 Patrick A. Ott
Patrick A. Ott Nina Bhardwaj
Nina Bhardwaj- 1Dana Farber Cancer Institute, Harvard Medical School, Boston, MA, USA
- 2Tisch Cancer Institute, Mount Sinai School of Medicine, New York, NY, USA
Constitutive upregulation of the MAPK pathway by a BRAFV600 mutation occurs in about half of melanomas. This leads to increased oncogenic properties such as tumor cell invasion, metastatic potential, and resistance to apoptosis. Blockade of the MAPK pathway with highly specific kinase inhibitors induces unprecedented tumor response rates in patients with advanced BRAFV600 mutant melanoma. Immune checkpoint blockade with monoclonal antibodies targeting cytotoxic T-lymphocyte antigen 4 and programed death-1/PD-L1 has also demonstrated striking anti-tumor activity in patients with advanced melanoma. Tumor responses are likely limited by multiple additional layers of immune suppression in the tumor microenvironment. There is emerging preclinical and clinical evidence suggesting that MAPK inhibition has a beneficial effect on the immunosuppressive tumor microenvironment, providing a strong rationale for combined immunotherapy and MAPK pathway inhibition in melanoma. The T cell response has been the main focus in the studies reported to date. Since dendritic cells (DCs) are important in the induction of tumor-specific T cell responses, the impact of MAPK pathway activation in melanoma on DC function is critical for the melanoma directed immune response. BRAFV600E melanoma cells modulate DCs through the MAPK pathway because its blockade in melanoma cells can reverse suppression of DC function. As both MEK/BRAF inhibition and immune checkpoint blockade have recently taken center stage in the treatment of melanoma, a deeper understanding of how MAPK pathway inhibition affects the tumor immune response is needed.
Introduction
Melanoma incidence rates have been increasing for at least 30 years. It is estimated that 76,690 individuals will be diagnosed in 2013 (1). The disease is usually curable when detected in its early stages (thin primary tumor, no lymph node involvement). For patients with unresectable or metastatic melanoma, recently emerged novel systemic treatment modalities such as Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) and Programed Death-1 (PD-1)/PD-L1 blockade as well as BRAF and MEK inhibition have expanded the spectrum of therapeutic options (2–13). The successes with immune checkpoint blocking antibodies in the treatment of patients with metastatic melanoma, with reported response rates of up to 50% are remarkable. Both CTLA-4 and PD-1/PD-L1 blockade can induce long lasting tumor responses in the absence of vaccination, suggesting that endogenous tumor-specific T cells exist in a substantial proportion of patients and that these T cells, once uncoupled from the inhibitory effect mediated by CTLA-4 and/or PD-1/PD-L1 can mediate effective tumor cell lysis (2, 3, 12–15). Multiple other immune suppressive mechanisms are at work in the tumor environment, including additional inhibitory molecules such as Tim-3 (16) and LAG-3 (17), regulatory T cells, myeloid derived suppressor cells (18), and soluble immunosuppressive mediators such as IDO (indoleamine 2,3-dioxygenase), arginase, prostaglandin E2 (PGE2), IL-6, IL-10, VEGF, TGF-β along with other suppressive cytokines and chemokines. Given the multitude of suppressive mechanisms, it is remarkable that a relatively high proportion of patients can achieve objective tumor responses by blockade of a single pathway, such as PD-1/PD-L1 or CTLA-4.
Approximately half of melanomas harbor a somatic point mutation of the BRAF oncogene at codon 600 (V600E and V600K). This mutation results in constitutive activation of the MAPK pathway and increased oncogenic behavior mediated through a variety of mechanisms such as increased apoptosis, invasiveness, and metastatic potential. The MAPK pathway is an important therapeutic target in melanoma: BRAF, MEK, and combined BRAF/MEK inhibition with small molecule kinase inhibitors are successful treatment strategies in patients with BRAF mutant metastatic melanoma (6–10). However, resistance to these treatments develops almost universally, limiting the median duration of treatment responses to 6–9 months. Investigation of resistance mechanisms and potential strategies to overcome resistance is a very active area of research; a number of different mechanisms have been identified, including the reactivation of MAPK signaling by other pathways (19–24).
Given the treatment successes with both kinase inhibition and immune checkpoint blockade in melanoma, there is considerable interest in combinatorial approaches. The promise is to combine the response durability that is characteristic for patients responding to immunotherapy with the high response rate seen with BRAF inhibition. The scientific rationale for such strategies is based on the interplay of the MAPK pathway and the tumor immune response in the microenvironment. Activation of signaling pathways in tumor cells have long been implicated in promoting suppressive immune networks in the tumor environment (25, 26). There is emerging evidence of a link between the MAPK pathway in melanoma and the tumor immune response. Preclinical and clinical observations indicate that inhibition of the MAPK pathway may have a favorable effect on the melanoma-specific immune response on the level of T cells, tumor cells, stromal cells, and dendritic cells (DCs) (Table 1; Figure 1).
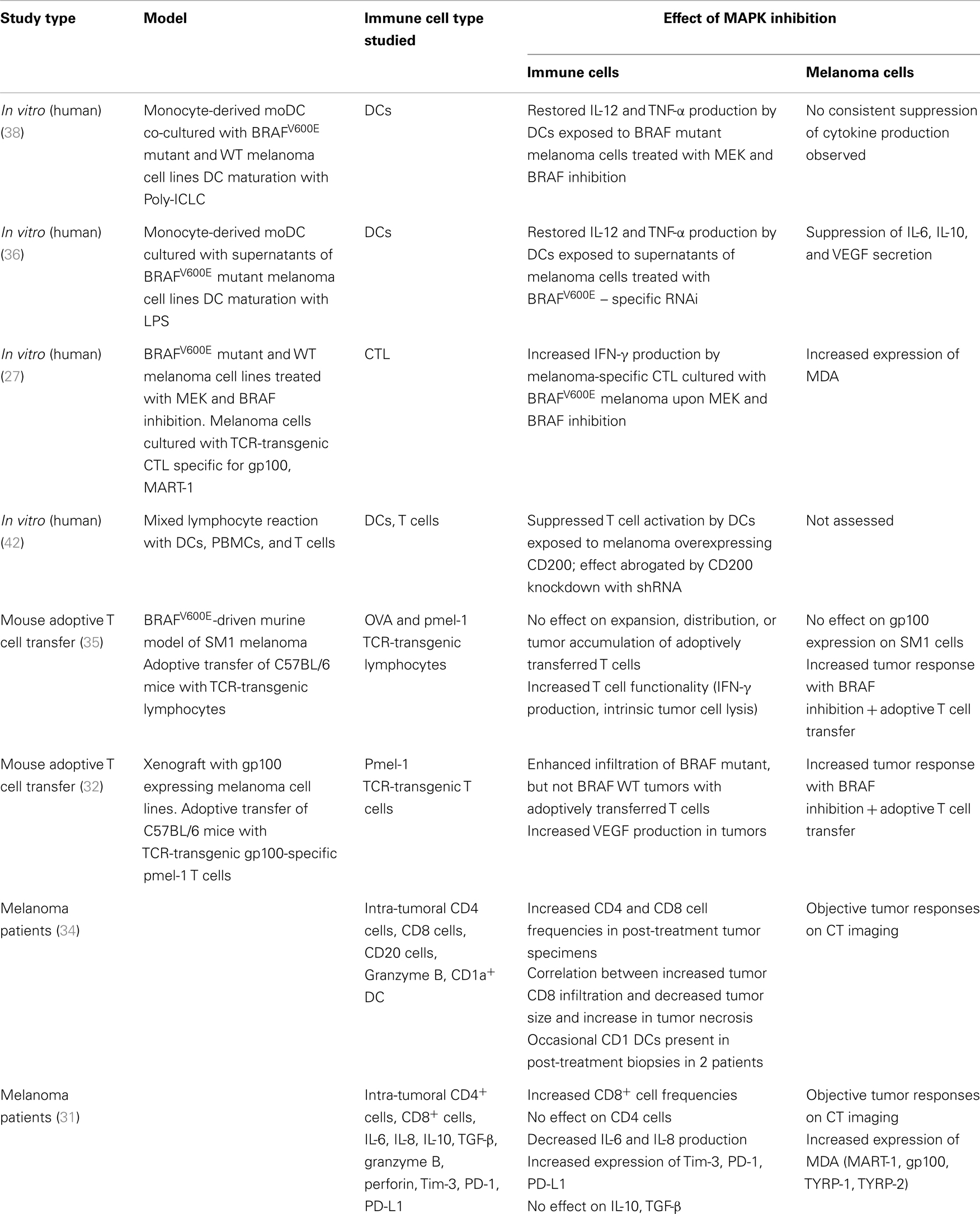
Table 1. Effects of MAPK inhibition on immune function and melanoma.
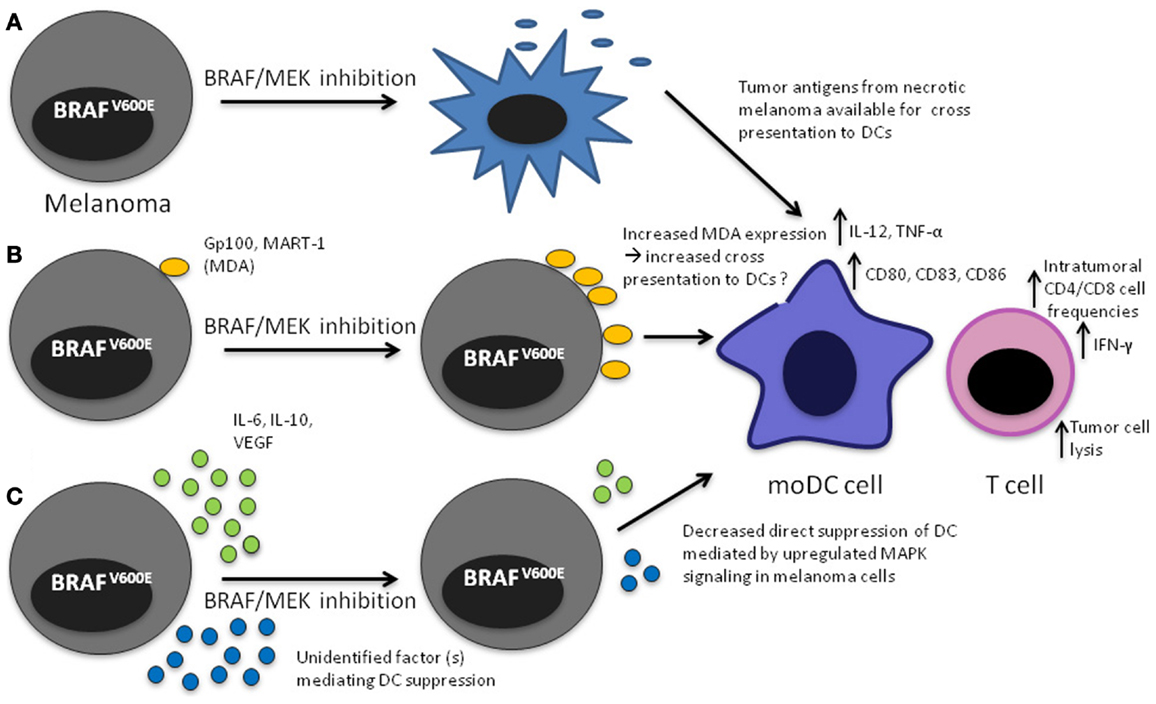
Figure 1. Mechanisms that may lead to increased DC function upon MAPK pathway blockade in the tumor microenvironment. (A) Apoptosis/necrosis of melanoma cells results in release of tumor antigens that will presumably be available to DCs for cross presentation; (B) Increased expression of MDA through direct effect of MAPK pathway inhibition, potentially making them available to DCs for cross presentation, (C) decreased direct inhibition of DCs leading to increased IL-12 and TNF-α production.
BRAF and MEK Inhibition in Melanoma Cell Lines Leads to Upregulation of Tumor Antigens and Increased Recognition by Melanoma-Specific T Cells In vitro
In melanoma cell lines, MEK and BRAF inhibition leads to increased expression of melanoma differentiation antigens (MDAs) such as gp100, MART-1, and tyrosinase on the mRNA and protein levels (27–29). The underlying mechanism of oncogenic BRAF-regulated MDA expression is unclear. It has been suggested that oncogenic BRAF suppresses MDA expression through microphthalmia-associated transcription factor. However additional pathways are likely involved and may account for the heterogeneity of MDA induction observed across different cell lines including mutant and wild-type cell lines (30). Increased expression of gp100 and MART-1 leads to improved antigen recognition by T cells as measured by IFN-γ production (27). Upregulation of gp100 and MART-1 was seen in both BRAF mutant and WT melanoma cell lines. BRAF inhibition did not negatively impact lymphocyte function, whereas MEK inhibition negatively affected T cell proliferative potential, viability, and IFN-γ production. These data were recently confirmed in vivo in patients with metastatic melanoma (31). Increased MART, TYRP-1, TYRP-2, and gp100 expression was found in metastatic melanoma specimens obtained from patients after treatment with BRAF and/or MEK inhibition. Interestingly, melanoma antigen expression in metastatic tumors was decreased at the time of tumor progression in patients treated with a BRAF inhibitor and partially restored upon initiation of dual MEK and BRAF blockade.
Increased Frequency of Tumor Infiltrating Lymphocytes after BRAF Inhibition
In an adoptive T cell transfer (ACT) model, frequencies of gp100 specific luciferase expressing pmel-1 T cells were markedly increased in gp100 expressing melanoma lesions after treatment with vemurafenib (32) and this was associated with improved tumor response compared to either vemurafenib or ACT alone. This observation was specific to BRAF mutant tumors and independent of BRAF inhibition-mediated upregulation of MDA. In this model, the increased intra-tumoral T cell frequencies were attributed to decreased VEGF in the tumor. It was previously shown that VEGF/VEGFR-2 inhibition can upregulate endothelial adhesion molecules in tumor vessels, which can in turn increase the infiltration of leukocytes in tumors (33). Wilmott et al. confirmed the observations of increased intra-tumoral T cell frequencies in melanoma patients who were treated with a BRAF inhibitor (34). Increased frequencies of CD4 and CD8 cells were seen in both intra-tumoral and peritumoral regions of metastatic tumor specimens obtained between 3 and 15 days after treatment initiation. The increase in lymphocyte numbers inversely correlated with tumor size, but not with clinical objective responses. Notably, intra-tumoral lymphocyte frequencies returned to pre-treatment levels at the time of tumor progression. Similar increases in tumor infiltration by CD8 cells (but not CD4 cells) and decrease upon tumor progression in melanoma patients treated with BRAF or dual BRAF/MEK inhibition was reported by Frederick et al. (31).
MAPK Inhibition Affects T Cell Functionality and Secretion of Immunosuppressive Cytokines in the Tumor Microenvironment
In an ACT model using the murine BRAFV600E mutant melanoma SM1 and transgenic T cells recognizing gp100 and ovalbumin (OVA), combined ACT and vemurafenib induced superior anti-SM1 tumor immune responses compared to either of the therapies alone. In this study, no difference in frequencies of adoptively transferred T cells was observed in tumors, lymph nodes, or spleen as assessed ex vivo by flow cytometry and immunofluorescence imaging and in vivo by tracking of the firefly luciferase transgene-labeled T cells using bioluminescence imaging when mice were treated with vemurafenib in addition to ACT. However, adoptively transferred T cells exhibited increased functionality as measured by IFN-γ production and their ability to lyse tumor cells (35) in mice treated with ACT and vemurafenib.
Cross-Talk between the MAPK Pathway in BRAF Mutant Melanoma and DCs
Sumimoto et al. demonstrated that BRAFV600E mutant cell lines can produce immunosuppressive cytokines such as VEGF, IL-6, and IL-10 and that MEK inhibition with U0126 and BRAF inhibition using BRAFV600E specific RNAi suppressed secretion of these cytokines. IL-12 and TNF-α production by DCs exposed to supernatant from the BRAF mutant A375 melanoma cell line prior to maturation by LPS was suppressed (36). This inhibitory effect was mediated by IL-6, IL-10, and VEGF and could be partially reversed by pre-treatment of the melanoma cells with BRAFV600E specific RNAi, indicating that constitutive activation of the MAPK pathway in melanoma cells may lead to compromised DC function and that this immune evasion may be overcome by MAPK inhibition. In a separate study, IL-10 expression in the melanoma line A375 was found to be induced by TGF-β, an effect that was mediated by cross-talk between the Smad, PI3K/AKT, and MAPK pathways (37).
We recently explored a potential link between constitutive MAPK pathway upregulation driven by a BRAFV600 mutation and DC function using a human melanoma-DC co-culture system (38). BRAFV600E mutant and wild-type melanoma cell lines were treated for 24 h with the MEK inhibitor U0126, the BRAF inhibitor vemurafenib, or respective controls (U0124 or DMSO). After removal of supernatant, monocyte-derived immature DC from healthy donors were added, cultured for 24 h and then stimulated with poly-ICLC. Poly-ICLC was chosen as the DC maturation stimulus because it induces the secretion of proinflammatory cytokines in the absence of IL-10 and is a potent TLR3 and MDA5 agonist (39). It has been widely used as a cancer vaccine adjuvant in clinical trials. We found that IL-12 and TNF-α production by DCs was inhibited when DCs were exposed to melanoma cells treated with vehicle control. Notably, the secretion of both cytokines could be partially or completely restored with both MEK and BRAF inhibition in BRAFV600E mutant, but not wild-type cell lines. Furthermore, CD80, CD83, and CD86 expression on DC was decreased upon co-culture with melanoma cells and could be partially restored with BRAF inhibition in BRAFV600E mutant melanoma cell lines. The inhibition of IL-12 and TNF-α secretion by DCs was not cell-contact dependent. In contrast to the study by Sumimoto, a soluble factor responsible for mediating the suppressive effect could not be identified in our investigations. It is possible that continuous local production of small amounts of soluble mediators by melanoma cells in close proximity to DCs accounts for the inhibitory effect observed in the melanoma cell/DC co-culture experiments in our study.
CD200, a type I membrane-associated glycoprotein and member of the immunoglobulin superfamily is highly expressed on melanoma cells and was found to be regulated by ERK activation (40). CD200 mRNA expression levels were found to be positively correlated with tumor progression. Moreover, MEK inhibition with U0126 and knockdown of mutant BRAF resulted in reduced expression of CD200 mRNA in melanoma cell lines. Of note, through interaction with the CD200 receptor, which is expressed on macrophages and DC, CD200 mediates an inhibitory signal (41). In mixed lymphocyte reactions with T cells, DCs, and melanoma cells, T cells produced larger amounts of IL-2 when CD200 in melanoma cells was knocked down with shRNA specifically targeting the CD200 ligand (42). These data suggest a link between MAPK/ERK activation in melanoma and the ability of DCs to activate T cells.
Direct Impact of MAPK Inhibition on DCs
Since there is a strong clinical interest in combined immunotherapy and BRAF/MEK inhibition in melanoma, the direct impact of MAPK pathway inhibition on immune cells is of great interest. BRAF inhibition, even at high concentrations, does not appear to directly compromise T cell function, and there is emerging data showing that low doses of RAF inhibition may even enhance T cell activation (43, 44). Furthermore, frequencies of DCs, monocytes, T cells, B cells, NK cells, and regulatory T cells in peripheral blood from metastatic melanoma patients were not affected by BRAF inhibition (45).
There is some controversy about the direct impact of signaling through the MAPK pathway on DC maturation. In LPS and TNF-α-matured DCs, MEK inhibition leads to upregulation of co-stimulation molecules, increased IL-12 secretion and enhanced ability to activate T cells (46), whereas activation of ERK in DCs leads to immune suppression, mediated by TGF-β and Treg cells (47). Only minimal or no effect of MEK inhibition on DC function was shown in other studies (48–51). Differences in the maturation stimuli may account for some of the inconsistencies observed in these investigations.
In monocyte-derived DC from healthy donors, MEK inhibition lead to reduced IL-12 and TNF-α secretion, whereas BRAF inhibition had no effect on cytokine production over a wide range of doses (38). The expression of CD40, CD80, CD83, and MHCI was also reduced by direct MEK inhibition, whereas it was unaffected by BRAF inhibition. In addition, DC viability was reduced with MEK, but not BRAF inhibition and the ability of DCs to induce T cell proliferation in an MLR was reduced with MEK, but not BRAF inhibition. The impact of MEK inhibitors currently used in the clinic on APC function in vivo remains to be determined.
MAPK Inhibition may Enhance DC Function in the Tumor Microenvironment by Several Mechanisms
Restoration of DC Function Compromised by Melanoma Cells
Our studies and earlier investigations by Sumimoto suggest that suppression of IL-12 and TNF-α production by DCs in the tumor microenvironment of a BRAFV600 mutant melanoma is mediated at least partially by constitutive activation of the MAPK pathway. These data also indicate that BRAF and MEK inhibition, by blocking the MAPK pathway in melanoma cells and thereby restoring IL-12 and TNF-α production in DCs, leads to improved DC function, presumably leading to better activation of melanoma-specific T cells. Notably in our studies there was none or only minimal apoptosis in BRAFV600E mutant and WT melanoma cell lines after 48 h of MEK or BRAF inhibition. This is consistent with prior studies showing an anti-proliferative effect, rather than apoptosis, during the first few days of treatment with these kinase inhibitors (52, 53). It is therefore unlikely that the reversal of compromised DC function mediated by melanoma cells in the in vitro experiments is mediated by melanoma cell death. Similarly, in the experiments by Sumimoto, no significant cell death was observed after treatment of the melanoma line A375 with the MEK inhibitor U0126, indicating that decreased IL-10, IL-6, and VEGF production was a direct effect of MAPK pathway inhibition rather than mere death of the melanoma cells. Taken together, these data suggest that MAPK pathway activation in BRAFV600 mutant melanoma cells has a direct suppressive effect on the capacity of DC to activate T cells.
Increased Cross Presentation of Overexpressed Melanoma Differentiation Antigens by DCs in the Tumor or Lymph Node?
Inhibition of the MAPK pathway in BRAF mutant melanoma leads to increased expression of MDAs (gp100, Mart-1, Tyrp-1, and Tyrp-2), resulting in improved antigen-specific recognition by gp100 and MART-1 specific TCR-transgenic CTL as measured by increased IFN-γ production in vitro (27). In patients with BRAFV600 mutant metastatic melanoma, MART-1 expression was upregulated in metastatic tumors after treatment with BRAF inhibition. Increased infiltration of metastatic tumors with both CD4 and CD8 cells in one study, and of CD8 cells, but not CD4 cells in another study was observed after treatment with BRAF inhibition. A correlation between intra-tumoral infiltration with CD8 cells and tumor necrosis was found in post-treatment biopsies in one study (34). In addition to the direct effect on CTL function shown in vitro, it is possible that MDA overexpression on melanoma cells in the tumor in vivo may lead to increased cross presentation of these antigens to DCs and thus further enhance the tumor-specific T cell response.
Increased Cross Presentation of Tumor Antigens Derived from Apoptotic Tumor Cells after MAPK Inhibition
As outlined above, in short-term (48–72 h) in vitro experiments using BRAF mutant melanoma cell lines, apoptosis or necrosis does not have a significant role in mediating the effects of melanoma MAPK pathway inhibition on DCs. BRAF inhibition does however eventually induce apoptosis and necrosis as evident by the fact that tumors shrink markedly in the majority of patients. Tumor necrosis/apoptosis likely leads to the release of antigens, which may be available for DCs either residing in the tumor or in draining lymph nodes to be taken up, processed, and cross-presented to T cells. Cross presentation may be one of the mechanisms mediating the synergy observed with ACT and BRAF inhibition in melanoma mouse models, although no direct evidence was provided in the reported studies (32, 35).
Conclusion
Constitutive upregulation of the MAPK pathway in BRAFV600 mutant melanoma appears to directly impact DC function as evident by partial restoration of IL-12 and TNF-α secretion upon treatment of melanoma cells with MEK or BRAF inhibition. These effects have so far been shown only in vitro. The beneficial effects of MAPK blockade on the tumor immune microenvironment shown in vivo in mouse models and melanoma patients argue for a broader impact of these treatments on the tumor-specific immune response, including increased T cell frequencies and improved function and changes in cytokine secretion patterns. Several mechanisms that may account for the improved immune response have been described such as increased MDA expression on melanoma cells and decreased intra-tumoral VEGF production, others remain speculative, such as increased cross presentation to DCs resulting from BRAF/MEK inhibition-mediated necrosis/apoptosis of melanoma cells. These observations reinforce the rationale for clinical trials assessing MEK/BRAF inhibition and immunotherapy in combination in patients with melanoma. Further studies are needed to delineate the phenotype and function of DCs in patients treated with BRAF/MEK inhibition. Because of superior efficacy and potentially improved tolerability, combined BRAF-MEK inhibition will likely replace BRAF inhibitor monotherapy. Defining the impact of both BRAF and MEK inhibition on the immune response will therefore be critical.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin (2013) 63(1):11–30. doi: 10.3322/caac.21166
2. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi:10.1056/NEJMoa1302369
3. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (Anti-PD-1) in melanoma. N Engl J Med (2013) 369(2):134–44. doi:10.1056/NEJMoa1305133
4. Flaherty KT, Hodi FS, Fisher DE. From genes to drugs: targeted strategies for melanoma. Nat Rev Cancer (2012) 12(5):349–61. doi:10.1038/nrc3218
5. Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol (2012) 13(8):773–81. doi:10.1016/S1470-2045(12)70270-X
6. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med (2011) 364(26):2507–16. doi:10.1056/NEJMoa1103782
7. Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol (2012) 13(8):782–9. doi:10.1016/S1470-2045(12)70269-3
8. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med (2012) 367(18):1694–703. doi:10.1056/NEJMoa1210093
9. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med (2010) 363(9):809–19. doi:10.1056/NEJMoa1002011
10. Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med (2012) 367(2):107–14. doi:10.1056/NEJMoa1203421
11. Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med (2012) 366(8):707–14. doi:10.1056/NEJMoa1112302
12. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi:10.1056/NEJMoa1200694
13. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
14. Robert C, Thomas L, Bondarenko I, O’Day S, M D JW, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364(26):2517–26. doi:10.1056/NEJMoa1104621
15. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
16. Anderson AC. Tim-3, a negative regulator of anti-tumor immunity. Curr Opin Immunol (2012) 24(2):213–6. doi:10.1016/j.coi.2011.12.005
17. Goldberg MV, Drake CG. LAG-3 in cancer immunotherapy. Curr Top Microbiol Immunol (2011) 344:269–78. doi:10.1007/82_2010_114
18. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi:10.1038/nri2506
19. Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature (2010) 468(7326):968–72. doi:10.1038/nature09627
20. Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell (2010) 18(6):683–95. doi:10.1016/j.ccr.2010.11.023
21. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature (2010) 468(7326):973–7. doi:10.1038/nature09626
22. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature (2012) 487(7408):500–4. doi:10.1038/nature11183
23. Jiang CC, Lai F, Thorne RF, Yang F, Liu H, Hersey P, et al. MEK-independent survival of B-RAFV600E melanoma cells selected for resistance to apoptosis induced by the RAF inhibitor PLX4720. Clin Cancer Res (2011) 17(4):721–30. doi:10.1158/1078-0432.CCR-10-2225
24. Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol (2011) 29(22):3085–96. doi:10.1200/JCO.2010.33.2312
25. Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med (2004) 10(1):48–54. doi:10.1038/nm976
26. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol (2007) 7(1):41–51. doi:10.1038/nri1995
27. Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res (2010) 70(13):5213–9. doi:10.1158/0008-5472.CAN-10-0118
28. Kono M, Dunn IS, Durda PJ, Butera D, Rose LB, Haggerty TJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res (2006) 4(10):779–92. doi:10.1158/1541-7786.MCR-06-0077
29. Sapkota B, Hill CE, Pollack BP. Vemurafenib enhances MHC induction in BRAF homozygous melanoma cells. Oncoimmunology (2013) 2(1):e22890. doi:10.4161/onci.22890
30. Wellbrock C, Rana S, Paterson H, Pickersgill H, Brummelkamp T, Marais R. Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PLoS One (2008) 3(7):e2734. doi:10.1371/journal.pone.0002734
31. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res (2013) 19(5):1225–31. doi:10.1158/1078-0432.CCR-12-1630
32. Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res (2013) 19(2):393–403. doi:10.1158/1078-0432.CCR-12-1626
33. Dirkx AE, oude Egbrink MG, Castermans K, van der Schaft DW, Thijssen VL, Dings RP, et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J (2006) 20(6):621–30. doi:10.1096/fj.05-4493com
34. Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res (2012) 18(5):1386–94. doi:10.1158/1078-0432.CCR-11-2479
35. Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res (2012) 72(16):3928–37. doi:10.1158/0008-5472.CAN-11-2837
36. Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med (2006) 203(7):1651–6. doi:10.1084/jem.20051848
37. Díaz-Valdés N, Basagoiti M, Dotor J, Aranda F, Monreal I, Riezu-Boj JI, et al. Induction of monocyte chemoattractant protein-1 and interleukin-10 by TGFbeta1 in melanoma enhances tumor infiltration and immunosuppression. Cancer Res (2011) 71(3):812–21. doi:10.1158/0008-5472.CAN-10-2698
38. Ott PA, Henry T, Baranda SJ, Frleta D, Manches O, Bogunovic D, et al. Inhibition of both BRAF and MEK in BRAF(V600E) mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol Immunother (2013) 62(4):811–22. doi:10.1007/s00262-012-1389-z
39. Bogunovic D, Manches O, Godefroy E, Yewdall A, Gallois A, Salazar AM, et al. TLR4 engagement during TLR3-induced proinflammatory signaling in dendritic cells promotes IL-10-mediated suppression of antitumor immunity. Cancer Res (2011) 71(16):5467–76. doi:10.1158/0008-5472.CAN-10-3988
40. Shields JM, Thomas NE, Cregger M, Berger AJ, Leslie M, Torrice C, et al. Lack of extracellular signal-regulated kinase mitogen-activated protein kinase signaling shows a new type of melanoma. Cancer Res (2007) 67(4):1502–12. doi:10.1158/0008-5472.CAN-06-3311
41. Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science (2000) 290(5497):1768–71. doi:10.1126/science.290.5497.1768
42. Petermann KB, Rozenberg GI, Zedek D, Groben P, McKinnon K, Buehler C, et al. CD200 is induced by ERK and is a potential therapeutic target in melanoma. J Clin Invest (2007) 117(12):3922–9. doi:10.1172/JCI32163
43. Callahan M. Defining the Effects of MAPK Pathway Inhibition in Human Immune Cells. Perspectives in Melanoma XV. New York, NY: Imedex (2011).
44. Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, et al. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res (2010) 16(24):6040–8. doi:10.1158/1078-0432.CCR-10-1911
45. Hong DS, Vence L, Falchook G, Radvanyi LG, Liu C, Goodman V, et al. Braf(V600) inhibitor Gsk2118436 targeted inhibition of mutant Braf in cancer patients does not impair overall immune competency. Clin Cancer Res (2012) 18(8):2326–35. doi:10.1158/1078-0432.CCR-11-2515
46. Puig-Kröger A, Relloso M, Fernández-Capetillo O, Zubiaga A, Silva A, Bernabéu C, et al. Extracellular signal-regulated protein kinase signaling pathway negatively regulates the phenotypic and functional maturation of monocyte-derived human dendritic cells. Blood (2001) 98(7):2175–82. doi:10.1182/blood.V98.7.2175
47. Escors D, Lopes L, Lin R, Hiscott J, Akira S, Davis RJ, et al. Targeting dendritic cell signaling to regulate the response to immunization. Blood (2008) 111(6):3050–61. doi:10.1182/blood-2007-11-122408
48. Ardeshna KM, Pizzey AR, Devereux S, Khwaja A. The PI3 kinase, p38 SAP kinase, and NF-kappaB signal transduction pathways are involved in the survival and maturation of lipopolysaccharide-stimulated human monocyte-derived dendritic cells. Blood (2000) 96(3):1039–46.
49. Arrighi JF, Rebsamen M, Rousset F, Kindler V, Hauser C. A critical role for p38 mitogen-activated protein kinase in the maturation of human blood-derived dendritic cells induced by lipopolysaccharide, TNF-alpha, and contact sensitizers. J Immunol (2001) 166(6):3837–45.
50. Martínez D, Vermeulen M, von Euw E, Sabatté J, Maggíni J, Ceballos A, et al. Extracellular acidosis triggers the maturation of human dendritic cells and the production of IL-12. J Immunol (2007) 179(3):1950–9.
51. Yu Q, Kovacs C, Yue FY, Ostrowski MA. The role of the p38 mitogen-activated protein kinase, extracellular signal-regulated kinase, and phosphoinositide-3-OH kinase signal transduction pathways in CD40 ligand-induced dendritic cell activation and expansion of virus-specific CD8+ T cell memory responses. J Immunol (2004) 172(10):6047–56.
52. Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res (2010) 70(21):8736–47. doi:10.1158/0008-5472.CAN-10-0902
Keywords: melanoma, dendritic cell, T cell, BRAF, MEK, immunotherapy, kinase inhibitor
Citation: Ott PA and Bhardwaj N (2013) Impact of MAPK pathway activation in BRAFV600 melanoma on T cell and dendritic cell function. Front. Immunol. 4:346. doi: 10.3389/fimmu.2013.00346
Received: 23 September 2013; Paper pending published: 07 October 2013;
Accepted: 08 October 2013; Published online: 28 October 2013.
Edited by:
Timothy Bullock, University of Virginia, USAReviewed by:
Franca Ronchese, Malaghan Institute of Medical Research, New ZealandPedro Berraondo, Centro Para la Investigación Médica Aplicada, Spain
Copyright: © 2013 Ott and Bhardwaj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nina Bhardwaj, Division of Hematology and Oncology, Hess Center for Science and Medicine, Tisch Cancer Institute, Mount Sinai School of Medicine, 1470 Madison Avenue Room 116, New York, NY 10029, USA e-mail:bmluYS5iaGFyZHdhakBtc3NtLmVkdQ==