 Birthe Jessen1†
Birthe Jessen1† Tamara Kögl2†
Tamara Kögl2† Fernando E. Sepulveda3,4†
Fernando E. Sepulveda3,4† Genevieve de Saint Basile3,4,5#
Genevieve de Saint Basile3,4,5# Peter Aichele2#
Peter Aichele2# Stephan Ehl1#*
Stephan Ehl1#*- 1Centre of Chronic Immunodeficiency, University Medical Center Freiburg, University of Freiburg, Freiburg, Germany
- 2Department for Medical Microbiology and Hygiene, Institute of Immunology, University Medical Center Freiburg, University of Freiburg, Freiburg, Germany
- 3U768, INSERM, Paris, France
- 4Institut Imagine, Université Paris Descartes-Sorbonne Paris Cité, Paris, France
- 5Centre d’Etudes des Déficits Immunitaires, Assistance Publique-Hôpitaux de Paris, Hôpital Necker, Paris, France
Primary hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disease of hyperinflammation resulting from immune dysregulation due to inherited defects in the cytolytic machinery of natural killer and T cells. In humans, mutations in seven genes encoding proteins involved in cytolytic effector functions have so far been identified that predispose to HLH. However, although most affected patients develop HLH eventually, disease onset and severity are highly variable. Due to the genetic heterogeneity and variable time and nature of disease triggers, the immunological basis of these variations in HLH progression is incompletely understood. Several murine models of primary HLH have been established allowing to study HLH pathogenesis under more defined conditions. Here we directly compare the clinical HLH phenotype in six HLH-prone mouse strains with defects in the granule-dependent cytotoxic pathway. A severity gradient of HLH manifestations could be identified that is defined by the genetically determined residual lytic activity of cytotoxic T lymphocytes (CTL) and their ability to control lymphocytic choriomeningitis virus, which was used as a trigger for disease induction. Importantly, analysis of cohorts of HLH patients with severe bi-allelic mutations in the corresponding genes yielded a similar severity gradient in human HLH as reflected by the age at disease onset. Our findings define HLH as a threshold disease determined by subtle differences in the residual lytic activity of CTL.
Introduction
Primary hemophagocytic lymphohistiocytosis (HLH) is a rare life-threatening syndrome of hyperinflammation due to genetic defects in the perforin-dependent granule exocytosis pathway of natural killer (NK) and T cells (1–4). The HLH syndrome is characterized by uncontrolled inflammatory and immunopathological processes in various tissues as a result of infiltrating, excessively activated T cells, NK cells, and macrophages, accompanied by a massive cytokine production (IFN-γ, TNF, IL-6, IL-18) (5–7). Due to this loss in immune homeostasis, HLH patients present with prolonged fever, hepatosplenomegaly, severe cytopenia, and frequently with neurologic manifestations. In addition, elevated serum levels of ferritin, triglycerides, soluble CD25 (IL-2 receptor α chain), and liver enzymes, as well as hemophagocytosis in various tissues and reduced cytolytic activity of lymphocytes are characteristic criteria for HLH (Table 1) (8). Typically, patients with primary HLH develop disease in early childhood with a poor prognosis in the absence of therapeutic intervention (9–11).

Table 1. Diagnostic criteria for HLH.
Traditionally, “familial HLH” (FHL) has been defined as a genetic disease, in which the predisposition to HLH is the dominant feature (PERFORIN deficiency, MUNC13-4 deficiency, SYNTAXIN-11 deficiency, and MUNC18-2 deficiency) (12–17), while “immunodeficiencies with albinism” (Chediak–Higashi syndrome (CHS) or LYST deficiency, Griscelli syndrome type 2 (GS2) or RAB27A deficiency, and Hermansky–Pudlak syndrome type 2 (HPS2) or AP3b1 deficiency) (18–22) combine this predisposition with clinical manifestations of albinism and variable degrees of other immune cell and platelet dysfunction (23–28). From a pathophysiological viewpoint, this distinction is arbitrary. First, all genes mutated in these two groups of conditions are critically involved in the biogenesis, intracellular transport, release, and function of perforin-containing lytic granules of NK and T cells (1). Second, it becomes increasingly obvious that defects in platelets and other immune cells such as neutrophils or mast cells are also observed in diseases currently classified as FHL (29–33). Because the genetic predisposition to HLH is the dominant life-threatening clinical feature in all of these diseases, we prefer to classify them collectively as familial HLH syndromes (FHL syndromes).
While the overall pattern of clinical manifestations of HLH in patients with the different FHL syndromes is quite characteristic, onset of disease, severity of clinical symptoms, and duration of disease-free remission periods are highly variable (31, 34–36). This depends not only on the affected gene, but also on the nature of the mutation (null or hypomorphic) and the time point and nature of exposure to predominantly infectious triggers that can elicit HLH in predisposed individuals. In addition, in >60% of patients with FHL syndromes, no clear trigger for HLH can be identified and it is still a matter of debate whether an exogenous trigger is needed for disease induction at all (37–40). This variability makes it difficult to define the a priori risk of an individual patient to develop HLH in the different human FHL syndromes. A study of additional functional parameters may help to improve the predictability of HLH progression. For example, it is so far not clear, in what hierarchy the dysfunction of the different affected proteins becomes limiting for in vivo cytotoxicity.
In this context, animal models of FHL syndromes have proven useful to analyze the pathogenesis of HLH under more defined conditions. In 2004, Jordan et al. reported that following lymphocytic choriomeningitis virus (LCMV) infection as initial trigger, perforin-deficient (PKO) mice develop the full clinical picture of HLH as it is described for FHL2 patients (41). It was demonstrated that hyperactive cytotoxic T lymphocytes (CTL) and high levels of IFN-γ are the driving force behind the development of fatal HLH in PKO mice. Non-fatal HLH was observed after LCMV infection of Jinx mice (model for FHL3) (42), STX-11-deficient mice (model for FHL4) (43, 44), ashen mice (model for GS2) (45), souris mice (model for CHS) (46), and pearl mice (model for HPS2) (20). Although some of these strains were compared directly in these publications, the different mouse models were not analyzed in parallel under identical experimental conditions with standardized immunological and clinical criteria for HLH. Therefore, the relative risk for HLH development in these models in relation to the individual genetic defect and its consequences for cytotoxicity have not been fully defined. Moreover, the role of virus control and a potential contribution of the various proteins in processes other than cytotoxicity to the pathogenesis of HLH remain controversial.
In the present study we therefore performed a comprehensive comparative analysis of the clinical and immunological HLH phenotype in six different mouse models of FHL syndromes. In addition, recently published results on HLH severity (as determined by age at onset of HLH) in patients with FHL syndromes due to severe bi-allelic mutations were extended to additional genetic conditions (44). We discuss our results in the context of the overall value of LCMV-induced HLH in various murine cytotoxicity mutants for the understanding of human FHL syndromes and point out some key questions to be addressed in human and mouse models in the future.
Results
HLH Severity Differs in Various Mouse Models of FHL Syndromes
To analyze the impact of different defects in the cytotoxicity pathway of T and NK cells on HLH development, we assessed HLH parameters following intravenous LCMV infection in six established and previously described HLH-prone mouse models under identical experimental conditions with standardized read-out systems. The following mouse strains were used in this study: two mouse models for CHS carrying different mutations in the Lyst gene – beige and souris mice – (46), one mouse model for HPS2 deficient in AP-3 – pearl mice – (20), a Rab27a-deficient mouse model for GS2 – ashen mice – (45), one model for familial hemophagocytic lymphohistiocytosis (FHL) 2 deficient in perforin – PKO mice – (41, 47), and one model for FHL4 deficient in syntaxin-11 – STX-11KO mice – (43, 44). As previously described, none of these mutant mouse strains develop disease spontaneously. Infection with LCMV was used to induce disease.
Following LCMV infection, mice were weighed and ear temperature was taken daily. Mice were analyzed either at day 8 or at day 12 after infection and all eight criteria (Table 1) defined by the HLH study group of the Histiocyte Society for the diagnostic evaluation of patients with suspected HLH were determined (8). A drop in ear temperature due to circulatory centralization was taken as an equivalent of fever in humans. In addition, we measured lactate dehydrogenase (LDH) and glutamate dehydrogenase (GLDH) reflecting liver damage and IFN-γ serum levels, which have been shown to correlate well with HLH activity in mice (41, 48). Weight loss as a rough, but easily accessible measure of disease revealed a clear hierarchy of HLH severity in the six mutant mouse strains (Figure 1). While there was no weight loss in wild-type and beige mice, weight loss was transient until day 8 in pearl mice, progressive but moderate in souris mice and equally severe in STX-11KO, ashen, and PKO mice.
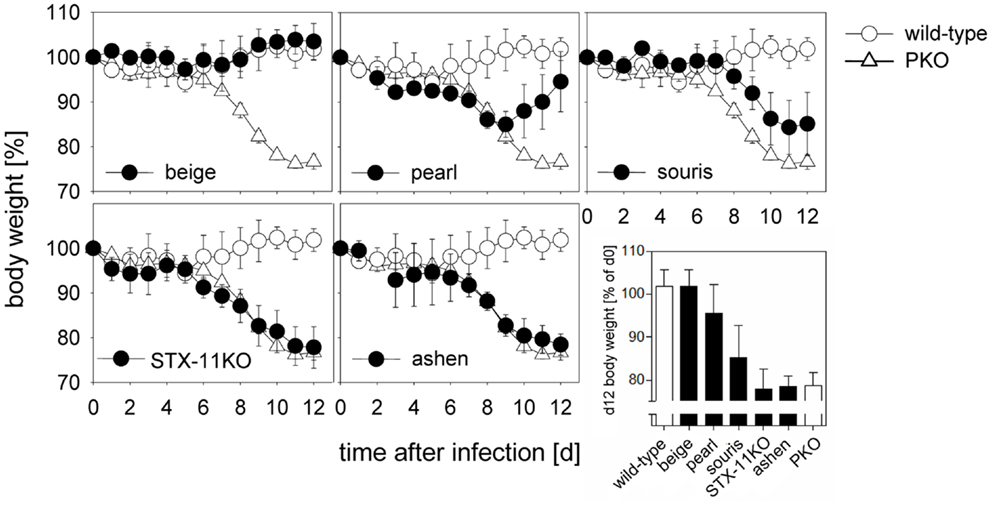
Figure 1. Degree of weight loss after LCMV infection of different HLH-prone mouse models depends on the affected gene. Mice were infected with 200 pfu LCMV i.v. Percent weight loss of initial body weight is depicted over the course of 12 days. Body weight of mutant mice (beige, pearl, souris, STX-11KO, ashen/filled circles) in comparison with wild-type (open circle) and PKO (open triangle) mice are shown. The graph depicted on the lower right illustrates a direct comparison of body weight loss of all mouse groups on day 12 after LCMV infection.
A similar hierarchy of disease severity was observed, when the formal diagnostic HLH criteria were evaluated on day 12 (Table 2). LCMV infection of wild-type mice led to splenomegaly and rare hemophagocytosis, but the other HLH criteria were not fulfilled. Beige mice, carrying a hypomorphic mutation in the lyst gene, in addition had low NK cell cytotoxicity, but no other HLH features (Table 2). Pearl mice in addition had cytopenia, elevated ferritin, liver enzymes, and IFN-γ and thus fulfilled 5/8 diagnostic criteria at day 8 after infection (Table A1 in Appendix), but – as reported – disease was transient and the criteria were not fulfilled at day 12 (Table 2). All other strains fulfilled 7/8 (apart from elevated triglycerides) or 8/8 diagnostic criteria at day 12 and additionally had elevated liver enzymes and IFN-γ levels, which were more pronounced at day 8 after infection. Nevertheless, some notable differences were observed: first, the drop in temperature and alterations in hemoglobin, ferritin, sCD25, and triglycerides were less severe in souris mice than in the other three mouse strains – STX-11KO, ashen, PKO – although IFN-γ levels were similar or even higher. Second, alterations in platelet counts, ferritin, and sCD25 were less pronounced in STX-11KO than in ashen or PKO mice. Third, levels of ferritin and IFN-γ on day 8 after infection were higher in PKO than in ashen mice. Finally, previous experiments assessing survival beyond day 12 have shown that disease is usually lethal in PKO mice, sometimes lethal in ashen mice and not lethal in any of the other investigated mutant mouse strains (data not shown). Overall, HLH severity as assessed by weight loss, survival, and HLH criteria showed the following hierarchy: wild-type < beige (no HLH) < pearl (transient HLH) < souris < STX-11KO < ashen < PKO (full HLH).
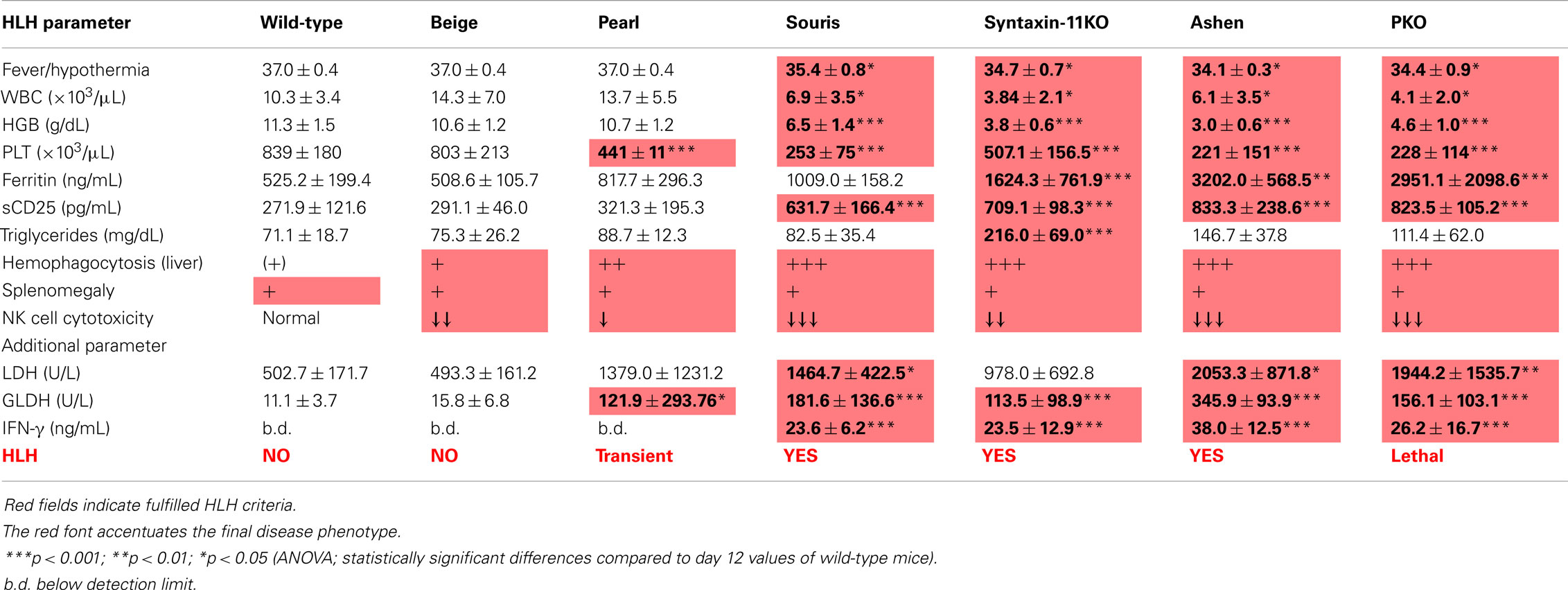
Table 2. Analysis of HLH diagnosis parameters in various HLH-prone mouse models on day 12 after LCMV infection.
Disease Development in HLH Mouse Models Correlates with Virus Persistence
A comparison of virus titers in the spleen of the different HLH-prone mouse strains confirmed that virus persistence is one of the key characteristics of HLH development. Mice that showed no signs of disease such as wild-type and beige mice were able to reduce virus titers until day 8 and no virus was detectable at day 12 (Figure 2). In pearl mice, the transient HLH at day 8 was associated with a delay in virus control at this time point. When these mice recovered eventually, virus elimination was achieved. All of the mice fulfilling the criteria of HLH – irrespective of disease severity – failed to reduce or eliminate the virus until day 12 (Figure 2) and were persistently infected with similar titers in the spleen. Thus, virus persistence appears to be a prerequisite for the development of HLH in the various mouse models of impaired cytotoxicity, but disease severity does not correlate with titers of persisting virus.
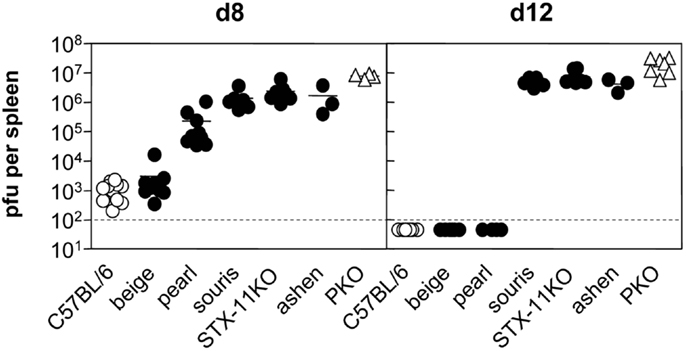
Figure 2. Virus control following LCMV infection differs between various HLH-prone mouse models. Mice were infected with 200 pfu LCMV i.v. On day 8 and day 12 after LCMV infection, viral titers in spleen of beige, pearl, souris, STX-11KO, and ashen mice (filled circle) in comparison to wild-type (open circle) and PKO (open triangle) mice were determined.
Graded Defects in CTL Cytotoxicity Determine Outcome of Disease
As extensively discussed before (47, 49–53), the cytotoxicity of CD8 T cells is the major factor in virus control following LCMV infection and the major determinant in preventing HLH development (41, 43, 45). Here, we directly compared CTL degranulation and CTL cytotoxicity in the six different HLH-prone mouse strains on day 8 after LCMV infection. First, we analyzed the degranulation capacity of virus-specific CTL in the different mouse strains by quantifying the expression of CD107a on IFN-γ positive CTL upon antigen-specific in vitro stimulation with gp33 peptide, the immunodominant epitope of LCMV. CTL from the four mutant mouse strains that developed the full picture of HLH – souris, STX-11KO, ashen, PKO – continuously produced high levels of IFN-γ even in the absence of stimulation (Figure 3A, upper panel), while this was not the case in the other strains. Interestingly, the grade of ex vivo IFN-γ expression of CTL correlated very well with IFN-γ levels in serum and disease severity (Figure 3C). In contrast, the grade of the degranulation defect did not completely reflect the observations on disease severity. The degranulation defect was more pronounced in beige (no HLH) than in pearl mice (transient HLH) and no difference could be found in the degranulation defect between STX-11KO and ashen mice. Unexpectedly, a mild reduction in degranulation was also observed in PKO mice (Figure 3B), although a role for perforin in the process of granule exocytosis has so far not been described. Analyzing the ex vivo cytolytic activities of CTL from the different mouse strains revealed a graded impairment of cytotoxicity from beige to PKO CTL that perfectly reflected distinct disease severity (Figure 3D). In addition, as reported previously, the cytotoxicity defect was more pronounced in PKO than in ashen or STX-11KO mice.
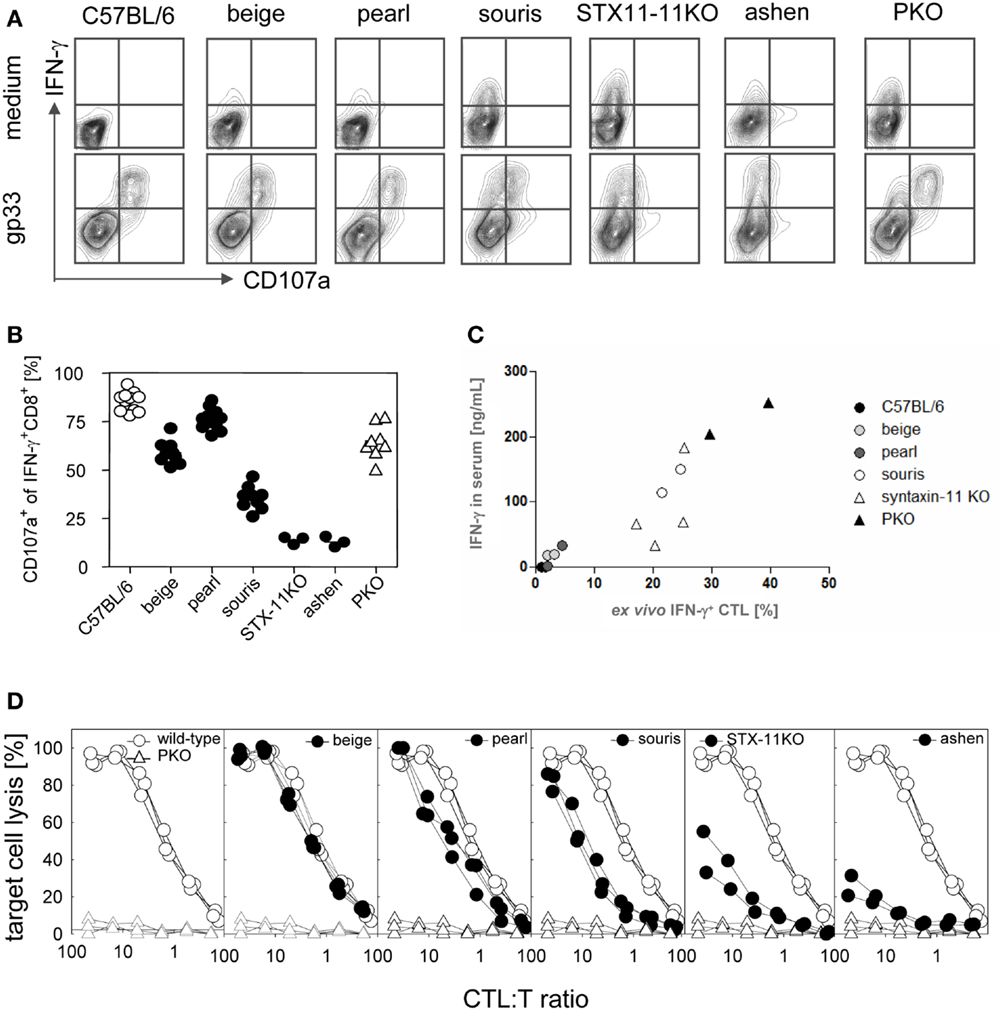
Figure 3. Cytotoxic T lymphocytes degranulation and cytotoxicity are differently impaired in various HLH-prone mouse models. Mice were infected with 200 pfu LCMV i.v. On day 8 after LCMV infection, spleen cells of mutant mice (beige, pearl, souris, STX-11KO, ashen/filled circles) and wild-type (open circle) and PKO (open triangle) mice were restimulated with gp33 peptide or were left in medium without peptide. Surface expression of CD107a of IFN-γ+CD8+ T cells was determined by flow cytometry as illustrated by (A) representative FACS plots. (B) Percentage of CD107a expressing IFN-γ+CD8+ T cells after restimulation with gp33 peptide. (C) Correlation of percentage of CTL expressing IFN-γ without restimulation ex vivo with the IFN-γ levels in sera of the various HLH-prone mutant mice. (D) CTL cytotoxicity was determined in a 51Cr-release assay by using LCMV-infected MC57 target cells.
In a next step, we functionally evaluated the cytotoxic activity of CTL derived from the different HLH-prone mouse strains in an in vivo assay of virus control (50). For this, CTL were adoptively transfered into wild-type mice that had been infected with LCMV 10 h previously (Figure 4). In this assay, CTL from wild-type mice eliminated the virus from the spleen within 18 h, while CTL from the four strains that developed the full picture of HLH – souris, STX-11KO, ashen, PKO – had no impact on viral titers and failed to clear LCMV. Despite a more pronounced defect in degranulation – but not in cytotoxicity – beige CTL cleared the virus, while pearl CTL had an intermediate effect on virus clearance. Taken together, when CTL from the different HLH-prone mice failed to eliminate LCMV in this short-term protection assay, the mice developed HLH independent from their residual cytolytic activity as determined in a 51Cr-release assay in vitro (Figure 3D). Thus, this in vivo virus protection assay is a better correlate for HLH susceptibility than cytolytic activity of CTL measured in vitro, since it translates a gradient of impaired cytotoxicity into the clinically observed “yes-no” decision for the development of HLH.
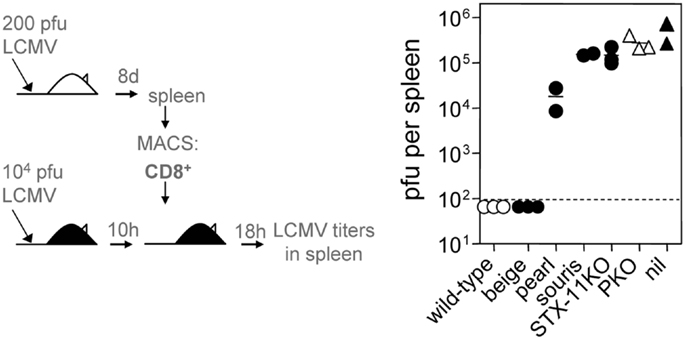
Figure 4. Subtle differences in CTL cytotoxicity of various HLH-prone mouse models determine virus control. On day 8 after 200 pfu of LCMV infection, splenic CD8+ T cells of mutant mice (beige, pearl, souris, STX-11KO, ashen/filled circles), wild-type (open circle), and PKO (open triangle) mice were MACS purified and 2 × 106 of MACS purified CD8+ T cells were transferred into wild-type C57BL/6 mice that had been infected with 104 pfu LCMV 10 h before. After 18 h of CD8+ T cell transfer, viral titers in spleen were determined (nil: infected wild-type C57BL/6 without cell transfer).
HLH Disease Severity in Patients with FHL Syndromes
To assess how these observations in the murine cytotoxicity mutants relate to human patients, we intended to determine HLH disease severity in cohorts of patients carrying the respective mutations. Disease severity in humans is, however, not only determined by the affected gene, but also by the nature of the mutation (complete vs. partial loss-of function), genomic heterogeneity, and environmental factors including infections. Since disease severity in human cytotoxicity mutants correlates with the age at onset of HLH (54, 55), we used this as a surrogate parameter. To control in part for the nature of the mutation, we selected only patients with predicted severe impairment of protein expression due to a null mutation, a large gene deletion, the introduction of a stop codon or a frame shift mutations leading to a stop codon in the corresponding genes. We recently published data on patients with severe bi-allelic mutations in the PERFORIN, the SYNTAXIN-11, and the RAB27A genes (44). For this study we added observations in a cohort of patients with mutations in the LYST gene that was identified from the literature. Since only one HPS2 patient with an AP3b1 mutation has been reported to have developed the full picture of HLH (at 5 years of age) (19), we did not include this group of HPS2 patients in our analysis.
As expected from our previous study, although there was a high variability in the age at onset for all four diseases, significant differences could be demonstrated between the different cohorts (Figures 5A,B). The mean age of HLH onset was 3.4 ± 5 months in PERFORIN-deficient patients, 13.4 ± 19 months in RAB27A-deficient patients, 27.3 ± 37 months in SYNTAXIN-11-deficient patients and 37.7 ± 41.9 months in LYST-deficient patients. Thus, this analysis reveals a gradient of HLH severity in humans that is identical to the mouse models: HPS2 < CHS < FHL4 < GS2 < FHL2.
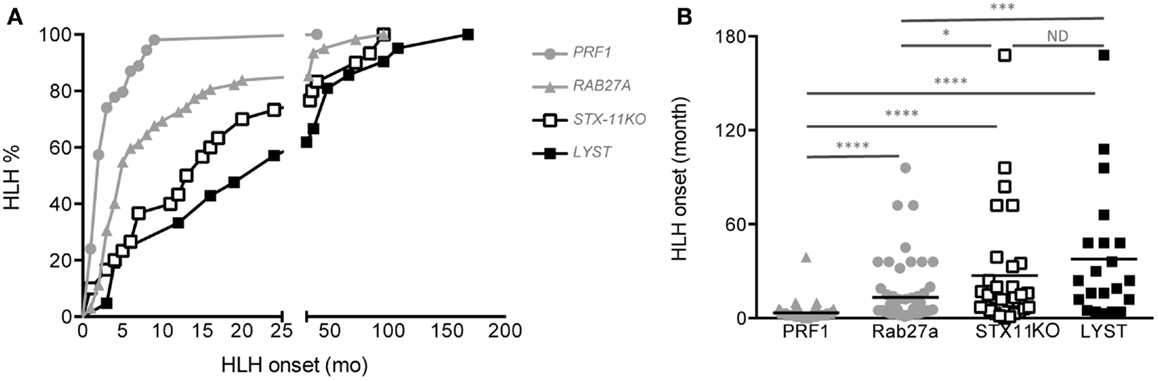
Figure 5. Delayed HLH onset in patients with SYNTAXIN-11 and LYST deficiency compared with RAB27A- and PRF1-deficient patients. (A) Cumulative incidence and (B) age at onset of HLH in FHL2 (PRF1 deficiency, gray circles; n = 72), GS2 (RAB27A deficiency, gray triangles; n = 61), FHL4 (STX11 deficiency, open squares; n = 30), and CHS (LYST deficiency, black squares; n = 21) patients carrying null bi-allelic mutations, as detailed in Table 3. The incidence of HLH was analyzed with a log-rank test; *p < 0.05 for FHL4 vs. GS2 patients; ***p < 0.001 for GS2 vs. CHS patients; ****p < 0.0001 for FHL2 vs. GS2, FHL2 vs. FHL4 and FHL2 vs. CHS; ND = no statistical differences were observed between FHL4 vs. CHS patients. The onset of HLH was analyzed with a one-way ANOVA. *p < 0.05; ***p < 0.001; ***p < 0.0001. Mutations in PRF1, RAB27A, and STX11 are detailed in Ref. (44), and mutations in CHS1/LYST are detailed in Table 3.
Discussion
This study provides a comprehensive comparative analysis of HLH disease severity in mice and humans with mutations in different genes involved in cellular cytotoxicity. While the study builds on a number of previously published observations comparing individual strains of mice or patient cohorts, it is the first study to directly compare a large number of mutant mouse strains, in which HLH is induced and assessed with a single experimental protocol. Notably, we related the clinical HLH symptoms not only to the affected gene, but also to immunobiological parameters such as the degree of the degranulation/cytotoxicity defect of T cells and the ability to provide control of the triggering viral infection.
The overall result is a clear hierarchy of HLH severity among the different genetic defects predisposing to HLH that is surprisingly consistent between humans and mice. This is an important validation of using LCMV infection of murine cytotoxicity mutants for the study of human genetic defects in cytotoxicity and FHL development. While beige mice did not develop HLH upon LCMV infection, pearl mice showed transient HLH and souris, STX-11KO, ashen, and PKO mice exhibited the full picture of the disease. Moreover, among the latter four strains a gradient of disease severity could be established when considering the individual HLH associated parameters (Table 2). Souris mice had the least weight loss and drop in temperature, while PKO mice had the most severe and frequently lethal HLH progression (Figure 1). Finally, we could confirm our previous observation that the disease course was more severe in ashen (GS2) than in STX-11KO (FHL4) mice. Thus, the gradient of HLH severity in mice was: wild-type < HPS2 < CHS < FHL4 < GS2 < FHL2 (Figure 6). Interestingly, the parameters best reflecting this gradient were ferritin and sCD25, while IFN-γ serum levels did not correlate as good, at least when analyzed at day 12 post infection.
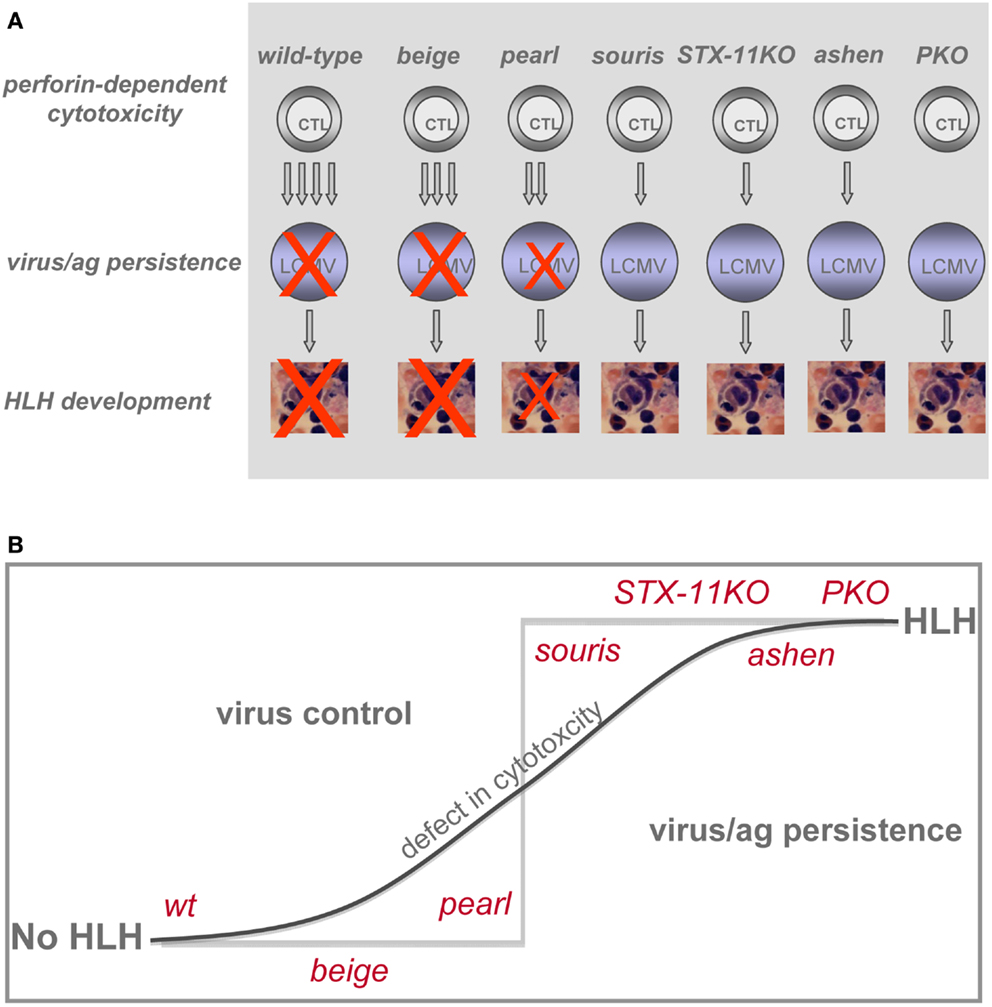
Figure 6. Impact of various genetic defects on threshold of HLH development. (A) Differences in CTL cytotoxicity and their impact on LCMV control determine whether or not HLH develops. (B) Pronounced impairment of CTL cytotoxicity results in loss of virus control and development of HLH.
In humans, disease severity as assessed by age at onset of HLH followed exactly the same pattern. However, in all cohorts, there were patients who developed HLH in the first months of life, confirming that the affected gene cannot predict disease onset in the individual patient. The different ages at onset probably rather reflect the likelihood of loosing control of perforin-mediated immune homeostasis in response to triggers of different intensity that are encountered by all children.
As expected, none of the mouse strains developed disease spontaneously. Infection with LCMV was required to trigger disease, while other infections including respiratory syncytical virus (RSV) and pneumonia virus of mice (PVM) failed to induce disease, even in the most severe mutant lacking perforin (unpublished observations). Of note, mouse cytomegalovirus (MCMV) is able to induce HLH in PKO (56, 57), but not in jinx mice (42). No data on HLH are so far available for the other HLH-prone mouse strains, although a higher susceptibility of beige mice to MCMV infection has been reported (58). The potency of LCMV to induce HLH might in part be explained by the fact that it infects antigen-presenting cells such as dendritic cells (63), leading to direct stimulation of the T cell response without the need for cross-presentation. This contributes to the fact that it is a better stimulator of CD8 T cell responses in the mouse than any of the other infections. Of note, this is a property that is also shared by EBV infection in humans, where infected B cells can also serve as APC and will lead to prolonged antigenic stimulation, if they are not eliminated by NK or T cells (64). Overall, a very potent CTL stimulation is required to provoke the impaired immune homeostasis characteristic of HLH in mice. This is in apparent contrast to early-onset HLH in patients with cytotoxicity defects, where in the majority of cases no pathogen trigger can be identified. This could reflect variable and incomplete infectious disease work-up of the patients, which may not only relate to known infections, but could also point to a role for so far unknown novel viruses. Alternatively, it may indicate that in humans, different from mice, perforin-mediated cytotoxicity also plays a role in T cell homeostasis under resting conditions, as it is described for the Fas/FasL pathway (65, 66). Thus, T cell–T cell interactions or T/NK cell–APC interactions involving perforin could be relevant for maintaining immune homeostasis even in the absence of infections or other obvious immune stimulations (67–73). Further research in HLH patients will be needed to address this important issue, since treatment of a triggering infection can be an important component of successful therapy for HLH (37, 74, 75).
What then is the role of the virus in the murine disease models? Our data and previous experiments suggest that it is not just a trigger of a pathogenetic sequence that – once initiated – becomes self-perpetuating and independent of the virus. This is probably best illustrated by the phenotype of pearl mice. Within the first 8 days after infection, these mice could not control virus replication and developed HLH symptoms. However, virus elimination was eventually achieved and this led to full recovery from HLH symptoms. Complementary to these findings, chronic antigen application (gp33 peptide injections in 12 h intervals) induced HLH-like symptoms in LCMV-infected wild-type mice indicating the critical role of antigen persistence and prolonged antigen presentation for disease development (41). Thus, persistence of cells presenting viral antigen (usually in the context of persisting virus) is the decisive factor for HLH induction in all animal models studied so far. A previous study has suggested that perforin may have an immunomodulatory function that is independent from its role in the clearance of virus and killing of APCs presenting viral antigen, but related to a role of perforin in modulating antigen presentation by DC (76). In this view, HLH development is not dependent on virus persistence per se but determined by an enhanced antigen presentation in the context of perforin deficiency. Further experiments are needed to decide whether it is possible to functionally separate antigen persistence from enhanced antigen presentation in this context.
While virus/antigen persistence was associated with perpetuated disease in all HLH-prone mouse strains analyzed, various factors may determine the hierarchy of disease severity in mice with different genetic defects in cellular cytotoxicity. First, early virus control based on differences in residual cytotoxic activity of T and NK cells may be an important factor in determining the severity of initial disease manifestation. The graded virus load in the spleen at day 8 after infection paralleled very well HLH severity in beige, pearl, souris, and STX-11KO mice. However, differences in splenic virus titers on day 8 could not explain the different HLH severity in STX-11KO, ashen, and PKO mice. Thus, a so far unsolved question is why differences in HLH severity are observed in these mice, although all of them exhibited virus persistence, which should lead to the same extent of chronic T cell stimulation by presented viral antigens. Early virus spread to other, non-lymphoid organs could be an important factor determining HLH severity. Differences in the spread of virus to key organs like the liver or the brain with subsequent recruitment of highly activated CTL may explain the fact that PKO mice die, while the other mutant mouse strains survive (77). Analysis of the early kinetics of virus spread to other organs may help to resolve this issue. Second, differences in the residual cytotoxic activity may not only affect early virus control and spread, but additionally influence effector cell homeostasis via a more or less efficient killing of distinct APC populations. The elimination of certain APC populations may critically determine the activation status and survival of the hyperreactive T cells and act as a rheostat to limit T cell responses (73, 78–81). As recently demonstrated, the elimination of a rare, antigen-presenting DC population by CD8 T cells in a negative feedback loop critically determines the magnitude of the T cell response in a perforin-dependent way after LCMV infection (82). Third, the proteins affected in the various cytotoxicity mutants are involved in different steps of lysosomal trafficking [as discussed for syntaxin-11 and Rab27a (26, 44)], which could also contribute to the quality of antigen presentation and hence indirectly determine T cell activity and HLH progression. This may also be the case for AP3b1 known to regulate several processes involved in antigen recognition/processing/presentation, i.e., CD1b presentation of phagocytosed antigens and TLR recruitment to phagosome (83, 84). Along the same line, defects in proteins involved in perforin-mediated cytotoxicity may have additional functions in other immune cell types like platelets, neutrophils, and mast cells, which can be relevant for LCMV specific immune responses and thus modulate HLH pathogenesis (33, 85, 86). Fourth, different susceptibility to T cell exhaustion in the various cytotoxicity mutants can modify HLH progression and determine survival. As recently demonstrated, effector T cells chronically exposed to antigenic stimulation showed a variable extent of exhaustive differentiation in different HLH-prone mouse strains. Initially, STX-11KO mice developed all diagnostic symptoms of HLH after LCMV infection comparable to PKO mice. However, in STX-11KO mice with more extensive T cell exhaustion the HLH disease course was attenuated and the mice survived, whereas PKO mice developed lethal HLH (43). T cell exhaustion in STX-11KO mice was characterized by sustained expression of inhibitory receptors, step-wise loss of effector functions, and finally deletion of the disease-mediating T cells. Thus, T cell exhaustion can be an important disease-modifying parameter in HLH.
In summary, following a defined viral stimulus, the degree of impairment of CTL cytotoxicity was the best predictor of HLH development in the described animal models, but other host factors contributed. HLH appeared to be a threshold disease. Up to a certain degree of impaired cytotoxicity, disease was mild and transient, but resolved once the delayed virus control had been achieved. However, subtle differences in CTL cytotoxicity allowing the establishment of viral persistence led to the full picture of HLH and persistent disease (Figure 6A). Thus, the graded differences in cytotoxicity translated into a “yes-no” phenotype (Figure 6B) with respect to HLH. In HLH patients it is still a matter of debate to which extent CTL or NK cells contribute to disease induction. Hence, this concept may apply in a more general form to cytotoxic lymphocytes. Depending on the initial trigger CTL and/or NK cells may play the critical role in HLH induction and progression. While restoration of cytotoxicity and virus control appear to be key variables in the causal treatment of the disease, a further investigation of these factors in humans and mice may point to additional treatment approaches for this highly aggressive syndrome.
Materials and Methods
Patients
All patients with FHL or GS2 diagnosis were previously reported [summarized in Ref. (44)]. Patients with CHS diagnosis were previously published (as referenced in Table 3).
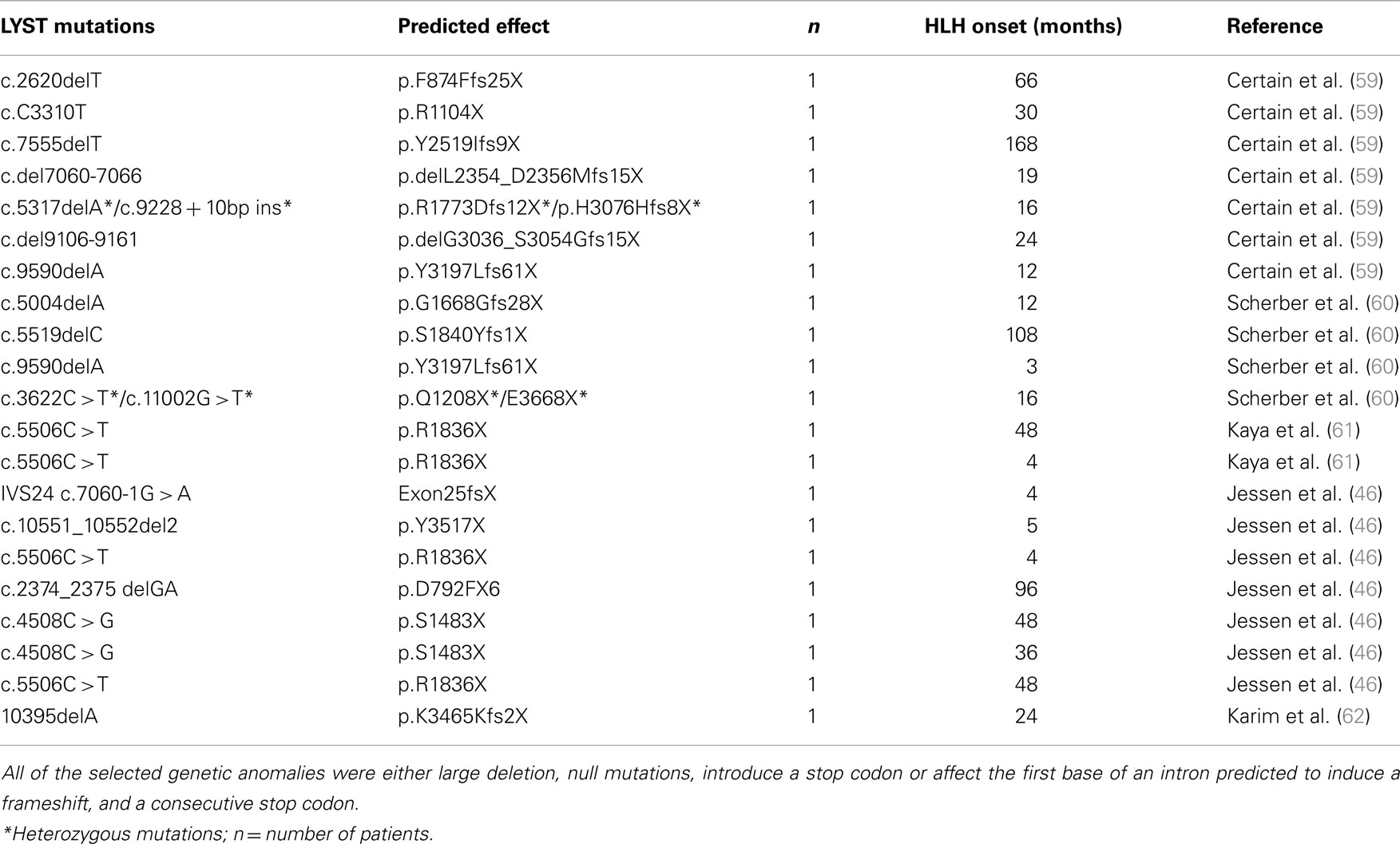
Table 3. Genotype and age at HLH onset of previously published patients carrying severe bi-allelic mutations in CHS1/LYST.
Mice and Virus
C57BL/6 (wild-type, wt) mice were purchased from Charles River Laboratories (Sulzfeld, Germany). C57BL/6J-LystbgJ/J (beigeJ; stock no. 000629) and B6Pin.C3-Ap3b1pe/J (pearl, stock no. 003215) mice were purchased from the Jackson Laboratory (Bar Harbor, USA), and C57BL/6-Lystbg-Btlr/Mmcd (souris; stock no. 010470-UCD) mice originally generated by Dr. B. Beutler and colleagues (Scripps Research Institute, La Jolla, CA, USA) were obtained from the Mutant Mouse Regional Resource Center (University of California, Davis, CA, USA). Syntaxin-11-deficient (STX-11KO) mice were generated by Dr. U. zur Stadt (Hamburg) on a C57BL/6 background by deletion of the only coding exon. C3H/HeSn-Rab27aash/J mice were purchased from the Jackson Laboratory and backcrossed to the C57BL/6 background for 10 generations (C57BL/6J-Rab27aash/j; ashen). Perforin-deficient C57BL/6-Prf1tm1Sdz (PKO) mice were obtained from Dr. H. Hengartner (Zurich). Mice were kept under specific pathogen-free conditions. All mouse experiments were approved by the Regierungspraesidium Freiburg. The lymphocytic choriomeningitis virus WE (LCMV-WE) was grown on MC57G fibroblasts and stored at −80°C until use. Mice were injected intravenously with 200 pfu (plaque forming units). To quantify virus in organs from infected mice a focus forming assay was used as described (87). Temperatures were obtained using a digital infrared ear thermometer (Braun, ThermoScan type 6022).
HLH Biomarkers in Mice
Blood counts were determined by a Sysmex KX-21 hematology analyzer. Serum levels of ferritin, triglycerides, LDH, and GLDH were analyzed by the Department of Clinical Chemistry using the Roche Modular Analytics Evo. Levels of sCD25 were determined by using the mouse IL-2Ralpha DuoSet kit (R&D systems) according to the instructions of the manufacturer. The IFN-γ ELISA was performed as described before (46).
Histology
To evaluate hemophagocytic macrophages, immunohistochemistry on paraffin-embedded liver sections was performed as previously described (46).
Antibodies, Intracellular Staining, Degranulation, and Cytotoxicity Assay
Antibodies were purchased from eBioscience or BD Biosciences. Surface expression of CD107a and intracellular IFN-γ of CD8+CD3+ CTL was determined after 4 h of restimulation with the immunodominant CTL epitope gp33-41 (PolyPeptide) or medium in the presence of monensin (BD Biosciences). For fixation and permeabilization of spleen cells the Cytofix/Cytoperm kit (BD Biosciences) was used. CTL cytotoxicity was determined in a 5 h 51chromium-release assay by incubating spleen cells as effectors with LCMV-infected MC57 target cells. In order to calculate the CTL to target ratio, CD8 T cells were quantified by antibody staining and flow cytometry.
Adoptive Transfer Experiment
Splenic CD8 T cells from mice that had been infected with 200 pfu LCMV-WE 8 days earlier were MACS purified using the MACS CD8a+ T Cell Isolation Kit II (Miltenyi Biotec). Purity was determined by flow cytometry and was above 90% in all experiments. About 2 × 106 purified CD8 T cells were transferred intravenously into C57BL/6 wild-type mice that had been infected with 104 pfu LCMV 10 h before. After 18 h of adoptive cell transfer, splenic virus titers were determined.
Statistical Analysis
Tests were performed using the GaphPad InStat software version 3.06. The comparison between data was evaluated with a one-way ANOVA (Analysis Of Variance) with posttest. Differences were considered significant at a p-value below 0.05.
Authors Contribution
Birthe Jessen, Tamara Kögl, and Fernando E. Sepulveda performed experiments; Stephan Ehl, Peter Aichele, Genevieve de Saint Basile designed the study and supervised the project; Stephan Ehl, Peter Aichele, Genevieve de Saint Basile, and Birthe Jessen wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Thyssen Stiftung, the European Union (FP7 CURE-HLH grant agreement 201461), the German Federal Ministry of Education and Research (BMBF 01 EO 0803), the French National Institute of Health and Medical Research (INSERM) and the Agence National de la Recherche (ANR HLH-cytotox/ANR-12-BSV1-0020-01). Fernando E. Sepulveda is funded by a post-doctoral fellowship from the Association pour la Recherche sur le Cancer (ARC) and Becas Chile.
References
1. de Saint Basile G, Menasche G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol (2010) 10(8):568–79. doi: 10.1038/nri2803
2. Filipovich AH. Hemophagocytic lymphohistiocytosis and related disorders. Curr Opin Allergy Clin Immunol (2006) 6(6):410–5. doi:10.1097/01.all.0000246626.57118.d9
3. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr (2007) 166(2):95–109. doi:10.1007/s00431-006-0258-1
4. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med (2012) 63:233–46. doi:10.1146/annurev-med-041610-134208
5. Henter JI, Elinder G, Soder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood (1991) 78(11):2918–22.
6. Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood (1997) 89(11):4100–3.
7. Takada H, Ohga S, Mizuno Y, Suminoe A, Matsuzaki A, Ihara K, et al. Oversecretion of IL-18 in haemophagocytic lymphohistiocytosis: a novel marker of disease activity. Br J Haematol (1999) 106(1):182–9. doi:10.1046/j.1365-2141.1999.01504.x
8. Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer (2007) 48(2):124–31. doi:10.1002/pbc.21039
9. Henter JI, Samuelsson-Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood (2002) 100(7):2367–73. doi:10.1182/blood-2002-01-0172
10. Janka GE. Hemophagocytic syndromes. Blood Rev (2007) 21(5):245–53. doi:10.1016/j.blre.2007.05.001
11. Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood (2011) 118(15):4041–52. doi:10.1182/blood-2011-03-278127
12. Bryceson YT, Rudd E, Zheng C, Edner J, Ma D, Wood SM, et al. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood (2007) 110(6):1906–15. doi:10.1182/blood-2007-02-074468
13. Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell (2003) 115(4):461–73. doi:10.1016/S0092-8674(03)00855-9
14. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science (1999) 286(5446):1957–9. doi:10.1126/science.286.5446.1957
15. zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet (2005) 14(6):827–34. doi:10.1093/hmg/ddi076
16. Cote M, Menager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest (2009) 119(12):3765–73. doi:10.1172/JCI40732
17. zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet (2009) 85(4):482–92. doi:10.1016/j.ajhg.2009.09.005
18. Barbosa MD, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature (1996) 382(6588):262–5. doi:10.1038/382262a0
19. Enders A, Zieger B, Schwarz K, Yoshimi A, Speckmann C, Knoepfle EM, et al. Lethal hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type II. Blood (2006) 108(1):81–7. doi:10.1182/blood-2005-11-4413
20. Jessen B, Bode SF, Ammann S, Chakravorty S, Davies G, Diestelhorst J, et al. The risk of hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type 2. Blood (2013) 121(15):2943–51. doi:10.1182/blood-2012-10-463166
21. Menasche G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet (2000) 25(2):173–6. doi:10.1038/76024
22. Nagle DL, Karim MA, Woolf EA, Holmgren L, Bork P, Misumi DJ, et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet (1996) 14(3):307–11. doi:10.1038/ng1196-307
23. Badolato R, Parolini S. Novel insights from adaptor protein 3 complex deficiency. J Allergy Clin Immunol (2007) 120(4):735–41. doi:10.1016/j.jaci.2007.08.039
24. Benson KF, Li FQ, Person RE, Albani D, Duan Z, Wechsler J, et al. Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat Genet (2003) 35(1):90–6. doi:10.1038/ng1224
25. Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell (1999) 3(1):11–21. doi:10.1016/S1097-2765(00)80170-7
26. Jancic C, Savina A, Wasmeier C, Tolmachova T, El-Benna J, Dang PM, et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat Cell Biol (2007) 9(4):367–78. doi:10.1038/ncb1552
27. Stinchcombe J, Bossi G, Griffiths GM. Linking albinism and immunity: the secrets of secretory lysosomes. Science (2004) 305(5680):55–9. doi:10.1126/science.1095291
28. Zhen L, Jiang S, Feng L, Bright NA, Peden AA, Seymour AB, et al. Abnormal expression and subcellular distribution of subunit proteins of the AP-3 adaptor complex lead to platelet storage pool deficiency in the pearl mouse. Blood (1999) 94(1):146–55.
29. D’Orlando O, Zhao F, Kasper B, Orinska Z, Muller J, Hermans-Borgmeyer I, et al. Syntaxin 11 is required for NK and CD8(+) T-cell cytotoxicity and neutrophil degranulation. Eur J Immunol (2013) 43(1):194–208. doi:10.1002/eji.201142343
30. Martin-Verdeaux S, Pombo I, Iannascoli B, Roa M, Varin-Blank N, Rivera J, et al. Evidence of a role for Munc18-2 and microtubules in mast cell granule exocytosis. J Cell Sci (2003) 116(Pt 2):325–34. doi:10.1242/jcs.00216
31. Meeths M, Entesarian M, Al-Herz W, Chiang SC, Wood SM, Al-Ateeqi W, et al. Spectrum of clinical presentations in familial hemophagocytic lymphohistiocytosis type 5 patients with mutations in STXBP2. Blood (2010) 116(15):2635–43. doi:10.1182/blood-2010-05-282541
32. Sandrock K, Nakamura L, Vraetz T, Beutel K, Ehl S, Zieger B. Platelet secretion defect in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL-5). Blood (2010) 116(26):6148–50. doi:10.1182/blood-2010-08-302943
33. Ye S, Karim ZA, Al Hawas R, Pessin JE, Filipovich AH, Whiteheart SW. Syntaxin-11, but not syntaxin-2 or syntaxin-4, is required for platelet secretion. Blood (2012) 120(12):2484–92. doi:10.1182/blood-2012-05-430603
34. Pachlopnik Schmid J, Cote M, Menager MM, Burgess A, Nehme N, Menasche G, et al. Inherited defects in lymphocyte cytotoxic activity. Immunol Rev (2010) 235(1):10–23. doi:10.1111/j.0105-2896.2010.00890.x
35. Rudd E, Goransdotter Ericson K, Zheng C, Uysal Z, Ozkan A, Gurgey A, et al. Spectrum and clinical implications of syntaxin 11 gene mutations in familial haemophagocytic lymphohistiocytosis: association with disease-free remissions and haematopoietic malignancies. J Med Genet (2006) 43(4):e14. doi:10.1136/jmg.2005.035253
36. Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood (2011) 118(22):5794–8. doi:10.1182/blood-2011-07-370148
37. Beutel K, Gross-Wieltsch U, Wiesel T, Stadt UZ, Janka G, Wagner HJ. Infection of T lymphocytes in Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in children of non-Asian origin. Pediatr Blood Cancer (2009) 53(2):184–90. doi:10.1002/pbc.22037
38. Booth C, Gilmour KC, Veys P, Gennery AR, Slatter MA, Chapel H, et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood (2011) 117(1):53–62. doi:10.1182/blood-2010-06-284935
39. Henter JI, Ehrnst A, Andersson J, Elinder G. Familial hemophagocytic lymphohistiocytosis and viral infections. Acta Paediatr (1993) 82(4):369–72. doi:10.1111/j.1651-2227.1993.tb12699.x
40. Suzuki N, Morimoto A, Ohga S, Kudo K, Ishida Y, Ishii E. Characteristics of hemophagocytic lymphohistiocytosis in neonates: a nationwide survey in Japan. J Pediatr (2009) 155(2):235.e–8.e. doi:10.1016/j.jpeds.2009.02.050
41. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood (2004) 104(3):735–43. doi:10.1182/blood-2003-10-3413
42. Crozat K, Hoebe K, Ugolini S, Hong NA, Janssen E, Rutschmann S, et al. Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J Exp Med (2007) 204(4):853–63. doi:10.1084/jem.20062447
43. Kögl T, Muller J, Jessen B, Schmitt-Graeff A, Janka G, Ehl S, et al. Hemophagocytic lymphohistiocytosis in syntaxin-11-deficient mice: T-cell exhaustion limits fatal disease. Blood (2013) 121(4):604–13. doi:10.1182/blood-2012-07-441139
44. Sepulveda FE, Debeurme F, Menasche G, Kurowska M, Cote M, Pachlopnik Schmid J, et al. Distinct severity of HLH in both human and murine mutants with complete loss of cytotoxic effector PRF1, RAB27A, and STX11. Blood (2013) 121(4):595–603. doi:10.1182/blood-2012-07-440339
45. Pachlopnik Schmid J, Ho CH, Diana J, Pivert G, Lehuen A, Geissmann F, et al. A Griscelli syndrome type 2 murine model of hemophagocytic lymphohistiocytosis (HLH). Eur J Immunol (2008) 38(11):3219–25. doi:10.1002/eji.200838488
46. Jessen B, Maul-Pavicic A, Ufheil H, Vraetz T, Enders A, Lehmberg K, et al. Subtle differences in CTL cytotoxicity determine susceptibility to hemophagocytic lymphohistiocytosis in mice and humans with Chediak-Higashi syndrome. Blood (2011) 118(17):4620–9. doi:10.1182/blood-2011-05-356113
47. Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature (1994) 369(6475):31–7. doi:10.1038/369031a0
48. Pachlopnik Schmid J, Ho CH, Chretien F, Lefebvre JM, Pivert G, Kosco-Vilbois M, et al. Neutralization of IFNgamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol Med (2009) 1(2):112–24. doi:10.1002/emmm.200900009
49. Barber DL, Wherry EJ, Ahmed R. Cutting edge: rapid in vivo killing by memory CD8 T cells. J Immunol (2003) 171(1):27–31.
50. Ehl S, Klenerman P, Aichele P, Hengartner H, Zinkernagel RM. A functional and kinetic comparison of antiviral effector and memory cytotoxic T lymphocyte populations in vivo and in vitro. Eur J Immunol (1997) 27(12):3404–13. doi:10.1002/eji.1830271240
51. Fung-Leung WP, Kundig TM, Zinkernagel RM, Mak TW. Immune response against lymphocytic choriomeningitis virus infection in mice without CD8 expression. J Exp Med (1991) 174(6):1425–9. doi:10.1084/jem.174.6.1425
52. Lehmann-Grube F, Lohler J, Utermohlen O, Gegin C. Antiviral immune responses of lymphocytic choriomeningitis virus-infected mice lacking CD8+ T lymphocytes because of disruption of the beta 2-microglobulin gene. J Virol (1993) 67(1):332–9.
53. Zinkernagel RM, Leist T, Hengartner H, Althage A. Susceptibility to lymphocytic choriomeningitis virus isolates correlates directly with early and high cytotoxic T cell activity, as well as with footpad swelling reaction, and all three are regulated by H-2D. J Exp Med (1985) 162(6):2125–41. doi:10.1084/jem.162.6.2125
54. Feldmann J, Le Deist F, Ouachee-Chardin M, Certain S, Alexander S, Quartier P, et al. Functional consequences of perforin gene mutations in 22 patients with familial haemophagocytic lymphohistiocytosis. Br J Haematol (2002) 117(4):965–72. doi:10.1046/j.1365-2141.2002.03534.x
55. Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol (2006) 6(12):940–52. doi:10.1038/nri1983
56. van Dommelen SL, Sumaria N, Schreiber RD, Scalzo AA, Smyth MJ, Degli-Esposti MA. Perforin and granzymes have distinct roles in defensive immunity and immunopathology. Immunity (2006) 25(5):835–48. doi:10.1016/j.immuni.2006.09.010
57. Chiossone L, Audonnet S, Chetaille B, Chasson L, Farnarier C, Berda-Haddad Y, et al. Protection from inflammatory organ damage in a murine model of hemophagocytic lymphohistiocytosis using treatment with IL-18 binding protein. Front immunol (2012) 3:239. doi:10.3389/fimmu.2012.00239
58. Shellam GR, Allan JE, Papadimitriou JM, Bancroft GJ. Increased susceptibility to cytomegalovirus infection in beige mutant mice. Proc Natl Acad Sci USA (1981) 78(8):5104–8. doi:10.1073/pnas.78.8.5104
59. Certain S, Barrat F, Pastural E, Le Deist F, Goyo-Rivas J, Jabado N, et al. Protein truncation test of LYST reveals heterogenous mutations in patients with Chediak-Higashi syndrome. Blood (2000) 95(3):979–83.
60. Scherber E, Beutel K, Ganschow R, Schulz A, Janka G, Stadt U. Molecular analysis and clinical aspects of four patients with Chediak-Higashi syndrome (CHS). Clin Genet (2009) 76(4):409–12. doi:10.1111/j.1399-0004.2009.01205.x
61. Kaya Z, Ehl S, Albayrak M, Maul-Pavicic A, Schwarz K, Kocak U, et al. A novel single point mutation of the LYST gene in two siblings with different phenotypic features of Chediak Higashi syndrome. Pediatr Blood Cancer (2011) 56(7):1136–9. doi:10.1002/pbc.22878
62. Karim MA, Suzuki K, Fukai K, Oh J, Nagle DL, Moore KJ, et al. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet (2002) 108(1):16–22. doi:10.1002/ajmg.10184.abs
63. Macal M, Lewis GM, Kunz S, Flavell R, Harker JA, Zuniga EI. Plasmacytoid dendritic cells are productively infected and activated through TLR-7 early after arenavirus infection. Cell Host Microbe (2012) 11(6):617–30. doi:10.1016/j.chom.2012.04.017
64. Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol (2007) 25:587–617. doi:10.1146/annurev.immunol.25.022106.141553
65. Bidere N, Su HC, Lenardo MJ. Genetic disorders of programmed cell death in the immune system. Annu Rev Immunol (2006) 24:321–52. doi:10.1146/annurev.immunol.24.021605.090513
66. Chen M, Felix K, Wang J. Critical role for perforin and Fas-dependent killing of dendritic cells in the control of inflammation. Blood (2012) 119(1):127–36. doi:10.1182/blood-2011-06-363994
67. Biron CA. Yet another role for natural killer cells: cytotoxicity in immune regulation and viral persistence. Proc Natl Acad Sci U S A (2012) 109(6):1814–5. doi:10.1073/pnas.1120528109
68. Davis DM. Intercellular transfer of cell-surface proteins is common and can affect many stages of an immune response. Nat Rev Immunol (2007) 7(3):238–43. doi:10.1038/nri2020
69. Lang PA, Lang KS, Xu HC, Grusdat M, Parish IA, Recher M, et al. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc Natl Acad Sci U S A (2012) 109(4):1210–5. doi:10.1073/pnas.1118834109
70. Marcenaro E, Ferranti B, Moretta A. NK-DC interaction: on the usefulness of auto-aggression. Autoimmun Rev (2005) 4(8):520–5. doi:10.1016/j.autrev.2005.04.015
71. Menasche G, Feldmann J, Fischer A, de Saint Basile G. Primary hemophagocytic syndromes point to a direct link between lymphocyte cytotoxicity and homeostasis. Immunol Rev (2005) 203:165–79. doi:10.1111/j.0105-2896.2005.00224.x
72. Su MW, Walden PR, Golan DB, Eisen HN. Cognate peptide-induced destruction of CD8+ cytotoxic T lymphocytes is due to fratricide. J Immunol (1993) 151(2):658–67.
73. Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature (2011) 481(7381):394–8. doi:10.1038/nature10624
74. Chellapandian D, Das R, Zelley K, Wiener SJ, Zhao H, Teachey DT, et al. Treatment of Epstein-Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol (2013) 162(3):376–82. doi:10.1111/bjh.12386
75. Imashuku S. Treatment of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis (EBV-HLH); update 2010. J Pediatr Hematol Oncol (2010) 33(1):35–9. doi:10.1097/MPH.0b013e3181f84a52
76. Lykens JE, Terrell CE, Zoller EE, Risma K, Jordan MB. Perforin is a critical physiologic regulator of T-cell activation. Blood (2011) 118(3):618–26. doi:10.1182/blood-2010-12-324533
77. Zinkernagel RM, Ehl S, Aichele P, Oehen S, Kundig T, Hengartner H. Antigen localisation regulates immune responses in a dose- and time-dependent fashion: a geographical view of immune reactivity. Immunol Rev (1997) 156:199–209. doi:10.1111/j.1600-065X.1997.tb00969.x
78. Badovinac VP, Tvinnereim AR, Harty JT. Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science (2000) 290(5495):1354–8. doi:10.1126/science.290.5495.1354
79. Odermatt B, Eppler M, Leist TP, Hengartner H, Zinkernagel RM. Virus-triggered acquired immunodeficiency by cytotoxic T-cell-dependent destruction of antigen-presenting cells and lymph follicle structure. Proc Natl Acad Sci U S A (1991) 88(18):8252–6. doi:10.1073/pnas.88.18.8252
80. Wong P, Pamer EG. Feedback regulation of pathogen-specific T cell priming. Immunity (2003) 18(4):499–511. doi:10.1016/S1074-7613(03)00081-5
81. Yang J, Huck SP, McHugh RS, Hermans IF, Ronchese F. Perforin-dependent elimination of dendritic cells regulates the expansion of antigen-specific CD8+ T cells in vivo. Proc Natl Acad Sci U S A (2006) 103(1):147–52. doi:10.1073/pnas.0509054103
82. Terrell CE, Jordan MB. Perforin deficiency impairs a critical immunoregulatory loop involving murine CD8(+) T cells and dendritic cells. Blood (2013) 121(26):5184–91. doi:10.1182/blood-2013-04-495309
83. Sasai M, Linehan MM, Iwasaki A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science (2010) 329(5998):1530–4. doi:10.1126/science.1187029
84. Sugita M, Cao X, Watts GF, Rogers RA, Bonifacino JS, Brenner MB. Failure of trafficking and antigen presentation by CD1 in AP-3-deficient cells. Immunity (2002) 16(5):697–706. doi:10.1016/S1074-7613(02)00311-4
85. Iannacone M, Sitia G, Isogawa M, Whitmire JK, Marchese P, Chisari FV, et al. Platelets prevent IFN-alpha/beta-induced lethal hemorrhage promoting CTL-dependent clearance of lymphocytic choriomeningitis virus. Proc Natl Acad Sci U S A (2008) 105(2):629–34. doi:10.1073/pnas.0711200105
86. Loria GD, Romagnoli PA, Moseley NB, Rucavado A, Altman JD. Platelets support a protective immune response to LCMV by preventing splenic necrosis. Blood (2013) 121(6):940–50. doi:10.1182/blood-2011-08-376822
87. Battegay M, Cooper S, Althage A, Banziger J, Hengartner H, Zinkernagel RM. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24- or 96-well plates. J Virol Methods (1991) 33(1–2):191–8. doi:10.1016/0166-0934(91)90018-U
Appendix
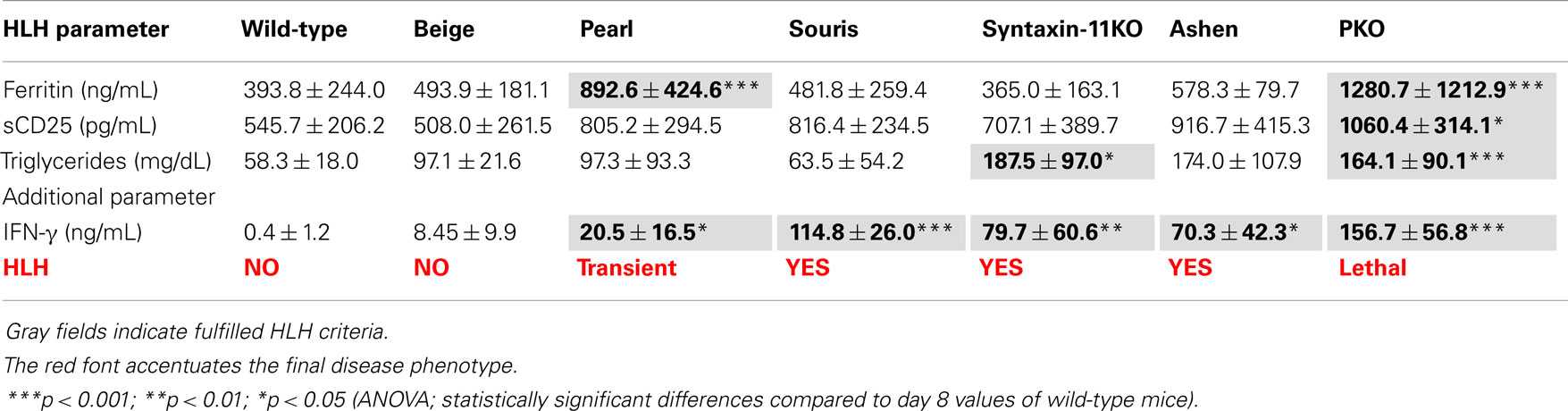
Table A1. Analysis of HLH biomarker on day 8 after LCMV infection.
Keywords: cytotoxicity, hemophagocytic lymphohistiocytosis, inflammation, CTL, virus persistence, antigen persistence
Citation: Jessen B, Kögl T, Sepulveda FE, de Saint Basile G, Aichele P and Ehl S (2013) Graded defects in cytotoxicity determine severity of hemophagocytic lymphohistiocytosis in humans and mice. Front. Immunol. 4:448. doi: 10.3389/fimmu.2013.00448
Received: 04 October 2013; Accepted: 26 November 2013;
Published online: 16 December 2013.
Edited by:
Yenan Bryceson, Karolinska Institutet, SwedenReviewed by:
Christine Susanne Falk, Hannover Medical School MHH, GermanyJoseph A. Trapani, Peter MacCallum Cancer Centre, Australia
Copyright: © 2013 Jessen, Kögl, Sepulveda, de Saint Basile, Aichele and Ehl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephan Ehl, Centre of Chronic Immunodeficiency, University Medical Center Freiburg, University of Freiburg, Breisacher Strasse 117, Freiburg D-79106, Germany e-mail:c3RlcGhhbi5laGxAdW5pa2xpbmlrLWZyZWlidXJnLmRl
†Birthe Jessen, Tamara Kögl and Fernando E. Sepulveda have contributed equally to this work.
#Joint senior authorship