 Lucia De Martino†
Lucia De Martino† Donatella Capalbo†
Donatella Capalbo† Nicola Improda
Nicola Improda Federica D’Elia
Federica D’Elia Raffaella Di Mase
Raffaella Di Mase Roberta D’Assante
Roberta D’Assante Ida D’Acunzo
Ida D’Acunzo Claudio Pignata
Claudio Pignata Mariacarolina Salerno*
Mariacarolina Salerno*- Pediatric Section, Department of Translational Medical Sciences, “Federico II” University, Naples, Italy
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) is a rare autosomal recessive disease, caused by mutations of a single gene named Autoimmune regulator gene (AIRE) which results in a failure of T-cell tolerance. Central tolerance takes place within the thymus and represents the mechanism by which potentially auto-reactive T-cells are eliminated through the negative selection process. The expression of tissue-specific antigens (TSAs) by medullary thymic epithelial cells (mTECs) in the thymus is a key process in the central tolerance and is driven by the protein encoded by AIRE gene, the transcription factor autoimmune regulator (AIRE). A failure in this process caused by AIRE mutations is thought to be responsible of the systemic autoimmune reactions of APECED. APECED is characterized by several autoimmune endocrine and non-endocrine manifestations and the phenotype is often complex. Although APECED is the paradigm of a monogenic autoimmune disorder, it is characterized by a wide variability of the clinical expression even between siblings with the same genotype, thus implying that additional mechanisms, other than the failure of Aire function, are involved in the pathogenesis of the disease. Unraveling open issues of the molecular basis of APECED, will help improve diagnosis, management, and therapeutical strategies of this complex disease.
Introduction
Autoimmune Polyglandular Syndrome Type 1 (APS-1), also called Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), is a rare autosomal recessive disease caused by mutations of the autoimmune regulator gene (AIRE). Immunologically, APECED is characterized by destruction of the target organs by a cellular- and/or antibody-mediated attack (1). In the past decade, much interest has been focused on the pathogenesis of this syndrome. Indeed, APECED represents a paradigm of genetically determined systemic autoimmunity. However, the great variability that characterizes APECED, irrespectively of the AIRE genotype, implies that several factors are involved in the disease phenotypic expression.
In this review, we will focus on the complex pathogenesis of APECED and on the potential interfering factors involved in the clinical expression of the disease.
The Basis of the Immunological Tolerance
Tolerance represents a state of immunologic non-responsiveness in the presence of a particular antigen. In this context, T-cell tolerance is crucial for the creation of a proper T-cell repertoire, able to respond to a huge number of foreign antigens, but preventing autoimmune reactions. Imposition and regulation of self-tolerance within the T-cell repertoire is exerted at two levels: (1) central tolerance (development and selection of T-cells in the thymus) and (2) peripheral tolerance (deletion, anergy of mature T-cells in lymphoid and non-lymphoid organs) (2).
T-cell central tolerance, established within the thymus, mostly relies on two main mechanisms: negative selection, also referred to as clonal deletion of maturing thymocytes and positive selection of maturing T-cells able to bind to a surface major histocompatibility complex (MHC) molecule with mild threshold of reactivity (Figure 1). The thymus provides the necessary environment for thymopoiesis and establishment and maintenance of self-tolerance (3–5). Thymus contains thymic epithelial cells (TECs) that form a complex three-dimensional network organized in cortical and medullary compartments (6). On entering the thymus, immature thymocytes promote the differentiation of precursor thymic epithelial cells (pTECs) into cortical TECs (cTECs) and medullary TECs (mTECs), playing an important role in the formation of the thymic microenvironment (7–9). During postnatal life, hematopoietic progenitors enter the thymus from the bloodstream (10) and cells committed to the T lineage undergo division, mostly within the double-negative (DN) stage of the T-cell development. The first checkpoint is the rearrangement of T-cell receptor (TCR) β and α locus. Expression of αβ TCR heterodimers on the cell surface allows DN thymocytes to progress to the double-positive (DP) CD4+CD8+ stage. At DP stage, the TCR affinity for self-peptide-MHC on mTECs within the thymus determines thymocyte’s fate. mTECs express a wide array of tissue-specific antigens (TSAs) in the context of MHC class II molecules; these TSAs include self-proteins derived from different organs in the body. DP thymocytes expressing TCRs that do not bind self-peptide-MHC complexes are programed to undergo “death by neglect” or apoptosis. Only about 5% of DP has a low affinity for self-peptide-MHC complexes and differentiate to CD4+CD8− or CD4−CD8+ single positive (SP) lineage (positive selection) (11–13). DP thymocytes with high-affinity TCR for MHC complexes represent a potential reservoir of auto-reactive lymphocytes and “clonal deletion” (negative selection) is the main mechanism in the thymus to preserve self-tolerance (14, 15). Compelling evidence indicates that an altered promiscuous thymic expression of TSAs leads to autoimmunity. In the autoimmune attack, T helper cells (Th) escaped to self-tolerance, produce pro-inflammatory cytokines able to begin inflammation and activate auto-reactive B-cells, resulting in autoantibodies production, which lead to tissue inflammation and damage (1). Some of the thymocytes that recognize self-peptide-MHC complexes with high-affinity express Foxp3 and through “clonal diversion” mature as regulatory T-cells (Tregs), which are able to suppress auto-reactive T-cells in the periphery (16–18). The central tolerance is not able alone to completely remove mature T-cells with self-antigens specificity, therefore additional mechanisms in the periphery are also needed to maintain immunological tolerance.

Figure 1. Positive and negative selection of immature thymocytes within thymus.
The peripheral tolerance recognizes as possible mechanisms the induction of functional anergy, deletion of auto-reactive clones, and the suppressive action of T-regulatory cells (Tregs). Anergy is a state of long-term hyporesponsiveness with inactivation of self-reactive T-cells in the presence of a TCR signal but in the absence of a second costimulatory signal, necessary to T-cell activation. Deletion of self-reactive lymphocytes is achieved in both the thymus and the periphery by apoptosis through interaction of Fas/FasL. The function of Tregs (Foxp3-expressing CD4 T-cells) is to suppress immune responses through numerous mechanisms including the production of anti-inflammatory cytokines, direct cell–cell contact, and by modulating the activation state and function of antigen-presenting cell (APC) (19). An additional mechanism involved in controlling reactivity to self in the periphery is NK cell activity.
AIRE and the Maintenance of Immunological Tolerance
Autoimmune regulator gene encodes for a transcription factor (Aire) involved in the maintenance of tolerance. In humans, the AIRE gene maps to chromosome 21q22.3 (20, 21). It consists of 14 exons spanning 11.9 kb of genomic DNA (22) and encodes a 545 amino acid protein with a molecular weight of 58 kDa that works as a “non-classical” transcriptional factor in immune-related organs. The highest level of AIRE expression has been detected within the thymus (23) in mTECs, followed by thymic dendritic cells (DCs). In addition to the thymus, low level of Aire seems to be expressed in secondary lymphoid organs, such as lymph nodes, fetal liver, and spleen (24, 25). The Aire protein, mostly localized in the cell nucleus, is composed by specific domains including the amino-terminal HSR domain, the nuclear localization signal (NLS), the Sp100, AIRE1, nucP41/75, DEAF 1 (SAND) domain, two plant homeodomain (PHD) type zinc fingers, and four LXXLL motifs (26) (Figure 2). The HSR region has been shown to be responsible for the dimerization of the polypeptides belonging to the Sp100 protein family (27). The SAND domain is important for AIRE transactivation capacity and subcellular localization. The PHD zinc fingers are often found in proteins involved in the regulation of transcription (28). The LXXLL motifs are found on coactivators nuclear receptors and proline-rich regions (PRR) and are also associated to transcription regulation (29). Although the precise molecular mechanism is still unclear, Aire seems to regulate the transcription process acting as a coactivator in a large transcriptional complex (30), and interacting with a large set of partners, divided into four main classes based on their function: nuclear transport, chromatin binding/structure, transcription, and pre-mRNA processing factors (31). The first protein reported to bind to AIRE was CREB-binding protein (CBP) (32). Its interaction with AIRE may lead to promotion of gene transcription through histone acetylation and the recruitment of chromatin-transcription factors (33). Other AIRE partners have been identified, such as DNA protein kinase (DNA-PK), SP-RING domain protein inhibitor of activated STAT1 (PIAS1), positive transcription elongation factor b (P-TEFb) (34–36). Moreover, it has been proposed a possible epigenetic control of the AIRE target genes since AIRE’s PHD1 finger domain appears to be able to bind histone three molecules with unmethylated lysine at position 4, generally associated with repressed genes (37). Overall, it is possible that Aire mediates the expression of TSA in mTECs through its co-transcriptional partners (38). The intriguing question is how the AIRE gene alone can influence the transcription of such a large number of TSA genes. Indeed, two models have been suggested to explain the action of Aire: transcription model and maturation model. In the transcription model, TSAs are considered to be the direct target genes of Aire’s transcriptional activity and the lack of Aire protein within the cell would result in the defective TSA gene expression, while the maturation program of mTECs would be in principle unaffected. The maturation model suggests that Aire may affect the thymic microenvironment more globally than through simple control of TSA expression levels. Consequently, in keeping with the latest model the regulation of TSA gene expression might not be the major defect of Aire-deficient mTECs responsible for impaired negative selection (39).
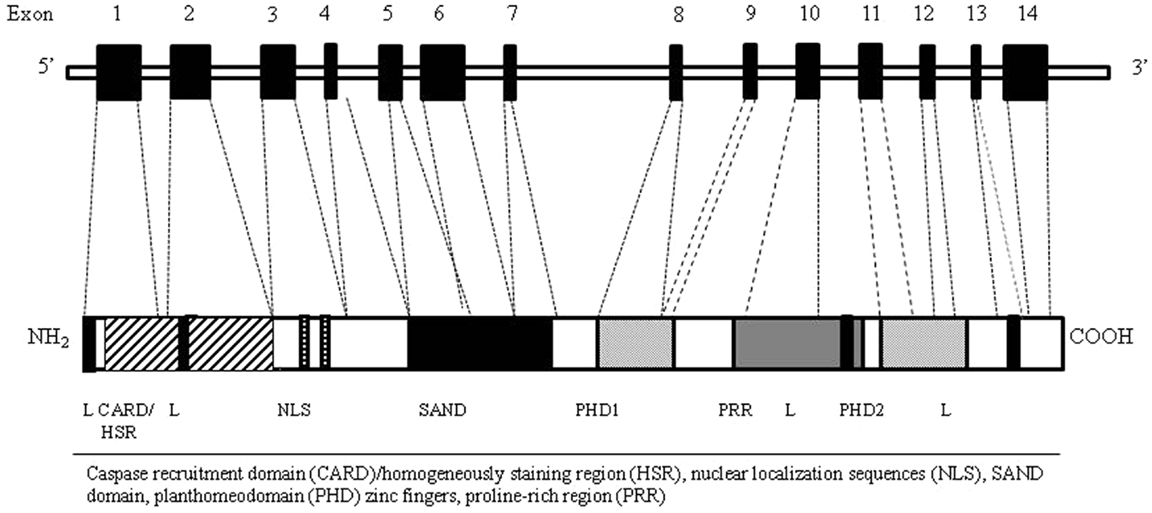
Figure 2. AIRE gene (top) and corresponding protein (bottom) with functional domains. The Aire protein is composed by nuclear localization sequences (NLS), two planthomeodomain (PHD) zinc fingers, a caspase recruitment domain (CARD)/homogeneously staining region (HSR), a SAND domain, and a proline-rich region (PRR).
Although the exact role of AIRE in controlling T-cell tolerance is still largely unclear, several mechanisms have been suggested. Functional alterations of AIRE may affect processing and/or presentation of self-antigens within the mTECs (40). The process of thymocyte maturation (41), the attraction of mature thymocytes to their final location for a proper negative selection (40, 42), the control of cross-presentation through alteration of the relationship between APCs and mTECs (43) may also represent potential mechanisms by which AIRE alterations may lead to functional abnormalities of the central tolerance. The alteration in the balance between negative selection and regulatory T-cell production (44) may also be implicated in the pathogenesis. In addition, Aire may also play a role in the proper differentiation of the thymic medullary epithelium, in the induction of apoptosis in end-stage terminally differentiated mTECs (39) as well as in mTECs’ differentiation program. In particular, evidence suggests that lack of Aire in mTECs results in an arrest of the differentiation program, with the cells remaining at the premature stage just before terminal differentiation (45, 46).
The Clinical Counterpart of AIRE Mutation: APECED
Genetic Background
Mutations in AIRE gene result in development of APECED, which represents the paradigm of a genetically determined failure of central tolerance leading to autoimmunity (46). APECED is a rare autoimmune syndrome, but it has been reported worldwide showing a relatively higher prevalence in genetically isolated populations such as Iranian Jews (1:9,000) (47), Finns (1:25,000) (48, 49), and Sardinians (1:14,400) (50). It is also quite frequent in Norway (1:90,000) (51) and in some regions of Italy (52–55). The most frequent model of inheritance is autosomal recessive, even though a dominant pattern has also been sporadically reported (56). So far, over 70 different mutations of AIRE have been documented (2). Due to the molecular organization and the complexity of intermolecular connection of Aire, it would be expected that different mutation in the molecule might imply different functional abnormalities, thus being associated with a variable phenotypic expression. Single nucleotide substitutions, small insertions, deletions, and mutations affecting splice consensus sequences have been identified along the entire coding region, and include either nonsense or frameshift mutations that result in truncated polypeptides, or missense mutations that result in single amino acid-changing (27). Most of these AIRE mutations lead to a change in its subcellular location altering the distribution of the protein between the nucleus and cytoplasm (27). Mutations of the predicted surface area of the HSR domain cause the protein accumulation in the nucleus blocking its cytoplasmic localization probably enhancing nuclear import or inhibiting nuclear export (57). Mutations of the SAND domain disturb the distribution of Aire between the nucleus and cytoplasm suggesting a role for the SAND domain in nuclear transport mechanisms (57). Moreover, since the six helix CARD domain is involved in homodimerization, missense mutations in this region often affect Aire multimerization or localization to nuclear bodies (58) while most of the missense mutations in PHD domains alter the zinc-finger fold and decrease Aire’s transcriptional activation capacity (38).
Some different mutations have been found to be peculiar to certain populations. R257X is the most common mutation among Finnish and other European patients (59–61). R257X is a nonsense mutation, which most probably results in a carboxy-terminally truncated, non-functional Aire protein leading to alterated subcellular localization and inhibition of the transactivation function and complex formation of Aire (27). The 1094–1106 del113 (or 967–979 del-13 bp) is the most common mutation in British (62), Irish (63), North American (64, 65), and Norwegian patients (51) leading to the truncation and loss of function of Aire. Y85C is the only missense mutation found among Iranian Jews (57). In Italy, typical mutations of AIRE have been detected in Sardinia (R139X on exon 3) (50, 54), where this nonsense mutation leads to a total absence of Aire and seems to be associated with a more severe phenotype. In Apulia, the missense mutation W78R on exon 2, and the nonsense mutation Q358X on exon 9 have been found. The mutation Q358X lies in the PRR resulting in a truncated protein which lacks the second PHD finger and thereby is most likely non-functional protein (53). In Sicily, the most frequent mutation is R203X on exon 5, and two novel mutations, S107C and Q108fs on exon 3, have been detected. The mutation S107C is a missense mutation, whilst Q108fs is a small deletion, both affecting the HSR domain of Aire protein and it is likely that the Aire protein loses its homodimerization properties (66, 67). In Venetian patients, the most frequent mutations are R257X on exon 6 and 979 del-13 bp on exon 8, that are analogous to those detected in Finnish and Anglo-saxon patients but different from Italian ones (55). No typical mutations have been identified neither in Calabria nor in Campania (52, 68) even though patients from Campania show a high frequency of mutations in the exon/intron 1 junction. Compared to other mutations, the R257X results in a total loss of function, whereas the less dramatic truncations of the AIRE protein and many missense mutations, especially the predicted surface mutations of the HSR domain and the mutations in the leucine zipper domain, seem to exert less severe effects on the function of the Aire protein (27). Therefore, despite considerable variations in the APECED genotype, correlations with specific phenotypic features are far from being well elucidated. Only in patients affected with Candida infection, a correlation has been proved. In fact, candidiasis was significantly less prevalent in patients homozygous for 967–979del-13bp than in patients carrying the R257X or R139X, suggesting that AIRE truncation upstream the SAND domain promotes the susceptibility to this infection (69).
Diagnosis of APECED
The onset of APECED usually occurs during childhood. The clinical diagnosis is based on the presence of two of the three classical components: chronic mucocutaneous candidiasis (CMC), chronic hypoparathyroidism (CH), and Addison’s disease (AD). The presence of only one of these features is sufficient for the diagnosis, when a sibling is affected. Molecular analysis of AIRE may help to confirm the clinical diagnosis, in particular in those cases with an atypical presentation (70, 71). Neutralizing autoantibodies against IFN-ω and IFN-α may represent a precocious biomarker detectable in the majority of patients and, thus they have been recently included in the diagnostic criteria of APECED (72).
Clinical Expression, Autoantibodies Profile, and Susceptibility Factors
APECED is characterized by a highly variable pattern of destructive autoimmune reaction, mainly mediated by specific autoantibodies toward different endocrine and non-endocrine organs. Virtually, all tissues and organs may represent the target of the autoimmune attacks, thus leading to a wide spectrum of clinical features. As already mentioned, the three main components of APECED are CMC, CH, and AD. CMC is, generally, the first component to develop, often followed by CH, before the age of 10 years and later by adrenal insufficiency (73, 74). In addition to the main components, the spectrum of minor manifestations may include ectodermal dystrophy, other endocrinopathies, such as hypergonadotripic hypogonadism, insulin-dependent diabetes, autoimmune thyroiditis, and pituitary dysfunction. Moreover, gastrointestinal disorders (chronic atrophic gastritis, pernicious anemia, malabsorption, autoimmune hepatitis and cholelithiasis), skin diseases (vitiligo and alopecia), keratoconjunctivitis, immunological defects, asplenia may be present (70). More rare manifestations of the disease include immune-mediated central and peripheral neurological manifestations, such as chronic inflammatory demyelinating polyneuropathy (54) and posterior reversible encephalopathy syndrome (PRES) (75), tubulointerstitial nephritis, autoimmune bronchiolitis, reversible metaphyseal dysplasia, hypokalemia, and hypertension (72).
The majority of APECED components have been correlated with specific autoantibodies that may represent an useful tool for the diagnosis and the follow-up of patients (Table 1). Autoantibodies’ profile may parallel clinical expression even though a strong correlation with the phenotype and the severity of the disease is not always present. Indeed, only some autoantibodies are highly predictive of specific organ’s failure, being detectable years before the onset of the overt clinical manifestations.
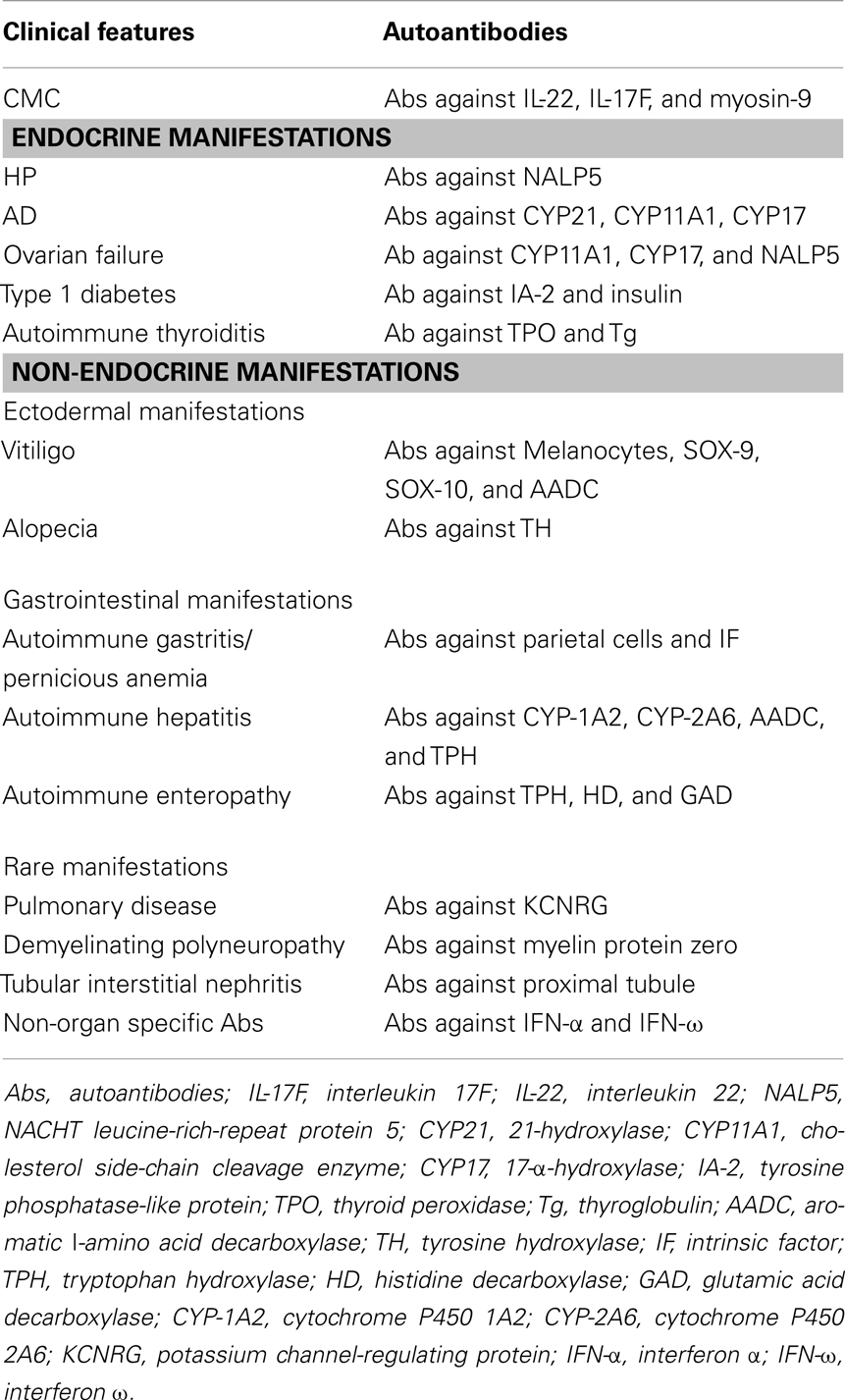
Table 1. Clinical counter part of autoantibodies profile in APECED [modified by Capalbo et al. (73)].
APCED-related CMC has been associated with the presence of specific autoantibodies against the Th17-related cytokines interleukin- (IL-) 22 and IL-17F (76, 77). A parathyroid-specific autoantigen called NACHT leucine-rich-repeat protein 5 (NALP5), which is expressed in the cytoplasm of the main cell type in the parathyroid glands (78), has been recently proposed as the immunological hallmark of APECED-related CH.
Antibodies against the enzyme 21-hydroxylase (CYP21) are strongly associated and highly predictive for the development of AD in patients with CH and/or CMC (79, 80). Steroidogenic enzymes such as Cholesterol side-chain cleavage enzyme (CYP11A1) and 17α-hydroxylase/17,20-lyase (CYP17) represent a further targets of autoimmune reaction against adrenal cortex, moreover they are highly correlated with ovarian insufficiency due to lymphocytic oophoritis and can precede the clinical onset of the component (81, 82). Autoimmune gastritis is associated with the presence of autoantibodies against parietal cells and intrinsic factor (IF), the latter being involved in the development of pernicious anemia (83). The presence of autoantibodies against tryptophan hydroxylase (TPH), an enzyme involved in the synthesis of neurotransmitters in the nervous system and in the gastrointestinal endocrine cells correlates with Autoimmune enteropathy (84–87). Moreover, autoantibodies against both histidine decarboxylase (HD), an enzyme expressed in entero-chromaffin-like cells, and GAD (88, 89) have been associated with an autoimmune intestinal involvement. AH is mainly associated with the presence of autoantibodies against cytochrome P4501A2 (CYP-1A2), CYP-2A6, and aromatic l-amino acid decarboxylase (AADC), even though other types of autoantibodies, such as those directed against TPH, have been correlated with the AH component of the APECED phenotype (54, 90–92). Complement-fixing melanocyte autoantibodies and antibodies against transcription factors SOX-9, SOX-10, and AADC (83, 89) and tyrosine hydroxylase (TH) strongly correlate with the presence of vitiligo and alopecia (72, 83). Recently, several reports have confirmed an important role of autoantibodies against IFN-α and IFN-ω, which, although not tissue-specific, have been detected in the serum of almost all APECED patients (93, 94). Furthermore, they appear at a very early stage, often before the onset of any clinical manifestation. With this regard, their presence may be considered as an additional diagnostic marker of the disease, especially in those cases with an atypical presentation (94, 95). Although autoantibodies’ production seems to be a key-event in the development of the clinical disease, their role in the pathogenesis of APECED still remains to be defined.
APECED is a paradigmatic example of an autoimmune monogenic disease, however, the phenotypic presentation can widely vary from one patient to another (67, 70, 96, 97). Indeed, there are observations documenting a genotype-phenotype correlation only for specific traits (98, 99), but a clear genotype-phenotype correlation is lacking. We have, recently, reported on a family with an extremely wide intra-familial clinical variability despite the same mutation of AIRE (100). These observations suggest that genetic background is not able to explain alone the variability of the clinical expression and the severity of APECED and that, as for other monogenic diseases, the phenotypic variability of the syndrome may result from the complex interaction between several genetic, epigenetic, immunological, and/or environmental factors. The HLA class I and class II alleles have been reported to confer susceptibility to develop autoimmune diseases, such as Type 1 diabetes and autoimmune thyroid diseases (101). Only few studies investigated the association between the APECED phenotype and HLA genotypes, reporting conflicting results. In fact, although some studies did not find any significant association between HLA antigens class I or II and autoantibodies’ production or clinical expression of the disease (74, 102–104), other showed an increased frequency of specific HLA genotypes in APECED patients (105). However, in a more recent study on 18 Sardinian patients (54) autoimmune hepatitis, as well as LKM autoantibodies, have been found to be strongly associated with HLA-DRB1*0301/DQB1*0201. However, there is no evidence indicating that the HLA haplotype might be associated to a particular severity of the disease. Infectious agents are potent stimuli for the immune system, and thus both viruses and bacteria can be considered as trigger of an autoreaction via different mechanisms, such as molecular mimicry, bystander activation, and epitope spreading (106–111). Moreover, evidence suggests that a genetically determined susceptibility may favor the development of an autoimmune disorder after an infection. Many viruses have also been proposed as factors exacerbating several autoimmune processes (112). However, the role of the infectious triggers has not been sufficiently investigated in patients with APECED, and preliminary results did not show any significant effect of different infections on the phenotypic expression of the syndrome (100). As already mentioned, along with the central tolerance network, which is primarily involved in the pathogenesis of APECED, several peripheral mechanisms are capable of contributing to the control and regulation of the immune system. These factors are involved in maintenance of the homeostasis by controlling residual auto-reactive clones, which escape negative selection within the thymus and play a significant role in preventing or minimizing reactivity to self-antigens. The peripheral tolerance recognizes as possible mechanisms the induction of functional anergy with inactivation of self-reactive T-cells, deletion of auto-reactive clones by apoptosis, through Fas/FasL interaction, and the suppressive action of Tregs. An additional mechanism involved in controlling reactivity to self engages in the periphery is represented by NK cell activity. A possible role of altered peripheral tolerance in the pathogenesis and clinical expression of APECED might be hypothesized also considering that recent evidence suggesting that Aire may also be implicated in the control of peripheral mechanisms dedicated to the peripheral maintenance of self-tolerance. In the periphery, Aire is expressed in DCs and a specific population of extrathymic Aire-expressing cells (113, 114). As in the thymus, also in secondary lymphoid organs Aire is required for the expression of many TSAs. However, only few studies investigated the functionality of peripheral tolerance mechanisms in patients with APECED and the role of a failure in the peripheral mechanisms of Aire’s function is still poorly defined. Studies on animal models of APECED suggest that Aire does not influence per se Tregs as in Aire-KO mice the number of CD4+CD25+ cells are normal, and the functionality in in vitro suppression assays is normal as well (115, 116). However, the link between Aire and Treg cells is still not fully understood. Some recent studies suggest that Aire-expressing mTECs are involved in the generation of TSA-specific Foxp3+ Treg cells. A recent study supports this concept by showing that Aire-expressing mTECs, in addition to providing an antigen reservoir, also serve as APCs, thus enhancing the selection of Treg cells. The commitment of Tregs was shown to occur independently of Foxp3, and interaction of developing thymocytes with thymic stromal cells may drive the differentiation of a thymocyte subpopulation into the Treg cell lineage and, subsequently, trigger the expression of Foxp3 (117). Some adult APECED patients have lower proportion of Tregs (118), this finding being probably related to chronic infections, to the extent of autoimmune inflammation or therapy. Unfortunately, Tregs have been evaluated in only two children with APECED. Although in these children the number of Tregs was reduced in comparison to healthy controls, confirming the results obtained in adult patients, this reduction was not related to the severity of the disease, thus ruling out a potential role in modulating the clinical expression of the syndrome (100).
Closing Remarks
Although APECED is a monogenic autoimmune disease, the great variability of the clinical expression and the absence of a clear genotype-phenotype correlation implies that, beyond AIRE mutations, other susceptibility factors such as immunological and environmental factors may be involved in the pathogenesis of the disease. The evidence of a role of an impairment of central and peripheral tolerance and of other susceptibility factors in the phenotypic variability of APECED is limited and needs to be further investigated. So far, the reason of such variability still remains obscure. Unraveling the open issues of the molecular basis of APECED, will be extremely useful in improving the diagnosis, management, and therapeutical strategies of this complex disease. As for other Mendelian diseases, total exome sequencing could be a good perspective to analyze other genetic variations and to identify potential disease-modifying genes involved in the clinical expressivity of organ-specific autoimmunity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mathis D, Benoist C. Aire. Annu Rev Immunol (2009) 27:287–312. doi: 10.1146/annurev.immunol.25.022106.141532
2. Griesemer AD, Sorenson EC, Hardy MA. The role of the thymus in tolerance. Transplantation (2010) 90:465–74. doi:10.1097/TP.0b013e3181e7e54f
3. Manley NR, Blackburn CC. A developmental look at thymus organogenesis: where do the non-hematopoietic cells in the thymus come from? Curr Opin Immunol (2003) 15(2):225–32. doi:10.1016/S0952-7915(03)00006-2
4. Boehm T. Thymus development and function. Curr Opin Immunol (2008) 20:178–84. doi:10.1016/j.coi.2008.03.001
5. Anderson G, Lane PJ, Jenkinson EJ. Generating intrathymic microenvironments to establish T-cell tolerance. Nat Rev Immunol (2007) 7:954–63. doi:10.1038/nri2187
6. van Ewijk W, Wang B, Hollander G, Kawamoto H, Spanopoulou E, Itoi M, et al. Thymic microenvironments, 3-D versus 2-D? Semin Immunol (1999) 11(1):57–64. doi:10.1006/smim.1998.0158
7. Holländer GA, Wang B, Nichogiannopoulou A, Platenburg PP, van Ewijk W, Burakoff SJ. Developmental control point in induction of thymic cortex regulated by a subpopulation of prothymocytes. Nature (1995) 373(6512):350–3. doi:10.1038/373350a0
8. van Ewijk W, Hollander G, Terhorst C, Wang B. Stepwise development of thymic microenvironments in vivo is regulated by thymocyte subsets. Development (2000) 127:1583–91.
9. Klug DB, Carter C, Gimenez-Conti IB, Richie ER. Cutting edge: thymocyte-independent and thymocyte-dependent phases of epithelial patterning in the fetal thymus. J Immunol (2002) 169:2842–5.
10. Shortman K, Egerton M, Spangrude GJ, Scollay R. The generation and fate of thymocytes. Semin Immunol (1990) 2(1):3–12.
11. Cheroutre H. Starting at the beginning: new perspectives on the biology of mucosal T cells. Annu Rev Immunol (2004) 22:217–46. doi:10.1146/annurev.immunol.22.012703.104522
12. Franki AS, Van Beneden K, Dewint P, Meeus I, Veys E, Deforce D, et al. Lymphotoxin alpha 1 beta 2: a critical mediator in V alpha 14i NKT cell differentiation. Mol Immunol (2005) 2005:413–7. doi:10.1016/j.molimm.2004.07.020
13. Baldwin TA, Hogquist KA, Jameson SC. The fourth way? Harnessing aggressive tendencies in the thymus. J Immunol (2004) 173:6515–20.
14. McGargill MA, Derbinski JM, Hogquist KA. Receptor editing in developing T cells. Nature Immunol (2000) 1:336–41. doi:10.1038/79790
15. Huang CY, Sleckman BP, Kanagawa O. Revision of T cell receptor {alpha} chain genes is required for normal T lymphocyte development. Proc Natl Acad Sci U S A (2005) 102(40):14356–61. doi:10.1073/pnas.0505564102
16. Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol (2005) 6:331–7. doi:10.1038/ni1179
17. Liu H, Leung BP. CD4+CD25+ regulatory T cells in health and disease. Clin Exp Pharmacol Physiol (2006) 33:519–24. doi:10.1111/j.1440-1681.2006.04401.x
18. Cabarrocas J, Cassan C, Magnusson F, Piaggio E, Mars L, Derbinski J, et al. Foxp3+ CD25+ regulatory T cells specific for a neo-self-antigen develop at the double-positive thymic stage. Proc Natl Acad Sci U S A (2006) 30:8453–8. doi:10.1073/pnas.0603086103
19. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity (2009) 30(5):636–45. doi:10.1016/j.immuni.2009.04.010
20. Finnish-German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet (1997) 17(4):399–403. doi:10.1038/ng1297-399
21. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M. Positional cloning of the APECED gene. Nat Genet (1997) 17:393–8. doi:10.1038/ng1297-393
22. Aaltonen J, Horelli-Kuitunen N, Fan JB, Björses P, Perheentupa J, Myers R, et al. High-resolution physical and transcriptional mapping of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy locus on chromosome 21q22.3 by FISH. Genome Res (1997) 7:820–9.
23. Gotter J, Brors B, Hergenhahn M, Kyewski B. Medullary epithelial cells of the human thymus express a highly diverse selection of tissue-specific genes colocalized in chromosomal clusters. J Exp Med (2004) 199(2):155–66. doi:10.1084/jem.20031677
24. Heino M, Peterson P, Kudoh J, Nagamine K, Lagerstedt A, Ovod V, et al. Autoimmune regulator is expressed in the cells regulating immune tolerance in thymus medulla. Biochem Biophys Res Commun (1999) 257(3):821–5. doi:10.1006/bbrc.1999.0308
25. Halonen M, Pelto-Huikko M, Eskelin P, Peltonen L, Ulmanen I, Kolmer M. Subcellular location and expression pattern of autoimmune regulator (Aire), the mouse orthologue for human gene defective in autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED). J Histochem Cytochem (2001) 49(2):197–208. doi:10.1177/002215540104900207
26. Peterson P, Org T, Rebane A. Transcriptional regulation by AIRE: molecular mechanisms of central tolerance. Nat Rev Immunol (2008) 8:948–57. doi:10.1038/nri2450
27. Peterson P, Peltonen L. Autoimmune polyendocrinopathy syndrome type 1 (APS1) and AIRE gene: new views on molecular basis of autoimmunity. J Autoimmun (2005) 25(Suppl):49–55. doi:10.1016/j.jaut.2005.09.022
28. Bottomley MJ, Stier G, Pennacchini D, Legube G, Simon B, Akhtar A. NMR structure of the first PHD finger of autoimmune regulator protein (AIRE1). Insights into autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) disease. J Biol Chem (2005) 280(12):11505–12. doi:10.1074/jbc.M413959200
29. Savkur RS, Burris TP. The coactivator LXXLL nuclear receptor recognition motif. J Pept Res (2004) 63(3):207–12. doi:10.1111/j.1399-3011.2004.00126.x
30. Johnnidis JB, Venanzi ES, Taxman DJ, Ting JP, Benoist CO, Mathis DJ. Chromosomal clustering of genes controlled by the aire transcription factor. Proc Natl Acad Sci U S A (2005) 102(20):7233–8. doi:10.1073/pnas.0502670102
31. Abramson J, Giraud M, Benoist C, Mathis D. Aire’s partners in the molecular control of immunological tolerance. Cell (2010) 140:123–35. doi:10.1016/j.cell.2009.12.030
32. Pitkänen J, Doucas V, Sternsdorf T, Nakajima T, Aratani S, Jensen K. The autoimmune regulator protein has transcriptional transactivating properties and interacts with the common coactivator CREB-binding protein. J Biol Chem (2000) 275(22):16802–9. doi:10.1074/jbc.M908944199
33. Pitkänen J, Rebane A, Rowell J, Murumägi A, Ströbel P, Möll K. Cooperative activation of transcription by autoimmune regulator AIRE and CBP. Biochem Biophys Res Commun (2005) 333(3):944–53. doi:10.1016/j.bbrc.2005.05.187
34. Liiv I, Rebane A, Org T, Saare M, Maslovskaja J, Kisand K. DNA-PK contributes to the phosphorylation of AIRE: importance in transcriptional activity. Biochim Biophys Acta (2008) 1783(1):74–83. doi:10.1016/j.bbamcr.2007.09.003
35. Ilmarinen T, Kangas H, Kytömaa T, Eskelin P, Saharinen J, Seeler JS. Functional interaction of AIRE with PIAS1 in transcriptional regulation. Mol Immunol (2008) 45(7):1847–62. doi:10.1016/j.molimm.2007.10.045
36. Oven I, Brdicková N, Kohoutek J, Vaupotic T, Narat M, Peterlin BM. AIRE recruits P-TEFb for transcriptional elongation of target genes in medullary thymic epithelial cells. Mol Cell Biol (2007) 27(24):8815–23. doi:10.1128/MCB.01085-07
37. Org T, Chignola F, Hetényi C, Gaetani M, Rebane A, Liiv I. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep (2008) 9(4):370–6. doi:10.1038/sj.embor.2008.11
38. Koh AS, Kuo AJ, Park SY, Cheung P, Abramson J, Bua D. Aire employs a histone-binding module to mediate immunological tolerance, linking chromatin regulation with organ-specific autoimmunity. Proc Natl Acad Sci U S A (2008) 105(41):15878–83. doi:10.1073/pnas.0808470105
39. Matsumoto M, Nishikawa Y, Nishijima H, Morimoto J, Matsumoto M, Mouri Y. Which model better fits the role of aire in the establishment of self-tolerance: the transcription model or the maturation model? Front Immunol (2013) 22(4):210. doi:10.3389/fimmu.2013.00210
40. Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The cellular mechanism of Aire control of T cell tolerance. Immunity (2005) 23:227–39. doi:10.1016/j.immuni.2005.07.005
41. Li J, Li Y, Yao JY, Jin R, Zhu MZ, Qian XP. Developmental pathway of CD4+CD8− medullary thymocytes during mouse ontogeny and its defect in Aire−/− mice. Proc Natl Acad Sci U S A (2007) 104(46):18175–80. doi:10.1073/pnas.0708884104
42. Laan M, Kisand K, Kont V, Möll K, Tserel L, Scott HS, et al. Autoimmune regulator deficiency results in decreased expression of CCR4 and CCR7 ligands and in delayed migration of CD4+ thymocytes. J Immunol (2009) 183(12):7682–91. doi:10.4049/jimmunol.0804133
43. Lei Y, Ripen AM, Ishimaru N, Ohigashi I, Nagasawa T, Jeker LT, et al. Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory cell development. J Exp Med (2011) 208(2):383–94. doi:10.1084/jem.20102327
44. Hinterberger M, Aichinger M, Prazeres da Costa O, Voehringer D, Hoffmann R, Klein L. Autonomous role of medullary thymic epithelial cells in central CD4(+) T cell tolerance. Nat Immunol (2010) 11(6):512–9. doi:10.1038/ni.1874
45. Yano M, Kuroda N, Han H, Meguro-Horike M, Nishikawa Y, Kiyonari H, et al. Aire controls the differentiation program of thymic epithelial cells in the medulla for the establishment of self-tolerance. J Exp Med (2008) 205(12):2827–38. doi:10.1084/jem.20080046
46. Gallo V, Giardino G, Capalbo D, Palamaro L, Romano R, Santamaria F, et al. Alterations of the autoimmune regulator transcription factor and failure of central tolerance: Apeced as a model. Expert Rev Clin Immunol (2013) 9(1):43–51. doi:10.1586/Eci.12.88
47. Zlotogora J, Shapiro MS. Polyglandular autoimmune syndrome type I among Iranian Jews. J Med Genet (1992) 29:824–6. doi:10.1136/jmg.29.11.824
48. Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med (1990) 322(26):1829–36. doi:10.1056/NEJM199006283222601
49. Aaltonen J, Bjorses P, Sandkuijl L, Perheentupa J, Peltonen L. An autosomal locus causing autoimmune disease: autoimmune polyglandular disease type I assigned to chromosome 21. Nat Genet (1994) 8:83–7. doi:10.1038/ng0994-83
50. Rosatelli MC, Meloni A, Meloni A, Devoto M, Cao A, Scott HS, et al. A common mutation in Sardinian autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients. Hum Gen (1998) 103:428–34. doi:10.1007/s004390050846
51. Wolff AS, Erichsen MM, Meager A, Magitta NF, Myhre AG, Bollerslev J, et al. Autoimmune polyendocrine syndrome type 1 in Norway: phenotypic variation, autoantibodies, and novel mutations in the autoimmune regulator gene. J Clin Endocrinol Metab (2007) 92(2):595–603. doi:10.1210/jc.2006-1873
52. Capalbo D, Mazza C, Giordano R, Improda N, Arvat E, Cervato S, et al. Molecular background and genotype-phenotype correlation in autoimmune-polyendocrinopathy-candidiasis-ectodermal-dystrophy patients from Campania and in their relatives. J Endocrinol Invest (2012) 35:169–73. doi:10.3275/7677
53. Meloni A, Perniola R, Faà V, Corvaglia E, Cao A, Rosatelli MC. Delineation of the molecular defects in the AIRE gene in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients from Southern Italy. J Clin Endocrinol Metab (2002) 87:841–6. doi:10.1210/jc.87.2.841
54. Meloni A, Willcox N, Meager A, Atzeni M, Wolff AS, Husebye ES, et al. Autoimmune polyendocrine syndrome type 1: an extensive longitudinal study in Sardinian patients. J Clin Endocr Metab (2012) 97:1114–24. doi:10.1210/jc.2011-2461
55. Cervato S, Mariniello B, Lazzarotto F, Morlin L, Zanchetta R, Radetti G, et al. Evaluation of the autoimmune regulato (AIRE) gene mutations in a cohort of Italian patients with autoimmune-polyendocrinopathy-candidiasis-ectoderaml-dystrophy (APECED) and in their relatives. Clin Endocrinol (2009) 70:421–8. doi:10.1111/j.1365-2265.2008.03318.x
56. Cetani F, Barbesino G, Borsari S, Pardi E, Cianferotti L, Pinchera A, et al. A novel mutation of the autoimmune regulator gene in an Italian kindred with autoimmune polyendocrinopathy-candidiasis-ectoderma dystrophy, acting in a dominant fashion and strongly consegregating with hypothyroid autoimmune thyroiditis. J Clin Endocrinol Metab (2001) 86:4747–52. doi:10.1210/jc.86.10.4747
57. Björses P, Halonen M, Palvimo JJ, Kolmer M, Aaltonen J, Ellonen P, et al. Mutations in the AIRE gene: effects on subcellular location and transactivation function of the autoimmune polyendocrine-candidiasis-ectodermal dystrophy protein. Am J Hum Genet (2000) 66:378–92. doi:10.1086/302765
58. Ferguson BJ, Alexander C, Rossi SW, Liiv I, Rebane A, Worth CL. AIRE’s CARD revealed, a new structure for central tolerance provokes transcriptional plasticity. J Biol Chem (2008) 283(3):1723–31. doi:10.1074/jbc.M707211200
59. Scott HS, Heino M, Peterson P, Mittaz L, Lalioti MD, Betterle C. Common mutations in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients of different origins. Mol Endocrinol (1998) 12:1112–9. doi:10.1210/me.12.8.1112
60. Podkrajsek KT, Bratanic N, Krzisnik C, Battelino T. Autoimmune regulator-1 messenger ribonucleic acid analysis in a novel intronic mutation and two additional novel AIRE gene mutations in a cohort of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients. J Clin Endocrinol Metab (2005) 90:4930–5. doi:10.1210/jc.2005-0418
61. Stolarski B, Pronicka E, Korniszewski L, Pollak A, Kostrzewa G, Rowinska E, et al. Molecular background of polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome in a Polish population: novel AIRE mutations and an estimate of disease prevalence. Clin Genet (2006) 70:348–54. doi:10.1111/j.1399-0004.2006.00690.x
62. Pearce SH, Cheetham T, Imrie H, Vaidya B, Barnes ND, Bilous RW. A common and recurrent 13-bp deletion in the autoimmune regulator gene in British kindreds with autoimmune polyendocrinopathy type 1. Am J Hum Genet (1998) 63:1675–84. doi:10.1086/302145
63. Dominguez M, Crushell E, Ilmarinen T, McGovern E, Collins S, Chang B, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in the Irish population. J Pediatr Endocrinol Metab (2006) 19:1343–52. doi:10.1515/JPEM.2006.19.11.1343
64. Wang CY, Davoodi-Samiromi A, Huang W, Connor E, Shi JD, She JX. Characterization of mutations in patients with autoimmune polyglandular syndrome type 1 (APS1). Hum Genet (1998) 103:681–5. doi:10.1007/s004390050891
65. Heino M, Scott HS, Chen Q, Peterson P, Mäebpää U, Papasavvas MP, et al. Mutation analyses of North American APS-1 patients. Hum Mutat (1999) 13:69–74. doi:10.1002/(SICI)1098-1004(1999)13:1<69::AID-HUMU8>3.0.CO;2-6
66. Giordano C, Modica R, Allotta ML, Guarnotta V, Cervato S, Masiero S. Autoimmune polyendocrinopathy-candidiasis-ectodermal-dystrophy (APECED) in Sicily: confirmation that R203X is the peculiar AIRE gene mutation. J Endocrinol Invest (2012) 35:384–8. doi:10.3275/7965
67. Valenzise M, Wasniewska M, Mirabelli S, De Luca F, Cervato S, Betterle C. Identification of two novel mutations in the first Sicilian APECED patient with no R203X mutation in AIRE gene and review of Italian APECED genotypes. Gene (2012) 499:343–6. doi:10.1016/j.gene.2012.03.032
68. Betterle C, Ghizzoni L, Cassio A, Baronio F, Cervato S, Garelli S, et al. Autoimmune-polyendocrinopathy-candidiasis-ectodermal-dystrophy in Calabria: clinical, immunological and genetic patterns. J Endocrinol Invest (2012) 35(10):877–81. doi:10.3275/8109
69. Kisand K, Peterson P. Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy: known and novel aspects of the syndrome. Ann N Y Acad Sci (2011) 1246:77–91. doi:10.1111/j.1749-6632.2011.06308.x
70. Capalbo D, De Martino L, Giardino G, Di Mase R, Di Donato I, Parenti G, et al. Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy: insights into genotype-phenotype correlation. Int J Endocrinol (2012) 2012:353250. doi:10.1155/2012/353250
71. Mazza C, Buzi F, Ortolani F, Vitali A, Notarangelo LD, Weber G, et al. Clinical heterogeneity and diagnostic delay of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome. Clin Immunol (2011) 139(1):6–11. doi:10.1016/j.clim.2010.12.021
72. Husebye ES, Perheentupa J, Rautemaa R, Kšmpe O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J Intern Med (2009) 265:514–29. doi:10.1111/j.1365-2796.2009.02090.x
73. Capalbo D, Improda N, Esposito A, De Martino L, Barbieri F, Betterle C, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy from the pediatric perspective. J Endocrinol Invest (2013). [Epub ahead of print]
74. Betterle C, Greggio NA, Volpato M. Autoimmune polyglandular syndrome type 1. J Clin Endocr Metab (1998) 83:1049–55. doi:10.1210/jc.83.4.1049
75. Capalbo D, Elefante A, Spagnuolo MI, Mazza C, Betterle C, Pignata C, et al. Posterior reversible encephalopathy syndrome in a child during an accelerated phase of a severe APECED phenotype due to an uncommon mutation of AIRE. Clin Endocrinol (Oxf) (2008) 69:511–3. doi:10.1111/j.1365-2265.2008.03206.x
76. Kisand K, Bøe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med (2010) 207(2):299–308. doi:10.1084/jem.20091669
77. Puel A, Döffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneouscandidiasis and autoimmune polyendocrine syndrome type I. J Exp Med (2010) 207(2):291–7. doi:10.1084/jem.20091983
78. Alimohammadi M, Björklund P, Hallgren A, Pöntynen N, Szinnai G, Shikama N, et al. Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen. N Engl J Med (2008) 358:1018–28. doi:10.1056/NEJMoa0706487
79. Betterle C, Dal Pra C, Mantero F, Zanchetta R. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev (2002) 23:327–64. doi:10.1210/er.23.3.327
80. Coco G, Dal Pra C, Presotto F, Albergoni MP, Canova C, Pedini B, et al. Estimated risk for developing autoimmune Addison’s disease in patients with adrenal cortex autoantibodies. J Clin Endocrinol Metab (2006) 91:1637–45. doi:10.1210/jc.2005-0860
81. Hoek A, Schoemaker J, Drexhage HA. Premature ovarian failure and ovarian autoimmunity. Endocr Rev (1997) 18:107–34. doi:10.1210/er.18.1.107
82. Reato G, Morlin L, Chen S, Furmaniak J, Smith BR, Masiero S, et al. Premature ovarian failure in patients with autoimmune Addison’s disease: clinical, genetic, and immunological evaluation. J Clin Endocrinol Metab (2011) 96:e1255–61. doi:10.1210/jc.2011-0414
83. Betterle C, Zanchetta R. Update on autoimmune polyendocrine syndrome (APS). Acta Biomed (2003) 74:9–33. doi:10.1007/s13317-012-0034-3
84. Ekwall O, Hedstrand H, Grimelius L, Haavik J, Perheentupa J, Gustafsson J, et al. Identification of tryptophan hydroxylase as an intestinal autoantigen. Lancet (1998) 352:279–83. doi:10.1016/S0140-6736(97)11050-9
85. Dal Pra C, Chen S, Betterle C, Zanchetta R, McGrath V, Furmaniak J, et al. Autoantibodies to human tryptophan hydroxylase and aromatic l-amino acid decarboxylase. Eur J Endocrinol (2004) 150:313–21. doi:10.1530/eje.0.1500313
86. Gianani R, Eisenbarth GS. Autoimmunity to gastrointestinal endocrine cells in autoimmune polyendocrine syndrome type I. J Clin Endocrinol Metab (2003) 88:1442–4. doi:10.1210/jc.2003-030247
87. Scarpa R, Alaggio R, Norberto L, Furmaniak J, Chen S, Smith BR, et al. Tryptophan hydroxylase autoantibodies as markers of a distinct autoimmune gastrointestinal component of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab (2013) 98:704–12. doi:10.1210/jc.2012-2734
88. Skoldberg F, Portela-Gomes GM, Grimelius L, Nilsson G, Perheentupa J, Betterle C, et al. Histidine decarboxylase, a pyridoxal phosphate-dependent enzyme, is an autoantigen of gastric enterochromaffin-like cells. J Clin Endocrinol Metab (2003) 88:1445–52. doi:10.1210/jc.2002-021761
89. Söderbergh A, Myhre AG, Ekwall O, Gebre-Medhin G, Hedstrand H, Landgren E, et al. Prevalence and clinical associations of 10 defined autoantibodies in autoimmune polyendocrine syndrome type I. J Clin Endocrinol Metab (2004) 89:557–62. doi:10.1210/jc.2003-030279
90. Clemente MG, Obermayer-Straub P, Meloni A, Strassburg CP, Arangino V, Tukey RH, et al. Cytochrome P450 1A2 is a hepatic autoantigen in autoimmune polyglandular syndrome type 1. J Clin Endocrinol Metab (1997) 82:1353–61. doi:10.1210/jc.82.5.1353
91. Clemente MG, Meloni A, Obermayer-Straub P, Frau F, Manns MP, De Virgiliis S. Two cytochromes P450 are major hepatocellular autoantigens in autoimmune polyglandular syndrome type 1. Gastroenterology (1998) 114:324–8. doi:10.1016/S0016-5085(98)70484-6
92. Gebre-Mendhin G, Husebye ES, Gustafsson J, Winqvist O, Goksøyr A, Rorsman F, et al. Cytocrome P459IA2 and aromatic l-amino acid decarboxylase are hepatic autoantigens in autoimmune polyendocrine syndrome type I. FEBS Lett (1997) 412:439–45. doi:10.1016/S0014-5793(97)00797-7
93. Meager A, Visvalingam K, Peterson P, Möll K, Murumägi A, Krohn K, et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med (2006) 3:e289. doi:10.1371/journal.pmed.0030289
94. Meloni A, Furcas M, Cetani F, Marcocci C, Falorni A, Perniola R, et al. Autoantibodies against type I interferon sas an additional diagnostic criterion for autoimmune polyendocrine syndrome type I. J Clin Endocrinol Metab (2008) 93:4389–97. doi:10.1210/jc.2008-0935
95. Proust-Lemoine E, Saugier-Veber P, Wémeau JL. Polyglandular autoimmune syndrome type I. Presse Med (2012) 41(12 P 2):e651–62. doi:10.1016/j.lpm.2012.10.005
96. Ishii T, Suzuki Y, Ando N, Matsuo N, Ogata T. Novel mutations of the autoimmune regulator gene in two siblings with autoimmune polyendocrinopathy-candidiasis ectodermal dystrophy. J Clin Endocrinol Metab (2000) 8:2922–6. doi:10.1210/jc.85.8.2922
97. Capalbo D, Giardino G, De Martino L, Palamaro L, Romano R, Gallo V, et al. Genetic basis of altered central tolerance and autoimmune diseases: a lesson from AIRE mutations. Int Rev Immunol (2012) 31:344–62. doi:10.3109/08830185.2012.697230
98. Gylling M, Tuomi T, Björses P, Kontiainen S, Partanen J, Christie MR, et al. Ss-cell autoantibodies, human leukocyte antigen II alleles, and type 1 diabetes in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab (2000) 85(12):4434–40. doi:10.1210/jc.85.12.4434
99. Halonen M, Eskelin P, Myhre AG, Perheentupa J, Husebye ES, Kämpe O, et al. AIRE mutations and human leukocyte antigen genotypes as determinants of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy phenotype. J Clin Endocrinol Metab (2002) 87(6):2568–74. doi:10.1210/jc.87.6.2568
100. Capalbo D, Fusco A, Aloj G, Improda N, Vitiello L, Dianzani U, et al. High intrafamilial variability in autoimmune polyendocrinopathy- candidiasis-ectodermal dystrophy: a case study. J Endocrinol Invest (2012) 35:77–81. doi:10.3275/8055
101. Concannon P, Erlich HA, Julier C, Morahan G, Nerup J, Pociot F, et al. Type 1 diabetes: evidence for susceptibility loci from four genome-wide linkage scans in 1,435 multiplex families. Diabetes (2005) 54:2995–3001. doi:10.2337/diabetes.54.10.2995
102. MacLaren N, Riley W. Inherited susceptibility to autoimmune Addison’s disease is linked to human leukocyte antigens-DR3 and/or DR4, except when associated with type 1 autoimmune polyglandular syndrome. J Clin Endocrinol Metab (1986) 62:455–9. doi:10.1210/jcem-62-3-455
103. Ahonen P, Koskimies S, Lokki ML, Tiilikainen A, Perheentupa J. The expression of autoimmune polyglandular disease type I appears associated with several HLA-A antigens but not with HLA-DR J. Clin Endocrinol Metab (1988) 66:1152–7. doi:10.1210/jcem-66-6-1152
104. Huang W, Connor E, Rosa TD, Muir A, Schatz D, Silverstein J, et al. Although DR3-DQB1*0201 may be associated with multiple component diseases of the autoimmune polyglandular syndromes, the human leukocyte antigen DR4-DQB1*0302 haplotype is implicated only in beta-cell autoimmunity. J Clin Endocrinol Metab (1996) 81:2559–63. doi:10.1210/jc.81.7.2559
105. Betterle C, Volpato M, Rees Smith B, Furmaniak J, Chen S, Greggio NA. Adrenal cortex and steroid 21-hydroxylase autoantibodies in adult patients with organ-specific autoimmune diseases: markers of low progression to clinical Addison’s disease. J Clin Endocrinol Metab (1997) 82(3):932–8. doi:10.1210/jc.82.3.932
106. von Herrat MG, Dockter J, Oldstone MB. How virus induces a rapid or slow onset insulin-dependent diabetes mellitus in a transgenic model. Immunity (1994) 1:231–42. doi:10.1016/1074-7613(94)90101-5
107. Panoutsakopoulou V, Sanchirico ME, Huster KM, Jansson M, Granucci F, Shim DJ. Analysis of the relationship between viral infection and autoimmune disease. Immunity (2001) 15:137–47. doi:10.1016/S1074-7613(01)00172-8
108. Chen HD, Fraire AE, Joris I, Welsh RM, Selin LK. Specific history of heterologous virus infections determines anti-viral immunity and immunopathology in the lung. Am J Pathol (2003) 163:1341–55. doi:10.1016/S0002-9440(10)63493-1
109. Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med (1998) 4:781–5. doi:10.1038/nm0798-781
110. Mena I, Fischer C, Gebhard JR, Perry CM, Harkins S, Whitton JL. Coxsackievirus infection of the pancreas: evaluation of receptor expression, pathogenesis, and immunopathology. Virology (2000) 271:276–88. doi:10.1006/viro.2000.0332
111. Carl PL, Temple BR, Cohen PL. Most nuclear systemic autoantigens are extremely disordered proteins: implications for the etiology of systemic autoimmunity. Arthritis Res Ther (2005) 7:1360–74. doi:10.1186/ar1832
112. Pender MP. Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol (2003) 24:584–8. doi:10.1016/j.it.2003.09.005
113. Ramsey C, Hässler S, Marits P, Kämpe O, Surh CD, Peltonen L, et al. Increased antigen presenting cell-mediated T cell activation in mice and patients without the autoimmune regulator. Eur J Immunol (2006) 36(2):305–17. doi:10.1002/eji.200535240
114. Gardner JM, Devoss JJ, Friedman RS, Wong DJ, Tan YX, Zhou X. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science (2008) 321:843–7. doi:10.1126/science.1159407
115. Kuroda N, Mitani T, Takeda N, Ishimaru N, Arakaki R, Hayashi Y, et al. Development of autoimmunity against transcriptionally unrepressed target antigen in the thymus of Aire-deficient mice. J Immunol (2005) 174(4):1862–70.
116. Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire regulates negative selection of organ-specific T cells. Nat Immunol (2003) 4(4):350–4. doi:10.1038/ni906
117. Aschenbrenner K, D’Cruz LM, Vollmann EH, Hinterberger M, Emmerich J, Swee LK, et al. Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat Immunol (2007) 8:351–8. doi:10.1038/ni1444
Keywords: autoimmune polyglandular syndrome type 1, APECED, autoimmune regulator gene, phenotypic variability, tolerance
Citation: De Martino L, Capalbo D, Improda N, D’Elia F, Di Mase R, D’Assante R, D’Acunzo I, Pignata C and Salerno M (2013) APECED: a paradigm of complex interactions between genetic background and susceptibility factors. Front. Immunol. 4:331. doi: 10.3389/fimmu.2013.00331
Received: 12 July 2013; Accepted: 30 September 2013;
Published online: 23 October 2013.
Edited by:
Ana E. Sousa, Universidade de Lisboa, PortugalReviewed by:
Masanobu Satake, Tohoku University, JapanKoji Yasutomo, University of Tokushima, Japan
Copyright: © 2013 De Martino, Capalbo, Improda, D’Elia, Di Mase, D’Assante, D’Acunzo, Pignata and Salerno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariacarolina Salerno, Pediatric Endocrinology Unit, Department of Translational Medical Sciences, “Federico II” University, Street Sergio Pansini 5, Naples 80131, Italy e-mail:c2FsZXJub0B1bmluYS5pdA==
†Lucia De Martino and Donatella Capalbo have contributed equally to this work.