 Alessandra De Riva
Alessandra De Riva Robert Busch
Robert Busch- Department of Medicine, University of Cambridge, Cambridge, UK
Major histocompatibility complex class II (MHCII) proteins are loaded with endosomal peptides and reside at the surface of antigen-presenting cells (APCs) for a time before being degraded. In vitro, MHCII protein levels and turnover are affected by peptide loading and by rates of ubiquitin-dependent internalization from the cell surface, which is in turn affected by APC type and activation state. Prior work suggested that fast turnover of disease-associated MHCII alleles may contribute to autoimmunity. We recently developed novel stable isotope tracer techniques to test this hypothesis in vivo. In non-obese diabetic (NOD) mice, a model of type 1 diabetes (T1D), MHCII turnover was affected by APC type, but unaffected by disease-associated structural polymorphism. Differences in MHCII turnover were observed between NOD colonies with high and low T1D incidence, but fast turnover was dispensable for autoimmunity. Moreover, NOD mice with gene knockouts of peptide loading cofactors do not develop T1D. Thus, fast turnover does not appear pathogenic, and conventional antigen presentation is critical for autoimmunity in NOD mice. However, shared environmental factors may underpin colony differences in MHCII protein turnover, immune regulation, and pathogenesis.
Introduction
Major histocompatibility complex class II (MHCII) molecules present peptides at cell surfaces for recognition by CD4+ T lymphocytes. They shape a functional, self-tolerant T-cell repertoire via positive and negative thymocyte selection and support peripheral CD4+ T-cell homeostasis (1). Presentation of pathogen-derived peptides activates specific CD4 T cells, initiating adaptive immunity (2). Extensive polymorphism in the peptide-binding groove, shaping the presented peptide repertoire, is a key characteristic of MHCII proteins (3). These polymorphisms also control susceptibility to autoimmune diseases.
The regulation of MHCII protein expression is critical to these functions. Here, we discuss MHCII protein turnover, a critical but poorly understood determinant of steady-state expression. We describe recent methodological advances in measuring MHCII protein turnover in vivo, which enabled us to examine the postulated role of MHCII protein turnover in autoimmune pathogenesis in a mouse model.
Functional Significance of MHCII Protein Turnover
Major histocompatibility complex class II proteins are expressed constitutively by antigen-presenting cells [APCs: B cells, dendritic cells (DCs), macrophages, thymic epithelial cells] and inducibly by other cell types. MHCII gene transcription is controlled by the master regulator, class II transactivator (CIITA) (4). Post-translational regulation fine-tunes MHCII expression levels and determines antigen persistence. For example, in DCs, CIITA, and MHCII gene transcription are shut down following activation by bacterial lipopolysaccharide, yet MHCII surface levels rise, because MHCII protein degradation is also shut down (5). As a result, long-lived MHCII/peptide complexes are able to survive during DC migration to lymph nodes (6), where they provide persistent stimuli for CD4+ T-cell priming (7). In other APC types, cytokine regulation of MHCII turnover also affects expression (8, 9), and stimuli can increase turnover, rather than shutting it down.
Determinants of MHCII Protein Turnover
Major histocompatibility complex class II degradation in non-activated DCs begins with internalization from the cell surface, which is triggered by ubiquitination of the cytoplasmic tail of the MHCII β chain (10) by the ubiquitin E3 ligase, membrane-associated RING-CH 1 (MARCH-1) (11, 12). The α chain is also ubiquitinated to a small extent (13). When activated by proinflammatory stimuli, conventional DCs downregulate MARCH-1, terminating MHCII protein turnover; in contrast, activated plasmacytoid DCs maintain this process (14). MARCH-1 also mediates cytokine regulation of MHCII turnover in monocytes and B cells (8, 9, 15). Other MARCH family members may be involved, as well (16).
The internalized MHCII molecules are targeted for lysosomal degradation by unknown proteases. Studies using inhibitors suggest a role for cysteine proteases, although the effects are small (17) and MHCII molecules resist proteolysis by many cysteine cathepsins in vitro (18). The serine protease, Cathepsin G, cleaves MHCII molecules at a specific membrane-proximal site in vitro, but this does not detectably contribute to MHCII degradation in APCs (18).
Major histocompatibility complex class II protein degradation is also influenced by the preceding assembly and maturation steps, which are reviewed elsewhere in this series (Figure 1) (19). Briefly, αβ heterodimers assemble with invariant chain (Ii) in the endoplasmic reticulum (ER) and travel to endosomes. There, Ii is cleaved, leaving class II-associated Ii peptides (CLIP) in the peptide-binding groove, which must be released to enable loading with endosomal peptides. A co-factor, DM (HLA-DM in humans, H2-DM/H2-M in mice), releases CLIP, “edits” peptides to select stable binders, and stabilizes empty MHCII molecules (“chaperoning”). Finally, MHCII/peptide complexes travel from endosomes to the plasma membrane. Defects in these maturation steps accelerate MHCII protein degradation, sometimes by use of alternative degradation pathways. In Ii-deficient cells, a significant proportion of MHCII molecules are retained in the ER and degraded (20), presumably by the proteasome following retrotranslocation to the cytosol. In DM-deficient cells, the turnover of some MHCII alleles in post-Golgi compartments is accelerated, likely because MHCII acquire loosely bound peptides and are not chaperoned following spontaneous CLIP release (21). Moreover, peptide occupancy appears to protect MHCII molecules from degradation even in normal APCs: exposure of splenocytes to high doses of exogenous antigen protects MHCII molecules from degradation, increases their resistance to disruptive detergents (SDS), and raises their surface levels (22).
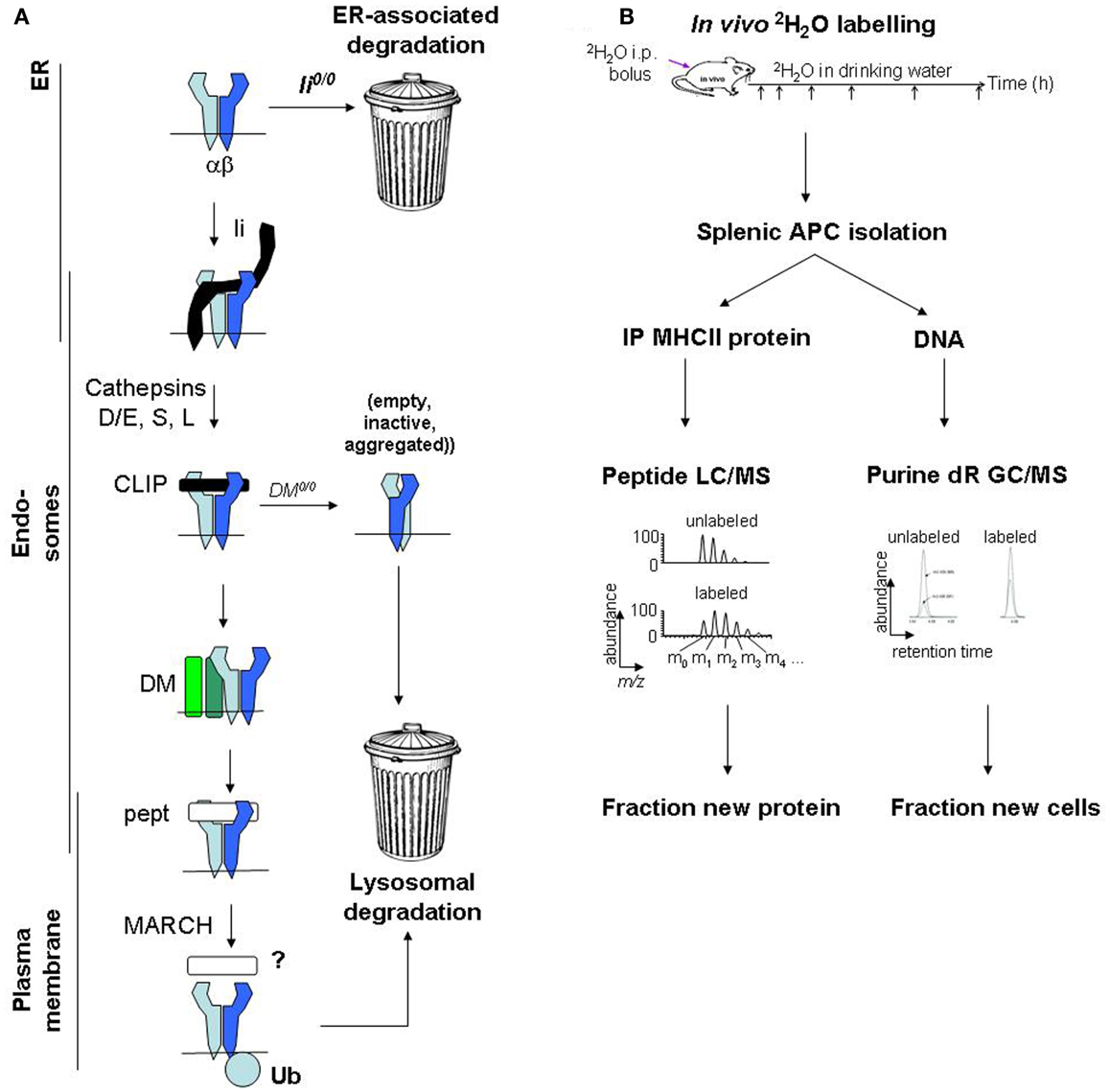
Figure 1. Major histocompatibility complex class II protein turnover: molecular regulation and mass spectrometric measurement. (A) Regulation of MHCII protein turnover in APCs. (B) Work flow for SINEW measurement of MHCII protein turnover in mice.
Many details of these mechanisms remain ill-defined. Moreover, while polymorphisms may affect MHCII fate in DM-deficient cells, via their impact on CLIP affinity, it is unclear to what extent MHCII allelic variants may differ in their turnover rates in normal APCs.
Could Accelerated MHCII Protein Turnover Promote Autoimmunity?
A role for MHCII protein turnover in autoimmunity was first proposed in non-obese diabetic (NOD) mice, which develop autoimmune beta-islet cell destruction leading to type 1 diabetes (T1D) under complex genetic, environmental, and developmental control. The sole expressed MHCII gene product, H2-Ag7, is critical for T1D development [recently reviewed in detail by Busch et al. (23)]. Ag7 has a unique β chain, differing by 17 amino acids from Aβd; its α chain is identical to Aαd. Ag7 shares structural features with HLA-DQ alleles associated with T1D in humans, including a key non-Asp57β polymorphism, affecting the P9 specificity pocket.
One reason to suspect that Ag7 polymorphisms confer a broader structural deficit is that Ag7-restricted autoreactivity in NOD mice is not islet-specific. Crossing NOD mice with KRN T-cell receptor transgenic mice serendipitously creates reactivity to a ubiquitously expressed, Ag7-presented self peptide, triggering autoimmune arthritis (24). Moreover, NOD mice exhibit spontaneous autoreactivity (25), and immunization with foreign antigens elicits Ag7-restricted bystander responses to self peptides (26). Numerous unusual biochemical features of Ag7 might contribute to the tolerance deficits (Figure 2A):
• Numerous peptides bind weakly to, or dissociate rapidly from, Ag7 (27–30). Peptide binding to Ag7 may be unusually promiscuous (31).
• Ag7 has low CLIP affinity at endosomal pH (32). Paradoxically, CLIP levels on NOD splenocytes are high (33), suggesting inefficient DM editing in vivo, which could diminish the levels or stability of Ag7 complexes with self peptides and accelerate turnover (21, 34). The reason is unclear, as Ag7 seems to interact productively with DM in transfectants (35). Unstable peptide binding may be determined extrinsically, rather than by Ag7 structural polymorphism.
• One study has shown unusually fast turnover of Ag7 in NOD splenocytes in vitro (27).
• Ag7 heterodimers dissociate at room temperature in the presence of SDS detergent (27), more so than other H-2A alleles. This feature is shared by human DQ T1D risk alleles (36). Although its significance is unclear, SDS instability could correlate with some of the other biochemical features described above.
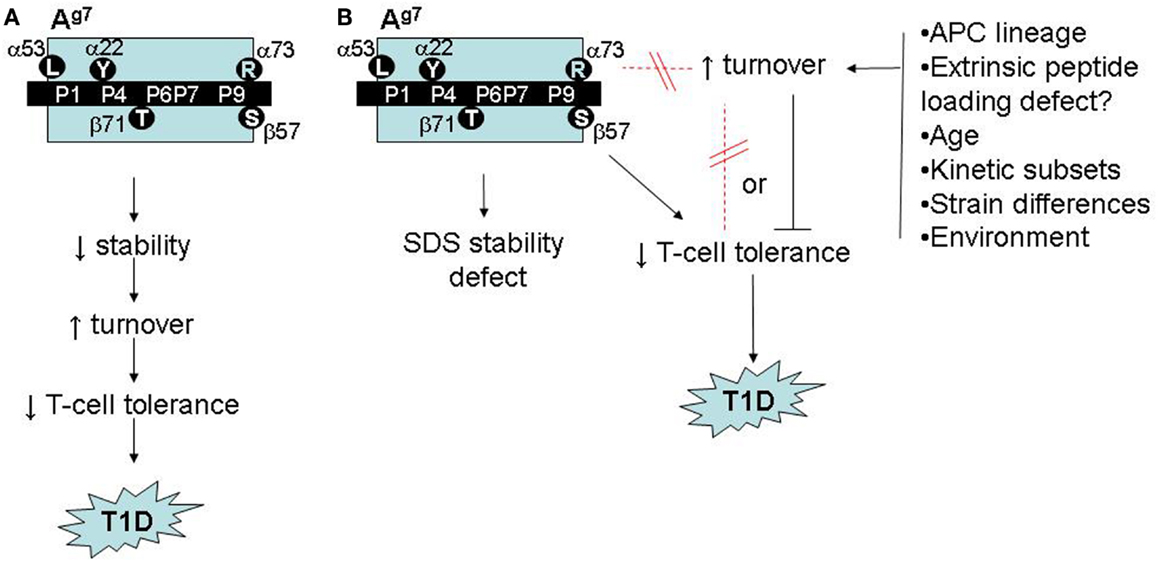
Figure 2. Possible roles of accelerated Ag7 protein turnover in autoimmune pathogenesis in NOD mice. (A) Model adapted from (26) and (27), linking Ag7 polymorphism to intrinsic stability defects, accelerated turnover, impaired T-cell tolerance, and increased risk of autoimmunity. (B) Revised model, based on (53), showing accelerated Ag7 turnover in vivo to be regulated, independently of structural polymorphisms and SDS instability, by unknown environmental factors, with either a neutral or inhibitory role in autoimmune pathogenesis.
Other studies, however, questioned the relevance of these findings to pathogenesis. The SDS instability of Ag7 may represent the lower range of normal variation (37). Not all studies found defects in peptide binding selectivity or life span of Ag7 (37, 38). Studies of soluble recombinant Ag7 provided no evidence for stability defects (39, 40). Lastly, the biochemistry of Ag7 differs from that of the homologous human HLA-DQ T1D risk alleles, DQ2 and DQ8, in key details (e.g., the mechanism of CLIP retention (41), so these details may not be critical for pathogenesis.
Peptide-specific mechanisms for Ag7-restricted autoreactivity have also been proposed. Ag7 selects peptides similar to those binding human DQ T1D risk alleles (38). Neo-self determinants may be generated by post-translational modification of peptides by transglutaminases, resulting in increased binding to Ag7 (42), analogous to mechanisms explaining DQ associations with celiac disease in humans (23). Both peptide-dependent and -independent mechanisms could contribute to Ag7 associations with autoimmunity.
In vivo Measurements of Protein Turnover: A Methodological Excursion
Given these uncertainties, we re-examined whether Ag7 molecules exhibit unusually fast turnover in NOD mice, and what this meant for autoimmunity. We also quantified the baseline turnover of MHCII molecules in APCs of normal mice for the first time. This required the development of novel methods for measuring MHCII protein turnover in vivo.
Protein turnover is measured using tracers, which are biosynthetically incorporated into newly synthesized proteins and persist until the protein is degraded. In cell culture, newly synthesized proteins may be tagged with radiolabeled amino acids (e.g., [35S]-Met/Cys); during a subsequent chase, the fate of the labeled proteins is tracked by autoradiography (43). This approach is powerful but difficult to use in vivo, because substantial amounts of radioactivity would be required, even in small-animal models.
Stable isotope tracers, lacking radiation hazards, are easier to use in vivo. “SILAC” (stable isotope labeling of amino acids in cell culture), used for kinetics rather than quantitation, is one variant of this approach (44). Arg and Lys in culture media are replaced with all-[13C]-Arg/Lys, and their incorporation into newly synthesized proteins is quantified by mass spectrometric analysis of tryptic digests. The relative abundances of unlabeled and labeled peptides define the proportion of molecules that were synthesized during the labeling interval and survived until analysis (“fractional protein synthesis”). The approach has been adapted for in vivo use (45). However, label entry into amino acyl-tRNAs used for protein synthesis is not fully understood; pools of unlabeled amino acids may persist in animals even after long-term labeling.
An alternative strategy uses deuterated water (2H2O) as a biosynthetic label. 2H2O labeling is simple and inexpensive to perform in cell culture and in vivo (46). At levels used for metabolic labeling (1–5% in body water), 2H2O does not interfere with normal physiology. It rapidly equilibrates throughout the body following administration. Protons from water are used primarily in the (comparatively rapid) synthesis of non-essential amino acids, such as Ala, by cells (47). For proteins with turnover rates of a few hours or longer, protein synthesis is the rate-limiting step for 2H label incorporation from 2H2O (48, 49). This approach was named “SINEW” (stable isotope labeling of non-essential amino acids with heavy water). Like SILAC, SINEW tracks the proportions of unlabeled to labeled tryptic peptides by mass spectrometry. Although these species are less well resolved in SINEW experiments, precise measurements of fractional protein synthesis rates are possible (50–52).
New protein is synthesized not only to replace protein lost to turnover, but also to maintain protein levels as cells divide. For cell-associated proteins with slow turnover rates, the proliferative contribution can be considerable. During SINEW experiments, this contribution can be quantified accurately by measuring 2H incorporation from 2H2O into newly synthesized DNA (46). SINEW was therefore used to explore MHCII protein turnover (53).
Extrinsic Regulation of MHCII Protein Turnover In vivo
To quantify Ag7 protein turnover in vivo (53), we labeled young female prediabetic NOD mice with 2H2O; BALB/c mice (Ad, Ed) were used as controls. MHCII molecules were immunoprecipitated from splenic B cells and DCs (which together harbor most splenic MHCII molecules), resolved on SDS gels, digested with trypsin, and analyzed by mass spectrometry. MHCII-derived peptides were identified, and changes in their mass distributions from 2H2O labeling were used to quantify fractional protein synthesis (Figure 1B). MHCII protein synthesis greatly exceeded the rate required to support the turnover of the APCs themselves (quantified by analysis of 2H incorporation into DNA). Thus, fractional protein synthesis reflected MHCII protein turnover rates.
The turnover rates of MHCII proteins in vivo proved to be markedly affected by the cellular microenvironment: turnover half-lives (t1/2) were ≈10–12 h in B cells but twice as fast (t1/2 ≈5–6 h) in DCs (53). Thus, turnover rates measured in unseparated splenocytes (27) might be somewhat confounded by differences in APC composition, because NOD splenocytes contained a greater proportion of DCs, relative to B cells, than BALB/c splenocytes. Other APC types or locations of potential relevance to pathogenesis were not analyzed here.
Given that inflammatory stimuli influence MHCII protein turnover rates (5), it should be noted that these experiments were performed in specific pathogen-free mice, which exhibited no detectable activation of splenic APCs; conceivably, altered MHCII protein turnover in a few activated APCs might be relevant to pathogenesis. The difference in MHCII half-lives between APC types may reflect the greater extent of ubiquitination and internalization of MHCII molecules in DCs (54).
We also examined Ag7 turnover in transfected M12 B lymphoma cells (50, 53). These cells doubled approximately every 27 h and produced new Ag7 molecules at the same rate. Thus, Ag7 protein synthesis was attributable to cell growth, with only a small contribution from turnover. The implied long Ag7 half-life (>2 days) argues against an intrinsic stability deficit of this allele and confirms a substantial impact of the cellular microenvironment on MHCII turnover rates. These findings also indicated that physiological turnover rates are not necessarily recapitulated in model cell lines.
Within each cell type, MHCII alleles apparently differed in their dynamics (53): the half-life of Ag7 molecules in NOD B cells was significantly shorter than that of Ad in BALB/c B cells, albeit by only a small amount (13%). However, kinetic modeling indicated that that Ag7 turnover was heterogeneous: its 2H2O labeling curve was significantly better explained by a mixture of long- and short-lived molecules than by a uniform half-life.
The presence in NOD (but not BALB/c) B cells of some H2-A molecules with faster turnover could reflect strain differences in the cellular microenvironment. To address this, we bred (NOD × BALB/c) F1 mice, expressing both H2-A molecules in the same cells. In F1 APCs, the turnover of Ad and Ag7 was indistinguishable (53), so the cellular microenvironment is decisive. The fast-turnover component may be attributable to inefficient peptide loading in NOD splenocytes, although this is not the only possible explanation.
Accelerated Ag7 Turnover is Dispensable for Autoimmunity
Several observations suggested that fast-turnover Ag7 molecules do not correlate closely with T1D development (53). Ag7 turnover is indistinguishable between B cells from male and female NOD mice, yet T1D incidence is lower in males. Moreover, the fast-turnover subpopulation detectable in young female NOD mice becomes undetectable at 12 weeks of age, prior to the onset of T1D.
More compellingly, we initially observed the fast-turnover Ag7 molecules in one colony of NOD mice, which exhibited a relatively low incidence of T1D (22% diabetic females at 30 weeks of age). When we repeated the SINEW experiments in a second NOD colony, which develops a much higher T1D incidence (>80% at 30 weeks in females), its Ag7 molecules showed no accelerated turnover (53). Thus, fast Ag7 turnover appears dispensable for T1D development (Figure 2B).
This conclusion might seem disappointing, yet our findings may harbor a critical clue. Worldwide, NOD colonies differ widely in their disease incidence as a result of unknown environmental variables (55); our two colonies represent extremes on this spectrum. The environmental regulator(s) in our colony remain unknown but appear to be novel. Our results suggest, however, that key environmental influences may be sensed by B cells in secondary lymphoid organs, triggering changes in the dynamics of antigen presentation (Figure 2B). Dissection of these mechanisms may yield novel insights into mechanisms of gut microbe/immune interactions in autoimmunity.
Normal MHCII Peptide Loading Required for Autoimmunity in NOD Mice
Intact MHCII peptide loading pathways are critical to T1D pathogenesis in NOD mice. Invariant (Ii) chain-deficient NOD mice express reduced levels of Ag7 and are protected from T1D (56). Their effector CD4+ T cells have islet autoreactivity similar to Ii-sufficient NOD mice, but the frequency of regulatory T cells (Tregs) is increased (57). Similarly, DM-deficient NOD mice show reduced Ag7 surface expression and are protected from T1D development (58). These mice also have increased Treg frequencies but, unlike Ii-null NOD mice, do not develop autoreactive effector T cells, suggesting that presentation of islet autoantigens requires peptide editing. A more subtle defect was engineered into DCs of NOD mice by transgenic expression of HLA-DO, an inhibitor of DM, under control of the CD11c promoter (59). These mice also failed to develop T1D, indicating an important role of normal MHCII peptide editing in DCs during T1D pathogenesis. Because defects in MHCII peptide loading pathways accelerate Ag7 protein turnover (21, 34), the findings are consistent with our observations that fast-turnover Ag7 molecules are dispensable for autoimmunity (53). Indeed, the increased Treg frequencies caused by peptide loading defects raise the possibility that short-lived Ag7 might be protective.
Conclusion
Turnover rates of MHCII molecules in vivo depend strongly on APC type and are affected by environmental factors, but not substantially by the MHCII structural polymorphisms we have examined. Whether MHC protein turnover is generally insensitive to structural polymorphism in normal APCs is an interesting question, given the allelic differences previously observed in the presence of peptide loading defects. Work is ongoing to extend these observations, explore their functional and evolutionary significance, and examine the underlying molecular mechanisms. Accelerated Ag7 turnover in NOD spleens affects only a subset of Ag7 molecules and does not represent an intrinsic stability defect associated with this risk allele. Moreover, this phenotype is dispensable for autoimmune pathogenesis, and might even counteract autoimmunity (though the latter possibility will be difficult to prove). Surprisingly, colony differences in Ag7 turnover reveal novel potential connections between environmental regulators, B cells, and autoimmune pathogenesis, which are now being explored. Lastly, SINEW, used here to quantify MHCII protein turnover, has important advantages in quantifying the turnover of cell-associated proteins. SINEW shows promise for probing genotype/phenotype associations and normal and pathological immune responses.
Conflict of Interest Statement
Robert Busch holds stock in KineMed Inc., a biopharmaceutical company with related commercial interests and intellectual property, and is a former employee and consultant for this company. The other co-author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Robert Busch holds a Research Progression Award from Arthritis Research UK (Ref.: 20648). The work discussed herein was supported primarily by a Senior Research Fellowship (Ref.: 18543, to Robert Busch), and further by grants from Diabetes UK, Cambridge Arthritis Research Endeavor, and the Cambridge Biomedical Research Centre of the National Institute of Health Research. We thank Prof. Elizabeth D. Mellins, Dr. Timo Burster, Prof. Anne Cooke, and Dr. Mike Deery for collaborations.
References
2. MacLennan IC, Gulbranson-Judge A, Toellner KM, Casamayor-Palleja M, Chan E, Sze DM, et al. The changing preference of T and B cells for partners as T-dependent antibody responses develop. Immunol Rev (1997) 156:53–66. doi: 10.1111/j.1600-065X.1997.tb00958.x
3. Rammensee HG, Friede T, Stevanovic S. MHC ligands and peptide motifs: first listing. Immunogenetics (1995) 41(4):178–228. doi:10.1007/BF00172063
4. Krawczyk M, Reith W. Regulation of MHC class II expression, a unique regulatory system identified by the study of a primary immunodeficiency disease. Tissue Antigens (2006) 67(3):183–97. doi:10.1111/j.1399-0039.2006.00557.x
5. Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature (1997) 388(6644):782–7. doi:10.1038/42030
6. Martin-Fontecha A, Lanzavecchia A, Sallusto F. Dendritic cell migration to peripheral lymph nodes. Handb Exp Pharmacol (2009) 188:31–49. doi:10.1007/978-3-540-71029-5_2
7. Obst R, van Santen HM, Melamed R, Kamphorst AO, Benoist C, Mathis D. Sustained antigen presentation can promote an immunogenic T cell response, like dendritic cell activation. Proc Natl Acad Sci U S A (2007) 104(39):15460–5. doi:10.1073/pnas.0707331104
8. Galbas T, Steimle V, Lapointe R, Ishido S, Thibodeau J. MARCH1 down-regulation in IL-10-activated B cells increases MHC class II expression. Cytokine (2012) 59(1):27–30. doi:10.1016/j.cyto.2012.03.015
9. Thibodeau J, Bourgeois-Daigneault MC, Huppe G, Tremblay J, Aumont A, Houde M, et al. Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur J Immunol (2008) 38(5):1225–30. doi:10.1002/eji.200737902
10. Shin JS, Ebersold M, Pypaert M, Delamarre L, Hartley A, Mellman I. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature (2006) 444(7115):115–8. doi:10.1038/nature05261
11. Walseng E, Furuta K, Bosch B, Weih KA, Matsuki Y, Bakke O, et al. Ubiquitination regulates MHC class II-peptide complex retention and degradation in dendritic cells. Proc Natl Acad Sci U S A (2010) 107(47):20465–70. doi:10.1073/pnas.1010990107
12. De Gassart A, Camosseto V, Thibodeau J, Ceppi M, Catalan N, Pierre P, et al. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc Natl Acad Sci U S A (2008) 105(9):3491–6. doi:10.1073/pnas.07088741050708874105
13. Lapaque N, Jahnke M, Trowsdale J, Kelly AP. The HLA-DRα chain is modified by polyubiquitination. J Biol Chem (2009) 284(11):7007–16. doi:10.1074/jbc.M805736200
14. Young LJ, Wilson NS, Schnorrer P, Proietto A, ten Broeke T, Matsuki Y, et al. Differential MHC class II synthesis and ubiquitination confers distinct antigen-presenting properties on conventional and plasmacytoid dendritic cells. Nat Immunol (2008) 9(11):1244–52. doi:10.1038/ni.1665
15. Matsuki Y, Ohmura-Hoshino M, Goto E, Aoki M, Mito-Yoshida M, Uematsu M, et al. Novel regulation of MHC class II function in B cells. EMBO J (2007) 26(3):846–54. doi:10.1038/sj.emboj.7601556
16. Jahnke M, Trowsdale J, Kelly AP. Structural requirements for recognition of major histocompatibility complex class II by membrane-associated RING-CH (MARCH) protein E3 ligases. J Biol Chem (2012) 287(34):28779–89. doi:10.1074/jbc.M112.381541
17. Herrmann TL, Agrawal RS, Connolly SF, McCaffrey RL, Schlomann J, Kusner DJ. MHC Class II levels and intracellular localization in human dendritic cells are regulated by calmodulin kinase II. J Leukoc Biol (2007) 82(3):686–99. doi:10.1189/jlb.0107045
18. Burster T, Macmillan H, Hou T, Schilling J, Truong P, Boehm BO, et al. Masking of a cathepsin G cleavage site in vivo contributes to the proteolytic resistance of major histocompatibility complex class II molecules. Immunology (2010) 130(3):436–46. doi:10.1111/j.1365-2567.2010.03247.x
19. Busch R, Rinderknecht CH, Roh S, Lee AW, Harding JJ, Burster T, et al. Achieving stability through editing and chaperoning: regulation of MHC class II peptide binding and expression. Immunol Rev (2005) 207:242–60. doi:10.1111/j.0105-2896.2005.00306.x
20. Elliott EA, Drake JR, Amigorena S, Elsemore J, Webster P, Mellman I, et al. The invariant chain is required for intracellular transport and function of major histocompatibility complex class II molecules. J Exp Med (1994) 179(2):681–94. doi:10.1084/jem.179.2.681
21. Rinderknecht CH, Roh S, Pashine A, Belmares MP, Patil NS, Lu N, et al. DM influences the abundance of major histocompatibility complex class II alleles with low affinity for class II-associated invariant chain peptides via multiple mechanisms. Immunology (2010) 131(1):18–32. doi:10.1111/j.1365-2567.2010.03282.x
22. Germain RN, Rinker AG Jr. Peptide binding inhibits protein aggregation of invariant-chain free class II dimers and promotes surface expression of occupied molecules. Nature (1993) 363(6431):725–8. doi:10.1038/363725a0
23. Busch R, De Riva A, Hadjinicolaou AV, Jiang W, Hou T, Mellins ED. On the perils of poor editing: regulation of peptide loading by HLA-DQ and H2-A molecules associated with celiac disease and type 1 diabetes. Expert Rev Mol Med (2012) 14:e15. doi:10.1017/erm.2012.9S1462399412000099
24. Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell (1996) 87(5):811–22. doi:10.1016/S0092-8674(00)81989-3
25. Kanagawa O, Martin SM, Vaupel BA, Carrasco-Marin E, Unanue ER. Autoreactivity of T cells from nonobese diabetic mice: an I-Ag7-dependent reaction. Proc Natl Acad Sci U S A (1998) 95(4):1721–4. doi:10.1073/pnas.95.4.1721
26. Ridgway WM, Ito H, Fasso M, Yu C, Fathman CG. Analysis of the role of variation of major histocompatibility complex class II expression on nonobese diabetic (NOD) peripheral T cell response. J Exp Med (1998) 188(12):2267–75. doi:10.1084/jem.188.12.2267
27. Carrasco-Marin E, Shimizu J, Kanagawa O, Unanue ER. The class II MHC I-Ag7 molecules from non-obese diabetic mice are poor peptide binders. J Immunol (1996) 156(2):450–8.
28. Levisetti MG, Lewis DM, Suri A, Unanue ER. Weak proinsulin peptide-major histocompatibility complexes are targeted in autoimmune diabetes in mice. Diabetes (2008) 57(7):1852–60. doi:10.2337/db08-0068db08-0068
29. Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci U S A (2010) 107(24):10978–83. doi:10.1073/pnas.10065451071006545107
30. Munz C, Hofmann M, Yoshida K, Moustakas AK, Kikutani H, Stevanovic S, et al. Peptide analysis, stability studies, and structural modeling explain contradictory peptide motifs and unique properties of the NOD mouse MHC class II molecule H2-Ag7. Eur J Immunol (2002) 32(8):2105–16. doi:10.1002/1521-4141(200208)32:8<2105::AID-IMMU2105>3.0.CO;2-Q
31. Stratmann T, Apostolopoulos V, Mallet-Designe V, Corper AL, Scott CA, Wilson IA, et al. The I-Ag7 MHC class II molecule linked to murine diabetes is a promiscuous peptide binder. J Immunol (2000) 165(6):3214–25.
32. Hausmann DH, Yu B, Hausmann S, Wucherpfennig KW. pH-Dependent peptide binding properties of the type I diabetes-associated I-Ag7 molecule: rapid release of CLIP at an endosomal pH. J Exp Med (1999) 189(11):1723–34. doi:10.1084/jem.189.11.1723
33. Bhatnagar A, Milburn PJ, Lobigs M, Blanden RV, Gautam AM. Nonobese diabetic mice display elevated levels of class II-associated invariant chain peptide associated with I-Ag7 on the cell surface. J Immunol (2001) 166(7): 4490–7.
34. Rinderknecht CH, Lu N, Crespo O, Truong P, Hou T, Wang N, et al. I-Ag7 is subject to post-translational chaperoning by CLIP. Int Immunol (2010) 22(8):705–16. doi:10.1093/intimm/dxq056
35. Peterson M, Sant AJ. The inability of the nonobese diabetic class II molecule to form stable peptide complexes does not reflect a failure to interact productively with DM. J Immunol (1998) 161(6):2961–7.
36. Ettinger RA, Liu AW, Nepom GT, Kwok WW. Exceptional stability of the HLA-DQA1*0102/DQB1*0602 alpha beta protein dimer, the class II MHC molecule associated with protection from insulin-dependent diabetes mellitus. J Immunol (1998) 161(11):6439–45.
37. Reizis B, Eisenstein M, Bockova J, Konen-Waisman S, Mor F, Elias D, et al. Molecular characterization of the diabetes-associated mouse MHC class II protein, I-Ag7. Int Immunol (1997) 9(1):43–51. doi:10.1093/intimm/9.1.43
38. Suri A, Walters JJ, Gross ML, Unanue ER. Natural peptides selected by diabetogenic DQ8 and murine I-Ag7 molecules show common sequence specificity. J Clin Invest (2005) 115(8):2268–76. doi:10.1172/JCI25350
39. Corper AL, Stratmann T, Apostolopoulos V, Scott CA, Garcia KC, Kang AS, et al. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science (2000) 288(5465):505–11. doi:10.1126/science.288.5465.505
40. Latek RR, Suri A, Petzold SJ, Nelson CA, Kanagawa O, Unanue ER, et al. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity (2000) 12(6):699–710. doi:10.1016/S1074-7613(00)80220-4
41. Hou T, Macmillan H, Chen Z, Keech CL, Jin X, Sidney J, et al. An insertion mutant in DQA1∗0501 restores susceptibility to HLA-DM: implications for disease associations. J Immunol (2011) 187(5):2442–52. doi:10.4049/jimmunol.1100255
42. Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K. Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes (2012) 61(12):3239–46. doi:10.2337/db12-0112
43. Hou T, Rinderknecht CH, Hadjinicolaou AV, Busch R, Mellins E. Pulse-chase analysis for studies of MHC class II biosynthesis, maturation, and peptide loading. Methods Mol Biol (2012) 960:411–32. doi:10.1007/978-1-62703-218-6_31
44. Mann M. Functional and quantitative proteomics using SILAC. Nat Rev (2006) 7(12):952–8. doi:10.1038/nrm2067
45. Kruger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, et al. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell (2008) 134(2):353–64. doi:10.1016/j.cell.2008.05.033
46. Busch R, Neese RA, Awada M, Hayes GM, Hellerstein MK. Measurement of cell proliferation by heavy water labeling. Nat Protoc (2007) 2(12):3045–57. doi:10.1038/nprot.2007.420
47. Commerford SL, Carsten AL, Cronkite EP. The distribution of tritium among the amino acids of proteins obtained from mice exposed to tritiated water. Radiat Res (1983) 94(1):151–5. doi:10.2307/3575870
48. Busch R, Kim YK, Neese RA, Schade-Serin V, Collins M, Awada M, et al. Measurement of protein turnover rates by heavy water labeling of nonessential amino acids. Biochim Biophys Acta (2006) 1760(5):730–44. doi:10.1016/j.bbagen.2005.12.023
49. Herath K, Bhat G, Miller PL, Wang SP, Kulick A, Andrews-Kelly G, et al. Equilibration of 2H labeling between body water and free amino acids: enabling studies of proteome synthesis. Anal Biochem (2011) 415(2):197–9. doi:10.1016/j.ab.2011.04.031
50. De Riva A, Deery MJ, McDonald S, Lund T, Busch R. Measurement of protein synthesis using heavy water labeling and peptide mass spectrometry: discrimination between major histocompatibility complex allotypes. Anal Biochem (2010) 403(1-2):1–12. doi:10.1016/j.ab.2010.04.018S0003-2697(10)00255-1
51. Price JC, Holmes WE, Li KW, Floreani NA, Neese RA, Turner SM, et al. Measurement of human plasma proteome dynamics with 2H2O and liquid chromatography tandem mass spectrometry. Anal Biochem (2012) 420(1):73–83. doi:10.1016/j.ab.2011.09.007
52. Rachdaoui N, Austin L, Kramer E, Previs MJ, Anderson VE, Kasumov T, et al. Measuring proteome dynamics in vivo: as easy as adding water? Mol Cell Proteomics (2009) 8(12):2653–63. doi:10.1074/mcp.M900026-MCP200
53. De Riva A, Varley MC, Bluck LJ, Cooke A, Deery MJ, Busch R. Accelerated turnover of MHC class II molecules in nonobese diabetic mice is developmentally and environmentally regulated in vivo and dispensable for autoimmunity. J Immunol (2013) 190(12):5961–71. doi:10.4049/jimmunol.1300551
54. Ma JK, Platt MY, Eastham-Anderson J, Shin JS, Mellman I. MHC class II distribution in dendritic cells and B cells is determined by ubiquitin chain length. Proc Natl Acad Sci U S A (2012) 109(23):8820–7. doi:10.1073/pnas.12029771091202977109
55. Pozzilli P, Signore A, Williams AJ, Beales PE. NOD mouse colonies around the world – recent facts and figures. Immunol Today (1993) 14(5):193–6. doi:10.1016/0167-5699(93)90160-M
56. Koonce CH, Bikoff EK. Dissecting MHC class II export, B cell maturation, and DM stability defects in invariant chain mutant mice. J Immunol (2004) 173(5):3271–80.
57. Mellanby RJ, Koonce CH, Monti A, Phillips JM, Cooke A, Bikoff EK. Loss of invariant chain protects nonobese diabetic mice against type 1 diabetes. J Immunol (2006) 177(11):7588–98.
58. Morgan MA, Muller PS, Mould A, Newland SA, Nichols J, Robertson EJ, et al. The nonconventional MHC class II molecule DM governs diabetes susceptibility in NOD mice. PLoS One (2013) 8(2):e56738. doi:10.1371/journal.pone.0056738
Keywords: autoimmune pathogenesis, type 1 diabetes mellitus, major histocompatibility complex class II, antigen-presenting cell, protein turnover, mass spectrometry
Citation: De Riva A and Busch R (2013) MHC class II protein turnover in vivo and its relevance for autoimmunity in non-obese diabetic mice. Front. Immunol. 4:399. doi: 10.3389/fimmu.2013.00399
Received: 29 August 2013; Accepted: 08 November 2013;
Published online: 25 November 2013.
Edited by:
Laura Santambrogio, Albert Einstein College of Medicine, USAReviewed by:
Masaaki Murakami, Osaka University, JapanM. Karen Newell-Rogers, Texas A&M University, USA
Copyright: © 2013 De Riva and Busch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Busch, Department of Medicine, Addenbrooke’s Hospital, University of Cambridge, Box 157, Level 5, Hills Road, Cambridge CB2 0QQ, UK e-mail:cmI0NjhAbWVkc2NobC5jYW0uYWMudWs=
†Present address: Robert Busch, Department of Life Sciences, Whitelands College, University of Roehampton, Parkstead House, London SW15 4JD, UK e-mail:cm9iZXJ0LmJ1c2NoQHJvZWhhbXB0b24uYWMudWs=