142:3553–9.
84. Havlir DV, Barnes PF. Tuberculosis in patients with human immunodeficiency virus infection. N Engl J Med (1999) 340:367–73. doi:10.1056/NEJM199902043400507
85. Patel NR, Zhu J, Tachado SD, Zhang J, Wan Z, Saukkonen J, et al. HIV impairs TNF-alpha mediated macrophage apoptotic response to Mycobacterium tuberculosis. J Immunol (2007) 179:6973–80.
86. Diedrich CR, Mattila JT, Klein E, Janssen C, Phuah J, Sturgeon TJ, et al. Reactivation of latent tuberculosis in cynomolgus macaques infected with SIV is associated with early peripheral T cell depletion and not virus load. PLoS One (2010) 5:e9611. doi:10.1371/journal.pone.0009611
87. Lawn SD, Myer L, Edwards D, Bekker LG, Wood R. Short-term and long-term risk of tuberculosis associated with CD4 cell recovery during antiretroviral therapy in South Africa. AIDS (2009) 23:1717–25. doi:10.1097/QAD.0b013e32832d3b6d
88. Kumawat K, Pathak SK, Spetz AL, Kundu M, Basu J. Exogenous Nef is an inhibitor of Mycobacterium tuberculosis-induced tumor necrosis factor-alpha production and macrophage apoptosis. J Biol Chem (2010) 285:12629–37. doi:10.1074/jbc.M109.073320
89. Patel NR, Swan K, Li X, Tachado SD, Koziel H. Impaired M. tuberculosis-mediated apoptosis in alveolar macrophages from HIV+ persons: potential role of IL-10 and BCL-3. J Leukoc Biol (2009) 86:53–60. doi:10.1189/jlb.0908574
90. Blanc FX, Sok T, Laureillard D, Borand L, Rekacewicz C, Nerrienet E, et al. Earlier versus later start of antiretroviral therapy in HIV-infected adults with tuberculosis. N Engl J Med (2011) 365:1471–81. doi:10.1056/NEJMoa1013911
91. Lawn SD, Butera ST, Shinnick TM. Tuberculosis unleashed: the impact of human immunodeficiency virus infection on the host granulomatous response to Mycobacterium tuberculosis. Microbes Infect (2002) 4:635–46. doi:10.1016/S1286-4579(02)01582-4
92. Meintjes G, Lawn SD, Scano F, Maartens G, French MA, Worodria W, et al. Tuberculosis-associated immune reconstitution inflammatory syndrome: case definitions for use in resource-limited settings. Lancet Infect Dis (2008) 8:516–23. doi:10.1016/S1473-3099(08)70184-1
93. Oliver BG, Haddow LJ, Agarwal U, Kumar M, Easterbrook PJ, Moosa MY, et al. Pre-antiretroviral therapy plasma levels of CCL2 may aid in the prediction of tuberculosis-associated immune reconstitution inflammatory syndrome in HIV patients after they commence antiretroviral therapy. J Acquir Immune Defic Syndr (2013) 63:e72–4. doi:10.1097/QAI.0b013e31828e6182
94. Scanga CA, Mohan VP, Yu K, Joseph H, Tanaka K, Chan J, et al. Depletion of CD4(+) T cells causes reactivation of murine persistent tuberculosis despite continued expression of interferon gamma and nitric oxide synthase 2. J Exp Med (2000) 192:347–58. doi:10.1084/jem.192.3.347
95. Legrand N, Ploss A, Balling R, Becker PD, Borsotti C, Brezillon N, et al. Humanized mice for modeling human infectious disease: challenges, progress, and outlook. Cell Host Microbe (2009) 6:5–9. doi:10.1016/j.chom.2009.06.006
96. Sun Z, Denton PW, Estes JD, Othieno FA, Wei BL, Wege AK, et al. Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J Exp Med (2007) 204:705–14. doi:10.1084/jem.20062411
97. Shen Y, Shen L, Sehgal P, Huang D, Qiu L, Du G, et al. Clinical latency and reactivation of AIDS-related mycobacterial infections. J Virol (2004) 78:14023–32. doi:10.1128/JVI.78.24.14023-14032.2004
Keywords: HIV/Mtb co-infection, immuno-pathogenesis, CCL2, macrophages, granuloma, CD4+ T cells, viremia
Citation: Ansari AW, Kamarulzaman A and Schmidt RE (2013) Multifaceted impact of host C–C chemokine CCL2 in the immuno-pathogenesis of HIV-1/M. tuberculosis co-infection. Front. Immunol. 4:312. doi: 10.3389/fimmu.2013.00312
Received: 25 July 2013; Accepted: 17 September 2013;
Published online: 04 October 2013.
Edited by:
Guido Poli, Vita-Salute San Raffaele University, ItalyReviewed by:
Guido Poli, Vita-Salute San Raffaele University, ItalyAnna-Lena Spetz, Karolinska Institutet, Sweden
Copyright: © 2013 Ansari, Kamarulzaman and Schmidt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: A. Wahid Ansari, Faculty of Medicine, Centre of Excellence for Research in AIDS (CERiA), University of Malaya, Level-17 Wisma R&D, Jalan Pantai Baharu, Kuala Lumpur 59990, Malaysia e-mail:YXdhbnNhcmlAdW0uZWR1Lm15