Abstract
Sarcoidosis is a granulomatous disorder of unknown cause, affecting multiple organs, but mainly the lungs. The exact order of immunological events remains obscure. Reviewing current literature, combined with careful clinical observations, we propose a model for granuloma formation in pulmonary sarcoidosis. A tight collaboration between macrophages, dendritic cells, and lymphocyte subsets, initiates the first steps toward granuloma formation, orchestrated by cytokines and chemokines. In a substantial part of pulmonary sarcoidosis patients, granuloma formation becomes an on-going process, leading to debilitating disease, and sometimes death. The immunological response, determining granuloma sustainment is not well understood. An impaired immunosuppressive function of regulatory T cells has been suggested to contribute to the exaggerated response. Interestingly, therapeutical agents commonly used in sarcoidosis, such as glucocorticosteroids and anti-TNF agents, interfere with granuloma integrity and restore the immune homeostasis in autoimmune disorders. Increasing insight into their mechanisms of action may contribute to the search for new therapeutical targets in pulmonary sarcoidosis.
Introduction
Sarcoidosis is a granulomatous disorder of unknown cause, affecting multiple organs, but mainly the lungs. In 10–30% of the cases, sarcoidosis becomes chronic and progressive leading to debilitating disease and sometimes death (1). Its etiology is intriguing, since a part of its definition (i.e., unknown cause) makes it uniquely different from granulomatous disorders arising from exposure to a known chronically persisting antigen, such as tuberculosis, visceral leishmaniasis, and chronic beryllium disease (2, 3). Nevertheless, several observations support an antigen-induced disease etiology. First, epidemiological research identified environmental and occupational risk factors, such as exposure to musty odors and insecticides (4). Second, infectious agents, including Propionibacterium acnes (P. acnes) and Mycobacterium tuberculosis (Mtb), have been implicated, since genomes of these species are detected within sarcoid granulomas (5). A role for mycobacterial peptides is further supported by the presence of T lymphocytes that are highly responsive toward 6-kDa early secreted antigenic protein (ESAT-6) or catalase peroxidase (KatG) in the broncho-alveolar lavage fluid (BALF) of sarcoidosis patients (6– 8). Third, a limited clonality of CD4+ T cells, expressing the AV2S3 T cell receptor, was demonstrated within the lungs of HLA-DRB1*03 positive sarcoidosis patients, which is consistent with an antigenic response (9–12). Finally, evidence for an antigen-induced disease lies within the granulomatous reaction that is virtually indistinguishable from sarcoid granulomas and occurs in individuals with sarcoidosis upon subcutaneous injection of homogenates from allogeneic sarcoid spleen or lymph nodes (LNs), i.e., the Kveim–Siltzbach test (13, 14).
Genetic Risk Factors in Sarcoidosis
People all over the world suffer from sarcoidosis (15). Familial clustering (16), increased concordance in monozygotic twins (17) and variations in susceptibility and disease presentation among different ethnic groups (18), suggest the importance of genetic, next to environmental risk factors in the etiology.
Genome-wide association studies (GWAS) identified polymorphisms within genes coding for proteins involved in T cell activation, differentiation, proliferation, and survival, including NOTCH 4 and ANXA11 (19, 20). Additionally, GWAS and case-control studies identified important genetic risk factors within the antigen presentation locus at 6p21.3, which contains genes encoding proteins involved in both antigen presentation and T cell regulation, including human leukocyte antigen (HLA) and butyrophilin-like protein (BTNL)-2, respectively (20–23).
Specific HLA class II antigens are associated with certain sarcoidosis disease phenotypes. For example, the HLA-DRB1*03 and DQB1*0201 alleles have been associated with an acute disease onset, Löfgren syndrome and resolving disease, whereas in contrast HLA-DRB1*15 and DQB1*0601 are associated with chronic sarcoidosis (24–27). It is conceivable that both resolving and persistent sarcoidosis arise due to a unique combination of a specific genetic background and exposure to one or several environmental triggers (28). This unique combination might lead to persistent stimulation of the immune system, contributing to granuloma formation and sustainment.
In this article we review the current knowledge on the role of the immune activation in pulmonary sarcoidosis and propose a hypothesis on the origin of granuloma formation. Secondly, we aim to discuss granuloma integrity, highlighting areas for research into new therapeutical targets.
Granuloma Formation
A well-developed sarcoid granuloma consists of a tightly formed conglomerate of epithelioid- and multinucleated-giant cells (MGCs) encircled by lymphocytes, especially CD4+ T helper (Th) cells, but also rare CD8+ T cells and B cells (1). Both granuloma formation and integrity depend on the availability and supply of these different cells (29). The chronological order of immunological events and the exact role of these cells during the sarcoid granulomatous response remain obscure, due to the lack of an animal model for sarcoidosis. Nevertheless, careful clinical observations and in-depth research on functional properties of different cells involved provide essential information to unravel the cellular and molecular mechanisms of granuloma formation.
Clinical signs
Cardinal features of pulmonary sarcoidosis are mediastinal lymphadenopathy, parenchymal, and airway granulomas, giving rise to upper lobe nodules in a perilymphatic or bronchovascular distribution and signs of a CD4+ T cell alveolitis. An interstitial pneumonitis, found on open lung biopsy, is classically thought to represent a very early stage of granuloma formation (30). Spontaneous remission and reactivation of sarcoidosis makes it difficult to ascertain the exact sequence of these cardinal features, however several findings strongly suggest a certain order in the majority of patients, which may add to the hypothesis on granuloma formation as described below.
Although it is well known that patients do not go through all disease stages as described by Scadding (from I to IV) sequentially, arguably pulmonary sarcoidosis starts in the draining LN. As stage I (bihilar lymphadenopathy) is most often asymptomatic, it is conceivable that it precedes pulmonary involvement, seen in stage II and III. Additionally, progression of stage I to II disease is well known, while development of stage I after stage III is uncommon. Finally, a recent trial found an increased diagnostic sensitivity of LN-derived fine needle aspirates, compared with transbronchial lung biopsies (31). These data suggest that the first granulomas are formed within the mediastinal LN, only later followed by granuloma formation within the lungs.
Consequently, LN-specific immune reactions are important in early sarcoid granuloma formation, such as antigen presentation by dendritic cells (DCs). DCs are the only cells capable to pick up antigens and migrate to the LN where they present antigens to naïve T cells. Hereby they initiate highly specific clonal T cell differentiation and proliferation (32). Alternatively, LN-resident DCs may encounter antigenic particles, which we propose are submicroscopic and may therefore have passively migrated through the afferent lymph. The activated and differentiated Th cells migrate toward the site of inflammation, orchestrated by chemokines.
Macrophages contribute to early recognition of the putative sarcoid antigen in the lungs, thereby attracting mononuclear cells, including monocytes and LN-activated lymphocytes. The ensuing influx of cells leads to an interstitial pneumonitis, characterized by a mixed mononuclear cell infiltrate in the alveolar wall and CD4+ T cell alveolitis (30).
At the site of antigen encounter, antigen-presenting cells (APCs) induce persistent stimulation of the immune response, mediated by HLA-related proteins, leading to continuous recruitment and local expansion of lymphocytes and eventually granuloma formation. The central localization of macrophages within the final epithelioid aggregate supports an important role in antigen presentation at the site of granuloma formation. Alternatively, DCs may play a critical role in antigen presentation within the granuloma. Their capacity for antigen sampling within the lymph fluid makes them likely candidates to contribute to the induction of the perilymphatic localized granulomas (33, 34).
In the following paragraphs we describe the current knowledge on the role of macrophages, DCs, and lymphocytes in sarcoid granuloma formation in more detail, also summarized in Figure 1.
Figure 1
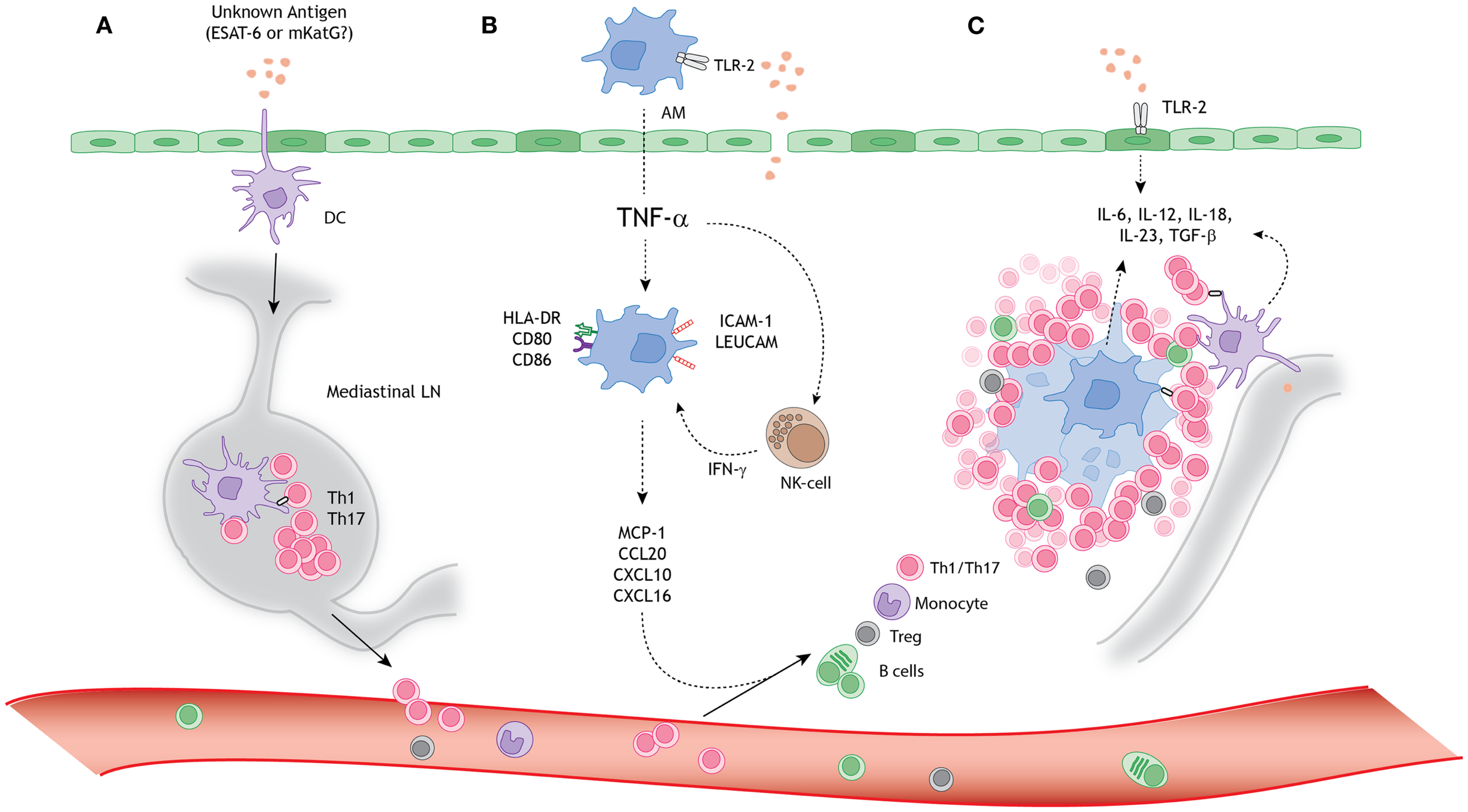
A schematic model for granuloma formation in pulmonary sarcoidosis. An unknown airborne-antigen activates (A) interstitial dendritic cells (DCs), (B) alveolar macrophages (AMs), and (C) alveolar epithelial cells type II (AEC-II) (dark green), simultaneously. This process is initiated by toll-like receptor-2 (TLR-2) ligands, possibly Mycobacterium tuberculosis-derived ESAT-6 or mKatG. (A) The interstitial DCs pick up the putative antigen and migrate toward the mediastinal lymph nodes (LNs), where they initiate differentiation and clonal expansion of T helper (Th)1 and 17 cells. (B) Simultaneously, AMs produce tumor necrosis factor-α (TNF-α), which initiates upregulation of activation (HLA-DR and CD80/86) and adhesion (ICAM-1 and LeuCAM) molecules. Macrophages produce chemokine ligands (MCP-1, CCL20, CXCL10, and CXCL16) under stimulation of both TNF-α and natural-killer (NK) cell-derived interferon-γ (INF-γ), thereby attracting Th1/17 cells, monocytes, regulatory T cells (Tregs), and B cells. (C) The lung environment is characterized by the presence of Th1 and Th17 favoring cytokines, such as IL-6, IL-12, IL-18, IL-23, and TGF-β, produced by macrophages, perilymphatic DCs, and AEC-II. Persistent stimulation, mediated by antigen presenting cells (APCs), leads to continuous cellular recruitment to the site of inflammation, which leads to granuloma formation. Tregs infiltrating the granuloma fail to diminish the exaggerated immune response, thereby contributing to granuloma persistence and integrity.
Macrophages
Upon activation, macrophages release nuclear factor (NF)-κB-dependent pro-inflammatory cytokines, such as interleukin (IL)-1 and tumor necrosis factor-α (TNF-α) (35). In sarcoidosis, BALF cells and monocytes highly express toll-like receptor (TLR)-2 (36, 37) and produce increased amounts of TNF-α, IL-1β, and IL-6 compared with controls, when stimulated with TLR-2 ligands, including ESAT-6 and KatG (7, 36–38). A role for TLR-2 in immune activation and granuloma formation in sarcoidosis is further supported by genetic and mouse studies (38, 39). Lately, continuous TLR-2 ligation by macrophage-derived serum amyloid A has been suggested to contribute to persistent stimulation of the immune response in sarcoidosis (37).
Intrinsically, unstimulated sarcoid-derived alveolar macrophages (AMs) produce increased amounts of IL-1 and TNF-α (40–43) and are highly activated (44, 45). The amounts of spontaneously produced TNF-α by BALF cells in vitro correlate with the presence of aggregates of AMs in the tissue (46, 47). Only AMs from patients with active and progressive disease produce increased amounts of TNF-α (48–51). These data highlight the role of TNF-α in granuloma formation and integrity, also supported by mouse studies (52–54).
Important mechanisms of action of TNF-α include macrophage activation, promotion of cellular migration toward the site of inflammation and leukocyte adhesion (52, 55, 56). In a mycobacterial-driven mouse model, TNF-α is responsible for the early production of chemokines that attract mononuclear cells to the site of inflammation, such as RANTES, MIP-1α, MIP-1β, MIP-2, and MCP-1 (55), of which increased amounts are found in sarcoidosis BALF (57–59). In active sarcoidosis, AMs produce high amounts of CCL20, when stimulated by TNF-α and IL-1β (60). CCL20 is a chemokine with high affinity for chemokine receptor CCR6, therefore attracting DCs, B cells, and specific T cell subsets toward the lungs (60, 61). Similarly, AM-derived CXCL10 and CXCL16 contribute to CXCR3+ and CXCR6+ CD4+ Th cell recruitment (62, 63).
In a mycobacterial-driven granuloma model, efficient cellular recruitment, mediated by AM-derived CXCL10 and CXCL16, depends on interferon-γ (INF-γ) (53). During the early innate response natural-killer (NK) cells are important producers of INF-γ, when stimulated by TNF-α, IL-1, and IL-12. In sarcoidosis, the size of a distinct NK cell subpopulation (CD56brightCD94highKIRlow) is increased in the BALF compared with controls (64). Furthermore, higher proportions of NK cells were found to correlate with a poor outcome (65).
Once recruited, TNF-α is needed for leukocyte adhesion, since an abrogation of tightly formed granulomas in TNF-α-deficient mice is observed following mycobacterial infection (55). In sarcoidosis, TNF-α induced the expression of intracellular adhesion molecule-1 (ICAM-1) on AMs, leading to cellular aggregation (66). Additionally, leukocyte adhesion molecule (LeuCAM) expression, such as CD11a/b/c and CD18 (67), is increased in sarcoid AMs compared with controls.
Following adhesion, epithelioid histiocytes and monocyte-derived DCs (moDCs) can fuse to MGCs when stimulated by local cytokines, such as TNF-α, GM-CSF, IL-17A, CCL20, and INF-γ (68, 69). Patient-derived macrophages and monocytes show an enhanced potential to form MGCs in vitro, compared with healthy controls and other granulomatous diseases (70).
Importantly, sarcoid-derived AMs have an increased accessory function on autologous blood- and lung-derived T lymphocytes, when compared with controls (71–73). Macrophages are not capable to migrate to the LN to induce naive T cell activation, making them weak APCs. Nonetheless, in sarcoidosis, macrophages might contribute to local antigen presentation, enhancing proliferation of chemokine-recruited memory Th cells.
In summary, macrophages are important for the initial accumulation, aggregation, and fusion of the cellular building blocks needed for granuloma formation. This process is mediated by the strong immune modulatory capacities of TNF-α and assisted by NK cells, which produce INF-γ.
Dendritic cells
Only a few studies investigated the role of DCs in sarcoid granuloma formation (47, 74). Our group has shown that granuloma formation surrounding intravenously injected antigen-loaded beads trapped in the lung vasculature is dependent on DC-initiated Th cell proliferation within the mediastinal LN (75). In sarcoidosis, an accumulation of mature (Fascin+HLA-DR+DC-LAMP+) DCs is found surrounding LN granulomas, adjacent to CD3+ lymphocytes, suggesting DC-T cell interaction at this site (76). Mature (CD11c+CD86+) DCs are found surrounding granulomas in sarcoid-derived mucosal biopsies (77), further supporting a role for DCs in airway and parenchymal granuloma formation.
An impaired accessory function of ex vivo blood-derived myeloid DCs (mDCs) has been suggested to contribute to granuloma formation, as clearance of the putative antigen may be ineffective and the immune system turns to granuloma formation as a default immunological response (78). In contrast, our group isolated BALF mDCs of sarcoidosis patients and found them to be immunocompetent, initiating proliferation of allogeneic, naïve T lymphocytes comparable with mDCs from healthy controls (77). Similarly, in vitro cultured moDCs showed a comparable accessory capacity as controls, although they are intrinsically prone to produce TNF-α (77, 79). Hence, it is most likely that DCs are involved in granuloma formation, instead of displaying diminished antigen-presenting capacities.
Differentiation of T lymphocytes depends on the local cytokines surrounding the initiating APC (80). Although it is very likely that LN-specific interactions, mediated by DCs, are responsible for the initial T cell polarization toward a Th1 and Th17 phenotype as observed in sarcoidosis, direct evidence is still lacking.
Lymphocytes
Sarcoidosis is characterized as a Th1- (81) and more recently a Th17-mediated disease (61, 82), based on the accumulation of INF-γ, IL-2, and IL17-producing Th cells in the lungs of patients with active sarcoidosis (44, 61, 82–84).
Th1 differentiation depends on IL-12 and IL-18, which are increased in BALF of sarcoidosis patients (85, 86). Alveolar epithelial cells type II (AEC-II) may contribute to this Th1-favoring environment, since patient-derived AEC-II produce IL-18 upon TLR-2 stimulation (87, 88). Additionally, AEC-II may contribute to CXCR3+ Th1 cell recruitment by production of CXCL10 (89).
Th17 differentiation is driven by IL-6 and TGF-β, both produced by sarcoid-derived BALF cells (90, 91), whereas survival and proliferation of this subset is IL-23-dependent (92–94). Increased expression of the IL-23-receptor and IL-17, both expressed by Th17 cells, is found in blood-, lung-, and LN-derived lymphocytes of active sarcoidosis patients, and not in inactive disease (61, 82). Recently, ESAT-6-specific Th17 cells in the BALF of sarcoidosis patients were found (95). Additionally, IL-17A is essential for granuloma formation in the lung during mycobacterial infection (96) or in chronic granulomatous disease (97).
We recently found that the proportions of circulating IL-17A/IFN-γ and IL-17A/IL-4 double-producing cells are significantly increased in the peripheral blood of patients and are present in substantial numbers in BALF (82). Findings in several autoimmune diseases have indicated the pathogenic potential of CD4+ Th cells producing both IL-17 and IFN-γ (98, 99). Processes underlying Th17 cell induction in sarcoidosis remain obscure, but the presence of these cells can suggest a role for autoimmune responses in sarcoidosis. B lymphocytes and plasma cells are found surrounding sarcoid granulomas (100). Additionally, active sarcoidosis patients have increased serum levels of B-cell-activating factor (BAFF) (101). Since B cell maturation and function depends on BAFF, its aberrant expression can initiate defective selection of autoreactive B cells, leading to autoantibody production (101, 102). In sarcoidosis, approximately 30–60% of the patients exhibit antinuclear antibody (ANA) positivity (101, 103).
A SNP in the IL-23 receptor gene has been associated with chronic sarcoidosis (104), which may contribute to Th17 cell development in sarcoidosis. Since IL-23 is a heterodimer of the p19 subunit and the p40 subunit of IL-12 (105) the Th1- and Th17-promoting cytokines share a common therapeutical target. Ustekinumab, a neutralizing antibody against the IL-12/IL-23 p40, was shown to be successful in the Th1/Th17-mediated diseases psoriasis and Crohn’s disease (CD) (106, 107), but not in chronic pulmonary or skin sarcoidosis (108, 109).
Granuloma Integrity
In the majority of the sarcoidosis patients, granulomas spontaneous resolve within several years, without need for therapy. However, a substantial proportion of the patients develop chronic progressive disease, whereby granulomas persist and form fibrotic lesions, leading to debilitating disease, and sometimes death (1, 110). The immunological response, determining granuloma sustainment is not well understood.
Regulatory T cells
Regulatory T cells (Tregs) play an important role in diminishing Th cell specific responses and are pivotal for maintenance of self-tolerance and immune homeostasis (111). An impaired immunosuppressive function of sarcoid-derived Tregs has been suggested to contribute to the on-going, exaggerated immune response, since sarcoid blood-derived (CD4+CD25high) Tregs fail to inhibit granuloma growth in an in vitro granuloma culture model (112, 113). Subsequently, an impaired immunosuppressive function of both blood- and BALF-derived sarcoidosis Tregs has repetitively been described on autologous and allogeneic healthy Th cell proliferation (114–116). These studies also show that sarcoid-derived Tregs fail to inhibit production of TNF-α, INF-γ, and IL-2, contributing to granuloma formation, rather than diminishing the immune response (112, 113, 116). It remains unknown what mechanism(s) underlies this impaired function.
Active and persisting sarcoidosis was recently associated with a global CD4+ T cell subset dysfunction (116). Notably, both Th anergy and Treg malfunctioning were restored in patients with disease resolution (116). These results highlight the complex interplay between pro-inflammatory and anti-inflammatory responses needed for granuloma integrity. This fine balance may explain contradictory results with regard to reported Treg numbers in the BALF (112–119) (Table 1). Low BALF Tregs (i.e., less immunosuppression) in patients have been associated with a favorable prognosis in a Scandinavian population (118). In contrast, a German study reported decreased BALF Treg numbers in sarcoidosis patients who develop chronic (active) disease, when compared with controls and patients who develop spontaneous resolution (115). Similarly, CD1d-restricted natural-killer T (NKT) cells with immunoregulatory function are greatly reduced in the peripheral blood of all sarcoidosis patients, except Löfgren patients (120).
Table 1
| Study | Methods |
Proportions |
Function |
Remarks | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Population | Treg definition | Technique | Blood | BALF | LN | Blood | BALF | LN | ||
| Miyara et al. (112) | Active disease | CD4+CD25+ | FC/IHC | ↑ | ↑ b | ↑ | ↓ ∧ | =b | = | ∧Blood-derived Tregs reduce autologous T cell proliferation similarly as controls, but do not inhibit the release of TNF-alpha and INF-y |
| % of CD4+ | ||||||||||
| Idali et al. (117) | Active disease | CD4+FoxP3+ | FC/PCR | ↓ | ↓ | BALF Treg proportions are significantly higher than blood Treg proportions in both healthy controls and patients | ||||
| % of CD4+ | ||||||||||
| Taflin et al. (113) | Active disease | CD4+CD45RA−FoxP3++ | FC/IHC | ↑ | ↑ b | ↓ | FoxP3+ Tregs in the sarcoid LN are highly proliferative (Ki67+) | |||
| % of CD4+ | ||||||||||
| Prasse et al. (115) | Pre-treatment patients | CD4+CD25+CD127−% of CD4+ | FC | ↓ ∧ | ↓,#c,# | ∧BALF Treg proportions are decreased in patients who develop active chronic disease, defined after 1 year follow-up | ||||
| #Vasoactive intestinal peptide (VIP) inhalation increased the number of BALF Tregs and the immunosuppressive function | ||||||||||
| Rappl et al. (114) | Unknown | CD25+CD7−% of CD4+ CD45RO+ FoxP3+CD127− | FC | ↑ | ↓ | Increased proportions of CD4+ FoxP3+ CD127−Tregs are CD7-, compared with healthy controls | ||||
| Wiken et al. (118) | Active diseasea | FoxP3+ | FC | ↓ d | BALF Tregs proportions are siginificantly decreased in HLA-DRB1*0301 positive patients, which are mostly (82%) Lofgren patients | |||||
| % of CD4+CD45RO+ CD27+ | ||||||||||
| Darlington et al. (119) | Active disease | CD4+FoxP3+ | FC | =b | % FoxP3 expressing CD4+ T cells is inversely correlated with% T cells with AV2S3 >10% in BALF | |||||
| % of CD4+ | ||||||||||
| Oswald-Richter et al. (116) | Active disease | CD4+CD45RO+ CD25high | FC | ↑ | ↓ | Treg malfunctioning restored during disease resolution | ||||
| % of CD4+ | ||||||||||
An overview of studies reporting regulatory T cell (Treg) proportions and functional properties in pulmonary sarcoidosis.
All results are compared with healthy controls, unless specified otherwise. Flowcytometry (FC), Immunohistochemistry (IHC), Polymerase chain reaction (PCR), broncho-alveolar lavage fluid (BALF), lymph node (LN).
aHLA-DRB1*0301 positive sarcoidosis patients were analyses vs. HLA-DRB1*0301 negative sarcoidosis patients.
bCompared with diseased controls.
cCompared with post-treatment.
dCompared with HLA-DRB1*0301 negative patients.
Taken together, these studies imply different roles for immune regulatory cells in sarcoidosis, either contributing to or preventing an on-going, exaggerated immune response. Arguably, whereas in the early sarcoid response there may be no need for Tregs to inhibit an effective immune response, during persistent stimulation immune regulatory cells should function as a natural brake on the exaggerated response to prevent immunopathology and autoimmunity.
Interfering with granuloma integrity
Effective treatment agents used for sarcoidosis interfere with granuloma integrity and would ideally prevent fibrogenesis. Glucocorticosteroids (GCs), the main stay of sarcoidosis therapy, partially exert their beneficial effect by repression of NF-κB-related cytokine gene transcription and induction of lymphocyte apoptosis (121, 122). Using a mouse model, Tregs are found to be less sensitive to GC-induced apoptosis compared with Th cells, favoring an anti-inflammatory milieu (123, 124). Similarly, anti-TNF agents induce monocyte and lymphocyte apoptosis (125–127), while improving Treg numbers (123). Interestingly, infliximab, which blocks membrane-bound TNF-α, is uniquely associated with a high risk of reactivation of latent Mtb infection, whereas etanercept, solely blocking secreted TNF-α, is not (29). This phenomenon implies a critical role of membrane-bound TNF-α signaling in granuloma integrity (29), which is further supported by mouse studies (128).
Whether GCs and anti-TNF agents interfere with the delicate Th/Treg balance in pulmonary sarcoidosis, remains to be elucidated. Research into this field will shed more light on the role of Tregs in sarcoid pathology and whether Treg induction holds a promising new therapeutical strategy. Finally, an interplay between anergic Th cells, IL-10, alternatively activated macrophages (M2), CCL18 and lung fibroblasts has recently been suggested to contribute to fibrotic remodelling of the lung in chronic sarcoidosis (129). These insights yield new therapeutical targets to prevent irreversible organ damage in chronic pulmonary sarcoidosis patients.
Conclusion
Sarcoidosis is an intriguingly complex granulomatous disorder, characterized by an exaggerated Th1/17 immune response, initiated by APCs, and maintained due to malfunctioning of Tregs. Refining insight into immunological events that determine granuloma fate may help identify new therapeutical targets and patients who will benefit such therapy in the future.
Statements
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Hunninghake GW Costabel U Ando M Baughman R Cordier JF du Bois R et al ATS/ERS/WASOG statement on sarcoidosis. American thoracic society/European respiratory society/World Association of sarcoidosis and other granulomatous disorders. Sarcoidosis Vasc Diffuse Lung Dis (1999) 16(2):149.
2
Guirado E Schlesinger LS . Modeling the Mycobacterium tuberculosis granuloma-the critical battlefield in host immunity and disease. Front Immunol (2013) 4:98.10.3389/fimmu.2013.00098
3
Moore JWJ Moyo D Beattie L Andrews PS Timmis J Kaye PM . Functional complexity of the Leishmania granuloma and the potential of in silico modeling. Front Immunol (2013) 4:98.10.3389/fimmu.2013.00035
4
Newman LS Rose CS Bresnitz EA Rossman MD Barnard J Frederick M et al A case control etiologic study of sarcoidosis: environmental and occupational risk factors. Am J Respir Crit Care Med (2004) 170(12):1324–30.10.1164/rccm.200402-249OC
5
Eishi Y Suga M Ishige I Kobayashi D Yamada T Takemura T et al Quantitative analysis of mycobacterial and propionibacterial DNA in lymph nodes of Japanese and European patients with sarcoidosis. J Clin Microbiol (2002) 40(1):198–204.10.1128/JCM.40.1.198-204.2002
6
Chen ES Wahlström J Song Z Willett MH Wikén M Yung RC et al T cell responses to mycobacterial catalase-peroxidase profile a pathogenic antigen in systemic sarcoidosis. J Immunol (2008) 181(12):8784–96.
7
Oswald-Richter KA Culver DA Hawkins C Hajizadeh R Abraham S Shepherd BE et al Cellular responses to mycobacterial antigens are present in bronchoalveolar lavage fluid used in the diagnosis of sarcoidosis. Infect Immun (2009) 77(9):3740–8.10.1128/IAI.00142-09
8
Wiken M Ostadkarampour M Eklund A Willett M Chen E Moller D et al Antigen-specific multifunctional T-cells in sarcoidosis patients with Lofgren’s syndrome. Eur Respir J (2012) 40(1):110–21.10.1183/09031936.00166110
9
Grunewald J Berlin M Olerup O Eklund A . Lung T-helper cells expressing T-cell receptor AV2S3 associate with clinical features of pulmonary sarcoidosis. Am J Respir Crit Care Med (2000) 161(3):814–8.10.1164/ajrccm.161.3.9906001
10
Grunewald J Wahlstrom J Berlin M Wigzell H Eklund A Olerup O . Lung restricted T cell receptor AV2S3+ CD4+ T cell expansions in sarcoidosis patients with a shared HLA-DRbeta chain conformation. Thorax (2002) 57(4):348–52.10.1136/thorax.57.4.348
11
Grunewald J . Review: role of genetics in susceptibility and outcome of sarcoidosis. Semin Respir Crit Care Med (2010) 31(4):380–9.10.1055/s-0030-1262206
12
Grunewald J Janson CH Eklund A Öhrn M Olerup O Persson U et al Restricted Vα2.3 gene usage by CD4+ T lymphocytes in bronchoalveolar lavage fluid from sarcoidosis patients correlates with HLA-DR3. Eur J Immunol (1992) 22(1):129–35.10.1002/eji.1830220120
13
Siltzbach LE . The Kveim test in sarcoidosis. A study of 750 patients. JAMA (1961) 178:476–82.10.1001/jama.1961.03040440028006
14
Kveim A . Some remarks on the aetiology of Boeck’s sarcoid. Acta Derm Venereol (1948) 28(2):169.
15
Rybicki BA Iannuzzi MC , editors. Epidemiology of sarcoidosis: recent advances and future prospects. Seminars in Respiratory and Critical Care Medicine. New York: Thieme Medical Publishers (2007). c1994 p.
16
Rybicki BA Kirkey KL Major M Maliarik MJ Popovich J Chase GA et al Familial risk ratio of sarcoidosis in African-American sibs and parents. Am J Epidemiol (2001) 153(2):188–93.10.1093/aje/153.2.188
17
Sverrild A Backer V Kyvik KO Kaprio J Milman N Svendsen CB et al Heredity in sarcoidosis: a registry-based twin study. Thorax (2008) 63(10):894–6.10.1136/thx.2007.094060
18
Rybicki BA Major M Popovich J Maliank MJ . Racial differences in sarcoidosis incidence: a 5-year study in a health maintenance organization. Am J Epidemiol (1997) 145(3):234–41.10.1093/oxfordjournals.aje.a009096
19
Hofmann S Franke A Fischer A Jacobs G Nothnagel M Gaede KI et al Genome-wide association study identifies ANXA11 as a new susceptibility locus for sarcoidosis. Nat Genet (2008) 40(9):1103–6.10.1038/ng.198
20
Adrianto I Lin CP Hale JJ Levin AM Datta I Parker R et al Genome-wide association study of African and European Americans implicates multiple shared and ethnic specific loci in sarcoidosis susceptibility. PLoS One (2012) 7(8):e43907.10.1371/journal.pone.0043907
21
Valentonyte R Hampe J Huse K Rosenstiel P Albrecht M Stenzel A et al Sarcoidosis is associated with a truncating splice site mutation in BTNL2. Nat Genet (2005) 37(4):357–64.10.1038/ng0605-652a
22
Rybicki BA Walewski JL Maliarik MJ Kian H Iannuzzi MC . The BTNL2 gene and sarcoidosis susceptibility in African Americans and Whites. Am J Hum Genet (2005) 77(3):491–9.10.1086/444435
23
Rossman MD Thompson B Frederick M Maliarik M Iannuzzi MC Rybicki BA et al HLA-DRB1*1101: a significant risk factor for sarcoidosis in blacks and whites. Am J Hum Genet (2003) 73(4):720–35.10.1086/378097
24
Berlin M Fogdell-Hahn A Olerup O Eklund A Grunewald J . HLA-DR predicts the prognosis in Scandinavian patients with pulmonary sarcoidosis. Am J Respir Crit Care Med (1997) 156(5):1601–5.10.1164/ajrccm.156.5.9704069
25
Sato H Grutters JC Pantelidis P Mizzon AN Ahmad T van Houte A-J et al HLA-DQB1* 0201: a marker for good prognosis in British and Dutch patients with sarcoidosis. Am J Respir Cell Mol Biol (2002) 27(4):406–12.10.1165/rcmb.4782
26
Grunewald J Eklund A Olerup O . Human leukocyte antigen class I alleles and the disease course in sarcoidosis patients. Am J Respir Crit Care Med (2004) 169(6):696–702.10.1164/rccm.200303-459OC
27
Grunewald J Eklund A . Lofgren’s syndrome: human leukocyte antigen strongly influences the disease course. Am J Respir Crit Care Med (2009) 179(4):307–12.10.1164/rccm.200807-1082OC
28
Wennerström A Pietinalho A Lasota J Salli K Surakka I Seppänen M et al Major histocompatibility complex class II and BTNL2 associations in sarcoidosis. Eur Respir J (2013) 42(2):550–3.10.1183/09031936.00035213
29
Wallis RS Ehlers S . Tumor necrosis factor and granuloma biology: explaining the differential infection risk of etanercept and infliximab. Semin Arthritis Rheum (2005) 34(5):34–8.10.1016/j.semarthrit.2005.01.009
30
Rosen Y Athanassiades TJ Moon S Lyons HA . Nongranulomatous interstitial pneumonitis in sarcoidosis. Relationship to development of epithelioid granulomas. Chest (1978) 74(2):122–5.10.1378/chest.74.2.122
31
von Bartheld MB Dekkers OM Szlubowski A Eberhardt R Herth FJ de Jong YP et al Endosonography vs conventional bronchoscopy for the diagnosis of sarcoidosis: the GRANULOMA randomized clinical trial. JAMA (2013) 309(23):2457–64.10.1001/jama.2013.5823
32
Banchereau J Steinman RM . Dendritic cells and the control of immunity. Nature (1998) 392(6673):245–52.10.1038/32588
33
Roozendaal R Mebius RE Kraal G . The conduit system of the lymph node. Int Immunol (2008) 20(12):1483–7.10.1093/intimm/dxn110
34
Kambouchner M Pirici D Uhl J-F Mogoanta L Valeyre D Bernaudin J-F . Lymphatic and blood microvasculature organisation in pulmonary sarcoid granulomas. Eur Respir J (2011) 37(4):835–40.10.1183/09031936.00086410
35
Bianchi ME . DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol (2007) 81(1):1–5.10.1189/jlb.0306164
36
Wiken M Grunewald J Eklund A Wahlstrom J . Higher monocyte expression of TLR2 and TLR4, and enhanced pro-inflammatory synergy of TLR2 with NOD2 stimulation in sarcoidosis. J Clin Immunol (2009) 29(1):78–89.10.1007/s10875-008-9225-0
37
Chen ES Song Z Willett MH Heine S Yung RC Liu MC et al Serum amyloid A regulates granulomatous inflammation in sarcoidosis through toll-like receptor-2. Am J Respir Crit Care Med (2010) 181(4):360.10.1164/rccm.200905-0696OC
38
Gabrilovich MI Walrath J van Lunteren J Nethery D Seifu M Kern JA et al Disordered toll-like receptor 2 responses in the pathogenesis of pulmonary sarcoidosis. Clin Exp Immunol (2013) 173(3):512–22.10.1111/cei.12138
39
Veltkamp M Wijnen PAHM Van Moorsel CHM Rijkers GT Ruven HJT Heron M et al Linkage between toll like receptor (TLR) 2 promotor and intron polymorphisms: functional effects and relevance to sarcoidosis. Clin Exp Immunol (2007) 149(3):453–62.10.1111/j.1365-2249.2007.03428.x
40
Hunninghake GW . Release of interleukin-1 by alveolar macrophages of patients with active pulmonary sarcoidosis. Am Rev Respir Dis (1984) 129(4):569–72.
41
Bachwich PR Lynch JP . Tumor necrosis factor production by human sarcoid alveolar macrophages. Am J Pathol (1986) 125(3):421.
42
Baughman RP Strohofer SA Buchsbaum J Lower EE . Release of tumor necrosis factor by alveolar macrophages of patients with sarcoidosis. J Lab Clin Med (1990) 115(1):36.
43
Müller-Quernheim J Pfeifer S Männel DN Strausz J Ferlinz R . Lung-restricted activation of the alveolar macrophage/monocyte system in pulmonary sarcoidosis. Am Rev Respir Dis (1992) 145(1):187–92.10.1164/ajrccm/145.1.187
44
Hoshino T Itoh K Gouhara R Yamada A Tanaka Y Ichikawa Y et al Spontaneous production of various cytokines except IL-4 from CD4+ T cells in the affected organs of sarcoidosis patients. Clin Exp Immunol (1995) 102(2):399–405.
45
Wahlstrom J Berlin M Skold CM Wigzell H Eklund A Grunewald J . Phenotypic analysis of lymphocytes and monocytes/macrophages in peripheral blood and bronchoalveolar lavage fluid from patients with pulmonary sarcoidosis. Thorax (1999) 54(4):339–46.10.1136/thx.54.4.339
46
Fehrenbach H Zissel G Goldmann T Tschernig T Vollmer E Pabst R et al Alveolar macrophages are the main source for tumour necrosis factor-α in patients with sarcoidosis. Eur Respir J (2003) 21(3):421–8.10.1183/09031936.03.00083002
47
Zissel G Prasse A Müller-Quernheim J , editors. Immunologic response of sarcoidosis. Seminars in Respiratory and Critical Care Medicine. New York: Thieme Medical Publishers, Inc (2010). p. 390–403.10.1055/s-0030-1262208
48
Muller-Quernheim J Pfeifer S Mannel D Strausz J Ferlinz R . Lung-restricted activation of the alveolar macrophage/monocyte system in pulmonary sarcoidosis. Am Rev Respir Dis (1992) 145(1):187–92.10.1164/ajrccm/145.1.187
49
Zheng L Teschler H Guzman J Hubner K Striz I Costabel U . Alveolar macrophage TNF-alpha release and BAL cell phenotypes in sarcoidosis. Am J Respir Crit Care Med (1995) 152(3):1061–6.10.1164/ajrccm.152.3.7663784
50
Ziegenhagen MW Schrum S Zissel G Zipfel PF Schlaak M Muller-Quernheim J . Increased expression of proinflammatory chemokines in bronchoalveolar lavage cells of patients with progressing idiopathic pulmonary fibrosis and sarcoidosis. J Investig Med (1998) 46(5):223–31.
51
Ziegenhagen MW Rothe ME Zissel G Muller-Quernheim J . Exaggerated TNFalpha release of alveolar macrophages in corticosteroid resistant sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis (2002) 19(3):185–90.
52
Flynn JL Goldstein MM Chan J Triebold KJ Pfeffer K Lowenstein CJ et al Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity (1995) 2(6):561–72.10.1016/1074-7613(95)90001-2
53
Smith D Hansch H Bancroft G Ehlers S . T-cell-independent granuloma formation in response to Mycobacterium avium: role of tumour necrosis factor-alpha and interferon-gamma. Immunology (1997) 92(4):413–21.10.1046/j.1365-2567.1997.00384.x
54
Ehlers S Kutsch S Ehlers EM Benini J Pfeffer K . Lethal granuloma disintegration in mycobacteria-infected TNFRp55-/- mice is dependent on T cells and IL-12. J Immunol (2000) 165(1):483–92.
55
Roach DR Bean AG Demangel C France MP Briscoe H Britton WJ . TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol (2002) 168(9):4620–7.
56
Newton SM Mackie SL Martineau AR Wilkinson KA Kampmann B Fisher C et al Reduction of chemokine secretion in response to mycobacteria in infliximab-treated patients. Clin Vaccine Immunol (2008) 15(3):506–12.10.1128/CVI.00401-07
57
Capelli A Di Stefano A Lusuardi M Gnemmi I Donner CF . Increased macrophage inflammatory protein-1alpha and macrophage inflammatory protein-1beta levels in bronchoalveolar lavage fluid of patients affected by different stages of pulmonary sarcoidosis. Am J Respir Crit Care Med (2002) 165(2):236–41.10.1164/ajrccm.165.2.2106084
58
Car BD Meloni F Luisetti M Semenzato G Gialdroni-Grassi G Walz A . Elevated IL-8 and MCP-1 in the bronchoalveolar lavage fluid of patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Am J Respir Crit Care Med (1994) 149(3 Pt 1):655–9.10.1164/ajrccm.149.3.8118632
59
Petrek M Kolek V Szotkowska J du Bois RM . CC and C chemokine expression in pulmonary sarcoidosis. Eur Respir J (2002) 20(5):1206–12.10.1183/09031936.02.00289902
60
Facco M Baesso I Miorin M Bortoli M Cabrelle A Boscaro E et al Expression and role of CCR6/CCL20 chemokine axis in pulmonary sarcoidosis. J Leukoc Biol (2007) 82(4):946–55.10.1189/jlb.0307133
61
Facco M Cabrelle A Teramo A Olivieri V Gnoato M Teolato S et al Sarcoidosis is a Th1/Th17 multisystem disorder. Thorax (2011) 66(2):144–50.10.1136/thx.2010.140319
62
Agostini C Cassatella M Zambello R Trentin L Gasperini S Perin A et al Involvement of the IP-10 chemokine in sarcoid granulomatous reactions. J Immunol (1998) 161(11):6413–20.
63
Agostini C Cabrelle A Calabrese F Bortoli M Scquizzato E Carraro S et al Role for CXCR6 and its ligand CXCL16 in the pathogenesis of T-cell alveolitis in sarcoidosis. Am J Respir Crit Care Med (2005) 172(10):1290–8.10.1164/rccm.200501-142OC
64
Katchar K Soderstrom K Wahlstrom J Eklund A Grunewald J . Characterisation of natural killer cells and CD56+ T-cells in sarcoidosis patients. Eur Respir J (2005) 26(1):77–85.10.1183/09031936.05.00030805
65
Tutor-Ureta P Citores MJ Castejon R Mellor-Pita S Yebra-Bango M Romero Y et al Prognostic value of neutrophils and NK cells in bronchoalveolar lavage of sarcoidosis. Cytometry B Clin Cytom (2006) 70(6):416–22.10.1002/cyto.b.20120
66
Sasaki M Namioka Y Ito T Izumiyama N Fukui S Watanabe A et al Role of ICAM-1 in the aggregation and adhesion of human alveolar macrophages in response to TNF-α and INF-γ. Mediators Inflamm (2001) 10(6):309–13.10.1080/09629350120102325
67
Schaberg T Rau M Stephan H Lode H . Increased number of alveolar macrophages expressing surface molecules of the CD11/CD18 family in sarcoidosis and idiopathic pulmonary fibrosis is related to the production of superoxide anions by these cells. Am Rev Respir Dis (1993) 147:1507–13.10.1164/ajrccm/147.6_Pt_1.1507
68
Coury F Annels N Rivollier A Olsson S Santoro A Speziani C et al Langerhans cell histiocytosis reveals a new IL-17A-dependent pathway of dendritic cell fusion. Nat Med (2008) 14(1):81–7.10.1038/nm1694
69
Van Maarsseveen TCM Vos W Van Diest PJ . Giant cell formation in sarcoidosis: cell fusion or proliferation with non-division?Clin Exp Immunol (2009) 155(3):476–86.10.1111/j.1365-2249.2008.03841.x
70
Mizuno K Okamoto H Horio T . Heightened ability of monocytes from sarcoidosis patients to form multi-nucleated giant cells in vitro by supernatants of concanavalin A-stimulated mononuclear cells. Clin Exp Immunol (2001) 126(1):151–6.10.1046/j.1365-2249.2001.01655.x
71
Lem VM Lipscomb MF Weissler JC Nunez G Ball EJ Stastny P et al Bronchoalveolar cells from sarcoid patients demonstrate enhanced antigen presentation. J Immunol (1985) 135(3):1766–71.
72
Venet A Hance AJ Saltini C Robinson BW Crystal RG . Enhanced alveolar macrophage-mediated antigen-induced T-lymphocyte proliferation in sarcoidosis. J Clin Invest (1985) 75(1):293–301.10.1172/JCI111688
73
Ina Y Takada K Yamamoto M Morishita M Miyachi A . Antigen-presenting capacity in patients with sarcoidosis. Chest (1990) 98(4):911–6.10.1378/chest.98.4.911
74
Zaba LC Smith GP Sanchez M Prystowsky SD . Dendritic cells in the pathogenesis of sarcoidosis. Am J Respir Cell Mol Biol (2010) 42(1):32–9.10.1165/rcmb.2009-0033TR
75
Willart MA Jan de Heer H Hammad H Soullie T Deswarte K Clausen BE et al The lung vascular filter as a site of immune induction for T cell responses to large embolic antigen. J Exp Med (2009) 206(12):2823–35.10.1084/jem.20082401
76
Ota M Amakawa R Uehira K Ito T Yagi Y Oshiro A et al Involvement of dendritic cells in sarcoidosis. Thorax (2004) 59(5):408–13.10.1136/thx.2003.006049
77
Ten Berge B KleinJan A Muskens F Hammad H Hoogsteden HC Hendriks RW et al Evidence for local dendritic cell activation in pulmonary sarcoidosis. Respir Res (2012) 13(1):33.10.1186/1465-9921-13-33
78
Mathew S Bauer KL Fischoeder A Bhardwaj N Oliver SJ . The anergic state in sarcoidosis is associated with diminished dendritic cell function. J Immunol (2008) 181(1):746–55.
79
Kulakova N Urban B McMichael AJ Ho LP . Functional analysis of dendritic cell-T cell interaction in sarcoidosis. Clin Exp Immunol (2010) 159(1):82–6.10.1111/j.1365-2249.2009.04046.x
80
Zou W Restifo NP . T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol (2010) 10(4):248–56.10.1038/nri2742
81
Moller DR . Cells and cytokines involved in the pathogenesis of sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis (1999) 16(1):24–31.
82
Ten Berge B Paats MS Bergen IM van den Blink B Hoogsteden HC Lambrecht BN et al Increased IL-17A expression in granulomas and in circulating memory T cells in sarcoidosis. Rheumatology (Oxford) (2012) 51(1):37–46.10.1093/rheumatology/ker316
83
Robinson BW McLemore TL Crystal RG . Gamma interferon is spontaneously released by alveolar macrophages and lung T lymphocytes in patients with pulmonary sarcoidosis. J Clin Invest (1985) 75(5):1488–95.10.1172/JCI111852
84
Kriegova E Fillerova R Tomankova T Hutyrova B Mrazek F Tichy T et al T-helper cell type-1 transcription factor T-bet is upregulated in pulmonary sarcoidosis. Eur Respir J (2011) 38(5):1136–44.10.1183/09031936.00089910
85
Moller DR Forman JD Liu MC Noble PW Greenlee BM Vyas P et al Enhanced expression of IL-12 associated with Th1 cytokine profiles in active pulmonary sarcoidosis. J Immunol (1996) 156(12):4952–60.
86
Shigehara K Shijubo N Ohmichi M Takahashi R Kon S Okamura H et al IL-12 and IL-18 are increased and stimulate IFN-gamma production in sarcoid lungs. J Immunol (2001) 166(1):642–9.
87
Pechkovsky DV Goldmann T Vollmer E Muller-Quernheim J Zissel G . Interleukin-18 expression by alveolar epithelial cells type II in tuberculosis and sarcoidosis. FEMS Immunol Med Microbiol (2006) 46(1):30–8.10.1111/j.1574-695X.2005.00013.x
88
Droemann D Goldmann T Branscheid D Clark R Dalhoff K Zabel P et al Toll-like receptor 2 is expressed by alveolar epithelial cells type II and macrophages in the human lung. Histochem Cell Biol (2003) 119(2):103–8.10.1007/s00418-003-0497-4
89
Pechkovsky DV Goldmann T Ludwig C Prasse A Vollmer E Muller-Quernheim J et al CCR2 and CXCR3 agonistic chemokines are differently expressed and regulated in human alveolar epithelial cells type II. Respir Res (2005) 6:75.10.1186/1465-9921-6-75
90
Zissel G Homolka J Schlaak J Schlaak M Muller-Quernheim J . Anti-inflammatory cytokine release by alveolar macrophages in pulmonary sarcoidosis. Am J Respir Crit Care Med (1996) 154(3 Pt 1):713–9.10.1164/ajrccm.154.3.8810610
91
Idali F Wiken M Wahlström J Mellstedt H Eklund A Rabbani H et al Reduced Th1 response in the lungs of HLA-DRB1* 0301 patients with pulmonary sarcoidosis. Eur Respir J (2006) 27(3):451–9.10.1183/09031936.06.00067105
92
Ivanov II McKenzie BS Zhou L Tadokoro CE Lepelley A Lafaille JJ et al The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126(6):1121–33.10.1016/j.cell.2006.07.035
93
Bettelli E Carrier Y Gao W Korn T Strom TB Oukka M et al Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441(7090):235–8.10.1038/nature04753
94
Veldhoen M Hocking RJ Atkins CJ Locksley RM Stockinger B . TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24(2):179–89.10.1016/j.immuni.2006.01.001
95
Richmond BW Ploetze K Isom J Chambers-Harris I Braun NA Taylor T et al Sarcoidosis Th17 cells are ESAT-6 antigen specific but demonstrate reduced IFN-γ expression. J Clin Immunol (2013) 33(2):446–55.10.1007/s10875-012-9817-6
96
Yoshida YO Umemura M Yahagi A O’Brien RL Ikuta K Kishihara K et al Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J Immunol (2010) 184(8):4414–22.10.4049/jimmunol.0903332
97
Romani L Fallarino F De Luca A Montagnoli C D’Angelo C Zelante T et al Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature (2008) 451(7175):211–5.10.1038/nature06471
98
Duhen R Glatigny S Arbelaez CA Blair TC Oukka M Bettelli E . Cutting edge: the pathogenicity of IFN-gamma-producing Th17 cells is independent of T-bet. J Immunol (2013) 190(9):4478–82.10.4049/jimmunol.1203172
99
Kebir H Ifergan I Alvarez JI Bernard M Poirier J Arbour N et al Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol (2009) 66(3):390–402.10.1002/ana.21748
100
Kamphuis LS van Zelm MC Lam KH Rimmelzwaan GF Baarsma GS Dik WA et al Perigranuloma localization and abnormal maturation of B cells: emerging key players in sarcoidosis? Am J Respir Crit Care Med (2013) 187(4):406–16.10.1164/rccm.201206-1024OC
101
Ueda-Hayakawa I Tanimura H Osawa M Iwasaka H Ohe S Yamazaki F et al Elevated serum BAFF levels in patients with sarcoidosis: association with disease activity. Rheumatology (Oxford) (2013) 52(9):1658–66.10.1093/rheumatology/ket186
102
Saussine A Tazi A Feuillet S Rybojad M Juillard C Bergeron A et al Active chronic sarcoidosis is characterized by increased transitional blood B cells, increased IL-10-producing regulatory B cells and high BAFF levels. PLoS One (2012) 7(8):e43588.10.1371/journal.pone.0043588
103
Weinberg I Vasiliev L Gotsman I . Anti-dsDNA antibodies in sarcoidosis. Semin Arthritis Rheum (2000) 29(5):328–31.10.1016/S0049-0172(00)80019-0
104
Fischer A Nothnagel M Franke A Jacobs G Saadati HR Gaede KI et al Association of inflammatory bowel disease risk loci with sarcoidosis, and its acute and chronic subphenotypes. Eur Respir J (2011) 37(3):610–6.10.1183/09031936.00049410
105
Steinman L . Mixed results with modulation of TH-17 cells in human autoimmune diseases. Nat Immunol (2010) 11(1):41–4.10.1038/ni.1803
106
Krueger GG Langley RG Leonardi C Yeilding N Guzzo C Wang Y et al A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N Eng J Med (2007) 356(6):580–92.10.1056/NEJMoa062382
107
Sandborn WJ Feagan BG Fedorak RN Scherl E Fleisher MR Katz S et al A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology (2008) 135(4):1130–41.10.1053/j.gastro.2008.07.014
108
Judson MA . Safety and efficacy of treatment with ustekinumab or golimumab in patients with chronic sarcoidosis. Am J Respir Crit Care Med (2013) 187(A1065).
109
Baughman RP . Efficacy of treatment with ustekinumab or golimumab in patients with chronic skin sarcoidosis. Am J Respir Crit Care Med (2013) 187(A1066).
110
Baughman RP Lower EE . Who dies from sarcoidosis and why?Am J Respir Crit Care Med (2011) 183(11):1446–7.10.1164/rccm.201103-0409ED
111
Sakaguchi S Miyara M Costantino CM Hafler DA . FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10(7):490–500.10.1038/nri2785
112
Miyara M Amoura Z Parizot C Badoual C Dorgham K Trad S et al The immune paradox of sarcoidosis and regulatory T cells. J Exp Med (2006) 203(2):359–70.10.1084/jem.20050648
113
Taflin C Miyara M Nochy D Valeyre D Naccache J-M Altare F et al FoxP3+ regulatory T cells suppress early stages of granuloma formation but have little impact on sarcoidosis lesions. Am J Pathol (2009) 174(2):497–508.10.2353/ajpath.2009.080580
114
Rappl G Pabst S Riemann D Schmidt A Wickenhauser C Schütte W et al Regulatory T cells with reduced repressor capacities are extensively amplified in pulmonary sarcoid lesions and sustain granuloma formation. Clin Immunol (2011) 140(1):71–83.10.1016/j.clim.2011.03.015
115
Prasse A Zissel G Lutzen N Schupp J Schmiedlin R Gonzalez-Rey E et al Inhaled vasoactive intestinal peptide exerts immunoregulatory effects in sarcoidosis. Am J Respir Crit Care Med (2010) 182(4):540–8.10.1164/rccm.200909-1451OC
116
Oswald-Richter KA Richmond BW Braun NA Isom J Abraham S Taylor TR et al Reversal of global CD4+ subset dysfunction is associated with spontaneous clinical resolution of pulmonary sarcoidosis. J Immunol (2013) 190(11):5446–53.10.4049/jimmunol.1202891
117
Idali F Wahlström J Müller Suur C Eklund A Grunewald J . Analysis of regulatory T cell associated forkhead box P3 expression in the lungs of patients with sarcoidosis. Clin Exp Immunol (2008) 152(1):127–37.10.1111/j.1365-2249.2008.03609.x
118
Wikén M Grunewald J Eklund A Wahlström J . Multiparameter phenotyping of T-cell subsets in distinct subgroups of patients with pulmonary sarcoidosis. J Intern Med (2012) 271(1):90–103.10.1111/j.1365-2796.2011.02414.x
119
Darlington P Haugom-Olsen H von Sivers K Wahlstrom J Runold M Svjatoha V et al T-cell phenotypes in bronchoalveolar lavage fluid, blood and lymph nodes in pulmonary sarcoidosis – indication for an airborne antigen as the triggering factor in sarcoidosis. J Intern Med (2012) 272(5):465–71.10.1111/j.1365-2796.2012.02543.x
120
Ho LP Urban BC Thickett DR Davies RJ McMichael AJ . Deficiency of a subset of T-cells with immunoregulatory properties in sarcoidosis. Lancet (2005) 365(9464):1062–72.10.1016/S0140-6736(05)71143-0
121
Planey SL Litwack G . Glucocorticoid-induced apoptosis in lymphocytes. Biochem Biophys Res Commun (2000) 279(2):307–12.10.1006/bbrc.2000.3922
122
Baumann S Dostert A Novac N Bauer A Schmid W Fas SC et al Glucocorticoids inhibit activation-induced cell death (AICD) via direct DNA-dependent repression of the CD95 ligand gene by a glucocorticoid receptor dimer. Blood (2005) 106(2):617–25.10.1182/blood-2004-11-4390
123
Chen X Murakami T Oppenheim JJ Howard OM . Differential response of murine CD4+ CD25+ and CD4+ CD25-T cells to dexamethasone-induced cell death. Eur J Immunol (2004) 34(3):859–69.10.1002/eji.200324506
124
Huang Z Yang B Shi Y Cai B Li Y Feng W et al Anti-TNF-alpha therapy improves Treg and suppresses Teff in patients with rheumatoid arthritis. Cell Immunol (2012) 279(1):25–9.10.1016/j.cellimm.2012.09.001
125
ten Hove T Van Montfrans C Peppelenbosch MP Van Deventer SJH . Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn’s disease. Gut (2002) 50(2):206–11.10.1136/gut.50.2.206
126
Sieper J van den Brande J . Diverse effects of infliximab and etanercept on T lymphocytes. Semin Arthritis Rheum (2005) 34(5 Suppl 1):23–7.10.1016/j.semarthrit.2005.01.006
127
van den Brande JMH Braat H van den Brink GR Versteeg HH Bauer CA Hoedemaeker I et al Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn’s disease. Gastroenterology (2003) 124(7):1774–85.10.1016/S0016-5085(03)00382-2
128
Olleros ML Guler R Corazza N Vesin D Eugster H-P Marchal G et al Transmembrane TNF induces an efficient cell-mediated immunity and resistance to Mycobacterium bovis bacillus Calmette-Guerin infection in the absence of secreted TNF and lymphotoxin-α. J Immunol (2002) 168(7): 3394–401.
129
Zissel G Prasse A Muller-Quernheim J . Immunologic response of sarcoidosis. Semin Respir Crit Care Med (2010) 31(4):390–403.10.1055/s-0030-1262208
Summary
Keywords
pulmonary sarcoidosis, granuloma, formation, integrity, dendritic cells, T helper 1 cells, T helper 17 cells, regulatory T cells
Citation
Broos CE, van Nimwegen M, Hoogsteden HC, Hendriks RW, Kool M and van den Blink B (2013) Granuloma Formation in Pulmonary Sarcoidosis. Front. Immunol. 4:437. doi: 10.3389/fimmu.2013.00437
Received
03 October 2013
Accepted
23 November 2013
Published
10 December 2013
Volume
4 - 2013
Edited by
Dov Lewis Boros, Wayne State University School of Medicine, USA
Reviewed by
Daisuke Kamimura, Osaka University, Japan; Hideki Ogura, Osaka University, Japan
Copyright
© 2013 Broos, van Nimwegen, Hoogsteden, Hendriks, Kool and van den Blink.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernt van den Blink, Department of Pulmonary Medicine, Erasmus MC, s-Gravendijkwal 230, 3015 CE, Rotterdam, Netherlands e-mail: b.vandenblink@erasmusmc.nl
This article was submitted to Inflammation, a section of the journal Frontiers in Immunology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.