
- 1Department of Microbiology and Immunology, McGill University, Montreal, QC, Canada
- 2Department of Medicine, McGill University, Montreal, QC, Canada
Nucleotide-binding and oligomerization domain NOD-like receptors (NLRs) are highly conserved cytosolic pattern recognition receptors that perform critical functions in surveying the intracellular environment for the presence of infection, noxious substances, and metabolic perturbations. Sensing of these danger signals by NLRs leads to their oligomerization into large macromolecular scaffolds and the rapid deployment of effector signaling cascades to restore homeostasis. While some NLRs operate by recruiting and activating inflammatory caspases into inflammasomes, others trigger inflammation via alternative routes including the nuclear factor-κB, mitogen-activated protein kinase, and regulatory factor pathways. The critical role of NLRs in development and physiology is demonstrated by their clear implications in human diseases. Mutations in the genes encoding NLRP3 or NLRP12 lead to hereditary periodic fever syndromes, while mutations in CARD15 that encodes NOD2 are linked to Crohn’s disease or Blau’s syndrome. Genome-wide association studies (GWASs) have identified a number of risk alleles encompassing NLR genes in a host of diseases including allergic rhinitis, multiple sclerosis, inflammatory bowel disease, asthma, multi-bacillary leprosy, vitiligo, early-onset menopause, and bone density loss in elderly women. Animal models have allowed the characterization of underlying effector mechanisms in a number of cases. In this review, we highlight the functions of NLRs in health and disease and discuss how the characterization of their molecular mechanisms provides new insights into therapeutic strategies for the management of inflammatory pathologies.
Introduction
The mammalian immune system encompasses an ancient genome-encoded innate immune system and a more recently acquired adaptive immune system capable of combating pathogens with exquisite specificity and long-term memory. The innate immune system remains a pivotal player in controlling host resistance. This system is equipped with an arsenal of pattern recognition receptors (PRRs) that translate microbial and danger sensing into immediate host defenses as well as provides signals to prime the adaptive immune response for long-lasting protection (1, 2). Nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) are a group of evolutionarily conserved intracellular PRRs that play a vital role in innate immunity and host physiology, as reflected by their prevalence among living organisms of both the plant and animal kingdoms (3–9). In humans there are 22 known NLRs, and the association of mutations and single nucleotide polymorphisms (SNPs) in their genes with human diseases reflect their vital role in host defense. The function of NLRs is not restricted to immunity, as they also play important roles in reproduction and embryonic development (10–12).
The characteristic feature of NLRs is a central NOD (or NACHT) domain, required for oligomerization, an N-terminal homotypic protein–protein interaction domain and a C-terminal series of leucine-rich repeats (LRRs) involved in agonist sensing or ligand binding. Mammalian NLRs are sub-divided into four subfamilies based on the variation in their N-terminal domain: NLRA or Class II transactivator (CIITA) contains an acid transactivation domain, NLRBs or neuronal apoptosis inhibitor proteins (NAIPs) possess a baculovirus inhibitor of apoptosis protein repeat (BIR), NLRCs have a caspase-recruitment domain (CARD), and NLRPs a pyrin domain (PYD). NLRX1 contains a CARD-related X effector domain (Figure 1). Upon ligand binding, the auto-inhibitory LRR undergoes a conformational change, which exposes the N-terminal domain allowing interaction with downstream signaling adaptors or effectors and formation of an oligomeric complex (13, 14). NLR platforms that recruit and activate the inflammatory protease caspase-1 are referred to as inflammasomes. Caspase-1 is required for the processing and maturation of inflammatory cytokines IL-1β and IL-18 and the induction of an inflammatory form of cell death termed pyroptosis (15, 16). Among the NLRs, NLRP1, NLRP3, NLRP6, NLRP7, NLRP12, NLRC4, and NAIP have been reported to operate via inflammasomes (Figure 2). Other NLRs such as NOD1, NOD2, NLRP10, NLRX1, NLRC5, and CIITA do not directly engage the inflammatory caspases, but instead activate nuclear factor-κB (NF-κB), mitogen-activated protein kinases (MAPKs), and interferon (IFN) regulatory factors (IRFs) to stimulate innate immunity. Below, we discuss the different NLRs along with their mechanisms of activation and diseases associated with defects in their activities (Figure 3).
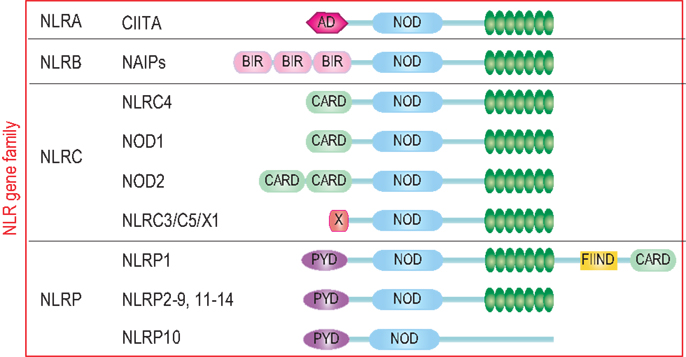
Figure 1. The human NLR gene family. The human NLR gene family consists of 22 members that share a tripartite structure, consisting of an N-terminal signaling domain, a central nucleotide-binding and oligomerization domain, and a C-terminal agonist sensing/ligand-binding domain. The NLR family is sub-divided into four sub-groups NLRA, NLRB, NLRC, and NLRP based on the nature of the N-terminal domain consisting respectively of an acidic transactivation domain, a baculovirus IAP repeat (BIR), a caspase-recruitment and activation domain (CARD), and a Pyrin domain (PYD).
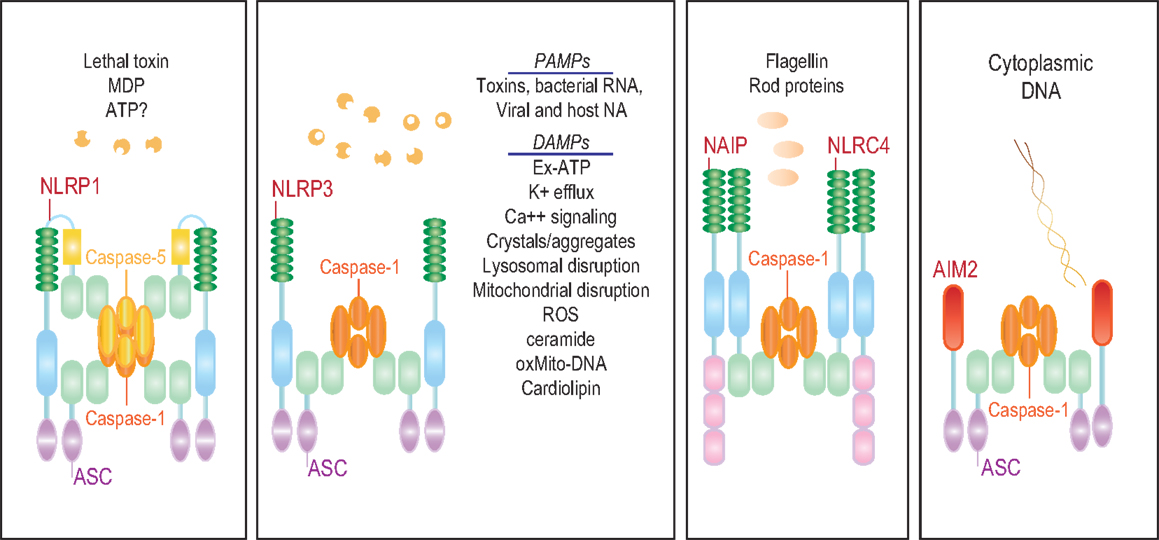
Figure 2. The NLR inflammasomes. The three biochemically characterized inflammasomes are depicted. The NLRP1 inflammasome consists of NLRP1, ASC, and caspases-1 and -5. Little is known about the agonists that activate NLRP1. Anthrax lethal toxin, MDP, and decreased cytosolic ATP have been reported to stimulate this inflammasome. NAIP and NLRC4 form a caspase-1 inflammasome in response to bacterial flagellin and T3SS rod proteins. NLRP3, on the other hand, is activated by a wide range of agonists including a number of MAMPs and DAMPs.

Figure 3. NOD-like receptors and disease. NLRs have been implicated in a plethora of diseases. Genetic studies have uncovered a number of variants in genes encoding NLRs or their signaling mediators associated with human diseases. Animal models have served as a key discovery platform to characterize the underlying functions and molecular mechanisms of NLRs in these diseases and associated pathologies. Together, these efforts have led to therapeutic success in the clinic for a subset of NLR-dependent auto-inflammatory diseases. When available, the mutations and SNPs linked to disease are listed and animal phenotypes are presented.
Inflammasome-Forming NLRs
In 2001, the causative mutation of Muckle–Wells Syndrome (MWS), a rare autosomal recessive auto-inflammatory disease, was mapped to NLRP3 (CIAS1) (17). In 2002, Tschopp and colleagues were the first to characterize the inflammasome, defining it biochemically as a complex consisting of an NLR (NLRP1), the bipartite adaptor protein ASC (which contains both a CARD and a PYD), and the two inflammatory caspases, caspases-1 and -5 (18). In 2004, the discovery of the links between the NLRP3 mutations, NLRP3 inflammasome hyper-activation, and excessive production of IL-1β has set the stage for the use of IL-1 blockade strategies, such as recombinant IL-1 receptor antagonist (anakinra) or anti-IL-1β antibodies (canakinumab), to cure patients inflicted with hereditary periodic fever syndromes [reviewed in Ref. (19)]. Concurrently, Dixit and colleagues reported the generation of the first inflammasome knockouts, namely mice deficient in IPAF (NLRC4) or the adaptor ASC, and showed that macrophages from these mice had a defect in IL-1β production following infection with flagellated bacteria (20). As more inflammasome-forming NLRs are continuously being characterized and studied, their importance in activating immune responses and consequently in conferring host resistance is becoming evident.
NLRP1
The NLRP1 protein has a unique structure amongst other NLRs. Human NLRP1 contains a PYD on the N-terminus and a CARD on the C-terminus, with ZU5 and UPA domains in the internal region which confers proteolytic activity upon the protein (21). Three murine NLRP1 homologs – Nlrp1a, Nlrp1b, and Nlrp1c – have been identified, although they lack the N-terminal PYD domain present in human NLRP1. Few ligands have been found for NLRP1 to date, and include bacterial products such as lethal toxin (LT) produced by Bacillus anthracis which activates murine NLRP1b (22), muramyl dipeptide (MDP), a component of bacterial peptidoglycan that activates human NLRP1; and reduced levels of cytosolic ATP (23–27).
Defects in NLRP1 have been linked to a variety of autoimmune disorders. Candidate gene analysis and Genome-wide association studies (GWAS) have shown a significant association of polymorphic variants in the extended promoter and/or coding regions of NLRP1 with familial cases of generalized vitiligo (28, 29), celiac disease (30), Addison’s disease and type 1 diabetes (31, 32), autoimmune thyroid disorders (AITDs) (33), systemic lupus erythematosus (SLE) (34), systemic sclerosis and giant cell arteritis (35, 36), congenital toxoplasmosis (37), rheumatoid arthritis (38), and Alzheimer’s disease (39) (Figure 3). A novel missense mutation M77T in NLRP1, which destabilizes the protein, has been recently shown to cause corneal intraepithelial dyskeratosis (40).
In a recent analysis of high-risk haplotypes with substitutions in human NLRP1, Levandowski et al. demonstrated that peripheral blood monocytes from heterozygous carriers of the haplotype 2A (which contains three non-synonymous substitutions: L155H-V1059M-M1184V) process significantly greater amounts of pro-IL-1β into mature IL-1β under basal conditions. It was thus proposed that the enhanced production of IL-1β predisposes carriers to a wide spectrum of autoimmune diseases (41). Consistently, patients diagnosed with vitiligo commonly suffer from other autoimmune disorders such as SLE (28, 42). However, the molecular mechanisms underlying the link between NLRP1 genetic variations and these disorders are still unknown. It is plausible that deregulation of an NLRP1 inflammasome effector function is at the basis of the autoimmunity phenotypes. This is consistent with recent results from mice. Masters et al. have recently reported that mice with an activating mutation in Nlrp1a exhibited increased T-cell progenitor death (pyroptosis) at the steady state, which rendered them cytopenic (43). In contrast, Nlrp1a-deficient mice, which may experience less pyroptosis, develop an over-exuberant immune response (43). However, while Masters et al. demonstrated that the inflammatory disease in Nlrp1a mutant mice was dependent on caspase-1, additional proof is needed to show that Nlrp1a formed an inflammasome complex in vivo (43). While anakinra has been shown to be successful in treating patients with SLE in preliminary studies, IL-1 blockade strategies have not been tested to date for other autoimmune diseases such as vitiligo or celiac disease (42, 44).
NLRP3
The NLRP3 inflammasome is arguably the most studied inflammasome to date. NLRP3 is predominantly expressed in splenic neutrophils, macrophages, monocytes, and conventional dendritic cells, and its expression is inducible in response to inflammatory stimuli (45). There is evidence suggesting that a two-step process is required for NLRP3 activation. The first, or priming signal, converges on the activation of NF-κB and transcriptional induction of inflammasome components including NLRP3 itself and pro-IL-1β. The second, or activating signal, in the form of a microbial or danger signal, is then able to directly activate inflammasome assembly (46). NLRP3 is able to recognize a wide variety of exogenous and endogenous stimuli such as microbial agonists, ATP, and particulate matters (47, 48). There is, however, scarce evidence that NLRP3 binds directly to its activators. Instead its activation is thought to be triggered by signaling intermediates (46). For instance, Shenoy et al. proposed that guanylate binding protein 5 (GBP5) may play a vital role in activating inflammasome assembly and promoting caspase-1 processing in response to live bacteria and bacterial cell wall components (49). A recent study by Zhong et al. suggested that particulate stimuli might induce mitochondrial production of reactive oxygen species (ROS), which triggers a calcium influx mediated by transient receptor potential melastatin 2 (TRPM2) to activate NLRP3 (50). The role of ROS in NLRP3 activation is consistent with earlier results by Zhou et al. who showed that ROS also leads to the dissociation of thioredoxin-interacting protein (TXNIP) from thioredoxin, freeing it to interact with and activate NLRP3 (51). In addition, it has been reported that NLRP3 activators are able to disrupt the mitochondria, resulting in the release of oxidized mitochondrial DNA and/or cardiolipin, which can bind to and activate NLRP3 (52, 53). Alternatively, it was argued that mitochondrial disruption is not required for NLRP3 activation; instead K+ efflux is sufficient to stimulate this NLR (54). NLRP3 inflammasome activation may involve at least two adaptors, ASC and the mitochondria-associated adaptor MAVS. It was recently shown that MAVS recruits NLRP3 to the mitochondria for activation in response to non-crystalline activators (55) and that microtubule-driven trafficking of the mitochondria is necessary for NLRP3-ASC complex assembly and activation (56).
Gain-of-function mutations in the NLRP3 gene were first associated with cryopyrin-associated periodic fever syndromes (CAPS), which are a group of rare hereditary auto-inflammatory diseases including familial cold urticaria, MWS, and neonatal onset multisystem inflammatory disease [reviewed in Ref. (19)]. Mutations in NLRP3 were reported to induce an overproduction of IL-1β that triggers the subsequent development of severe inflammation (57, 58). A knock-in mouse model of MWS have validated the observations made in human patients, and showed that equivalent mutations in murine Nlrp3 lead to the production of massive amounts of IL-1β, which mediates the disease (59, 60). IL-1 blockade therapies are frequently used to treat auto-inflammatory diseases. Anakinra and canakinumab, for example, have been used to treat CAPS patients with great success, as several groups have reported long-lasting clinical responses as well as the restoration of IL-1β production levels to normal amounts in patients after treatment (61, 62).
NLRP3 was also linked to gout, which is a result of uric acid crystal deposition in the joints as a consequence of a rich diet high in purines (63). The exact mechanism of NLRP3 activation by uric acid crystals is still unknown, but monosodium urate and calcium pyrophosphate dihydrate crystals were found to induce NLRP3 and caspase-1 activation and the subsequent processing of IL-1β and IL-18 (64). Since uric acid can also be released from dying cells as a DAMP (65), there has been speculation that NLRP3 may also detect danger signals released from dying cells (66).
Single nucleotide polymorphisms in the NLRP3 locus have been associated with a wide range of disorders, including type 1 diabetes (67), celiac disease (67), psoriasis (68), and increased susceptibility to HIV-1 infections (69). While no SNP in the NLRP3 region is directly associated with inflammatory bowel disease (IBD), a SNP downstream of NLRP3 has been previously identified as a risk allele in Crohn’s disease (70). Lewis and colleagues, however, were unable to reproduce this result, as they found no significant association between NLRP3 SNPs and Crohn’s disease (71). A recent GWAS meta-analysis has shown that SNPs that affect receptors downstream of NLRP3 such as IL18R1, IL1R1, IL1RL1, IL1RL2, and IL1R2, are associated with susceptibility to IBD (72). Thus, although there is conflicting data regarding the effects of NLRP3 variants in IBD, defects in inflammasome signaling likely play a role in IBD pathogenesis. Consistently, Nlrp3−/− mice or mice deficient in inflammasome components were found to be significantly more susceptible to experimental models of colitis compared to wild-type mice (73–76). Together, these studies indicate that NLRP3 may be involved in intestinal tissue repair mechanisms following injury.
The NLRP3 inflammasome has also been implicated in different metabolic pathologies. For instance, the NLRP3 inflammasome has been linked to obesity, insulin resistance, atherosclerosis, and Alzheimer’s disease. It has been shown that activation of caspase-1 and IL-1β processing downstream of NLRP3 lead to inhibition of adipocyte differentiation and contributes to high fat diet-induced obesity (77). Several studies have also shown that the NLRP3 inflammasome may play a crucial role in insulin resistance and the potential development of type 2 diabetes (51, 77, 78). Consistently, Nlrp3−/− or Asc−/− mice were reported to have improved glucose tolerance and insulin sensitivity when fed a high fat diet. Ceramide, a specific product from the metabolism of long-chain saturated fatty acids, and the saturated free fatty acid, palmitate, have been shown to induce IL-1β in an NLRP3-dependent fashion [Ref. (78) and reviewed in Ref. (63)]. IL-1β produced downstream of the NLRP3 inflammasome, which is also stimulated by islet amyloid polypeptide (79), promotes beta-cell dysfunction, and cell death (80), linking NLRP3 activation to insulin resistance. Crystalline cholesterol was proposed to cause atherosclerosis by acting as a danger signal and initiating inflammation through the NLRP3 inflammasome. Consistently, Duewell et al. observed that mice deficient in components of the NLRP3 inflammasome did not undergo acute inflammation after the injection of cholesterol crystals, and had markedly decreased atherosclerosis compared to wild-type animals (81). There is, however, some controversy in this area, as Menu et al. reported no differences in disease progression in Nlrp3−/−mice compared to wild-type animals (82). In Alzheimer’s disease, amyloid-β aggregates were shown to activate NLRP3 ex vivo in primary macrophages and microglia (83). This was supported by in vivo results by Heneka et al. who demonstrated that NLRP3 deficiency protected mice with familial Alzheimer’s disease mutations from memory loss (84).
NLRP6
Preliminary immunofluorescence data has proposed that the formation of the NLRP6 inflammasome is dependent on the recruitment of NLRP6 to ASC specks in the cytosol via its N-terminal PYD (85, 86). While much of its functions still remain unknown, recent studies have demonstrated that NLRP6 is important in the self-renewal and integrity of the intestinal epithelium, as Nlrp6−/− mice exhibited insufficient wound healing after injury (87, 88) and were more susceptible to carcinogen-induced tumor development compared with wild-type mice (89, 90). While the precise mechanisms by which NLRP6 protects against tumorigenesis is not clear. It is known that Nlrp6−/− mice are able to sustain increases in intestinal epithelial proliferative activity over longer periods of time along with the observed lower efficiency in wound repair – in other words, the repair mechanism in Nlrp6−/−mice fails to promote wound healing but is still able to promote general cell proliferation, leading to higher incidents of dysplasia and tumorigenesis (91).
In a study by Elinav et al., Nlrp6−/− mice showed an altered gut microbiota with an increase in colitogenic bacterial strains such as Prevotellaceae and TM7, which are also found in increased numbers in IBD patients, indicating a role for NLRP6 in the regulatory sensing system in the gut as well (92). The authors speculated that NLRP6 may act as a “gate keeper” by sensing bacterial products or cell damage and promoting the production of IL-18 during homeostasis which in turn supports the normal microbial flora in the gut to prevent dysbiosis (92). While this proposal raised some interesting points as to the functions of NLRP6 in the gut, a conclusion cannot be drawn as to whether the altered microbiota in Nlrp6−/− animals is due to the absence of the protein, as no littermate analysis or maternal microbiota transfer experiments were conducted by the authors to further their hypothesis. A study by Henao-Mejia et al. later linked changes in Nlrp6−/− mice microbiota to metabolic diseases (93). Namely, NLRP6 seems to negatively regulate the progression from non-alcoholic fatty liver disease to non-alcoholic steatohepatitis by preventing the increase in colitogenic bacteria (93). Additionally, Anand et al. observed that Nlrp6−/− mice were highly resistant to a variety of pathogens including Listeria monocytogenes and Escherichia coli (94). Nlrp6−/−mice had increased numbers of immune cells in their circulation, as well as enhanced activation of MAPK and NF-κB signaling, though Toll-like receptor (TLR) activation, suggesting that NLRP6 may suppress TLR pathways after the recognition of pathogens to prevent amplified inflammatory pathology (94). The exact mechanisms of how NLRP6 functions, however, still remain to be studied.
NLRP7
NLRP7, a human NLR with no murine orthologs, is characterized by an N-terminal PYD along with a NACHT domain and a C-terminal LRR region. Mutations in the NLRP7 gene are associated with recurrent hydatidiform moles and reproductive wastage (substitutions R693W, R693P, and N913S) (11, 95–97). Furthermore, NLRP7 expression is increased in certain type of cancers such as testicular (98) and endometrial (99) cancers. However, the mechanisms underlying these phenotypes are not clear. Messaed et al. showed that PBMCs from patients with NLRP7 mutations (at G118X, G380R, C399Y, R693W, A719V) secreted significantly lower levels of IL-1β and TNF in response to LPS despite high intracellular levels of pro-IL-1β and unimpaired pro-IL-1β processing. The authors concluded that NLRP7 might play a role in cytokine trafficking and secretion from the cell (100). Conversely, others have shown that overexpression of NLRP7 inhibited pro-IL-1β synthesis and secretion (88, 101). Moreover, it was recently reported that bacterial acylated lipopeptides (acLP) activated NLRP7 and stimulated formation of an NLRP7-ASC-caspase-1 inflammasome (102). Thus, further studies are needed to clarify NLRP7 mechanisms of actions and functions in reproduction and immunity.
NLRP12
NLRP12 was previously reported to form an inflammasome as well as function in modulating NF-κB signaling (see below). A recent study by Vladimer et al. has shown that the NLRP12 inflammasome has a key role in controlling IL-1β and IL-18 production after Yersinia pestis infection, where NLRP12-deficient mice were more susceptible to infection compared to the controls (103). Other pathogens such as Klebsiella pneumoniae and Mycobacterium tuberculosis and bacterial components such as LPS do not seem to depend on NLRP12 for infection or pathology (104).
While many of the functions and activators of NLRP12 remain unknown, mutations in the NLRP12 gene have been associated with auto-inflammatory diseases such as atopic dermatitis (105) and hereditary periodic fever syndromes (106, 107). Anti-IL-1 therapies, similar to those administered to patients with NLRP3 mutations, have been conducted on patients with NLRP12 mutations with limited success. Patients treated with anakinra showed improvements early on during the treatment process, but developed resistance to the drug within months and suffered from severe myalgia as a side effect (107). Similarly, levels of IL-1β in these patients returned to pre-treatment levels after 14 months of treatment (107). Further clinical studies are needed before conclusions are drawn regarding the efficacy of anti-IL-1 agents in the treatment of diseases associated with NLRP12.
There is currently much debate as to the role of NLRP12 in inflammation, and both stimulatory and inhibitory functions have been proposed. Some studies have suggested that NLRP12 may negatively regulate the NF-κB pathway (86, 108, 109). Nlrp12−/−mice were found to be more susceptible to colitis and colon cancer, and polyps isolated from these mice showed significantly higher non-canonical activation of NF-κB with an increased expression of inflammation and cancer-related genes (109, 110). Conversely, Arthur et al. demonstrated in a murine model of allergic dermatitis that proinflammatory cytokine production was unaffected in Nlrp12−/−mice (111). Instead, dendritic cells in Nlrp12−/− mice exhibited a much-reduced migratory capacity, and neither peripheral dendritic cells nor neutrophils in Nlrp12−/−mice responded to chemotaxic signals or chemokines in in vitro experiments (111). Yet another function for NLRP12 was proposed by Jéru et al., who discovered that mutations in NLRP12 did not affect NF-κB activation, but rather increased ASC speck formation and caspase-1 activation (112). Altogether, these results suggest that NLRP12 plays a role in suppressing NF-κB while stimulating the inflammasome and assisting in the migration of immune cells.
NLRC4 and NAIPs
NLRC4 possesses an N-terminal CARD that allows direct interaction with caspase-1 independently of ASC (113, 114). A recent study by Qu et al., showed that the phosphorylation of Ser533 in NLRC4 by PCKδ was crucial for the activation of the NLRC4 inflammasome (115). NAIPs, members of the NLRB sub-family, have been identified as critical components of the NLRC4 inflammasome. They are required for the recognition of bacterial components, as well as the scaffolding of the NAIP-NLRC4 inflammasome. Activators of this inflammasome include bacterial flagellin and components of the bacterial type III secretion system (T3SS) (113, 116, 117). Notably, murine Naip5 and Naip6 were shown to recognize bacterial flagellin and subsequently bind to NLRC4 to trigger the formation of the inflammasome, whereas Naip2 and human NAIP serve as receptors for the rod and the needle components, of the bacterial T3SS (118, 119).
NLRC4 plays an essential role in host survival and pathogen clearance following host infection with pathogens such as Legionella pneumophila (120, 121), Candida albicans (122), and Burkholderia pseudomallei (123). More recently, Cai et al. showed that, upon K. pneumoniae infections, Nlrc4−/−mice exhibited decreased survival compared to wild-type animals (124). Similarly, Franchi et al. reported that Nlrc4−/− mice were highly susceptible to orogastric Salmonella infections (125). Interestingly, Nlrc4−/−mice do not develop spontaneous colitis in response to the commensal microbiota (126), likely due to low soluble flagellin levels in the gut and a primary role of TLR5 in dealing with gut flagellated bacteria. NLRC4 thus serves as an additional sentinel against pathogenic enteric infections (126, 127). NLRC4 has been previously been shown to act in sync with NLRP3 during Salmonella infection (114). More recently, it was demonstrated that both NLRs play non-redundant roles in B. pseudomallei infection and melioidosis, where NLRC4 is critical for pyroptosis and NLRP3 for the production of IL-1β and IL-18 (123). Ceballos-Olvera et al. demonstrated that while IL-18 and pyroptosis are both essential for host resistance, the production of IL-1β by NLRP3 was deleterious, as it triggered excessive neutrophil recruitment and exacerbated the disease (123). Thus NLRC4 seems to act synergistically with both TLR5 and NLRP3, but its contributions to their functions seems to be secondary.
Non-Inflammasome-Forming NLRs
NOD1/2
NOD1 and 2, have been studied primarily in the context of their signaling activity following recognition of the peptidoglycan components diaminopimelic acid (DAP) and MDP from Gram-negative and Gram-positive bacteria (128–132). Despite this focus, much of the nature of the NOD1 and 2 interaction with these structures remains unknown, although recent findings suggest that NOD2 directly binds MDP with high affinity (133), with the N-glycosylated form specific to the mycobacterial cell wall triggering an exceptionally strong immunogenic response compared to N-acetyl MDP (134). The possibility of a role for NOD2 in non-bacterial infections has also been suggested, with NOD2 having been shown to induce an IFNβ-driven antiviral response following recognition of single-stranded viral RNA (135). Indeed, viral ssRNA from respiratory syncytial virus (RSV), vesicular stomatitis virus (VSV), and influenza virus has been shown to trigger a non-canonical NOD2-directed signaling pathway that requires mitochondrial antiviral signaling protein (MAVS) and induces IRF3 activity, leading to the production of type I IFNs (135) (Figure 4). However, it is still unclear whether lack of NOD2 results in susceptibility to viral infection in humans.
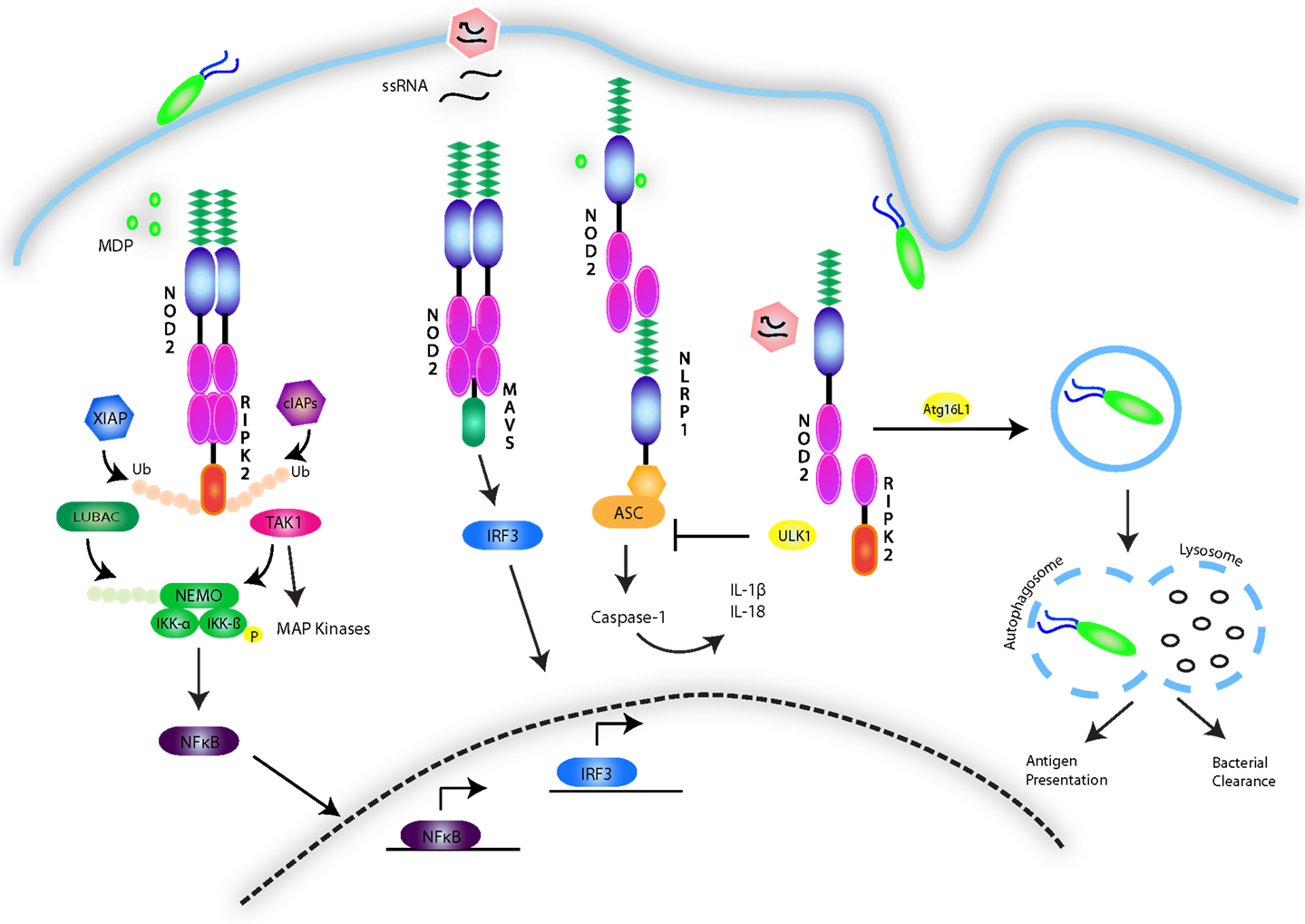
Figure 4. NOD1/2 signaling pathways. The NOD1 and 2 receptors recognize the bacterial peptidoglycan derivatives DAP and MDP. The events involved in signal transduction are depicted and involve the formation of a nodosome complex that is stabilized through a series of ubiquitin scaffolds mediated by a number of E3 ligases including cIAP1/2, XIAP, LUBAC, and ITCH. These scaffolds serve to engage effector kinases, including TAK1 and the IKK complex to activate NF-κB and MAPK pathways. NOD2 is additionally activated by single-stranded RNA viruses and stimulates an antiviral innate immune response by engaging MAVS and activating IRF3. The NOD receptors have also been shown to synergize with NLRP sensors to activate the inflammasome. Conversely, they have also been implicated in triggering autophagy though association with ATG16L1, and in response to viral infection, to inhibit the inflammasome by upregulating ULK1-dependent mitophagy.
NOD1 and 2 are encoded by the CARD4 and CARD15 genes respectively, and as NLRCs, both contain the shared NOD and LRR domains in addition to an amino-terminal CARD. Despite the strong similarities between the two receptors, differences exist; NOD1 contains one CARD domain, while NOD2 contains two (136) and expression of NOD1 is detected in a wide variety of cell types, whereas NOD2 expression is restricted to myeloid cells (136–138), keratinocytes (139) and intestinal, lung, and oral epithelial cells (140–142).
Activation of NOD1 and 2 follows the cytosolic recognition of peptidoglycan ligands that triggers oligomerization of the receptors via their NOD domain and the recruitment of mediators needed to form a signaling complex referred to as the nodosome (143). The nodosome is directed to the point of bacterial entry on the plasma membrane of polarized epithelial cells by the regulatory protein FRMBP2 (144). NOD1 and 2 both interact with RIPK2, via a CARD–CARD homotypic interaction (145–148). This association results in the recruitment of a number of E3 ubiquitin ligases, including TNF receptor-associated factors (TRAFs) (149), cellular inhibitor of apoptosis (cIAP)1 and cIAP2 (150), X-linked inhibitor of apoptosis (XIAP) (151, 152), and ITCH (153). K63-linked ubiquitination of RIPK2 has been established as a means to construct protein scaffolds that transduce downstream signaling. In a step-wise fashion, ubiquitination of RIPK2 leads to activation and recruitment of the TAK1 complex, consisting of TAK1 in association with TAK1-binding protein (TAB)2 and 3. The kinase activity of TAK1 leads to phosphorylation events that activate AP-1 and NF-κB. In parallel to cIAP-induced ubiquitination of RIPK2, XIAP’s enzymatic activity results in the formation of polyubiquitin chains on RIPK2, serving as a platform to engage another E3 ligase complex known as the Linear Ubiquitin Assembly Complex (LUBAC) (152, 154). LUBAC attaches linear ubiquitin chains to the regulatory protein NEMO, allowing for activation of the IKK complex. The kinase activity of IKKβ results in the phosphorylation and degradation of the inhibitor of NF-κB (IκB), allowing for NF-κB dimers to translocate to the nucleus and induce proinflammatory gene expression (155). Besides activating NF-κB, NOD1 and NOD2 have also been shown to activate the p38, JNK, and ERK MAPK pathways (147, 156, 157) and to interact with other NLRs such NLRP1 and NLRP12 (158, 159) (Figure 4).
NOD1 and 2 have been implicated in a number of chronic inflammatory diseases. Mutations and SNPs in CARD15 in particular, have been linked to a multitude of inflammatory diseases including Crohn’s disease (160, 161), Blau Syndrome (162), asthma (163, 164), atopic eczema (165), atopic dermatitis (105), arthritis (166, 167), and sarcoidosis (168). In the context of Crohn’s disease, the most common mutation that confers susceptibility is a frameshift mutation in the LRR region of the receptor (160), while the mutations conferring susceptibility to Blau syndrome occur in the NOD region (162). While the contribution of these mutations to disease is unknown, further work on understanding NOD2 function could unveil the link between the gene and the disease, as well as allow for the creation of new therapies for these chronic and often devastating diseases. Several NOD2 loss-of-function mouse lines have been generated in an attempt to elucidate its role in Crohn’s disease. Pauleau and Murray generated the first NOD2 knockout mice (156). Surprisingly, these mice lacked symptoms associated with spontaneous intestinal inflammation, although stimulation of primary macrophages from these animals with MDP failed to trigger inflammatory responses, confirming loss of NOD2 activity (156). Other NOD2 mouse mutants were later generated to express common Crohn’s disease susceptibility mutations (157, 169). While these mice did not develop any gut inflammation resembling that of Crohn’s disease patients, they did display increased susceptibility to bacterial infection, and were shown to produce decreased amounts of β-defensins when challenged with L. monocytogenes (157, 170). Similarly, mice deficient in NOD1, NOD2, or RIPK2 also exhibited enhanced susceptibility to bacteria including Helicobacter pylori (171–173), Chlamydophila pneumoniae (174), L. pneumophila (172, 175, 176), and B. anthracis (177). This susceptibility often resulted from an inability to control bacterial burden, possibly due to a reduced ability to recruit neutrophils as well as a decrease in the production of proinflammatory and antimicrobial molecules (176, 177). Despite the prevalence of NOD2-deficient models, there remains controversy as to whether Crohn’s disease-linked mutations in NOD2 diminish or enhance its activity in the context of the disease. Common Crohn’s disease-associated NOD2 variants expressed in HEK293T kidney cells are unable to detect MDP and activate NF-κB (178) and monocytes from Crohn’s disease patients with the 1007fs variant displayed defects in the secretion of TNFα, IL-6, IL-8, and IL-10 (179, 180) and many of these NOD2 variants seem to act recessively (181). While these findings point to a loss-of-function effect of the mutations, Karin and colleagues have argued that the models used in these studies lack resemblance to the natural course of the disease in humans and that the Crohn’s disease-associated NOD2 mutations may in fact result in a gain-of-function. Indeed, Crohn’s disease has been associated with the presence of activated NF-κB and inflammatory NF-κB target gene products in epithelial cells and lamina propria macrophages (182, 183) rather than in circulating blood monocytes used in studies with cultured cells, and results from experiments using tissue samples have differed from those using monocytes (179).
The study of NOD2 linkage to Crohn’s disease has been extended to encompass a key role of NOD1 and 2 in the regulation of autophagy. Autophagy is a housekeeping process in which organelles or other cellular components are degraded and recycled into nutrients during times of starvation or stress. This process results in the formation of a double membrane vacuole known as the autophagosome, which fuses with lysosomes to degrade its contents (184). The role of NOD1 and 2 in this process was initially proposed following GWAS findings of an association between a key component of the autophagy process, ATG16L1, and susceptibility to Crohn’s disease (185, 186). Not only have NOD1 and 2 been shown to interact with ATG16L1 (187), but murine Paneth cells expressing the ATG16L1 mutation associated with Crohn’s disease were unable to produce antimicrobial peptides despite NOD2 stimulation (188). Additionally, the autophagic machinery is involved in loading antigen onto MHC Class II, a process that has been observed to be defective in Crohn’s disease (189). Recently, Lupfer et al. further substantiated the link between NOD2 and autophagy by demonstrating a role for NOD2-RIPK2 signaling in the regulation of the NLRP3 inflammasome following infection with influenza A virus. By triggering the phosphorylation of the autophagy inducer ULK1, RIPK2 induces autophagy of disrupted mitochondria (mitophagy), preventing the accumulation of ROS and NLRP3 inflammasome activation. Mice lacking Nod2, Ripk2, or Ulk1 were hypersusceptible to influenza A infection due to a hyperactive NLRP3 inflammasome and excessive IL-18 levels (190) (Figure 4). Collectively, these studies provide evidence for a key role of NOD2 in autophagy-associated processes such as xenophagy, antigen presentation, antimicrobial peptide secretion, and mitophagy.
Other diseases have also been associated with genetic variants in loci encompassing the genes encoding NOD1 and/or 2. GWAS have linked SNPs in CARD15 to prostate (191) and endometrial (192) cancer, as well as to gastric lymphoma induced by H. pylori infection (173). Similarly, SNPs in CARD15 were linked to susceptibility to leprosy (193, 194) and tuberculosis (195, 196). The observation that CARD15, RIPK2, and NF-κB have been linked to leprosy (193), tuberculosis (195, 197, 198), and IBD (150, 199) by GWAS and other genetic studies in humans and mice, has led to speculation of a common etiology between mycobacterial diseases and Crohn’s disease (197, 200, 201).
NLRP10
NLRP10 was discovered based on its homology to NLRP3 and APAF1 (202). Lack of LRRs in NLRP10 may indicate a role for this protein as a signaling adaptor rather than an NLR sensor. NLRP10 has been found in human and murine skin (203), colon, kidney, and testis (204), with mRNA and protein expressed in epithelial cells (202, 205) and hematopoietic cells (206). NLRP10 was previously proposed as a negative regulator of NF-κB, cell death, and IL-1β release (202). These results have been supported by NLRP10 over-expression studies in Nlrp10 knock-in mice and in in vitro studies. In the murine model, Nlrp10 knock-in mice were found to be resistant to LPS-induced endotoxic shock, due to a decreased release of inflammatory cytokines (203). This was consistent with the observation that cells from these animals secreted reduced amounts of IL-1β following infection with Salmonella or TLR7 stimulation (203). However, another group proposed a role for NLRP10 in augmenting the NOD1 immune response to Shigella flexneri, indicating the possibility of an inflammasome-independent function for NLRP10 (205). While this mechanism is still poorly understood, the ability of NLRP10 to interact with NOD1 as well as its signaling targets RIPK2, TAK1, and NEMO, suggests that NLRP10 may be involved in optimizing cytokine release following bacterial infections. Furthermore, Flavell and colleagues reported a role of NLRP10 in adaptive immunity. Using NLRP10 knockout mice, this group examined T-cell responses to ovalbumin and aluminum hydroxide, complete Freund’s adjuvant with myelin oligodendrocyte glycoprotein, and LPS. Interestingly, Nlrp10−/− mice displayed major defects in TH2, TH17, and TH1 responses, potentially due to a defect in the ability of dendritic cells to transport antigen to draining lymph nodes (207). These findings, as well as those of another group that reported hematopoietic compartment-dependent susceptibility of Nlrp10−/− mice to C. albicans (206), highlight a role for NLRP10 in bridging innate and adaptive immunity. Despite these findings, understanding the role of NLRP10 in immunity is still in its infancy, and applications of this protein to human diseases are limited while the function of NLRP10 remains largely uncharacterized. However, GWAS have linked NLRP10 to atopic dermatitis (105, 165, 208), an interesting find considering the abundant expression of NLRP10 in the skin.
NLRX1
NLRX1 is unique among NLRs in that it contains an N-terminal mitochondrial targeting sequence (209, 210). The protein is broadly expressed in the mitochondria, although it is yet unclear whether it is localized to the matrix or to the outer membrane (209, 211). NLRX1 has been shown to enhance ROS production when it is overexpressed (212), following Chlamydia (213) and Shigella infection, as well as in response to TNFα and poly(I:C) (212). Like NOD2, NLRX1 has been implicated in host antiviral responses following viral RNA detection (135, 212) and has been shown to directly bind both single and double stranded viral RNA via its LRRs (210). Moore and colleagues initially characterized NLRX1 as a negative regulator of MAVS and antiviral signaling (211). Recently, Lei et al. demonstrated a role for NLRX1 and the mitochondrial protein TUFM in enhancing autophagy and reducing type I IFNs following viral infection (214, 215). However, these findings have been contested and the generation of NLRX1-deficient and Nlrx1 knockdown mice by several groups has produced conflicting results. In some laboratories, the NLRX1 mutant mice did not display any differences in MAVS antiviral signaling compared to wild-type controls (216–219). In contrast, another group’s findings supported their original claim of a role for NLRX1 in inhibiting MAVS signaling pathways (216, 218). Zhao et al. recently associated a missense mutation in the LRR of NLRX1 with susceptibility to chronic hepatitis B infection in human patients. The replacement of the highly conserved Arg707 with a cysteine between a α helix and a β strand was hypothesized to interfere with the electrostatic potential of the region and consequently modulate the activity of the protein (220). Lastly, a group characterizing the molecular signature of SIV-induced gastrointestinal dysfunction found an increase in NLRX1 expression in rhesus macaques 90 days following SIV infection (221). These findings highlight the role of NLRX1 in antiviral defense, but more research is needed to elucidate the precise mechanism.
NLRC5
One of the newest additions to the NLR family, NLRC5 has been shown to have a similar structure to other NLRs, although the CARD domain has been found to be structurally distinct from CARD domains expressed in other NLRs. The protein is most similar to CIITA, both in structure and activity. NLRC5 has been shown to be able to enter the nucleus, and its main function is believed to be as a MHC Class I transactivator, forming the basis of an enhanceosome for MHC Class I transcription (222). Accordingly, NLRC5 is expressed constitutively in both humans and mice, unlike the more restricted expression of CIITA (223, 224) and knockdown of NLRC5 in cells using siRNA and in knockout mice resulted in a decrease in MHC Class I expression, without significantly affecting expression of MHC Class II (222, 225, 226). Despite the widespread expression of NLRC5, however, there is a distinct upregulation of NLRC5 in lymphocytes compared to other hematopoietic and somatic cells (227, 228). As a key regulator of MHC Class I transcription, NLRC5 expression has been shown to be induced by a number of signals, including IFNβ, poly(I:C), VSV, and LPS (222– 225, 227, 228). However, the most efficient activator of NLRC5 known is IFNγ, which several of the aforementioned signals are known to induce. IFNγ functions via signal transducer and activator of transcription 1 (STAT1) and cannot induce NLRC5 expression in the absence of STAT1 (225, 228).
The effect of NLRC5 on human health and disease has yet to be extensively studied. However, inferences can be made based on NLRC5’s role in MHC Class I presentation and phenotypes observed in NLRC5-deficient mice. Yao et al. observed extreme immunodeficiency in these mice, with the animals unable to mount an effective CD8+ T-cell response when challenged with L. monocytogenes. Interestingly, NLRC5 deficiency also seemed to result in a decrease in NLRP3 inflammasome activation, suggesting that NLRC5 may play a role in the regulation of this pathway (226). Murine and cellular models of NLRC5 deficiency have also implicated NLRC5 in the negative regulation of TLR signaling (223, 229, 230), as well as in RIG-I-like receptor signaling(229). However, other groups have disputed these findings (222, 230), and more research needs to be done in order to gain a more comprehensive understanding of the functions of NLRC5.
Class II Transactivator (CIITA)
MHC CIITA was discovered in 1993 as the genetic basis of hereditary major histocompatibility complex Class II deficiency, or bare lymphocyte syndrome (BLS), a disease characterized by severe immunodeficiency due to a lack of MHC Class II expression (231). Its detection via complementation cloning marked the discovery of the first NLR family member, although the classification of NLRs was only later introduced, following the discovery of NOD1. Although CIITA retains the tripartite structure consistent across the NLR family, it contains an additional acidic domain and a proline/serine/threonine (PST)-rich domain at its N-terminus. Unlike other NLRs, the function of CIITA lies in transcriptional regulation of MHC Class II. The previously mentioned additions to the structure of CIITA do not allow it to bind DNA, but provide a platform for the recruitment and interaction of proteins required for the transcription of MHC Class II in leukocytes or other cells following IFNγ stimulation (232–234). Accordingly, CIITA contains nuclear localization signals and nuclear export signals (235–237). As its role suggests, CIITA is expressed in cells that express MHC Class II, mainly lymphocytes, dendritic cells, macrophages, and other professional antigen presenting cells.
In addition to BLS, CIITA has been linked to a number of other human diseases. GWAS and patient exome sequencing studies have linked SNPs in CIITA to celiac disease (238, 239), susceptibility to myocardial infarction (240), rheumatoid arthritis (240–242), multiple sclerosis (240, 242), primary adrenal insufficiency (243), SLE (244), and type 1 diabetes (245, 246), although these results have not always been replicated in subsequent studies (247–249). Gyllenberg et al. suggested that age-dependent variation in the gene encoding CIITA could be responsible for false associations in GWAS (250). Interestingly, women over 75 years of age expressing the rs3087456(G) allele were found to have a higher average bone mineral density and a decrease in bone fractures compared to controls, although the association was not observed in women aged 25 years (251). Ulrich Streidl and colleagues recently used RNA sequencing to identify a novel and frequently expressed CIITA-BX648577 gene fusion product in the KM-H2 Hodgkin lymphoma cell-line which was associated with decreased HLA Class II expression and increased programed cell death 1 (PDL1) on the surface of affected cells (252). Genomic CIITA breaks were found to occur frequently in B-cell lymphoma patients; 38% of primary mediastinal B-cell lymphoma patients and 15% of classical Hodgkin’s lymphoma patients displayed them. The group also observed a decrease in survival in B-cell lymphoma patients expressing genomic CIITA breaks compared to control. The role of CIITA gene fusion products in B-cell lymphomas remains a field of considerable interest. Understanding the effect of these genomic breaks could lead to novel therapies for a highly treatment-evasive cancer. At the very least, the discovery of these abnormal gene products could lead to the discovery of new biomarkers, which aid clinicians in stratifying patients according to prognosis and predicted therapeutic response.
Conclusion
NOD-like receptors have been described as master regulators of innate immunity, and research performed on the functions and signaling pathways of these proteins continues to support this claim. NLRs are essential in recognition of microbial- and pathogen-associated molecular patterns (MAMPs and PAMPs), and have the ability to initiate and support robust immune responses through the formation of inflammasomes and the activation of NF-κB, IRF, and MAPK pathways. Functions such as the enhancement of MHC transcription and presentation implicate NLRs in adaptive immunity, and their roles in reproduction, indicate a broader responsibility of this gene family than previously suspected. The potency of NLRs in inducing immune defenses is vital for the host, but can also provide serious problems when dysregulation or malfunction occurs. GWAS have found many SNPs in NLR genes associated with a plethora of inflammatory and autoimmune pathologies. Research is vastly expanding contemporary knowledge on the functions and roles of NLRs, but several NLRs still remain poorly characterized and understood. Specifically, it remains unclear how NLRs can interact with various and structurally diverse ligands. It is hypothesized that upstream receptors or effectors dictate the activation of NLRs. Alternatively, NLRs might employ co-receptors or dimerize with additional sensors to achieve their functions. Further description of the roles of NLRs in initiating and perpetuating human disease, as well as the role of NLRs at the steady state, will prove vital to gaining a comprehensive understanding of many human pathologies and will provide novel targets and therapies for patients afflicted with these diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol (1989) 54(Pt 1):1–13. doi: 10.1101/SQB.1989.054.01.003
2. Medzhitov R. Approaching the asymptote: 20 years later. Immunity (2009) 30:766–75. doi:10.1016/j.immuni.2009.06.004
3. Rast JP, Smith LC, Loza-Coll M, Hibino T, Litman GW. Genomic insights into the immune system of the sea urchin. Science (2006) 314:952–6. doi:10.1126/science.1134301
4. Sodergren E, Weinstock GM, Davidson EH, Cameron RA, Gibbs RA, Angerer RC, et al. The genome of the sea urchin Strongylocentrotus purpuratus. Science (2006) 314:941–52. doi:10.1126/science.1133609
5. Huang S, Yuan S, Guo L, Yu Y, Li J, Wu T, et al. Genomic analysis of the immune gene repertoire of amphioxus reveals extraordinary innate complexity and diversity. Genome Res (2008) 18:1112–26. doi:10.1101/gr.069674.107
6. Putnam NH, Butts T, Ferrier DE, Furlong RF, Hellsten U, Kawashima T, et al. The amphioxus genome and the evolution of the chordate karyotype. Nature (2008) 453:1064–71. doi:10.1038/nature06967
7. Zhang Q, Zmasek CM, Dishaw LJ, Mueller MG, Ye Y, Litman GW, et al. Novel genes dramatically alter regulatory network topology in amphioxus. Genome Biol (2008) 9:R123. doi:10.1186/gb-2008-9-8-r123
8. Lange C, Hemmrich G, Klostermeier UC, Lopez-Quintero JA, Miller DJ, Rahn T, et al. Defining the origins of the NOD-like receptor system at the base of animal evolution. Mol Biol Evol (2011) 28:1687–702. doi:10.1093/molbev/msq349
9. Maekawa T, Kracher B, Vernaldi S, Ver Loren van Themaat E, Schulze-Lefert P. Conservation of NLR-triggered immunity across plant lineages. Proc Natl Acad Sci U S A (2012) 109:20119–23. doi:10.1073/pnas.1218059109
10. Tong ZB, Gold L, Pfeifer KE, Dorward H, Lee E, Bondy CA, et al. Mater, a maternal effect gene required for early embryonic development in mice. Nat Genet (2000) 26:267–8. doi:10.1038/81547
11. Murdoch S, Djuric U, Mazhar B, Seoud M, Khan R, Kuick R, et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet (2006) 38:300–2. doi:10.1038/ng1740
12. Fernandes R, Tsuda C, Perumalsamy AL, Naranian T, Chong J, Acton BM, et al. NLRP5 mediates mitochondrial function in mouse oocytes and embryos. Biol Reprod (2012) 86(138):131–110. doi:10.1095/biolreprod.111.093583
13. Inohara N, Koseki T, Del Peso L, Hu Y, Yee C, Chen S, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem (1999) 274:14560–7. doi:10.1074/jbc.274.21.14560
14. Said-Sadier N, Ojcius DM. Alarmins, inflammasomes and immunity. Biomed J (2012) 35:437–49. doi:10.4103/2319-4170.104408
15. Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J (2001) 353:411–6. doi:10.1042/0264-6021:3530411
16. Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, et al. NLRP3 (NALP3, cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol (2009) 183: 2008–15. doi:10.4049/jimmunol.0900138
17. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet (2001) 29:301–5. doi:10.1038/ng756
18. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi:10.1016/S1097-2765(02)00599-3
19. Hoffman HM, Wanderer AA. Inflammasome and IL-1beta-mediated disorders. Curr Allergy Asthma Rep (2010) 10:229–35. doi:10.1007/s11882-010-0109-z
20. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature (2004) 430:213–8. doi:10.1038/nature02664
21. D’Osualdo A, Weichenberger CX, Wagner RN, Godzik A, Wooley J, Reed JC. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLoS One (2011) 6:e27396. doi:10.1371/journal.pone.0027396
22. Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog (2012) 8:e1002638. doi:10.1371/journal.ppat.1002638
23. Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell (2007) 25:713–24. doi:10.1016/j.molcel.2007.01.032
24. Frew BC, Joag VR, Mogridge J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog (2012) 8:e1002659. doi:10.1371/journal.ppat.1002659
25. Hellmich KA, Levinsohn JL, Fattah R, Newman ZL, Maier N, Sastalla I, et al. Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS One (2012) 7:e49741. doi:10.1371/journal.pone.0049741
26. Chavarria-Smith J, Vance RE. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog (2013) 9:e1003452. doi:10.1371/journal.ppat.1003452
27. Liao KC, Mogridge J. Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect Immun (2013) 81:570–9. doi:10.1128/IAI.01003-12
28. Jin Y, Mailloux CM, Gowan K, Riccardi SL, Laberge G, Bennett DC, et al. NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med (2007) 356:1216–25. doi:10.1056/NEJMoa061592
29. Alkhateeb A, Qarqaz F. Genetic association of NALP1 with generalized vitiligo in Jordanian Arabs. Arch Dermatol Res (2010) 302:631–4. doi:10.1007/s00403-010-1064-1
30. Pontillo A, Vendramin A, Catamo E, Fabris A, Crovella S. The missense variation Q705K in CIAS1/NALP3/NLRP3 gene and an NLRP1 haplotype are associated with celiac disease. Am J Gastroenterol (2011) 106:539–44. doi:10.1038/ajg.2010.474
31. Magitta NF, Boe Wolff AS, Johansson S, Skinningsrud B, Lie BA, Myhr KM, et al. A coding polymorphism in NALP1 confers risk for autoimmune Addison’s disease and type 1 diabetes. Genes Immun (2009) 10:120–4. doi:10.1038/gene.2008.85
32. Zurawek M, Fichna M, Januszkiewicz-Lewandowska D, Gryczynska M, Fichna P, Nowak J. A coding variant in NLRP1 is associated with autoimmune Addison’s disease. Hum Immunol (2010) 71:530–4. doi:10.1016/j.humimm.2010.02.004
33. Alkhateeb A, Jarun Y, Tashtoush R. Polymorphisms in NLRP1 gene and susceptibility to autoimmune thyroid disease. Autoimmunity (2013) 46:215–21. doi:10.3109/08916934.2013.768617
34. Pontillo A, Girardelli M, Kamada AJ, Pancotto JA, Donadi EA, Crovella S, et al. Polimorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity (2012) 45:271–8. doi:10.3109/08916934.2011.637532
35. Dieude P, Guedj M, Wipff J, Ruiz B, Riemekasten G, Airo P, et al. NLRP1 influences the systemic sclerosis phenotype: a new clue for the contribution of innate immunity in systemic sclerosis-related fibrosing alveolitis pathogenesis. Ann Rheum Dis (2011) 70:668–74. doi:10.1136/ard.2010.131243
36. Serrano A, Carmona FD, Castaneda S, Solans R, Hernandez-Rodriguez J, Cid MC, et al. Evidence of association of the NLRP1 gene with giant cell arteritis. Ann Rheum Dis (2013) 72:628–30. doi:10.1136/annrheumdis-2012-202609
37. Witola WH, Mui E, Hargrave A, Liu S, Hypolite M, Montpetit A, et al. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect Immun (2011) 79:756–66. doi:10.1128/IAI.00898-10
38. Sui J, Li H, Fang Y, Liu Y, Li M, Zhong B, et al. NLRP1 gene polymorphism influences gene transcription and is a risk factor for rheumatoid arthritis in Han Chinese. Arthritis Rheum (2012) 64:647–54. doi:10.1002/art.33370
39. Pontillo A, Catamo E, Arosio B, Mari D, Crovella S. NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis Assoc Disord (2012) 26:277–81. doi:10.1097/WAD.0b013e318231a8ac
40. Soler VJ, Tran-Viet KN, Galiacy SD, Limviphuvadh V, Klemm TP, St Germain E, et al. Whole exome sequencing identifies a mutation for a novel form of corneal intraepithelial dyskeratosis. J Med Genet (2013) 50:246–54. doi:10.1136/jmedgenet-2012-101325
41. Levandowski CB, Mailloux CM, Ferrara TM, Gowan K, Ben S, Jin Y, et al. NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1beta processing via the NLRP1 inflammasome. Proc Natl Acad Sci U S A (2013) 110:2952–6. doi:10.1073/pnas.1222808110
42. Shaw PJ, McDermott MF, Kanneganti TD. Inflammasomes and autoimmunity. Trends Mol Med (2011) 17:57–64. doi:10.1016/j.molmed.2010.11.001
43. Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity (2012) 37: 1009–23. doi:10.1016/j.immuni.2012.08.027
44. Ostendorf B, Iking-Konert C, Kurz K, Jung G, Sander O, Schneider M. Preliminary results of safety and efficacy of the interleukin 1 receptor antagonist anakinra in patients with severe lupus arthritis. Ann Rheum Dis (2005) 64:630–3. doi:10.1136/ard.2004.025858
45. Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, et al. Differential expression of NLRP3 among hematopoietic cells. J Immunol (2011) 186:2529–34. doi:10.4049/jimmunol.1002720
46. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol (2012) 13:325–32. doi:10.1038/ni.2231
47. Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev (2009) 227:106–28. doi:10.1111/j.1600-065X.2008.00734.x
48. Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol (2011) 29:707–35. doi:10.1146/annurev-immunol-031210-101405
49. Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, et al. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science (2012) 336:481–5. doi:10.1126/science.1217141
50. Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, et al. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun (2013) 4:1611. doi:10.1038/ncomms2608
51. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11:136–40. doi:10.1038/ni.1831
52. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36:401–14. doi:10.1016/j.immuni.2012.01.009
53. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for nlrp3 inflammasome activation. Immunity (2013) 39:311–23. doi:10.1016/j.immuni.2013.08.001
54. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38:1142–53. doi:10.1016/j.immuni.2013.05.016
55. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell (2013) 153:348–61. doi:10.1016/j.cell.2013.02.054
56. Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol (2013) 14:454–60. doi:10.1038/ni.2550
57. Aksentijevich I, Putnam CD, Remmers EF, Mueller JL, Le J, Kolodner RD, et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum (2007) 56:1273–85. doi:10.1002/art.22491
58. Jesus AA, Silva CA, Segundo GR, Aksentijevich I, Fujihira E, Watanabe M, et al. Phenotype-genotype analysis of cryopyrin-associated periodic syndromes (CAPS): description of a rare non-exon 3 and a novel CIAS1 missense mutation. J Clin Immunol (2008) 28:134–8. doi:10.1007/s10875-007-9150-7
59. Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity (2009) 30:875–87. doi:10.1016/j.immuni.2009.05.005
60. Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity (2009) 30:860–74. doi:10.1016/j.immuni.2009.04.012
61. Lachmann HJ, Lowe P, Felix SD, Rordorf C, Leslie K, Madhoo S, et al. In vivo regulation of interleukin 1beta in patients with cryopyrin-associated periodic syndromes. J Exp Med (2009) 206:1029–36. doi:10.1084/jem.20082481
62. Imagawa T, Nishikomori R, Takada H, Takeshita S, Patel N, Kim D, et al. Safety and efficacy of canakinumab in Japanese patients with phenotypes of cryopyrin-associated periodic syndrome as established in the first open-label, phase-3 pivotal study (24-week results). Clin Exp Rheumatol (2013) 31:302–9.
63. Wen H, Ting JP, O’Neill LA. A role for the NLRP3 inflammasome in metabolic diseases – did Warburg miss inflammation? Nat Immunol (2012) 13:352–7. doi:10.1038/ni.2228
64. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature (2006) 440:237–41. doi:10.1038/nature04516
65. Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol (2010) 28:321–42. doi:10.1146/annurev-immunol-030409-101311
66. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol (2010) 10:210–5. doi:10.1038/nri2725
67. Pontillo A, Brandao L, Guimaraes R, Segat L, Araujo J, Crovella S. Two SNPs in NLRP3 gene are involved in the predisposition to type-1 diabetes and celiac disease in a pediatric population from northeast Brazil. Autoimmunity (2010) 43:583–9. doi:10.3109/08916930903540432
68. Carlstrom M, Ekman AK, Petersson S, Soderkvist P, Enerback C. Genetic support for the role of the NLRP3 inflammasome in psoriasis susceptibility. Exp Dermatol (2012) 21:932–7. doi:10.1111/exd.12049
69. Pontillo A, Brandao LA, Guimaraes RL, Segat L, Athanasakis E, Crovella S. A 3’UTR SNP in NLRP3 gene is associated with susceptibility to HIV-1 infection. J Acquir Immune Defic Syndr (2010) 54:236–40. doi:10.1097/QAI.0b013e3181dd17d4
70. Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet (2009) 41:71–6. doi:10.1038/ng.285
71. Lewis GJ, Massey DC, Zhang H, Bredin F, Tremelling M, Lee JC, et al. Genetic association between NLRP3 variants and Crohn’s disease does not replicate in a large UK panel. Inflamm Bowel Dis (2011) 17:1387–91. doi:10.1002/ibd.21499
72. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491:119–24. doi:10.1038/nature11582
73. Allen IC, Tekippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med (2010) 207:1045–56. doi:10.1084/jem.20100050
74. Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, Leblanc PM, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity (2010) 32: 367–78. doi:10.1016/j.immuni.2010.02.012
75. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32:379–91. doi:10.1016/j.immuni.2010.03.003
76. Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis (2011) 17:1359–72. doi:10.1002/ibd.21478
77. Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab (2010) 12:593–605. doi:10.1016/j.cmet.2010.11.011
78. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med (2011) 17:179–88. doi:10.1038/nm.2279
79. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol (2010) 11:897–904. doi:10.1038/ni.1935
80. Mandrup-Poulsen T, Pickersgill L, Donath MY. Blockade of interleukin 1 in type 1 diabetes mellitus. Nat Rev Endocrinol (2010) 6:158–66. doi:10.1038/nrendo.2009.271
81. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464:1357–61. doi:10.1038/nature08938
82. Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, et al. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis (2011) 2:e137. doi:10.1038/cddis.2011.18
83. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9:857–65. doi:10.1038/ni.1636
84. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature (2013) 493:674–8. doi:10.1038/nature11729
85. Grenier JM, Wang L, Manji GA, Huang WJ, Al-Garawi A, Kelly R, et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-kappaB and caspase-1. FEBS Lett (2002) 530:73–8. doi:10.1016/S0014-5793(02)03416-6
86. Wang L, Manji GA, Grenier JM, Al-Garawi A, Merriam S, Lora JM, et al. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem (2002) 277:29874–80. doi:10.1074/jbc.M203915200
87. Herrera VL, Bagamasbad P, Didishvili T, Decano JL, Ruiz-Opazo N. Overlapping genes in Nalp6/PYPAF5 locus encode two V2-type vasopressin isoreceptors: angiotensin-vasopressin receptor (AVR) and non-AVR. Physiol Genomics (2008) 34:65–77. doi:10.1152/physiolgenomics.00199.2007
88. Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci U S A (2011) 108:9601–6. doi:10.1073/pnas.1100981108
89. Andersen CL, Christensen LL, Thorsen K, Schepeler T, Sorensen FB, Verspaget HW, et al. Dysregulation of the transcription factors SOX4, CBFB and SMARCC1 correlates with outcome of colorectal cancer. Br J Cancer (2009) 100:511–23. doi:10.1038/sj.bjc.6604884
90. Kim SY, Dunn IF, Firestein R, Gupta P, Wardwell L, Repich K, et al. CK1epsilon is required for breast cancers dependent on beta-catenin activity. PLoS One (2010) 5:e8979. doi:10.1371/journal.pone.0008979
91. Chen GY, Liu M, Wang F, Bertin J, Nunez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol (2011) 186:7187–94. doi:10.4049/jimmunol.1100412
92. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145:745–57. doi:10.1016/j.cell.2011.04.022
93. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature (2012) 482:179–85. doi:10.1038/nature10809
94. Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature (2012) 488:389–93. doi:10.1038/nature11250
95. Kou YC, Shao L, Peng HH, Rosetta R, Del Gaudio D, Wagner AF, et al. A recurrent intragenic genomic duplication, other novel mutations in NLRP7 and imprinting defects in recurrent biparental hydatidiform moles. Mol Hum Reprod (2008) 14:33–40. doi:10.1093/molehr/gam079
96. Deveault C, Qian JH, Chebaro W, Ao A, Gilbert L, Mehio A, et al. NLRP7 mutations in women with diploid androgenetic and triploid moles: a proposed mechanism for mole formation. Hum Mol Genet (2009) 18:888–97. doi:10.1093/hmg/ddn418
97. Hayward BE, De Vos M, Talati N, Abdollahi MR, Taylor GR, Meyer E, et al. Genetic and epigenetic analysis of recurrent hydatidiform mole. Hum Mutat (2009) 30:E629–39. doi:10.1002/humu.20993
98. Okada K, Hirota E, Mizutani Y, Fujioka T, Shuin T, Miki T, et al. Oncogenic role of NALP7 in testicular seminomas. Cancer Sci (2004) 95:949–54. doi:10.1111/j.1349-7006.2004.tb03182.x
99. Ohno S, Kinoshita T, Ohno Y, Minamoto T, Suzuki N, Inoue M, et al. Expression of NLRP7 (PYPAF3, NALP7) protein in endometrial cancer tissues. Anticancer Res (2008) 28:2493–7.
100. Messaed C, Akoury E, Djuric U, Zeng J, Saleh M, Gilbert L, et al. NLRP7, a nucleotide oligomerization domain-like receptor protein, is required for normal cytokine secretion and co-localizes with Golgi and the microtubule-organizing center. J Biol Chem (2011) 286:43313–23. doi:10.1074/jbc.M111.306191
101. Kinoshita T, Wang Y, Hasegawa M, Imamura R, Suda T. PYPAF3, a PYRIN-containing APAF-1-like protein, is a feedback regulator of caspase-1-dependent interleukin-1beta secretion. J Biol Chem (2005) 280:21720–5. doi:10.1074/jbc.M410057200
102. Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity (2012) 36:464–76. doi:10.1016/j.immuni.2012.02.001
103. Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity (2012) 37:96–107. doi:10.1016/j.immuni.2012.07.006
104. Allen IC, McElvania-Tekippe E, Wilson JE, Lich JD, Arthur JC, Sullivan JT, et al. Characterization of NLRP12 during the in vivo host immune response to Klebsiella pneumoniae and Mycobacterium tuberculosis. PLoS One (2013) 8:e60842. doi:10.1371/journal.pone.0060842
105. Macaluso F, Nothnagel M, Parwez Q, Petrasch-Parwez E, Bechara FG, Epplen JT, et al. Polymorphisms in NACHT-LRR (NLR) genes in atopic dermatitis. Exp Dermatol (2007) 16:692–8. doi:10.1111/j.1600-0625.2007.00589.x
106. Jeru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A (2008) 105:1614–9. doi:10.1073/pnas.0708616105
107. Jeru I, Hentgen V, Normand S, Duquesnoy P, Cochet E, Delwail A, et al. Role of interleukin-1beta in NLRP12-associated autoinflammatory disorders and resistance to anti-interleukin-1 therapy. Arthritis Rheum (2011) 63:2142–8. doi:10.1002/art.30378
108. Lich JD, Ting JP. Monarch-1/PYPAF7 and other CATERPILLER (CLR, NOD, NLR) proteins with negative regulatory functions. Microbes Infect (2007) 9:672–6. doi:10.1016/j.micinf.2007.01.018
109. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity (2012) 36:742–54. doi:10.1016/j.immuni.2012.03.012
110. Ye Z, Lich JD, Moore CB, Duncan JA, Williams KL, Ting JP. ATP binding by monarch-1/NLRP12 is critical for its inhibitory function. Mol Cell Biol (2008) 28:1841–50. doi:10.1128/MCB.01468-07
111. Arthur JC, Lich JD, Ye Z, Allen IC, Gris D, Wilson JE, et al. Cutting edge: NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J Immunol (2010) 185:4515–9. doi:10.4049/jimmunol.1002227
112. Jeru I, Le Borgne G, Cochet E, Hayrapetyan H, Duquesnoy P, Grateau G, et al. Identification and functional consequences of a recurrent NLRP12 missense mutation in periodic fever syndromes. Arthritis Rheum (2011) 63:1459–64. doi:10.1002/art.30241
113. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol (2006) 7:569–75. doi:10.1038/ni1344
114. Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med (2010) 207:1745–55. doi:10.1084/jem.20100257
115. Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M, et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature (2012) 490:539–42. doi:10.1038/nature11429
116. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in Salmonella-infected macrophages. Nat Immunol (2006) 7:576–82. doi:10.1038/ni1346
117. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A (2010) 107:3076–80. doi:10.1073/pnas.0913087107
118. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477:592–5. doi:10.1038/nature10394
119. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477:596–600. doi:10.1038/nature10510
120. Pereira MS, Morgantetti GF, Massis LM, Horta CV, Hori JI, Zamboni DS. Activation of NLRC4 by flagellated bacteria triggers caspase-1-dependent and -independent responses to restrict Legionella pneumophila replication in macrophages and in vivo. J Immunol (2011) 187:6447–55. doi:10.4049/jimmunol.1003784
121. Jamilloux Y, Pierini R, Querenet M, Juruj C, Fauchais AL, Jauberteau MO, et al. Inflammasome activation restricts Legionella pneumophila replication in primary microglial cells through flagellin detection. Glia (2013) 61:539–49. doi:10.1002/glia.22454
122. Tomalka J, Ganesan S, Azodi E, Patel K, Majmudar P, Hall BA, et al. A novel role for the NLRC4 inflammasome in mucosal defenses against the fungal pathogen Candida albicans. PLoS Pathog (2011) 7:e1002379. doi:10.1371/journal.ppat.1002379
123. Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1beta is deleterious. PLoS Pathog (2011) 7:e1002452. doi:10.1371/journal.ppat.1002452
124. Cai S, Batra S, Wakamatsu N, Pacher P, Jeyaseelan S. NLRC4 inflammasome-mediated production of IL-1beta modulates mucosal immunity in the lung against gram-negative bacterial infection. J Immunol (2012) 188:5623–35. doi:10.4049/jimmunol.1200195
125. Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, Suzuki S, et al. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol (2012) 13:449–56. doi:10.1038/ni.2263
126. Carvalho FA, Nalbantoglu I, Aitken JD, Uchiyama R, Su Y, Doho GH, et al. Cytosolic flagellin receptor NLRC4 protects mice against mucosal and systemic challenges. Mucosal Immunol (2012) 5:288–98. doi:10.1038/mi.2012.8
127. Miao EA, Andersen-Nissen E, Warren SE, Aderem A. TLR5 and Ipaf: dual sensors of bacterial flagellin in the innate immune system. Semin Immunopathol (2007) 29: 275–88. doi:10.1007/s00281-007-0078-z
128. Inohara N, Ogura Y, Chen FF, Muto A, Nunez G. Human Nod1 confers responsiveness to bacterial lipopolysaccharides. J Biol Chem (2001) 276:2551–4. doi:10.1074/jbc.M009728200
129. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol (2003) 4:702–7. doi:10.1038/ni945
130. Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science (2003) 300:1584–7. doi:10.1126/science.1084677
131. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem (2003) 278:8869–72. doi:10.1074/jbc.C200651200
132. Girardin SE, Travassos LH, Herve M, Blanot D, Boneca IG, Philpott DJ, et al. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem (2003) 278:41702–8. doi:10.1074/jbc.M307198200
133. Grimes CL, Ariyananda Lde Z, Melnyk JE, O’Shea EK. The innate immune protein Nod2 binds directly to MDP, a bacterial cell wall fragment. J Am Chem Soc (2012) 134:13535–7. doi:10.1021/ja303883c
134. Coulombe F, Divangahi M, Veyrier F, de Leseleuc L, Gleason JL, Yang Y, et al. Increased NOD2-mediated recognition of N-glycolyl muramyl dipeptide. J Exp Med (2009) 206:1709–16. doi:10.1084/jem.20081779
135. Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol (2009) 10:1073–80. doi:10.1038/ni.1782
136. Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem (2001) 276:4812–8. doi:10.1074/jbc.M008072200
137. Marriott I, Rati DM, McCall SH, Tranguch SL. Induction of Nod1 and Nod2 intracellular pattern recognition receptors in murine osteoblasts following bacterial challenge. Infect Immun (2005) 73:2967–73. doi:10.1128/IAI.73.5.2967-2973.2005
138. Tada H, Aiba S, Shibata K, Ohteki T, Takada H. Synergistic effect of Nod1 and Nod2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect Immun (2005) 73:7967–76. doi:10.1128/IAI.73.12.7967-7976.2005
139. Voss E, Wehkamp J, Wehkamp K, Stange EF, Schroder JM, Harder J. NOD2/CARD15 mediates induction of the antimicrobial peptide human beta-defensin-2. J Biol Chem (2006) 281:2005–11. doi:10.1074/jbc.M511044200
140. Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF, et al. Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut (2003) 52:1591–7. doi:10.1136/gut.52.11.1591
141. Uehara A, Fujimoto Y, Fukase K, Takada H. Various human epithelial cells express functional toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol (2007) 44:3100–11. doi:10.1016/j.molimm.2007.02.007
142. Uehara A, Imamura T, Potempa J, Travis J, Takada H. Gingipains from Porphyromonas gingivalis synergistically induce the production of proinflammatory cytokines through protease-activated receptors with toll-like receptor and NOD1/2 ligands in human monocytic cells. Cell Microbiol (2008) 10:1181–9. doi:10.1111/j.1462-5822.2008.01119.x
143. Tattoli I, Travassos LH, Carneiro LA, Magalhaes JG, Girardin SE. The nodosome: Nod1 and Nod2 control bacterial infections and inflammation. Semin Immunopathol (2007) 29: 289–301. doi:10.1007/s00281-007-0083-2
144. Lipinski S, Grabe N, Jacobs G, Billmann-Born S, Till A, Hasler R, et al. RNAi screening identifies mediators of NOD2 signaling: implications for spatial specificity of MDP recognition. Proc Natl Acad Sci U S A (2012) 109:21426–31. doi:10.1073/pnas.1209673109
145. Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature (2002) 416:194–9. doi:10.1038/416194a
146. Lecine P, Esmiol S, Metais JY, Nicoletti C, Nourry C, McDonald C, et al. The NOD2-RICK complex signals from the plasma membrane. J Biol Chem (2007) 282:15197–207. doi:10.1074/jbc.M606242200
147. Park JH, Kim YG, Shaw M, Kanneganti TD, Fujimoto Y, Fukase K, et al. Nod1/RICK and TLR signaling regulate chemokine and antimicrobial innate immune responses in mesothelial cells. J Immunol (2007) 179: 514–21.
148. Nembrini C, Kisielow J, Shamshiev AT, Tortola L, Coyle AJ, Kopf M, et al. The kinase activity of Rip2 determines its stability and consequently Nod1- and Nod2-mediated immune responses. J Biol Chem (2009) 284:19183–8. doi:10.1074/jbc.M109.006353
149. Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Nunez G, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J (2008) 27: 373–83. doi:10.1038/sj.emboj.7601962
150. Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity (2009) 30:789–801. doi:10.1016/j.immuni.2009.04.011
151. Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci U S A (2009) 106:14524–9. doi:10.1073/pnas.0907131106
152. Damgaard RB, Nachbur U, Yabal M, Wong WW, Fiil BK, Kastirr M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell (2012) 46:746–58. doi:10.1016/j.molcel.2012.04.014
153. Tao M, Scacheri PC, Marinis JM, Harhaj EW, Matesic LE, Abbott DW. ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways. Curr Biol (2009) 19:1255–63. doi:10.1016/j.cub.2009.06.038
154. Ikeda F, Deribe YL, Skanland SS, Stieglitz B, Grabbe C, Franz-Wachtel M, et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature (2011) 471:637–41. doi:10.1038/nature09814
155. Hasegawa M, Yang K, Hashimoto M, Park JH, Kim YG, Fujimoto Y, et al. Differential release and distribution of Nod1 and Nod2 immunostimulatory molecules among bacterial species and environments. J Biol Chem (2006) 281:29054–63. doi:10.1074/jbc.M602638200
156. Pauleau AL, Murray PJ. Role of nod2 in the response of macrophages to toll-like receptor agonists. Mol Cell Biol (2003) 23:7531–9. doi:10.1128/MCB.23.21.7531-7539.2003
157. Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science (2005) 307:731–4. doi:10.1126/science.1104911
158. Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci U S A (2008) 105:7803–8. doi:10.1073/pnas.0802726105
159. Wagner RN, Proell M, Kufer TA, Schwarzenbacher R. Evaluation of Nod-like receptor (NLR) effector domain interactions. PLoS One (2009) 4:e4931. doi:10.1371/journal.pone.0004931
160. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411:599–603. doi:10.1038/35079107
161. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature (2001) 411:603–6. doi:10.1038/35079114
162. Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, et al. CARD15 mutations in Blau syndrome. Nat Genet (2001) 29:19–20. doi:10.1038/ng720
163. Hysi P, Kabesch M, Moffatt MF, Schedel M, Carr D, Zhang Y, et al. NOD1 variation, immunoglobulin E and asthma. Hum Mol Genet (2005) 14:935–41. doi:10.1093/hmg/ddi087
164. Duan Y, Learoyd J, Meliton AY, Clay BS, Leff AR, Zhu X. Inhibition of Pyk2 blocks airway inflammation and hyperresponsiveness in a mouse model of asthma. Am J Respir Cell Mol Biol (2010) 42:491–7. doi:10.1165/rcmb.2008-0469OC
165. Weidinger S, Klopp N, Rummler L, Wagenpfeil S, Novak N, Baurecht HJ, et al. Association of NOD1 polymorphisms with atopic eczema and related phenotypes. J Allergy Clin Immunol (2005) 116:177–84. doi:10.1016/j.jaci.2005.02.034
166. Joosten LA, Helsen MM, van de Loo FA, van den Berg WB. Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice: a comparative study using anti-TNFalpha, anti-IL-1alpha/beta and IL-1Ra. Arthritis Rheum (2008) 58:S110–22. doi:10.1002/art.23363
167. Vieira SM, Cunha TM, Franca RF, Pinto LG, Talbot J, Turato WM, et al. Joint NOD2/RIPK2 signaling regulates IL-17 axis and contributes to the development of experimental arthritis. J Immunol (2012) 188:5116–22. doi:10.4049/jimmunol.1004190
168. Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood (2005) 105: 1195–7. doi:10.1182/blood-2004-07-2972
169. Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, et al. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science (2005) 307:734–8. doi:10.1126/science.1103685
170. Kim YG, Park JH, Shaw MH, Franchi L, Inohara N, Nunez G. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to toll-like receptor ligands. Immunity (2008) 28:246–57. doi:10.1016/j.immuni.2007.12.012
171. Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol (2004) 5:1166–74. doi:10.1038/ni1131
172. Boughan PK, Argent RH, Body-Malapel M, Park JH, Ewings KE, Bowie AG, et al. Nucleotide-binding oligomerization domain-1 and epidermal growth factor receptor: critical regulators of beta-defensins during Helicobacter pylori infection. J Biol Chem (2006) 281:11637–48. doi:10.1074/jbc.M510275200
173. Rosenstiel P, Hellmig S, Hampe J, Ott S, Till A, Fischbach W, et al. Influence of polymorphisms in the NOD1/CARD4 and NOD2/CARD15 genes on the clinical outcome of Helicobacter pylori infection. Cell Microbiol (2006) 8:1188–98. doi:10.1111/j.1462-5822.2006.00701.x
174. Shimada K, Chen S, Dempsey PW, Sorrentino R, Alsabeh R, Slepenkin AV, et al. The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS Pathog (2009) 5:e1000379. doi:10.1371/journal.ppat.1000379
175. Berrington WR, Iyer R, Wells RD, Smith KD, Skerrett SJ, Hawn TR. NOD1 and NOD2 regulation of pulmonary innate immunity to Legionella pneumophila. Eur J Immunol (2010) 40:3519–27. doi:10.1002/eji.201040518
176. Frutuoso MS, Hori JI, Pereira MS, Junior DS, Sonego F, Kobayashi KS, et al. The pattern recognition receptors Nod1 and Nod2 account for neutrophil recruitment to the lungs of mice infected with Legionella pneumophila. Microbes Infect (2010) 12:819–27. doi:10.1016/j.micinf.2010.05.006
177. Loving CL, Osorio M, Kim YG, Nunez G, Hughes MA, Merkel TJ. Nod1/Nod2-mediated recognition plays a critical role in induction of adaptive immunity to anthrax after aerosol exposure. Infect Immun (2009) 77:4529–37. doi:10.1128/IAI.00563-09
178. Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T, et al. Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology (2003) 124:140–6. doi:10.1053/gast.2003.50019
179. Netea MG, Kullberg BJ, de Jong DJ, Franke B, Sprong T, Naber TH, et al. NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn’s disease. Eur J Immunol (2004) 34:2052–9. doi:10.1002/eji.200425229
180. van Heel DA, Ghosh S, Butler M, Hunt KA, Lundberg AM, Ahmad T, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet (2005) 365:1794–6. doi:10.1016/S0140-6736(05)66582-8
181. Economou M, Trikalinos TA, Loizou KT, Tsianos EV, Ioannidis JP. Differential effects of NOD2 variants on Crohn’s disease risk and phenotype in diverse populations: a metaanalysis. Am J Gastroenterol (2004) 99:2393–404. doi:10.1111/j.1572-0241.2004.40304.x
182. Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T, et al. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology (1998) 115:357–69. doi:10.1016/S0016-5085(98)70202-1
183. Schreiber S, Nikolaus S, Hampe J. Activation of nuclear factor kappa B inflammatory bowel disease. Gut (1998) 42:477–84. doi:10.1136/gut.42.4.477
184. Lu JV, Walsh CM. Programmed necrosis and autophagy in immune function. Immunol Rev (2012) 249:205–17. doi:10.1111/j.1600-065X.2012.01147.x
185. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet (2007) 39:207–11. doi:10.1038/ng1954
186. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet (2007) 39:596–604. doi:10.1038/ng2032
187. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11:55–62. doi:10.1038/ni.1823
188. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature (2008) 456:259–63. doi:10.1038/nature07416
189. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med (2010) 16:90–7. doi:10.1038/nm.2069
190. Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol (2013) 14:480–8. doi:10.1038/ni.2563
191. Kang MJ, Heo SK, Song EJ, Kim DJ, Han SY, Han JH, et al. Activation of Nod1 and Nod2 induces innate immune responses of prostate epithelial cells. Prostate (2012) 72:1351–8. doi:10.1002/pros.22483
192. Ashton KA, Proietto A, Otton G, Symonds I, McEvoy M, Attia J, et al. Polymorphisms in genes of the steroid hormone biosynthesis and metabolism pathways and endometrial cancer risk. Cancer Epidemiol (2010) 34:328–37. doi:10.1016/j.canep.2010.03.005
193. Zhang FR, Huang W, Chen SM, Sun LD, Liu H, Li Y, et al. Genomewide association study of leprosy. N Engl J Med (2009) 361:2609–18. doi:10.1056/NEJMoa0903753
194. Berrington WR, Macdonald M, Khadge S, Sapkota BR, Janer M, Hagge DA, et al. Common polymorphisms in the NOD2 gene region are associated with leprosy and its reactive states. J Infect Dis (2010) 201:1422–35. doi:10.1086/651559
195. Austin CM, Ma X, Graviss EA. Common nonsynonymous polymorphisms in the NOD2 gene are associated with resistance or susceptibility to tuberculosis disease in African Americans. J Infect Dis (2008) 197:1713–6. doi:10.1086/588384
196. Azad AK, Sadee W, Schlesinger LS. Innate immune gene polymorphisms in tuberculosis. Infect Immun (2012) 80:3343–59. doi:10.1128/IAI.00443-12
197. Fortin A, Abel L, Casanova JL, Gros P. Host genetics of mycobacterial diseases in mice and men: forward genetic studies of BCG-osis and tuberculosis. Annu Rev Genomics Hum Genet (2007) 8:163–92. doi:10.1146/annurev.genom.8.080706.092315
198. Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, Behr MA, et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog (2009) 5:e1000500. doi:10.1371/journal.ppat.1000500
199. Watanabe T, Asano N, Murray PJ, Ozato K, Tailor P, Fuss IJ, et al. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest (2008) 118:545–59. doi:10.1172/JCI33145
200. Schurr E, Gros P. A common genetic fingerprint in leprosy and Crohn’s disease? N Engl J Med (2009) 361:2666–8. doi:10.1056/NEJMe0910690
201. Lalande JD, Behr MA. Mycobacteria in Crohn’s disease: how innate immune deficiency may result in chronic inflammation. Expert Rev Clin Immunol (2010) 6:633–41. doi:10.1586/eci.10.29
202. Wang Y, Hasegawa M, Imamura R, Kinoshita T, Kondo C, Konaka K, et al. PYNOD, a novel Apaf-1/CED4-like protein is an inhibitor of ASC and caspase-1. Int Immunol (2004) 16:777–86. doi:10.1093/intimm/dxh081
203. Imamura R, Wang Y, Kinoshita T, Suzuki M, Noda T, Sagara J, et al. Anti-inflammatory activity of PYNOD and its mechanism in humans and mice. J Immunol (2010) 184:5874–84. doi:10.4049/jimmunol.0900779
204. Lech M, Avila-Ferrufino A, Skuginna V, Susanti HE, Anders HJ. Quantitative expression of RIG-like helicase, NOD-like receptor and inflammasome-related mRNAs in humans and mice. Int Immunol (2010) 22:717–28. doi:10.1093/intimm/dxq058
205. Lautz K, Damm A, Menning M, Wenger J, Adam AC, Zigrino P, et al. NLRP10 enhances Shigella-induced pro-inflammatory responses. Cell Microbiol (2012) 14:1568–83. doi:10.1111/j.1462-5822.2012.01822.x
206. Joly S, Eisenbarth SC, Olivier AK, Williams A, Kaplan DH, Cassel SL, et al. Cutting edge: Nlrp10 is essential for protective antifungal adaptive immunity against Candida albicans. J Immunol (2012) 189:4713–7. doi:10.4049/jimmunol.1201715
207. Eisenbarth SC, Williams A, Colegio OR, Meng H, Strowig T, Rongvaux A, et al. NLRP10 is a NOD-like receptor essential to initiate adaptive immunity by dendritic cells. Nature (2012) 484:510–3. doi:10.1038/nature11012
208. Hirota T, Takahashi A, Kubo M, Tsunoda T, Tomita K, Sakashita M, et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat Genet (2012) 44:1222–6. doi:10.1038/ng.2438
209. Arnoult D, Soares F, Tattoli I, Castanier C, Philpott DJ, Girardin SE. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J Cell Sci (2009) 122:3161–8. doi:10.1242/jcs.051193
210. Hong M, Yoon SI, Wilson IA. Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity (2012) 36:337–47. doi:10.1016/j.immuni.2011.12.018
211. Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature (2008) 451:573–7. doi:10.1038/nature06501
212. Tattoli I, Carneiro LA, Jehanno M, Magalhaes JG, Shu Y, Philpott DJ, et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep (2008) 9: 293–300. doi:10.1038/sj.embor.7401161
213. Abdul-Sater AA, Said-Sadier N, Lam VM, Singh B, Pettengill MA, Soares F, et al. Enhancement of reactive oxygen species production and chlamydial infection by the mitochondrial Nod-like family member NLRX1. J Biol Chem (2010) 285: 41637–45. doi:10.1074/jbc.M110.137885
214. Lei Y, Wen H, Yu Y, Taxman DJ, Zhang L, Widman DG, et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity (2012) 36:933–46. doi:10.1016/j.immuni.2012.03.025
215. Lei Y, Wen H, Ting JP. The NLR protein, NLRX1, and its partner, TUFM, reduce type I interferon, and enhance autophagy. Autophagy (2013) 9:432–3. doi:10.4161/auto.23026
216. Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity (2011) 34:854–65. doi:10.1016/j.immuni.2011.03.026
217. Rebsamen M, Vazquez J, Tardivel A, Guarda G, Curran J, Tschopp J. NLRX1/NOD5 deficiency does not affect MAVS signalling. Cell Death Differ (2011) 18:1387. doi:10.1038/cdd.2011.64
218. Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity (2011) 34:843–53. doi:10.1016/j.immuni.2011.02.022
219. Soares F, Tattoli I, Wortzman ME, Arnoult D, Philpott DJ, Girardin SE. NLRX1 does not inhibit MAVS-dependent antiviral signalling. Innate Immun (2012) 19(4):438–48. doi:10.1177/1753425912467383
220. Zhao Q, Peng L, Huang W, Li Q, Pei Y, Yuan P, et al. Rare inborn errors associated with chronic hepatitis B virus infection. Hepatology (2012) 56:1661–70. doi:10.1002/hep.25850
221. Mohan M, Kaushal D, Aye PP, Alvarez X, Veazey RS, Lackner AA. Focused examination of the intestinal lamina propria yields greater molecular insight into mechanisms underlying SIV induced immune dysfunction. PLoS One (2012) 7:e34561. doi:10.1371/journal.pone.0034561
222. Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A (2010) 107:13794–9. doi:10.1073/pnas.1008684107
223. Benko S, Magalhaes JG, Philpott DJ, Girardin SE. NLRC5 limits the activation of inflammatory pathways. J Immunol (2010) 185:1681–91. doi:10.4049/jimmunol.0903900
224. Kuenzel S, Till A, Winkler M, Hasler R, Lipinski S, Jung S, et al. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J Immunol (2010) 184:1990–2000. doi:10.4049/jimmunol.0900557
225. Tong Y, Cui J, Li Q, Zou J, Wang HY, Wang RF. Enhanced TLR-induced NF-kappaB signaling and type I interferon responses in NLRC5 deficient mice. Cell Res (2012) 22:822–35. doi:10.1038/cr.2012.53
226. Yao Y, Wang Y, Chen F, Huang Y, Zhu S, Leng Q, et al. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res (2012) 22:836–47. doi:10.1038/cr.2012.56
227. Neerincx A, Lautz K, Menning M, Kremmer E, Zigrino P, Hosel M, et al. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J Biol Chem (2010) 285: 26223–32. doi:10.1074/jbc.M110.109736
228. Staehli F, Ludigs K, Heinz LX, Seguin-Estevez Q, Ferrero I, Braun M, et al. NLRC5 deficiency selectively impairs MHC class I-dependent lymphocyte killing by cytotoxic T cells. J Immunol (2012) 188:3820–8. doi:10.4049/jimmunol.1102671
229. Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell (2010) 141:483–96. doi:10.1016/j.cell.2010.03.040
230. Kumar H, Pandey S, Zou J, Kumagai Y, Takahashi K, Akira S, et al. NLRC5 deficiency does not influence cytokine induction by virus and bacteria infections. J Immunol (2011) 186:994–1000. doi:10.4049/jimmunol.1002094
231. Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell (1993) 75:135–46. doi:10.1016/0092-8674(93)90685-J
232. van den Elsen PJ, Peijnenburg A, van Eggermond MC, Gobin SJ. Shared regulatory elements in the promoters of MHC class I and class II genes. Immunol Today (1998) 19:308–12. doi:10.1016/S0167-5699(98)01287-0
233. Masternak K, Muhlethaler-Mottet A, Villard J, Zufferey M, Steimle V, Reith W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev (2000) 14:1156–66. doi:10.1101/gad.14.9.1156
234. Beresford GW, Boss JM. CIITA coordinates multiple histone acetylation modifications at the HLA-DRA promoter. Nat Immunol (2001) 2:652–7. doi:10.1038/35088514
235. Spilianakis C, Papamatheakis J, Kretsovali A. Acetylation by PCAF enhances CIITA nuclear accumulation and transactivation of major histocompatibility complex class II genes. Mol Cell Biol (2000) 20:8489–98. doi:10.1128/MCB.20.22.8489-8498.2000
236. Cressman DE, O’Connor WJ, Greer SF, Zhu XS, Ting JP. Mechanisms of nuclear import and export that control the subcellular localization of class II transactivator. J Immunol (2001) 167:3626–34.
237. Raval A, Weissman JD, Howcroft TK, Singer DS. The GTP-binding domain of class II transactivator regulates its nuclear export. J Immunol (2003) 170:922–30.
238. Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet (2010) 42:295–302. doi:10.1038/ng.543
239. Szperl AM, Ricano-Ponce I, Li JK, Deelen P, Kanterakis A, Plagnol V, et al. Exome sequencing in a family segregating for celiac disease. Clin Genet (2011) 80:138–47. doi:10.1111/j.1399-0004.2011.01714.x
240. Swanberg M, Lidman O, Padyukov L, Eriksson P, Akesson E, Jagodic M, et al. MHC2TA is associated with differential MHC molecule expression and susceptibility to rheumatoid arthritis, multiple sclerosis and myocardial infarction. Nat Genet (2005) 37:486–94. doi:10.1038/ng1544
241. Iikuni N, Ikari K, Momohara S, Tomatsu T, Hara M, Yamanaka H, et al. MHC2TA is associated with rheumatoid arthritis in Japanese patients. Ann Rheum Dis (2007) 66:274–5. doi:10.1136/ard.2006.063347
242. Martinez A, Sanchez-Lopez M, Varade J, Mas A, Martin MC, de Las Heras V, et al. Role of the MHC2TA gene in autoimmune diseases. Ann Rheum Dis (2007) 66:325–9. doi:10.1136/ard.2006.059428
243. Skinningsrud B, Husebye ES, Pearce SH, McDonald DO, Brandal K, Wolff AB, et al. Polymorphisms in CLEC16A and CIITA at 16p13 are associated with primary adrenal insufficiency. J Clin Endocrinol Metab (2008) 93:3310–7. doi:10.1210/jc.2008-0821
244. Bronson PG, Goldstein BA, Ramsay PP, Beckman KB, Noble JA, Lane JA, et al. The rs4774 CIITA missense variant is associated with risk of systemic lupus erythematosus. Genes Immun (2011) 12:667–71. doi:10.1038/gene.2011.36
245. Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature (2007) 447(7145):661–78. doi:10.1038/nature05911
246. Gyllenberg A, Asad S, Piehl F, Swanberg M, Padyukov L, Van Yserloo B, et al. Age-dependent variation of genotypes in MHC II transactivator gene (CIITA) in controls and association to type 1 diabetes. Genes Immun (2012) 13:632–40. doi:10.1038/gene.2012.44
247. Eyre S, Bowes J, Spreckley K, Potter C, Ring S, Strachan D, et al. Investigation of the MHC2TA gene, associated with rheumatoid arthritis in a Swedish population, in a UK rheumatoid arthritis cohort. Arthritis Rheum (2006) 54:3417–22. doi:10.1002/art.22166
248. Harrison P, Pointon JJ, Farrar C, Harin A, Wordsworth BP. MHC2TA promoter polymorphism (-168*G/A, rs3087456) is not associated with susceptibility to rheumatoid arthritis in British Caucasian rheumatoid arthritis patients. Rheumatology (Oxford) (2007) 46: 409–11. doi:10.1093/rheumatology/kel300
249. Bronson PG, Ramsay PP, Seldin MF, Gregersen PK, Criswell LA, Barcellos LF. CIITA is not associated with risk of developing rheumatoid arthritis. Genes Immun (2011) 12:235–8. doi:10.1038/gene.2010.67
250. Asad S, Nikamo P, Gyllenberg A, Bennet H, Hansson O, Wierup N, et al. HTR1A a novel type 1 diabetes susceptibility gene on chromosome 5p13-q13. PLoS One (2012) 7:e35439. doi:10.1371/journal.pone.0035439
251. Swanberg M, McGuigan FE, Ivaska KK, Gerdhem P, Akesson K. Polymorphisms in the inflammatory genes CIITA, CLEC16A and IFNG influence BMD, bone loss and fracture in elderly women. PLoS One (2012) 7:e47964. doi:10.1371/journal.pone.0047964
Keywords: NLR, inflammation, autoimmunity, IBD, polymorphisms, reproduction, innate immunity, infection
Citation: Zhong Y, Kinio A and Saleh M (2013) Functions of NOD-like receptors in human diseases. Front. Immunol. 4:333. doi: 10.3389/fimmu.2013.00333
Received: 05 September 2013; Paper pending published: 26 September 2013;
Accepted: 02 October 2013; Published online: 16 October 2013.
Edited by:
Thomas A. Kufer, University of Cologne, GermanyReviewed by:
Ashley Mansell, Monash Institute of Medical Research, AustraliaPhilippe Georgel, Strasbourg University, France
Copyright: © 2013 Zhong, Kinio and Saleh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maya Saleh, McGill University, Life Sciences Complex, Bellini Pavilion, Room 364, 3649 Promenade Sir-William Osler, Montreal, QC H3G 0B1, Canada e-mail:bWF5YS5zYWxlaEBtY2dpbGwuY2E=
†Yifei Zhong and Anna Kinio have contributed equally to this work.