 Ali Dalloul
Ali Dalloul- INSERM U 996, Clamart, France
Solid organs have been transplanted for decades. Since the improvement in graft selection and in medical and surgical procedures, the likelihood of graft function after 1 year is now close to 90%. Nonetheless even well-matched recipients continue to need medications for the rest of their lives hence adverse side effects and enhanced morbidity. Understanding Immune rejection mechanisms, is of increasing importance since the greater use of living-unrelated donors and genetically unmatched individuals. Chronic rejection is devoted to T-cells, however the role of B-cells in rejection has been appreciated recently by the observation that B-cell depletion improve graft survival. By contrast however, B-cells can be beneficial to the grafted tissue. This protective effect is secondary to either the secretion of protective antibodies or the induction of B-cells that restrain excessive inflammatory responses, chiefly by local provision of IL-10, or inhibit effector T-cells by direct cellular interactions. As a proof of concept B-cell-mediated infectious transplantation tolerance could be achieved in animal models, and evidence emerged that the presence of such B-cells in transplanted patients correlate with a favorable outcome. Among these populations, regulatory B-cells constitute a recently described population. These cells may develop as a feedback mechanism to prevent uncontrolled reactivity to antigens and inflammatory stimuli. The difficult task for the clinician, is to quantify the respective ratios and functions of “tolerant” vs. effector B-cells within a transplanted organ, at a given time point in order to modulate B-cell-directed therapy. Several receptors at the B-cell membrane as well as signaling molecules, can now be targeted for this purpose. Understanding the temporal expansion of regulatory B-cells in grafted patients and the stimuli that activate them will help in the future to implement specific strategies aimed at fighting chronic allograft rejection.
Introduction
Kidneys have been the most frequently transplanted organs for decades. Since the improvements in graft selection and in medical and surgical procedures, the likelihood of graft function after 1 year is now close to 90%. Nonetheless even well-matched recipients continue to need medications for the rest of their lives hence adverse side effects and enhanced morbidity. The half-life of transplanted kidneys is still below 10 years indicating a continuous deterioration of the organ due to combined metabolic and immunologic rejection mechanisms. The later are of increasing importance since the greater use of living-unrelated donors and genetically unmatched individuals. Chronic rejection is not specific of kidney transplants but works equally in other grafts and depends on HLA matching. Chronic rejection is mediated by T-cells and often successfully prevented by immunosuppressive drugs aimed at inhibiting T-cell proliferation. However the role of B-cells in chronic rejection has been appreciated recently in patients and in experimental models. Furthermore, the finding that B-cells can also, be beneficial to the grafted tissue generated great interest. This “tolerizing effect” occurs either through the secretion of protective antibodies or through induction of – recently described – regulatory B-cells and secondary induction of regulatory T-cells.
The Role of B-Cells in Transplant Rejection
Acute and hyperacute rejection are mediated by preformed or by induced antibodies
Induced antibodies. MHC (HLA) antigens and minor antigens that behave like nominal antigens and are presented by HLA molecules to T-cells, are responsible for T-cell alloreactivity. These new determinants expressed on donor tissue ultimately lead to the formation of new antibodies directed at MHC antigens. This T-cell-dependent response generates IgM and then converts to IgG production. These Abs are not responsible for acute graft rejection, however it is now clear that they play a major role in chronic graft destruction (1). They are produced by T-dependent (TD) B2 B-cells with a highly diversified repertoire. This is in contrast with preformed antibodies: anti-HLA Abs occur also following multiple blood transfusions a condition frequently observed in patients on the waiting list for kidney transplantation. Less often they occur as a result of pregnancy or of previous organ transplantation, these Abs are high affinity IgG produced by B2 B-cells. Preformed Antibodies also include natural Abs directed against blood group carbohydrate determinants. These are mostly T-independent (TI) IgM in germline configuration and are produced by a CD5neg B1b B-cell population. Preformed Abs cause hyperacute rejection in vascularized organs such as kidneys and heart. IgM recognize blood groups not only on red cells but also on vascular endothelium, and bind to and activate complement proteins c5–c9, thereby forming the membrane attack complex. This complex activates endothelial cells leading to the loss of antithrombotic molecules and hemorrhage (2) well before cell lysis occurs. Several regulatory molecules such as CD55, CD46, and CD59 control unwanted complement activation (3), however if the affinity of the Abs to the antigen is high enough, it overcomes regulatory molecules of the complement cascade. This is best achieved with IgG anti-HLA Abs. At present there is no treatment of hyperacute rejection, however prevention can be achieved by avoiding blood group disparities, testing the recipient for anti-HLA Abs, so called “cross match” and by plasmapheresis.
Chronic rejection
This condition generally takes 5–10 years to develop even in patients treated with immunosuppressive drugs. Several observations have emerged from clinical practice: the process is associated with the presence of anti-donor antibodies and is refractory to increases in immunosuppressive therapy in contrast to acute rejection episodes. There is also a correlation between the onset of chronic rejection and a history of acute rejection episode. Importantly, pathological manifestations include narrowing of vessels associated with proliferation of endothelial cells and smooth muscle cells (4). The media is progressively destroyed, cellular proliferation halts and cells are replaced by concentric fibrosis. In parallel there is increased expression of adhesion molecules and of NOS, aFGF, IGF1, and endothelin. The process is TD and involves cross priming of recipient T-cells with donor peptides presented by recipient antigen presenting cells (APC) (5). However in experimental models alloantibodies in the absence of T-cells can induce the typical vascular changes and lesions can develop in T-cell deficient mice. In other models graft vasculopathy occurs in T-cell-deficient animal allografts but not isografts. Yet, other models imply IFN-gamma-producing T-cells (Th1) and IL-12-unresponsive (Th1 deficient) STAT4−/− animals show markedly reduced severity of graft vasculopathy (6). Furthermore the respective roles of Th1 vs. Th2 responses are far from clear as both populations may be profibrotic and produce TGFbeta. In addition, Th1-produced IFN-gamma has antiproliferative activity and even propapoptotic activity on T-cells.
B-Cells are Prominent Effectors of Allograft Rejection
B-cells are positive regulators of the immune response. They differentiate in Ab-producing plasma cells. They are APC and as such can stimulate CD4+ T-cells directly (7) although they fail to stimulate naïve T-cells by contrast with professional (dendritic cells) APC (8). B-cells may also stimulate T-cells by producing cytokines, including IL-4, IL-6, and IFN-gamma. CD4+ T-cell activation is indeed impaired in the absence of B-cells in multiple models where B-cell development is blocked since birth (9, 10). B-cells infiltrate chronically rejected allografts, and several groups have reported the presence of ectopic germinal centers within grafted tissues, so called tertiary lymphoid tissues (11–13). Thus, a likely scenario is that B-cells present antigens to T-cells in situ and become activated and proliferate thereby producing a variety of antibodies including anti-self Abs (14). The role of B-cells in chronic graft rejection is not limited to solid organs. In chronic GVHD, B-cells from the donor contribute to the clinical manifestations of the disease and accordingly, B-cell depletion by anti-CD20 MAb/Rituximab improved steroid-resistant chronic GVHD (15). Moreover, responses occurred before the decline in Ab titers again suggesting a cell-mediated effect of B-cells in this process (16). B-cell activation in this setting, is evidenced by elevated BAFF/CD257 levels in patients with active GVHD (17). Although the efficiency of Rituximab is a proof of concept for the role of B-cells in GVHD, the few studies conducted so far have shown limited effect in treating chronic kidney rejection furthermore Rituximab was even deleterious as an induction therapy for acute cellular rejection (18). This suggested that B-cells may be protective in this setting, and strengthened the emerging concept that B-cells within the grafted tissue can be associated with tolerance rather than rejection (19). The “tolerant” or “effector” status of graft-infiltrating B-cells is therefore worth investigating as a prognostic factor in organ transplantation, inasmuch as regulatory B-cells are now much better characterized.
The Concept of “Tolerizing” and Regulatory- Breg- B-Cells, Against Autoimmune Diseases may Now Apply to Allogenic Transplantation
The observation of immunosuppressive B-cells, able to suppress delayed type hypersensitivity (DTH) dates back to 1974 (20, 21). Since then, B-cells have been shown to inhibit inflammation and autoimmunity in various mouse models. Tolerizing B-cells inhibited effector T-cells whether they have been polarized toward effector Th1, Th17, or Th2 cells (22–24). Their effect was often attributed to local provision of IL-10 although in some instances TGFbeta was the prominent cytokine involved in tolerance induction (25).
The data from this abundant literature were ultimately translated into human studies. A CD19+CD24hiCD38hiIL-10-producing regulatory B-cell subset similar to the murine one was described (26). This subset is functionally impaired in human SLE (27) and possibly in multiple sclerosis (28).
The vast majority of the data implying a protective role for B-cells relates to autoimmune diseases however, increasing results imply B-cells as potential inhibitors of allograft rejection. Indeed a B-cell signature is associated with a positive outcome in renal transplantation patients, and is characterized by increased CD19+CD24hiCD38hiIL-10+ B-cells (29). This signature can be useful to detect renal transplant tolerance in humans (30). Thus in given – yet to be determined – conditions, Breg expand in transplanted patients and are able to slow or prevent graft destruction. The other difficulty pertains to the definition of Breg-cells. Although great strides have been made toward understanding their phenotype, IL-10-producing (so called B10) and Breg-cells are not synonymous although these populations clearly overlap.
Mechanisms of Breg Induced Tolerance/Immunosupression
The suppressive capacity of Breg on Th and inflammatory responses has been ascribed chiefly to their secretion of IL-10. This poses the question of how is IL-10 production optimally triggered? In the mouse, B-cells produce high amounts of IL-10 following culture with LPS+ PMA+ Ionomycin, which mimics a combined TLR4 and strong BCR engagement whereas LPS alone induced IL-10 production in plasma cells but not in CD5+CD1d+ Breg-cells (31). Several other studies demonstrated that BCR-signaling is an important component of IL-10 production. We have observed a direct correlation between the intensity of BCR stimulation and IL-10 secretion by a B-cell line (32). An important outcome of BCR stimulation is a very rapid rise in intracytosolic Ca2+ [c]. Mice with a B-cell restricted deficiency in the stromal interaction molecules STIM-1 and -2 Ca2+ sensors, failed to develop IL-10-producing Breg-cells (33). CD40 is a major effector of Breg activation and IL-10 production, indirectly by inducing the expression of CD5 (32) but also directly as shown by the ability of agonistic CD40 Abs to inhibit the Th1 response via IL-10 production (34). However IL-10 may be dispensable as tolerance induction by anti-CD45RB required B-cells but was only dependent on B-7, CD40, and ICAM-1 expression on B-cells but not of IL-10 production (35). Above all, the main effect of IL-10 may reside in the promotion of regulatory T-cells Treg (36, 37), through induction of Foxp3 (38, 39) although IL-10 may be insufficient in this respect and needs to be associated with cognate CD80/CD86 interactions (40).
The BCR interact with several transmembrane positive (CD19) and negative (CD5 and CD22) regulators of signaling. Among surface inhibitors, FcgRIIb/CD32 is of special interest as a tolerance inducer (41). Its inhibitory activity is chiefly based on its immunotyrosine based inhibitory motif ITIM (42, 43). Its low affinity ensures that it interacts with Immune complexes and not with monomeric ligands (41) thereby explaining the efficacy of aggregated intravenous Ig (IVIG). B-cell stimulation through the BCR can be arrested with colligation (physiologically by immune complexes) of FcgRIIb to the BCR. This results in phosphorylation of ITIM on tyrosine and recruitment of the inositol phosphatase SHIP-1 (44) and arrest of Ca2+ influx (45). Meanwhile the Src kinase Lyn is phosphorylated which results in B-cell tolerance (46). The advantage of CD32 is that it is the only Fc receptor present on B-cells (47), its role in the efficacy of intravenous Ig IVIG to treat idiopathic thrombopenic purpura ITP has been therefore suggested (48). Therefore, co-engagement of the BCR with an inhibitory receptor such as CD32b, CD22 (a molecule with an ITIM domain), or CD5 (a molecule with an atypical ITIM domain) is worth investigating. We have indeed previously shown that BCR cross-linked to an Fc-gammaRIIb-intracytosolic CD5 chimera, in a B-cell line downregulated BCR-mediated early signaling events, notably Ca2+ influx and MAP Kinase activation pathway (49).
Phenotype of Breg and Comparison with Other IL-10-Producing B-Cells
There is now a consensus regarding the phenotype of human Breg [for recent reviews, see (50, 51)]. Breg-cells are CD19+IgMhi CD24hiCD5+CD10+CD27+CD38+CD138+CD1dhi, and express intracellular IL-10. However they cannot be sorted based on their CD1dhi or CD24hi expression, which does not delineate discrete B-cell subsets (unpublished results). CD10 is a marker of pro/pre B-cells, whereas CD38+CD24hi expression marks transitional B-cells (52). Likewise, CD5+ B-cells are very heterogeneous and in the adult blood, they are a mixture of transitional B-cells, B1a like B-cells, and activated/memory B2-B-cells (53). In B2-B-cells, CD5 is optimally expressed by stimulation with a combination of CD40-ligand and anti-IgM Abs (32) and produce IL-10. In brief, CD5 is expressed on BCR-engaged B-cells and serves as a negative feed-back to prevent unwanted overreactivity, in part through the secretion of IL-10 and also by downregulating early BCR-signaling. However CD5 expression is transient on B2 B-cells, unless they are triggered permanently by a cognate antigen, such as in the mice double transgenic for Hen Egg Lysozyme HEL and anti-HEL Ig, these animals are tolerant to HEL, however tolerance is broken and autoantibodies produced when the animals are bred into a CD5-null background (54). In contrast, B1a cells which produce natural IgM and IL-10, express “naturally” CD5, however 25% of the recently identified human B1 B-cells do not express surface CD5 (55). Thus, until recently, there were no surface markers exclusive of Breg; the description of T-cell Ig domain and mucin domain TIM-1 on Breg may nevertheless change the picture, inasmuch as TIM-1 ligation induced Breg to promote tolerance in mice (56). The status of the T-cell inhibitory receptor PD-1 in Breg-cells is presently undetermined as it is for PD-Ligands and may be of potential interest since PD-1 is also expressed on B-cells and modulate the humoral immune response [reviewed in Ref. (57)]. Finally, it should be mentioned that although IL-21 is the major cytokine involved in the expansion of B10-cells (58), the level of IL-21R is apparently similar in B10 and non-B10-cells, and thus is by no means a marker for Breg-cells.
Strategies to Inhibit B-Cell-Mediated Rejection
The ultimate goal in transplantation is to achieve tolerance to an allograft; this implies that the transplanted organ remains functional in the absence of immunosuppressive regimen. This is almost never achieved in humans contrary to animal models were adoptively transferred lymphocytes can promote “infectious” tolerance. Current strategies (Table 1) can be summarized in two main objectives: (1) eliminating effector B-cells or inhibiting their differentiation in Ab-producing plasma cells, (2) expanding or stimulating IL-10-producing B-cells/Breg.
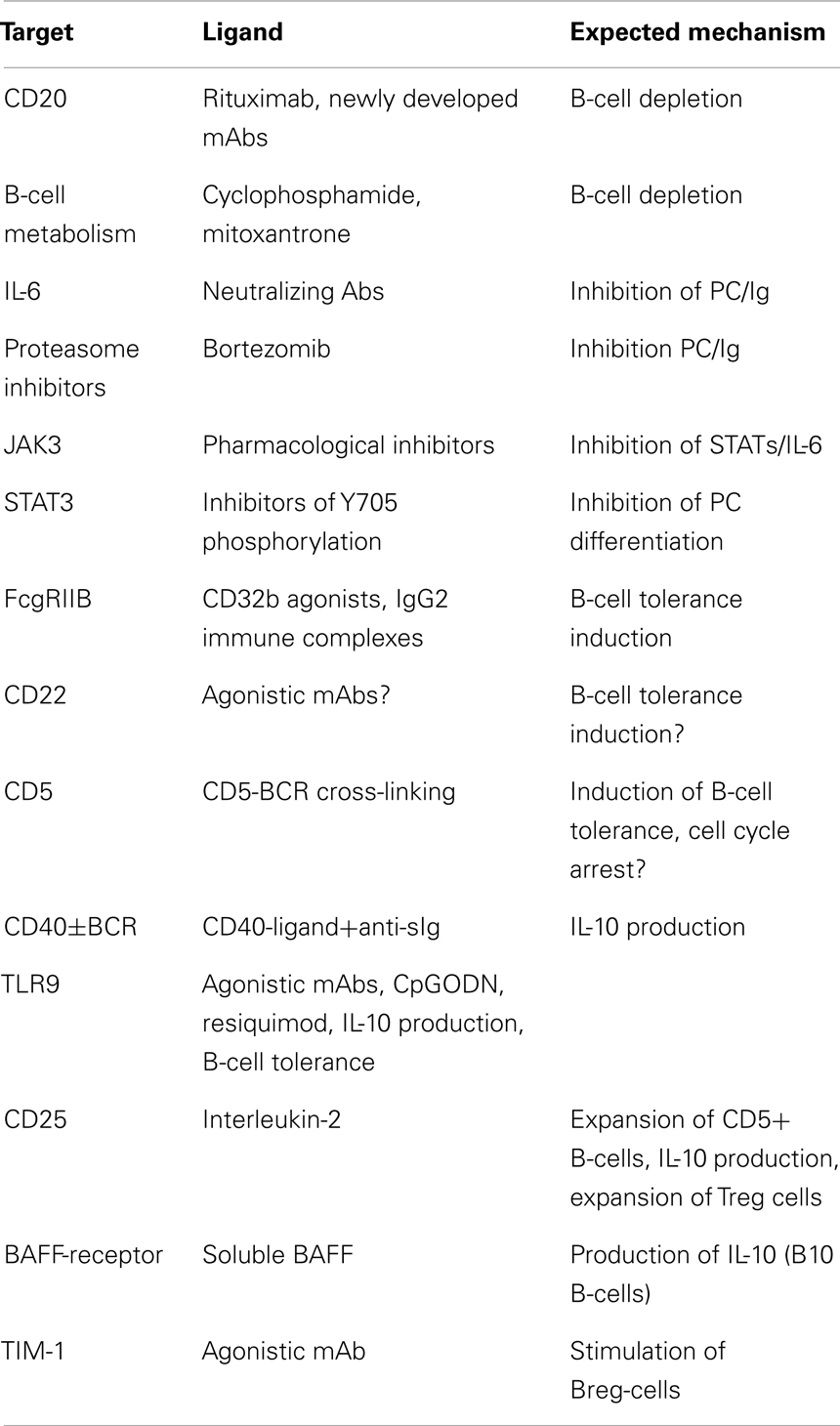
Table 1. Potential B-cell targets for the treatment of allograft rejection.
The first task appears difficult owing to the lack of specific target. Currently as stated above, there is no indication that Rituximab can be used to treat chronic graft rejection, however experimental data suggest that the depletion of alloreactive B-cells at the time of the transplantation may promote tolerance by expanding de novo B-cells, which become “accustomed” to the grafted organ since the beginning of their development (59). Similarly, a same strategy using cyclophosphamide, mycophenolate mofetil, and mitoxantrone can be implemented in order to achieve a mixed chimerism at the time of transplantation (60, 61). In view of the available data, the value of a B-cell-depleting conditioning regimen at the onset of the transplantation, to ameliorate long term organ acceptance should be considered as an option. In contrast the failure of B-cell depletion strategies once chronic rejection has started implies that redundant immune and inflammatory mechanisms involving various immune cells operate and cannot be halted by B-cell depletion. In this setting, expanding anti-inflammatory/regulatory lymphocytes is currently the best therapeutic option. Yet a strategy to inhibit plasma-cell differentiation in order to damp the production of anti-donor HLA Abs can be envisaged. The proteasome inhibitor bortezomib (62), and neutralizing anti-IL-6 Abs are currently studied as inhibitors of plasma-cell expansion; furthermore the anti-IL-6R MAb, Tocilizumab used in the treatment of Rheumatoid arthritis may be effective in transplantation (63). As STAT3 is a transcription factor needed for plasma-cell differentiation, inhibition of STAT3 (64, 65) may also be of interest. Drugs acting upstream STAT3, namely JAK3 inhibitors seem promising in this respect (66).
It is here worth mentioning that Abs may in some instances be protective. The finding that catalytic antibodies able to disrupt the amplification loop of the coagulation cascade (67) with anti-inflammatory properties (68) may instead pave the way for the selective use of IVIG (69) or immune complexes.
The Inhibitory B-cell signature observed in transplantations with favorable outcome (29, 30) included FcgRIIB (70) suggesting an in vivo stimulation of this inhibitory receptor. Another BCR-associated ITIM bearing transmembrane receptor CD22 (71), is a potential B-cell-specific target (72). Breg-cells can expand and be stimulated by CD40 agonists alone or in combination with anti-Ig (34). In addition, whereas cross-linking of CD40 by CD40-ligand induces the maturation of B-cells, prolonged CD40 stimulation inhibits Ig secretion and B-cell differentiation (73). Soluble BAFF was shown to stimulate B10-cells in a mouse model (74) and may be another attractive option. Among inducers of tolerance, the TLR agonists were extensively studied. Agonistic stimulators of TLR-4 or-9 were shown to suppress the course of autoimmune diseases (75–77), which may also explain why infection with salmonella triggered the expansion of IL-10-producing B-cells (78). The well established protective role of apoptotic cells (79) in inducing B10-Breg-cells (80) was highlighted by the observation that MZB and B1 B-cells recognize chromatin complexes and secrete IL-10 through TLR9 transduction pathway (81). As CD5+ B-cells express TLR9, secrete IL-10, and proliferate in response to IL-2 (53), expanding Breg-cells in vivo with IL-2 should be considered as a therapeutic option inasmuch as IL-2 also expands CD25+Treg cells (82). It is also inferred from these data that pharmacological inhibitors of NFAT-IL-2 pathway may be detrimental in chronic allograft rejection. The use of Rapamycin an inhibitor of TOR and an inducer of autophagy, instead of cyclosporine and FK506, should therefore be considered (83). Recently, the pivotal role of IL-21 in the expansion of murine B10-cells was demonstrated (58). Interestingly, B10-cells acquire their regulatory functions after they have been activated by IL-21-producing CD4+T-cells through CD40L/CD40 and CD4/MHC class-II interactions. This may pave the way for the selection and ex vivo expansion of Breg-cells for therapeutic purposes if it turns out that human Breg-cells can be expanded as murine ones.
Altogether, experimental data strongly suggest that some immunological conditions such as infections with bacterias or helminths may surprisingly improve tolerance to allografts by repressing harmful lymphocytes and expanding/activating protective cells among which, Breg-cells play a prominent role (Figure 1). As transplanted organs are connected de novo to the recipient blood stream but not lymphatics, is it likely that the tolerizing signals are brought to the transplanted tissue through the blood. Tolerant lymphocytes may also traffic, and induce tolerance into peripheral lymph nodes (Figure 1). A better knowledge of the grafted tissue microenvironment and a better appreciation of the local balance between inflammatory and regulatory cells and their temporal fate will undoubtedly lead to improvements in the treatment of chronic allograft rejection. Ultimately in situ immunomodulation may tip the balance of intra-graft B-cells toward Breg-cells and possibly expand them locally. Alternatively ex vivo expansion followed by injection of Breg-cells within the transplanted organ can be performed. These strategies may become therapeutic options in the future in the case of resistance to immunosuppressive drugs.
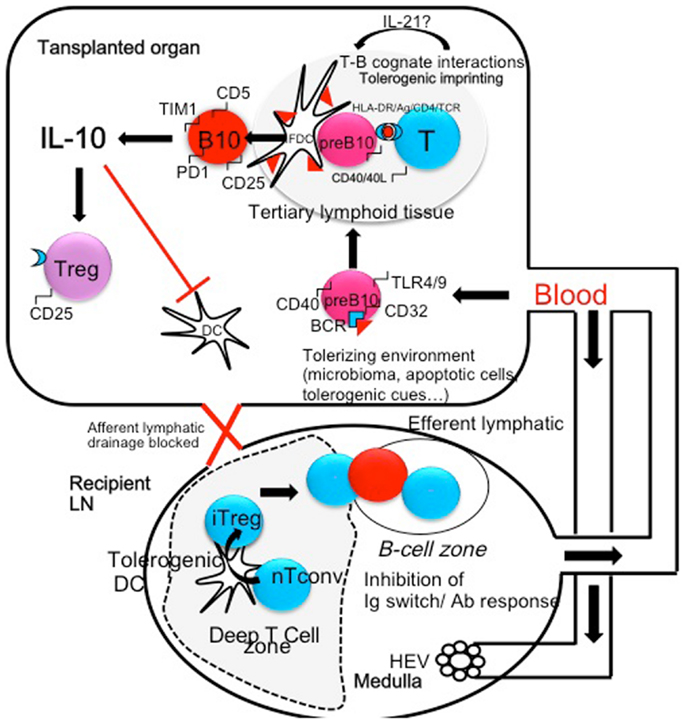
Figure 1. The microenvironment and/or the microbiome (helminthes, parasites, bacterias) can shift B-cells that recognize microbial antigen in the spleen or in the transplanted organ toward regulatory B-cells (pre B10). These cells may be generated in the spleen and colonize the transplanted organ. In turn activated B-cells may present donor MHC peptides to T-cells and undergo cognate T-B interactions in tertiary lymphoid organs through CD40/CD40L, HLA-DR/TCR, and HLA-DR/CD4 interactions. These interactions generate an amplification loop: in the presence of IL-21 secreted by T-cells, B-cells become IL-10-secreting cells, express CD5, TIM1, and PD-1 molecules. In turn, IL-10 inhibits antigen presentation by professional APC (Dendritic cells) and amplifies the production of Treg cells, themselves a potent source of local IL-10. Treg cells migrate to peripheral lymph node through the blood stream and concur to the inhibition of Th1 and Th2 polarization. In addition Treg migrate to the B-cell follicles where they inhibit Antibody secretion and Immunoglobulin isotype switching.
Potential Adverse Side Effects of B-Cell-Based Therapies: Infections
What would be the safety of such therapies? One might expect lesser drawbacks with B-cell-mediated therapies than with pharmacological immunosuppressive regimens especially those pertaining to drug toxicity. However there is no reason to believe that infectious complications similar to those induced by immune-suppression regimen (84) will be avoided. Indeed, activation of Treg cells secondary to that of Breg, may reactivate latent viral infections such as VZV, CMV, EBV, and polyoma (85). Treg cells were also shown to inhibit HCV-specific CD8+ T-cells (86). As mentioned above, infections and the microbiome control many aspects of alloimmunity in transplantation (87). Interestingly, B10 B-cells with a typical Breg phenotype were induced in an experimental model by infecting mice with the red blood cell parasite Babesia microti (88); this led to an enhanced susceptibility to infections and associated with a rise in Treg cells, and was not observed in B-cell-deficient mice. Altogether, it is likely that in vivo or ex vivo amplification of Breg-cells for therapy may lead to enhanced susceptibility to parasitic and possibly bacterial infections.
The role of Interleukin-10 in infections is complex, it may exacerbate T-cell dysfunctions and microbial infections. Indeed, increased circulating IL-10 concentrations have been associated with an adverse clinical outcome in patients with sepsis (89). In contrast some data are consistent with a protective role for IL-10 in kidney (90) lung (91) and heart transplant recipients (92). IL-10 has been shown to protect from septic shock in staphylococcal enterotoxin-induced shock (93). In an experimental model of peritonitis, intravenous injection of Mesenchymal Stem Cells into mice, albeit at a high concentration of 106cells/g body weight, protected animals by triggering IL-10 secretion by macrophages (94). Protection depended only on IL-10 but not of cellular interactions as MSC were trapped into the lungs and did not cross the blood barrier. IL-10 was also shown to control the onset of irreversible septic shock in mice (95). Altogether, strategies aimed at inducing IL-10 secretion by resident intra-graft lymphocytes should be promising and may obviate the adverse effects of systemic IL-10.
Conclusion
Recent progress in understanding the phenotype and functions of tolerant B- and T-lymphocytes rendered possible their analysis in biopsies of allograft tissues; Based on the discovery of new molecular targets in lymphocytes, implementing new strategies based on redirecting graft-infiltrating lymphocytes toward tolerance in order to fight chronic allograft rejection is becoming foreseeable.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Dr. Roman Krzysiek for carefully reading and criticizing the manuscript.
References
1. Loupy A, Hill GS, Jordan SC. The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nat Rev Nephrol (2012) 9:130–1. doi: 10.1038/nrneph.2012.81
3. Sahu A, Lambris JD. Complement inhibitors: a resurgent concept in anti-inflammatory therapeutics. Mol Immunol (2000) 49:133–48.
4. Libby P, Pober JS. Chronic rejection. Immunity (2001) 14:387–97. doi:10.1016/S1074-7613(01)00119-4
5. Shi C, Lee WS, He Q, Zhang D, Fletcher DL Jr, Newell JB, et al. Immunologic basis of transplantation-associated atherosclerosis. Proc Natl Acad Sci U S A (1996) 93:4501–6. doi:10.1073/pnas.93.9.4051
6. Koglin J, Glysing-Jensen T, Gadiraju S, Russell ME. Attenuated cardiac allograft vasculopathy in mice with targeted deletion of the transcription factor STAT4. Circulation (2000) 101:1034–9. doi:10.1161/01.CIR.101.9.1034
7. Ron Y, Sprent J. T cell priming in vivo: a major role for B cells in presenting antigen to T cells in lymph nodes. J Immunol (1987) 138:2848–56.
8. Ronchese F, Hausmann B. B lymphocytes in vivo fail to prime naive T cells but can stimulate antigen-experienced T lymphocytes. J Exp Med (1993) 177:679–90. doi:10.1084/jem.177.3.679
9. Ron Y, De Baetselier P, Gordon J, Feldman M, Segal S. Defective induction of antigen-reactive proliferating T cells in B-cell-deprived mice. Eur J Immunol (1981) 11:964–8. doi:10.1002/eji.1830111203
10. Kitamura D, Roes J, Kühn R, Rajewsky K. A B-cell deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature (1991) 350:423–6. doi:10.1038/350423a0
11. Baddoura FK, Nasr IW, Wrobel B, Li Q, Ruddle NH, Lakkis FG. Lymphoid neogenesis in murine cardiac allografts undergoing chronic rejection. Am J Transplant (2005) 5:510–6. doi:10.1111/j.1600-6143.2004.00714.x
12. Kerjaschki D, Regele HM, Moosberger I, Nagy-Bojarski K, Watschinger B, Soleiman A, et al. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphoid infiltrates. J Am Soc Nephrol (2004) 15:603–12. doi:10.1097/01.ASN.0000113316.52371.2E
13. Thaunat O, Field AC, Dai J, Louedec L, Patey N, Bloch MF, et al. Lymphoid neogenesis in chronic rejection: evidence for a local humoral alloimmune response. Proc Natl Acad Sci U S A (2005) 102:14723–8. doi:10.1073/pnas.0507223102
14. Motallebzadeh R, Rehakova S, Conlon TM, Win TS, Callaghan CJ, Goddard M, et al. Blocking lymphotoxin signaling abrogates the development of ectopic lymphoid tissue within cardiac allografts and inhibits effector antibody responses. FASEB J (2012) 26:51–62. doi:10.1096/fj.11-186973
15. Canninga-van Dijk MR, van der Straaten HM, Fijnheer R, Sanders CJ, van den Tweel JG, Verdonck LF. Anti-CD20 monoclonal antibody treatment in 6 patients with therapy-refractory chronic graft-versus-host disease. Blood (2004) 104:2603–6. doi:10.1182/blood-2004-05-1855
16. Cutler C, Miklos D, Kim HT, Treister N, Woo SB, Bienfang D, et al. Rituximab for steroid-refractory chronic versus graft host disease. Blood (2006) 108:756–62. doi:10.1182/blood-2006-01-0233
17. Kim SJ, Lee JW, Jung CW, Min CK, Cho B, Shin HJ, et al. Weekly rituximab followed by monthly rituximab treatment for steroid refractory chronic graft versus host disease: results from a multicenter phase II study. Haematologica (2010) 11:1935–42. doi:10.3324/haematol.2010.026104
18. Clatworthy MR, Watson CJ, Plotnek G, Bardsley V, Chaudhry AN, Bradley JA, et al. B-cell-depleting induction therapy and acute cellular rejection. N Engl J Med (2009) 360:2683–5. doi:10.1056/NEJMc0808481
19. Brown K, Sacks SH, Wong W. Tertiary lymphoid organs in renal allografts can be associated with donor-specific tolerance rather than rejection. Eur J Immunol (2011) 41:89–96. doi:10.1002/eji.201040759
20. Katz SI, Parker D, Turk JL. B cell suppression of delayed type hypersensitivity reactions. Nature (1974) 251:550. doi:10.1038/251550a0
21. Neta R, Salvin SB. Specific suppression of delayed hypersensitivity: the possible presence of a suppressor B-cell in delayed type hypersensitivity. J Immunol (1974) 113:1716–25.
22. Matsushita T, Fujimoto M, Hasegawa M, Komura K, Takehara K, Tedder TF, et al. Inhibitory role of CD19 in the progression of experimental autoimmune encephalomyelitis by regulating cytokine response. Am J Pathol (2006) 168:812–21. doi:10.2353/ajpath.2006.050923
23. Watanabe R, Fujimoto M, Ishiura N, Kuwano Y, Nakashima H, Yazawa N, et al. CD19 expression in B cells is important for suppression of contact hypersensitivity. Am J Pathol (2007) 171:560–70. doi:10.2353/ajpath.2007.061279
24. Hussain S, Delovitch TL. Intravenous transfusion of BCR-activated B cells protects NOD mice from type 1 diabetes in an IL-10-dependent manner. J Immunol (2007) 179:7225–32.
25. Gonnella PA, Chen YH, Waldner H, Weiner HL. Induction of oral tolerization in CD86 deficient mice: a role for CD86 and B cells in the up-regulation of TGF-beta. J Autoimmun (2006) 26:73–81.
26. Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. CD19+CD24hiCD38hi B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus. Immunity (2010) 32:129–40. doi:10.1016/j.immuni.2009.11.009
27. Iwata S, Saito K, Tokunaga M, Yamaoka K, Nawata M, Yukawa S, et al. Phenotypic changes of lymphocytes in patients with systemic lupus erythematosus who are in longterm remission after B cell depletion therapy with rituximab. J Rheumatol (2011) 38:633–41. doi:10.3899/jrheum.100729
28. Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H, et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implications in multiple sclerosis. J Immunol (2007) 178:6092–9.
29. Newell KA, Asare A, Kirk AD, Gisler TD, Bourcier K, Suthanthiran M, et al. Identification of a B cell signature associated with renal transplant tolerance in humans. J Clin Invest (2010) 120:1836–47. doi:10.1172/JCI39933
30. Sagoo P, Perucha E, Sawitzki B, Tomiuk S, Stephens DA, Miqueu P, et al. Development of a cross-platform biomarker signature to detect renal transplantation tolerance in humans. J Clin Invest (2010) 120:1848–61. doi:10.1172/JCI39922
31. Madan R, Demircik F, Surianarayanan S, Allen JL, Divanovic S, Trompette A, et al. Nonredundant roles for B cell-derived IL-10 in immune counter regulation. J Immunol (2009) 183:2312–20. doi:10.4049/jimmunol.0900185
32. Gary-Gouy H, Harriague J, Bismuth G, Platzer C, Schmitt C, Dalloul AH. Human CD5 promotes B-cell survival through stimulation of autocrine IL-10 production. Blood (2002) 100:4537–43. doi:10.1182/blood-2002-05-1525
33. Matsumoto M, Fujii Y, Baba A, Hikida M, Kurosaki T, Baba Y. The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin-10 production. Immunity (2011) 34:703–14. doi:10.1016/j.immuni.2011.03.016
34. Mauri C, Mars LT, Londei M. Therapeutic activity of agonistic monoclonal antibodies against CD40 in a chronic autoimmune inflammatory process. Nat Med (2000) 6:673–9. doi:10.1038/76251
35. Deng S, Moore DJ, Huang X, Lian MM, Mohiuddin M, Velededeoglu E, et al. Cutting edge: transplant tolerance induced by anti-CD45RB requires B-lymphocytes. J Immunol (2007) 178:6028–32.
36. Sun JB, Flach CF, Czerkinsky C, Holmgren JB. lymphocytes promote expansion of regulatory T cells in oral tolerance: powerful induction by antigen coupled to cholera toxin B subunit. J Immunol (2008) 181:8278–87.
37. Carter NA, Vasconcellos R, Rosser EC, Tulone C, Muñoz-Suano A, Kamanaka M, et al. Mice lacking endogenous IL-10-producing regulatory B-cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol (2011) 186:5569–79. doi:10.4049/jimmunol.1100284
38. Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol (2009) 10:1178–84. doi:10.1038/ni.1791
39. Darrasse-Jèze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med (2009) 206:1853–62. doi:10.1084/jem.20090746
40. Mann MK, Maresz K, Shriver LP, Tan Y, Dittel BN. B-cell regulation of CD4+CD25+ T regulatory cells and IL-10 via B7 is essential for recovery from experimental immune encephalitis. J Immunol (2007) 178:3447–56.
41. Ravetch JV, Lanier LL. Immune inhibitory receptors. Science (2000) 290:84–9. doi:10.1126/science.290.5489.84
42. Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13 amino-acid motif in the cytoplasmic domain of FcgRIIB modulates B-cell receptor signaling. Nature (1994) 369:340–3. doi:10.1038/369340a0
43. Daëron M, Latour S, Malbec O, Espinosa E, Pina P, Pasmans S, et al. The same tyrosine-based inhibition motif, in the intracytoplasmic domain of Fc gamma RIIB, regulates negatively BCR-, TCR-, and FcR-dependent cell activation. Immunity (1995) 3:635–46. doi:10.1016/1074-7613(95)90134-5
44. Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor FcgRIIB. Nature (1996) 383:263–6. doi:10.1038/383263a0
45. Choquet D, Partiseti M, Amigorena S, Bonnerot C, Fridman WH, Korn H. Cross-linking of IgG receptors inhibits membrane Immunoglobulin stimulated calcium influx in B lymphocytes. J Cell Biol (1993) 121:355–63. doi:10.1083/jcb.121.2.355
46. Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, et al. Multiple defects in the immune system of Lyn-deficient mice culminating in autoimmune disease. Cell (1995) 83:301–11. doi:10.1016/0092-8674(95)90171-X
47. Amigorena S, Bonnerot C, Drake JR, Choquet D, Hunziker W, Guillet JG, et al. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science (1992) 256:1808–12.
48. Samuelsson A, Towers TL, Ravetch JV. Anti inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science (2001) 291:484–6. doi:10.1126/science.291.5503.484
49. Gary-Gouy H, Bruhns P, Schmitt C, Dalloul A, Daëron M, Bismuth G. The pseudo immunoreceptor tyrosine-based activation motif of CD5 mediates its inhibitory action on B-cell receptor signaling. J Biol Chem (2000) 275:548–56. doi:10.1074/jbc.275.1.548
50. Bouaziz JD, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev (2008) 224:201–14. doi:10.1111/j.1600065X.2008.00661.x
51. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol (2012) 30:221–41. doi:10.1146/annurev-immunol-020711-074934
52. Carsetti R, Rosado MM, Wardmann H. Peripheral development of B cells in mouse and man. Immunol Rev (2004) 197:179–91. doi:10.1111/j.0105-2896.2004.0109.x
53. Dalloul A. CD5: a safeguard against autoimmunity and a shield for cancer cells. Autoimmun Rev (2009) 8:349–53. doi:10.1016/j.autrev.2008.11.007
54. Hippen KL, Tze LE, Behrens TW. CD5 maintains tolerance in anergic B-cells. J Exp Med (2000) 191:883–90.
55. Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70-. J Exp Med (2011) 208:67–80. doi:10.1084/jem.20101499. Erratum in: J Exp Med (2011) 208(4):871; J Exp Med (2011) 208(2):409; J Exp Med (2011) 208(1):67.
56. Ding Q, Yeung M, Camirand G, Zeng Q, Akiba H, Yagita H, et al. Regulatory B cells are identified by expression of TIM-1 and can be induced through TIM-1 ligation to promote tolerance in mice. J Clin Invest (2011) 121:3645–56. doi:10.1172/JCI46274
57. Kamphorst AQ, Ahmed R. Manipulating the PD-1 pathway to improve immunity. Curr Opin Immunol (2013) 25:381–8. doi:10.1016/j.coi.2013.03.003
58. Yoshizaki A, Miyagaki T, Di Lillo D, Matsushita T, Horikawa M, Kountikov E, et al. Regulatory B cells control T-cell autoimmunity through IL-21- dependent cognate interactions. Nature (2012) 491:264–8. doi:10.1038/nature11501
59. Parsons RF, Vivek K, Rostami SY, Zekavat G, Ziaie SM, Luo Y, et al. Acquisition of humoral transplantation tolerance upon de novo emergence of B lymphocytes. J Immunol (2011) 186:614–20. doi:10.4049/jimmunol.1002873
60. Snanoudj R, Beaudreuil S, Arzouk N, de Preneuf H, Durrbach A, Charpentier B. Immunological strategies targeting B cells in organ grafting. Transplantation (2005) 79:S33–6. doi:10.1097/01.TP.0000153298.48353.A4
61. Ashton-Chess J, Meurette G, Karam G, Petzold T, Minault D, Naulet J, et al. The study of mitoxantrone as a potential immunosuppressor in transgenic pig renal xenotransplantation in baboons: comparison with cyclophosphamide. Xenotransplantation (2004) 11:112–22. doi:10.1111/j.1399-3089.2004.00040.x
62. Walsh RC, Alloway RR, Girnita AL, Woodle ES. Proteasome inhibitor-based therapy for antibody-mediated rejection. Kidney Int (2012) 81:1067–74. doi:10.1038/ki.2011.502
63. Gergis U, Arnason J, Yantiss R, Shore T, Wissa U, Feldman E, et al. Effectiveness and safety of tocilizumab, an anti-interleukin-6 receptor monoclonal antibody, in a patient with refractory GI graft-versus-host disease. J Clin Oncol (2010) 28(30):e602–4. doi:10.1200/JCO.2010.29.1682
64. Diehl SA, Schmidlin H, Nagasawa M, van Haren SD, Kwakkenbos MJ, Yasuda E, et al. STAT3-mediated upregulation of BLIMP1 is coordinated with BCL2 downregulation to control human plasma cell differentiation. J Immunol (2008) 180:4805–15.
65. Maarof G, Bouchet-Delbos L, Gary-Gouy H, Durand-Gasselin I, Krzysiek R, Dalloul A. Interleukin-24 inhibits the plasma-cell differentiation program in human germinal center B-cells. Blood (2010) 115:1718–26. doi:10.1182/blood-2009-05-220251
66. West K. CP-690550, a JAK3 inhibitor as an immunosuppressant for the treatment of rheumatoid arthritis, transplant rejection, psoriasis and other immune-mediated disorders. Curr Opin Investig Drugs (2009) 10:491–504.
67. Wootla B, Nicoletti A, Patey N, Dimitrov JD, Legendre C, Christophe OD, et al. Hydrolysis of coagulation factors by circulating IgG is associated with a reduced risk for chronic allograft nephropathy in renal transplanted patients. J Immunol (2008) 180:8455–60.
68. Lacroix-Desmazes S, Bayry J, Kaveri SV, Hayon-Sonsino D, Thorenoor N, Charpentier J, et al. High levels of catalytic antibodies correlate with favorable outcome in sepsis. Proc Natl Acad Sci U S A (2005) 102:4109–13. doi:10.1073/pnas.0500586102
69. Vo AA, Peng A, Toyoda M, Kahwaji J, Cao K, Lai CH, et al. Use of intravenous immunoglobulins and rituximab for desensitization of highly HLA-sensitized patients awaiting kidney transplants. Transplantation (2010) 89:1095–102. doi:10.1097/TP.0b013e3181d21e7f
70. Le Texier L, Thebault P, Lavault A, Usal C, Merieau E, Quillard T, et al. Long-term allograft tolerance is characterized by the accumulation of B cells exhibiting an inhibited profile. Am J Transplant (2011) 11:429–38. doi:10.1111/j.1600-6143.2010.03336.x
71. Smith KG, Tarlinton DM, Doody GM, Hibbs ML, Fearon DT. Inhibition of B cells by CD22: a requirement for Lyn. J Exp Med (1998) 187:807–11. doi:10.1084/jem.187.5.807
72. Jordan SC, Kahwaji J, Toyoda M, Vo A. B cell immunotherapeutics: emerging role in solid organ transplantation. Curr Opin Organ Transplant (2011) 16:416–24. doi:10.1097/MOT.0b013e32834874f7
73. Miyashita T, McIlraith MJ, Grammer AC, Miura Y, Attrep JF, Shimaoka Y, et al. Bidirectional regulation of human B-cell responses by CD40-CD40 ligand interaction. J Immunol (1997) 158:4620–33.
74. Yang M, Sun L, Wang S, Ko KH, Xu H, Zheng BJ, et al. Novel function of B cell-activating factor in the induction of IL-10-producing regulatory B cells. J Immunol (2010) 184:3321–5. doi:10.4049/jimmunol.0902551
75. Buenafe AC, Bourdette DN. Lipopolysaccharide pretreatment modulate the disease course in experimental autoimmune encephalomyelitis. J Neuroimmunol (2007) 182:32–40. doi:10.1016/j.jneuroim.2006.09.004
76. Quintana FJ, Rotem A, Carmi P, Cohen IR. Vaccination with empty plasmid DNA or CpG oligonucleotide inhibits diabetes in nonobese mice: modulation of 60kVa heat shock protein autoimmunity. J Immunol (2000) 165:6148–55.
77. Wu HJ, Sawaya H, Binstadt B, Brickelmaier M, Blasius A, Gorelik L, et al. Inflammatory arthritis can be reined in by CpG-induced DC-NK cell cross talk. J Exp Med (2007) 204:1911–22. doi:10.1084/jem.20070285
78. Neves P, Lampropoulou V, Calderon-Gomez E, Roch T, Stervbo U, Shen P, et al. Signaling via the MyD88 adaptor protein in B-cells suppresses protective immunity Salmonella typhimurium during infection. Immunity (2010) 33:777–90. doi:10.1016/j.immuni.2010.10.016
79. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive properties of apoptotic cells. Nature (1997) 390:350–1. doi:10.1038/37022
80. Gray M, Miles K, Salter D, Gray D, Savill J. Apoptotic cells protect mice from autoimmune inflammation by the induction of regulatory B cells. Proc Natl Acad Sci U S A (2007) 104:14080–5. doi:10.1073/pnas.0700326104
81. Miles K, Heaney J, Sibinska Z, Salter D, Savill J, Gray D, et al. A tolerogenic role for Toll-like receptor 9 is revealed by B-cell interactions with DNA complexes expressed on apoptotic cells. Proc Natl Acad Sci U S A (2012) 109:887–92. doi:10.1073/pnas.1109173109
82. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self tolerance maintained by activated T cells expressing IL2 receptor a chains (CD25). J Immunol (1995) 155:1151–64.
83. Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol (2010) 12:842–6. doi:10.1038/ncb0910-842
84. Fishman JA. Infections in solid organ transplantation recipients. N Engl J Med (2007) 357:2601–14. doi:10.1056/NEJMra064928
85. Fishman JA. Opportunistic infections-coming to the limits of immunosupression. Cold Spring Harb Perspect Med (2013) 3(10):a015669. doi:10.1101/cshperspect.a015669
86. Spangenberg HC, Viazov S, Kersting N, Neumann-Haefelin C, McKinney D, Roggendorf N, et al. Intrahepatic CD8+ T-cell failure during chronic hepatitis C virus infection. Hepatology (2005) 42:828–37. doi:10.1002/hep.20856
87. Chong AS, Alegre ML. The impact of infection and tissue damage in solid organ transplantation. Nat Rev Immunol (2012) 12:459–71. doi:10.1038/nri3215
88. Jeong Y-I, Hong S-H, Cho S-H, Lee W-J, Lee S-E. Induction of IL-10-producing CD1dhiCD5+ regulatory B-cells following Babesia microti-infection. PLoS One (2013) 7:e46553. doi:10.1371/journal.pone.0046553
89. Oberholzer A, Oberholzer C, Moldawer LL. Interleukin-10: a complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med (2002) 30:S58–63. doi:10.1097/00003246-200201001-00008
90. Burke GW, Ciancio G, Cirocco R, MArkou M, Coker D, Roth D, et al. Association of interleukin-10 with rejection-sparing effect in septic kidney transplant recipients. Transplantation (1996) 61:1114–6. doi:10.1097/00007890-199604150-00023
91. Boehler A. The role of interleukin-10 in lung transplantation. Transpl Immunol (2002) 9:121–4. doi:10.1016/S0966-3274(02)00045-X
92. Fischbein MP, Yun J, Laks Irie Y, Oslund-Pinderski L, Fischbein MC, Bonavida B, et al. REgulated interleukin-10 expression prevents chronic rejection of transplanted hearts. J Thorac Cardiovasc Surg (2003) 126:216–23. doi:10.1016/S0022-5223(03)00026-6
93. Florquin S, Amraoui Z, Abramovicz D, Goldman M. Sustemic release and protective role of IL-10 in staphylococcal enterotoxin B-induced shock in mice. J Immunol (1994) 153:2618–23.
94. Németh K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med (2009) 15:42–9. doi:10.1038/nm.1905
Keywords: transplantation, chronic rejection, B-cells, regulatory B-cells, immunomodulation
Citation: Dalloul A (2013) B-cell-mediated strategies to fight chronic allograft rejection. Front. Immunol. 4:444. doi: 10.3389/fimmu.2013.00444
Received: 20 August 2013; Accepted: 26 November 2013;
Published online: 17 December 2013.
Edited by:
Seiamak Bahram, Université de Strasbourg, FranceCopyright: © 2013 Dalloul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali Dalloul, INSERM U 996, 32 Rue des Carnets, Clamart 92140, France e-mail:YWxpLmRhbGxvdWxAdS1wc3VkLmZy
†Alternative address: Ali Dalloul, Thérapie Cellulaire, CHU, Nancy, France e-mail:YWxpLmRhbGxvdWxAdW5pdi1sb3JyYWluZS5mcg==