 Ashley Mentlik James
Ashley Mentlik James Adam D. Cohen
Adam D. Cohen Kerry S. Campbell
Kerry S. Campbell- 1Immune Cell Development and Host Defense Program, The Research Institute at Fox Chase Cancer Center, Philadelphia, PA, USA
- 2Abramson Cancer Center at the University of Pennsylvania, Philadelphia, PA, USA
Natural killer (NK) cells are critical innate immune lymphocytes capable of destroying virally infected or cancerous cells through targeted cytotoxicity and further assisting in the immune response by releasing inflammatory cytokines. NK cells are thought to contribute to the process of tumor killing by certain therapeutic monoclonal antibodies (mAb) by directing antibody-dependent cellular cytotoxicity (ADCC) through FcγRIIIA (CD16). Numerous therapeutic mAb have been developed that target distinct cancer-specific cell markers and may direct NK cell-mediated ADCC. Recent therapeutic approaches have combined some of these cancer-specific mAb with additional strategies to optimize NK cell cytotoxicity. These include agonistic mAb targeting NK cell activating receptors and mAbs blocking NK cell inhibitory receptors to enhance NK cell functions. Furthermore, several drugs that can potentiate NK cell cytotoxicity through other mechanisms are being used in combination with therapeutic mAb. In this review, we examine the mechanisms employed by several promising agents used in combination therapies that enhance natural or Ab-dependent cytotoxicity of cancer cells by NK cells, with a focus on treatments for leukemia and multiple myeloma.
Introduction
Natural killer (NK) cells are generally known for the ability to mediate spontaneous “natural” cytotoxicity of major histocompatibility complex class I (MHC-I)-deficient tumor or virus-infected cells. NK cells kill target cells through the release of perforin and granzymes from cytolytic granules or the surface expression of Fas ligand or TNF-related apoptosis-inducing ligand (TRAIL). Activated NK cells are also a potent source of interferon (IFN)-γ, tumor necrosis factor (TNF)-α, and a variety of other cytokines and chemokines that contribute significantly to early immune responses.
The activation of NK cells is controlled by a balance of signals emanating from a collection of germline-encoded activating and inhibitory receptors. Several inhibitory receptors, including CD94/NKG2A, members of the highly polymorphic killer cell-Ig-like receptor (KIR) family and ILT2/LIR-1/CD85j, play critical roles in tolerizing NK cells toward healthy cells by binding to MHC-I ligands [human leukocyte antigen (HLA)-A, -B, and -C allotypes] expressed on virtually all healthy cells (1). When NK cells engage with MHC-I-expressing healthy cells, the inhibitory receptors transduce negative signaling that efficiently abrogate stimulatory signals from co-engaged activating receptors at the immunological synapse (2–4). Many tumors and virus-infected cells down-regulate their surface expression of MHC-I to avoid recognition by the antigen receptor on cytotoxic T cells, but these abnormal cells inherently become susceptible to NK cell-mediated attack in the absence of the tolerizing MHC-I ligands.
Activating receptors expressed on human NK cells include FcγRIIIA (CD16), activating forms of KIR, 2B4, NKG2D, and the natural cytotoxicity receptors (NCR), which are also known as NKp30, NKp44, and NKp46 (5, 6). Of these, the NCR and NKG2D are particularly important receptors for triggering NK cell responses toward tumor target cells (7). In contrast to inhibitory receptors, triggering of the activating receptor NKG2D is a key mechanism by which NK cells recognize stressed or diseased cells and destroy them (8). NKG2D specifically recognizes MHC chain-related (MIC) A, MICB, and UL16-binding proteins (ULBPs) in humans, which are HLA-related molecules lacking peptide presentation capacity that are not expressed on normal cells but are up-regulated on stressed cells, such as tumors (8, 9).
Thus, NK cells are important effectors in immune responses to tumors and viral infections, and increased understanding of the mechanisms controlling NK cell activation has led to the development of therapeutic agents that can improve their responsiveness. While these agents show promise, results are inconsistent between patients due to inherent differences in activity/function of an individual’s immune system, and expression of distinct biomarkers on cancers that can differentially influence NK cell responsiveness. A growing variety of treatment options can improve outcome for individual patients. Novel treatment regimens combining new and old therapies are even showing promise among relapsed and refractory patients. Here, we review some of the currently available therapies that are known to stimulate NK cell functions and how they are being used in combination with other agents to boost anti-tumor responses in cancer patients.
Antibody-Dependent Cellular Cytotoxicity
FcγRIIIA (CD16) provides NK cells with the capacity to mediate antibody-dependent cellular cytotoxicity (ADCC) upon recognition of the Fc segment of IgG bound to cell surfaces. This antigen-specific NK cell targeting mechanism appears to play an important role in tumor eradication by several therapeutic tumor-specific mAb, such as herceptin, rituximab, and elotuzumab. Interestingly, Deguine et al. recently suggested that NKG2D engagement might be crucial for NK cell-mediated ADCC responses. In that study, mouse tumors expressing NKG2D ligands induced enhanced ADCC degranulation, while responses were reduced toward tumors lacking NKG2D ligand. The results indicate that the mAb-FcR interaction mainly stabilized adhesion with the target cell to contribute contact stability, while NKG2D triggering provided the activation threshold needed to trigger NK cell degranulation (10). This concept is supported by earlier work in a mouse model of renal cancer that showed NKG2D ligation does not stimulate strong adhesion with tumor cells. In that report, target cells that did not express ICAM-1 were not killed in an NKG2D-dependent manner, but transfection of ICAM-1 into these cells restored NKG2D-mediated cytotoxicity (11).
An emerging strategy to enhance NK cell-mediated ADCC responsiveness is to inhibit the shedding of CD16 that occurs after NK cell activation (12, 13). Specifically, CD16 can be cleaved by metalloproteinases after being triggered by an IgG-opsinized target cell or treatment with IL-2, but if metalloproteinases are inhibited, CD16 is not shed. In fact, CD16 signaling is sustained and target cell killing and cytokine production are enhanced in the presence of metalloproteinase inhibitors (14). Furthermore, combining metalloproteinase inhibition with a bispecific antibody (against CD16 and CD33) resulted in a sustained and very specific anti-tumor response (15). Such a combination strategy has the potential to substantially increase NK cell responses to cancer.
An additional immunoglobulin-based strategy to target more immune responses toward tumor cells is to create bispecific or trispecific antibodies. The engineering of bispecific monoclonal antibodies (mAb) that create surrogate bridging between activating receptors on T or NK cells with tumor-specific antigens have shown therapeutic promise for decades (16). A common bispecific design theme to target NK cells has employed fusing antibody Fv domains that bind CD16 with Fv domains that bind tumor cell markers, such as CD30 (17), ErbB2 (18–20) or CD19 (21, 22). These constructs can trigger ADCC responses by NK cells and monocytes/macrophages through the CD16 FcR without the need for an incorporated Fc domain. As a cautionary tale for appropriate design, a bispecific incorporating a CD16-binding Fv domain in combination with a CD16-binding Fc domain was found in an early phase I study to induce toxicity through dimeric engagement of the FcR on the surface of monocytes and NK cells in the blood, which induced rapid release of cytokines, including TNF-α, IL-6, and IL-8 (18). Modern recombinant approaches fuse Fv domains into single chain constructs that have been termed bispecific or trispecific killer cell engagers (BiKEs or TriKEs), which lack Fc domains entirely. Importantly, these BiKEs and TriKEs can be highly effective at inducing NK cell ADCC and cytokine responses (22). In another approach, von Strandmann et al. created a bispecific protein (ULBP-BB4) that fused the NKG2D ligand, ULBP2, onto a single chain Fv targeting CD138, which is highly expressed on a number of malignancies, including multiple myeloma (MM). In xenograft mouse models injected with human tumor cells and human PBLs, ULBP-BB4 demonstrated potent anti-tumor activity while not significantly harming healthy cells (23). Many researchers have also developed trivalent antibodies composed of two tumor antigen-recognition domains and one monovalent domain that recognizes NK cells (19–21, 24). If properly manipulated for safety, bispecific and trispecific antibodies offer a very targeted approach to effective tumor therapy that can directly involve NK cell effector function.
The Use of NK Cell-Related Immune Therapies to Treat Multiple Myeloma
A common proving ground for testing immune therapies that stimulate NK cell activity has been MM. MM is a deadly hematologic cancer characterized by clonal expansion of malignant plasma cells that reside in the bone marrow and thrive on interactions with the bone marrow microenvironment (25). Despite advances in treatment strategies, MM remains an incurable disease, with about 20,000 newly diagnosed cases and over 10,000 deaths per year in the U.S. (26). Novel therapies have improved survival over the last decade, including autologous hematopoietic stem cell transplantation (HSCT) and the use of new drugs (27). Allogeneic stem cell transplantation can be curative, but is often associated with high transplantation-related mortality (28). Despite these advanced therapeutic options, median survival remains around 4–5 years in adults (29) and the development of better treatments is essential. Interestingly, evidence is accumulating that NK cells may play a prominent role in immune responses toward MM and can also contribute to graft-versus-myeloma responses in haploidentical HSCT (30), It is becoming clear that NK cells can elicit potent allogeneic and autologous responses to myeloma cells in vitro and in patients (30, 31). Given the importance of NK cells in immune responses toward MM, combination therapies that enhance NK cell functions are showing promise in treating this deadly disease, as will become evident in the following discussion.
Immunomodulatory Drugs (IMiDs®)
Thalidomide, lenalidomide, and pomalidomide form a new class of immunomodulatory drugs, referred to as IMiDs, which can broadly stimulate the functions of NK cells and T cells to treat cancer (32). Thalidomide is a glutamic acid derivative with a dark history as a therapeutic agent, since it caused severe birth defects when used to treat morning sickness in pregnant women in the late 1950s. Nonetheless, it was subsequently found to have anti-inflammatory, anti-angiogenic, anti-proliferative, and immunomodulatory properties that fostered further investigation (33–35). The anti-inflammatory properties of thalidomide are at least partially due to potent inhibition of the production of TNF-α by activated monocytes (35). Lenalidomide and pomalidomide are more potent thalidomide analogs that have since emerged (36), and pomalidomide is even more potent at co-stimulating T cells than lenalidomide (37). Since these IMiDs can enhance the functions of T cells and NK cells, suppress angiogenesis, inhibit TNF-α production, and directly repress tumor cell growth, they are potentially beneficial in treating cancer. To date, both lenalidomide and pomalidomide have been used to treat MM and a variety of other cancers.
The mechanism of immune stimulation by IMiDs is complex and not entirely established (32). Treatment of patients with lenalidomide has been shown to increase the overall frequency of NK cells in peripheral blood, suggesting that they either proliferate or migrate into the bloodstream (38–40). Lenalidomide does not appear to stimulate NK cells directly, however, but instead functions through effects on other leukocytes in peripheral blood (40). Stimulation of T cells by lenalidomide overcomes the need for signals from antigen presenting cells and induces increased proliferation and enhanced production of the type 1 cytokines, IL-2, and IFN-γ (37, 41, 42). At least part of the stimulatory effects of IMiDs on NK cells appears to be due to the T cell production of IL-2, which is a potent growth factor for NK cells (43, 44). Both lenalidomide and pomalidomide have also been shown to increase ADCC activity by NK cells (44, 45). At least part of this effect may result from an increased frequency of the CD56dim NK cells expressing CD16 and LFA-1 in peripheral blood, which are responsible for mediating ADCC (46). This ability of IMiDs to augment ADCC has been borne out in clinical studies, particularly in combination with the CD20-targeting antibody rituximab, where significant activity has been seen in relapsed/refractory B-cell lymphomas and chronic lymphocytic leukemia (47, 48). In MM, lenalidomide is usually used in combination with steroids (49, 50). However, the enhanced NK cell-mediated responses by lenalidomide can be reversed in combination with dexamethasone (40), suggesting that using steroids long-term in combination with lenalidomide may be counterproductive to its immune-stimulatory effects, and that steroid-free combinations should be explored. It should also be noted that tumor cell lines cultured in lenalidomide become more susceptible to NK cell-mediated lysis, due to their increased expression of ligands for NK cell activating receptors (38–40, 51). Taken together, NK cell-mediated anti-tumor responses can be stimulated in a variety of ways by IMiDs, and this enhanced function can be beneficial in treating cancer.
Bortezomib
Bortezomib is an inhibitor of the 26S proteasome that is currently used to treat MM and lymphoma. Inhibition of the proteasome has several direct negative impacts on tumor cells, including inhibiting proliferation and inducing apoptosis, but bortezomib-treated tumor cells also become more susceptible to attack by NK cells (52). Upon inhibition of the proteasome, tumor cells are incapable of processing and presenting proteolytic peptide fragments on MHC-I molecules on the plasma membrane. Consequently, bortezomib down-regulates the surface expression of MHC-I on tumor cells in vitro and in vivo (53), thereby reducing the levels of this important protein for NK cell tolerance and enhancing susceptibility to NK cell-mediated natural and antibody-dependent cytotoxicity (54, 55). Bortezomib treatment can augment the efficacy of adoptively transferred NK cells in murine tumor models (56), and this approach has now been translated to the clinic for cancer patients (57).
Elotuzumab
A promising new monoclonal antibody candidate for treatment of MM is elotuzumab (formerly HuLuc63). Elotuzumab is a fully humanized antibody that recognizes the SLAM family member CS1 (CRACC, SLAMF7, CD319), a surface glycoprotein normally expressed on NK cells, monocytes, mature dendritic cells, a subset of T cells, and stimulated B cells (58, 59). Normal plasma cells express high levels of CS1, which correlates with high expression on MM cells (60). CS1 is an attractive therapeutic ADCC target because the available clinical data indicate that expression persists on MM cells even after conventional treatments (61–63). CS1 was originally found to engage in homotypic interactions as a self-ligand, and pretreatment of a NK cell line with recombinant CS1-Ig fusion protein was shown to stimulate killing of K562 target cells, apparently by directly activating the NK cells via homotypic interaction (64). Two separate reports found high CS1 expression in most MM cases studied, and elotuzumab was found to significantly increase NK cell-mediated ADCC of primary MM cells (65, 66).
While initiation of NK cell-mediated ADCC upon engagement with CD16 is the best characterized function of elotuzumab, the exact mechanism(s) of action is unclear (67). Importantly, CS1 is also considered a co-stimulatory receptor on NK cells (64, 68), and recent evidence indicates that elotuzumab may also stimulate NK cells directly through direct interactions with CS1 on the NK cell surface (69). As an additional potential mechanism, elotuzumab may interfere with interactions of MM cells with the bone marrow microenvironment, which is a key requirement for tumor survival and proliferation (60, 67). MM cell interactions with bone marrow stromal cells, osteoclasts, and osteoblasts lead to bone deterioration, angiogenesis, and MM cell survival and proliferation (70), so disrupting the ability of MM cells to interact with the microenvironment could provide multiple benefits. All of these potential mechanisms of action continue to be explored, though so far the data continue to indicate that NK cells contribute to the therapeutic efficacy of this anti-CS1 monoclonal antibody.
Clinically, elotuzumab used alone was well-tolerated and led to disease stabilization in a subset of relapsed/refractory myeloma patients (63). More promising clinical activity was seen, however, when it was used in combination with lenalidomide and dexamethasone, with over 80% of relapsed patients responding in phase I and II trials, and progression-free survival significantly longer (median 26.9 months at the 10 mg/kg dose) than that previously observed for lenalidomide/dexamethasone alone (62). Elotuzumab-mediated ADCC of MM targets by NK cells can be enhanced in vitro by pretreatment with a proteasome inhibitor (54, 66), and a small combination study of elotuzumab and bortezomib in relapsed/refractory myeloma patients showed this combination to be safe, with preliminary efficacy observed (61). Currently, three randomized phase III trials are underway adding elotuzumab to either bortezomib or lenalidomide/dexamethasone, in both newly diagnosed and relapsed/refractory MM patients. These studies will more definitively assess if there is a benefit to adding elotuzumab to these standard therapies.
KIR-Blocking Monoclonal Antibody
Allogeneic HSCT has emerged as an effective treatment option for a variety of hematological cancers after chemotherapeutic ablation of the recipient’s immune cells (71). Because of the high polymorphic variability of KIR and MHC-I in the human population, variability of these receptor/ligand pairs should be considered in the context of transplantation. Velardi and colleagues first reported that donor allogenicity of NK cells in HSCT to treat acute myeloid leukemia (AML) resulted in a double benefit by enhancing anti-leukemia responses, while reducing graft-versus-host disease (72). In the absence of an HLA-identical sibling donor, haploidentical HSCT is commonly used, in which a mismatch exists in at least one HLA allele. This mismatch improves the odds that at least one inhibitory KIR in the donor NK cells lacks an MHC-I ligand in the transplant recipient. In this scenario, the donor-derived NK cells are less inhibited and considered to be more capable of triggering a graft-versus-leukemia effect. KIR/HLA mismatch in HSCT has resulted in improved outcomes by several groups, specifically in treating AML (73–75), and may play a role in myeloma as well (30, 76). It is believed that NK cell-mediated autoimmunity does not occur in these patients because healthy cells are less likely to up-regulate the stress ligands that trigger NK cells (77). It is important to note that recent evidence indicates that donors expressing activating KIR (especially donors expressing KIR2DS1, but lacking its ligand, HLA-C2) also contribute significantly to improved outcomes in HSCT to treat AML (78, 79). These results indicate that certain engineered mismatches of KIR/HLA interactions in the context of HSCT can significantly influence NK cell responses in AML patients and perhaps other cancers.
In addition to exploiting KIR/HLA ligand mismatch in the context of HSCT, monoclonal antibody-mediated blockade of the KIR/HLA interaction has emerged as a potential cancer immunotherapy to lower the threshold of NK cell activation. The in vitro use of mAb to block KIR function was first found to increase cytokine production in T cells in 1996 (2). To further test this concept in vitro, Binyamin et al. potentiated NK cell responses to autologous EBV-transformed B cells by combining a panel of mAbs to block numerous NK cell inhibitory receptors (KIR, CD94/NKG2A, and ILT2/LIR-1/CD85j) in combination with the B cell-specific anti-CD20 mAb rituximab to simultaneously reduce inhibitory signals and trigger ADCC, respectively (80). Importantly, NK cell-mediated cytotoxicity of the transformed target cells was not elevated by inhibitory receptor blockade alone, suggesting that other tolerizing mechanisms effectively prevent the attack of normal cells in the context of these inhibitory receptor-blocking conditions.
Based on this concept, a humanized KIR-blocking mAb IPH2101 (formerly 1-7F9) has been developed and is currently being tested in clinical trials. IPH2101 is a pan-specific anti-KIR antibody that binds KIR2DL1, -2 and -3, which are the most relevant inhibitory KIR family members, due to their combined capacity to recognize all allotypes of HLA-C. The antibody was shown to block the interaction between these inhibitory KIR2DL and HLA-C and also binds the activating receptors KIR2DS1 and KIR2DS2, although the functional impacts of these interactions have not been formally tested (77). In vitro preclinical studies demonstrated that IPH2101 mAb augments NK cell cytotoxicity of HLA-C-expressing tumor cells without targeting normal blood mononuclear cells, which is critical to assure that NK cells remain tolerant in treated patients (77). This was confirmed in a preclinical mouse model that was engineered to co-express KIR2DL3 and its ligand, HLA-Cw3, which was able to license or educate the mouse NK cells. When KIR was then blocked in vivo with IPH2101, the mouse NK cells were able to destroy HLA-Cw3-positive target cells without development of autoimmunity (81).
In view of the capacity of NK cells to respond to myeloma cells, phase I clinical trials have been initiated to treat MM patients with IPH2101 and the modified variant, IPH2102. When used alone in patients, the side effects of IPH2101 were minimal with no evidence of autoimmunity, and ex vivo functional assays showed enhanced NK cell cytotoxicity (82, 83). A trial of IPH2101 in combination with lenalidomide has since been initiated, based upon a variety of effects by these reagents that can potentially synergize to enhance NK cell responses. IPH2101 is expected to enhance NK cell killing by blocking inhibitory signals, while lenalidomide can stimulate general NK cell function and may even up-regulate triggering ligands on MM cells (51).
Importantly, the addition of KIR blocking mAbs may prove to be an asset in treating a variety of cancers, as a way to boost the potential of NK cells to kill stressed or cancerous cells, while retaining general NK cell tolerance toward normal cells. Nonetheless, it is possible that optimal clinical responses may require a combination therapy of multiple antibodies blocking a wider variety of inhibitory receptors expressed on NK cells, such as KIR3DL family members, CD94/NKG2A, and ILT2/LIR-1/CD85j, as well as the addition of an ADCC targeting mAb to stimulate tumor-specific cytotoxicity, as demonstrated by the in vitro studies of Binyamin et al. described earlier (80).
Agents Promoting the Expression of NKG2D Ligands on Tumor Cells
The human NKG2D ligands MICA, MICB, and ULBPs are commonly up-regulated on stressed or infected cells and thereby provide a key recognition element for NK cell-mediated attack of many cancer cells (8, 9). Several cancer types are able to shed NKG2D ligands into the sera as an immune evasive mechanism, and these soluble ligands have been shown to cause down-regulation of NKG2D on NK cells, which leads to a stunted immune response (84–87). Demonstrating the high frequency of shed ligands, Hilpert et al. recently found at least one soluble NKG2D ligand in the sera of 183 leukemia patients analyzed, and culture of NK cells in leukemia patient sera resulted in down-regulation of NKG2D expression (88). Shedding of the NKG2D ligand, MICA, by chronic lymphocytic leukemia cells can be induced upon translocation of the endoplasmic reticulum-resident proteins ERp5 and GRP78 to the tumor cell surface (89). Additionally, shedding of MICA/B has been attributed to proteolytic activity of the ADAM10 and ADAM17 metalloproteinases in some tumor cell lines (90). The MICA*008 allele can also be released into exosomes, which can subsequently down-regulate NKG2D expression and reduce NK cell-mediated cytotoxicity (90, 91). On the other hand, the expression of CEACAM1 in cancer cells can cause the intracellular retention of NKG2D ligands, thereby limiting NK cell detection through NKG2D (92). These observations have made the retention/upregulation of NKG2D ligands on tumor cells an attractive goal for cancer therapy.
Many drugs that were first considered for cancer therapy because they can alter gene expression in tumor cells were subsequently found to also increase the susceptibility of tumor cells to cytotoxicity by NKG2D-expressing NK cells. These drugs include those that promote gene upregulation (93–96), differentiation (97–99), and DNA or protein damage (100–103). For example, HDAC inhibitors cause the upregulation of NKG2D ligands, MICA/B and ULBPs, in tumor cells but not healthy cells. Treatment with the HDAC inhibitor valproic acid (VPA, valproate) leads to higher expression of NKG2D ligands at the transcriptional and translational levels by inducing acetylation of the histones bound to MICA and MICB gene promoters (104, 105). The relatively low toxicity and low occurrence of off target effects of VPA make it a reasonable means of boosting effector cell function. Furthermore, treating cultured cells with VPA and the DNA methylation inhibitor hydralazine was shown to increase dimethylated MICA/B gene promoters, thereby further stimulating transcription (106, 107). The addition of a metalloproteinase inhibitor has also been shown to block the proteolytic cleavage of NKG2D ligands, as a means to further decrease shedding into the sera when used in conjunction with VPA treatment (108). Lastly, spironolactone, a diuretic commonly used to treat heart failure and high blood pressure, has very recently been found to also upregulate NKG2D ligands and therefore increase NK cell cytotoxicity of colon cancer cell lines (109).
Glycogen synthase kinase (GSK)-3 has recently been discovered as a new target to promote MICA/B upregulation. GSK3 inhibitors have been widely used to suppress the proliferation of malignant lymphoid cells, but Fionda et al. recently showed that GSK3 inhibition also increased MICA expression at the protein and mRNA levels in MM cells (110). In conjunction with the increased MICA expression, the addition of the GSK3 inhibitor significantly enhanced NK cell-mediated cytotoxicity of tumor cells. Mechanistically, GSK3 inhibition correlated with the down-regulation of STAT3, which is a negative regulator of MICA expression (110). In addition, the combination of lenalidomide with GSK3 inhibition further enhanced MICA expression, which further supports the combined mechanistic benefit of these agents with current anti-tumor therapies.
Anti-CD137 Monoclonal Antibodies
CD137 (4-1BB, TNFRSF9) is an inducible member of the TNF receptor superfamily that functions as a co-stimulatory signaling molecule on the surface of activated T and NK cells. CD137 ligation further augments activation of these cells, increasing their proliferation, cytokine secretion, and effector function, and preventing activation-induced cell death (111, 112). Treatment with agonist anti-CD137 mAb can mimic this co-stimulatory signal, leading to regression of large tumors in multiple murine models, including B-cell lymphoma and myeloma (111, 113–115). This effect requires CD8+ T cells, but is also dependent upon the presence of NK cells (116, 117), implying an impact on these cells as well. Many diverse types of tumors express elevated levels of the CD137 ligand, CD137L, accompanied by increased expression of CD137 on immune cells within the tumor environment, while expression of either is negligible on healthy cells (115, 118–120).
There are conflicting data regarding the effects of anti-CD137 mAb on NK cells. CD137 engagement on mouse NK cells consistently results in activation, but this can be either activating or inhibitory in humans, depending on the setting or model used. In human leukemia cells, the CD137/CD137L interaction can also result in bidirectional signaling to suppress NK cell-mediated responses; CD137 recognition of CD137L on leukemic cells transmits inhibitory signals into the NK cell to impair cytokine production and cytotoxicity responses and CD137L engagement by CD137 induces the production of TNF and immunosuppressive interleukin (IL)-10 by the tumor cells (119). Inhibiting this interaction using soluble CD137-Fc or a “blocking” anti-CD137 mAb can restore NK cell cytotoxicity (119). Taking this rationale one step further, Buechele et al. suggested the potential merits of a dual strategy of blocking CD137/CD137L interaction and neutralizing immunosuppressive TNF (121). Lin et al. demonstrated that human NK cells up-regulate CD137 in vitro following Fc-receptor engagement, which promotes release of pro-inflammatory cytokines but decreased cytotoxicity against K562 targets. This implies a negative impact of CD137 expression and signaling in human NK cells, though the direct impact of CD137 ligation was not tested in this model (122, 123). In contrast, Kohrt et al. have shown that following FcR-induced CD137 up-regulation on NK cells, adding an agonist anti-CD137 mAb known to induce CD137 signaling actually enhances NK cell-mediated ADCC toward rituximab- or trastuzumab-coated target cells (124, 125). Whether this antibody may also be working by preventing CD137 on immune cells from binding to CD137L and inducing “reverse” signaling within the tumor cell was not explored in these studies. Nonetheless, these data have led to an ongoing clinical trial (NCT01775631) combining rituximab with agonist anti-CD137 mAb (BMS-663513, urelumab) in patients with relapsed/refractory B-cell malignancies.
These inconsistent findings on the impacts of manipulating CD137 in NK cells are likely the result of signaling differences between mice and men, the use of different CD137-targeting reagents, and even between different hematopoietic versus non-hematopoietic cancer models (120). For clinical relevance, it will be important to better understand the human mechanistic impact of CD137/CD137L interaction in order to properly exploit the potential treatment options. The role of CD137/CD137L interactions on interplay between NK cells and CD4+, CD8+, and regulatory T cells (Tregs) is also incompletely understood (126). For instance, the depletion of immunosuppressive Tregs has been shown to enhance the anti-tumor activity of anti-CD137 mAb (115). However, CD137 engagement on CD4+ and CD8+ T cells is believed to always be activating in mice and men (126). Nonetheless, while the exact mechanism is still unclear, the addition of the CD137 mAb along with neutralization of immunosuppressive cells and signals shows potential to boost immune function when added to current cancer therapies (121, 127).
Anti-GITR Monoclonal Antibodies
Glucocorticoid-induced TNF receptor (GITR, TNFRSF18) is another co-stimulatory member of the TNF receptor superfamily expressed on T cells, NK cells, and B cells, among other hematopoietic cell types (126). GITR expression is generally low in resting T and NK cells, is up-regulated after activation, and the receptor is expressed constitutively at high levels in CD4+Foxp3+ Tregs. GITR is an activating receptor in T cells, since in vitro or in vivo engagement with GITR ligand or agonist anti-GITR mAb has been reported to support the expansion of CD4+ and CD8+ T cells and renders T cells resistant to suppression by Tregs (128, 129). Treatment with agonist anti-GITR mAb leads to enhanced vaccine-induced and endogenous effector T cell responses and tumor rejection in multiple murine tumor models, and is associated with a marked reduction in Treg frequency within the tumor microenvironment, though the exact mechanism(s) by which GITR ligation modulates Tregs remains controversial (115, 130–133). Based on these preclinical findings, the first clinical trial of an agonist anti-human GITR mAb (TRX518) in patients with advanced melanoma or other solid tumors is now underway (NCT01239134).
Similar to CD137, there is conflicting evidence about whether GITR is activating or inhibitory in human NK cells, which may also reflect differential NK cell responses to GITR engagement between mice and men (120). While agonist anti-GITR mAb can augment murine NK cell proliferation and cytotoxicity, GITR engagement on human NK cells has been reported to block NF-κB activation, cause release of anti-inflammatory cytokines, suppress NK cell proliferation, and increase NK cell apoptosis (134). Furthermore, Baltz et al. found that soluble GITR ligand (sGITRL) is released by several hematologic malignancies, detectable in patient sera, and these patients display reduced NK cell cytotoxicity and IFN-γ production (135). In CLL, bidirectional GITR/GITRL signaling can support tumor cell growth by causing release of survival factors, such as IL-6, IL-8, and TNF, and interfering with rituximab-induced ADCC responses (136). However, an antagonistic anti-GITR mAb can block GITR-GITRL interaction, and restore NK cell-mediated ADCC responses. Finally, a dual strategy has been developed by Schmiedel et al. that has potential to enhance existing therapies for AML and CLL (137). An Fc-optimized GITR-Ig fusion protein was found to block the GITR/GITRL interaction and target GITRL-bearing cells for ADCC, thus enhancing NK cell-mediated cytotoxicity of cancer cells. Hence, like anti-CD137 mAb, anti-GITR mAb, and other GITR-targeting therapies have the potential to boost the effectiveness of current cancer therapies by virtue of pro-inflammatory and cytotoxic effects involving NK cells.
PD-1 or PD-L1 Blocking Monoclonal Antibodies
Programed death 1 (PD-1; CD279) is a member of the B7 family of co-signaling receptor that is up-regulated on activated T cells, NK cells, B cells, dendritic cells, and monocytes (138). The intracellular domain of PD-1 contains an immunoreceptor tyrosine-based inhibitory motif (ITIM), which can recruit the protein tyrosine phosphatases SHP-1 and SHP-2 to mediate inhibitory signaling (139). The engagement of PD-1 by its ligands, PD-L1 or PD-L2, blocks the immune response in both T and NK cells by inhibiting PI3K/Akt and Ras activation signaling (140–142). PD-1 expression also marks “exhaustion” in T cells, and engagement of PD-1 can cause apoptosis of CD8+ T cells and the differentiation of CD4+ T cells into immunosuppressive Tregs (143–146). In NK cells, PD-1 engagement impairs activation, conjugate formation, cytotoxicity, and cytokine production (145, 147, 148). In healthy tissue, PD-L1-induced inhibitory signaling minimizes damage to bystander cells and prevents excessive immune responses during acute infections (149). However, many tumor and viral models express PD-1 ligands as an immune evasion mechanism. In contrast, PD-L1 is expressed at low levels on healthy tissue, and resting NK cells express low levels of PD-1 (144, 145, 147, 148). IFN-γ can potently up-regulate PD-L1 expression (150), suggesting that localized cytokine production by NK cells and Th1 cells may actually promote PD-1-based immune evasion by tumors.
The disruption of PD-1/PD-1 ligand interactions can significantly potentiate immune responses to viral infections and cancer, and antibody-mediated blockade of these interactions has emerged as a prime target for immune therapies. In the case of viral persistence in the liver, the abrogation of PD-L1 by siRNA was shown to enhance the number of intrahepatic NK cells and CTL, thereby increasing cytotoxicity, cytokine production, viral clearance, and memory (148). During HIV infection, PD-1 levels increase on patient NK cells, and this has been shown to diminish NK cell proliferation (145). PD-L1 expression on tumors and PD-1 expression on tumor-infiltrating lymphocytes have been associated with poor outcome in renal cell carcinoma patients (151, 152). We have recently reported significantly increased expression of PD-1 on cytolytic NK cells in renal cell carcinoma patients, suggesting that these tumors can directly suppress tumor-infiltrating NK cells by this mechanism (153). Remarkably, the PD-1 expression on NK cells and other leukocytes was rapidly reduced after surgical resection of the primary renal tumor. Anti-PD-1 or anti-PD-L1 mAbs block the interaction of PD-1 on T and NK cells with its ligand, PD-L1 and this restores the function of exhausted cytolytic T cells, augments T cell proliferation, and enhances NK cell cytokine production and cytotoxicity responses, leading to enhanced anti-tumor effector responses and tumor regression in multiple murine models (146, 149). In clinical trials of anti-PD-1 and anti-PD-L1 antibodies to treat a variety of solid tumors, objective, often durable responses were seen in up to a third of patients, demonstrating proof of principle for this approach, and patients that responded to the treatment were shown to express PD-L1 in their tumors (154, 155). Further clinical trials of several candidate antibodies in both solid and hematologic cancers are ongoing.
In studies of MM patients, Benson et al. found upregulation of PD-1 on NK cells in conjunction with PD-L1 expression on MM cells, and in vitro treatment with an anti-PD-1 mAb enhanced NK cell conjugation with PD-L1-expressing MM target cells, resulting in enhanced cytotoxicity and IFN-γ production (147). In the same study, in vitro treatment with lenalidomide was shown to further enhance NK cell responsiveness by lowering PD-L1 expression on MM cells. In a mouse model of MM, Hallett et al. also saw increased levels of PD-L1 on MM cells, along with an exhausted phenotype in T cells, release of immunosuppressive IL-10 and expansion of Treg cells accompanied by increased levels of PD-1 on T and NK cells (144). Blockade of the PD-1/PD-L1 interaction with a PD-L1-specific mAb increased mouse survival by 40%. Like CS1, PD-1 expression persists after stem cell transplant and prior treatment, so PD-1 is a reliable target in relapsed or refractory cancer. Although PD-1 upregulation on human NK cells has only been reported in MM and renal cell carcinoma to date, its expression may be elevated in a variety of cancers as a mechanism to suppress anti-tumor responses. Therefore, the addition of PD-1/PD-L1 blocking mAb to an existing treatment regimen shows encouraging promise in boosting anti-tumor and anti-viral responses by several immune cell types, including NK cells, through counteracting a potent mechanism of immune evasion.
Conclusion
A variety of new therapeutic agents have recently become available to potentiate NK cell responses in cancer patients, and many of these drugs are either approved or undergoing clinical trials (see Table 1). Here we have discussed a representation of this expanding toolbox of agents that allows clinicians to potentiate NK cell functions and thereby enhance anti-tumor therapy. Major future goals will be to expand upon these therapeutic options, better define mechanisms of action for these agents, identify specific combination therapies that are most effective at boosting NK (and T) cell function, and identify biomarkers (e.g., PD-L1 expression on tumor cells) that may better predict which patients are most likely to respond to these immune therapies. Furthermore, while MM has served as an appropriate proving ground for testing the therapeutic efficacy of several of these agents (156), they will need to be tested on other cancers, as well as viral infections and other disease states, to expand their usefulness.
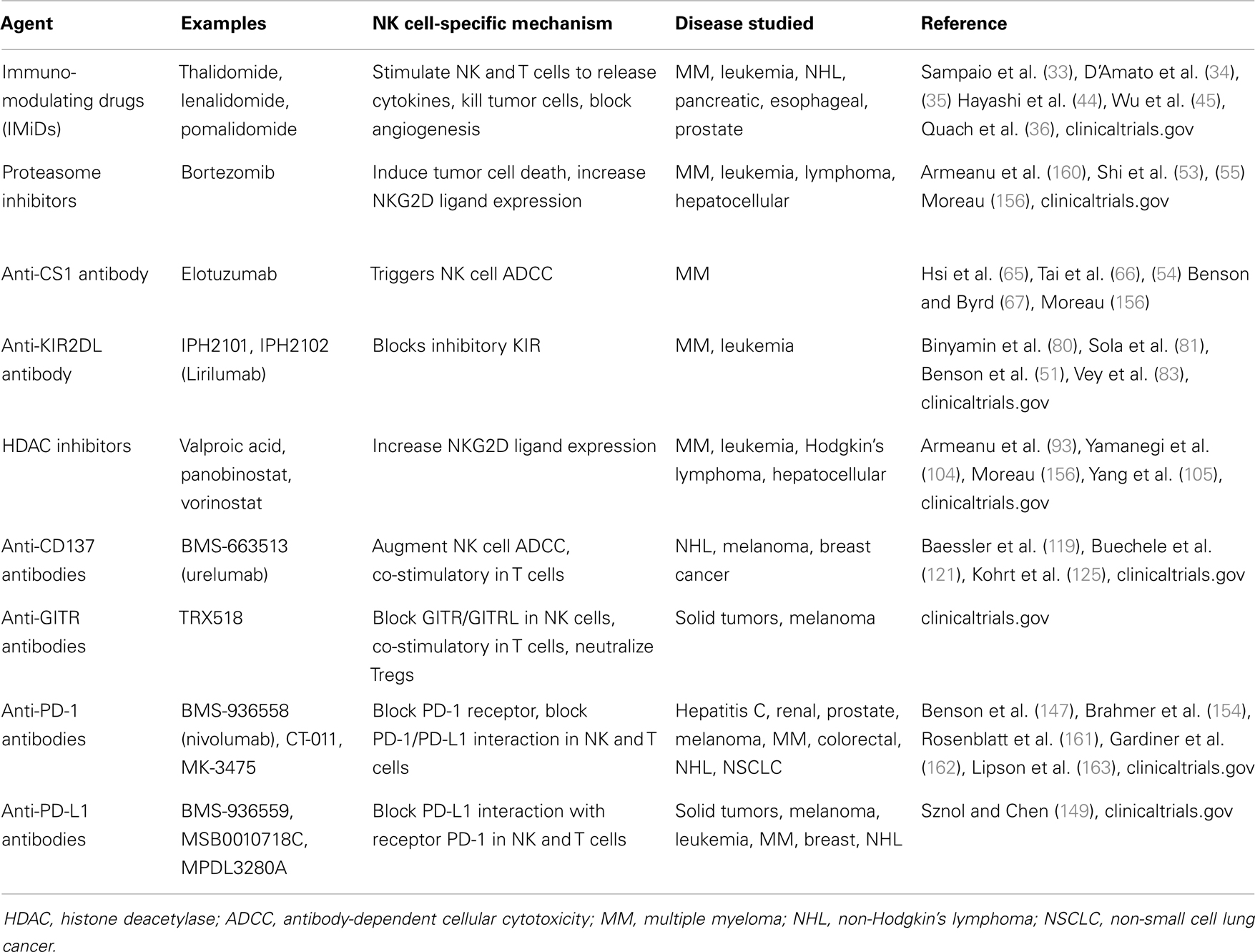
Table 1. Summary of some major therapeutic agents discussed in the text.
Many of these agents are already being tested in conjunction with other immune therapies; especially in combination with ADCC-inducing mAbs that target NK cells to attack tumors. Certainly any new therapy utilizing immunostimulatory mAbs must be carefully evaluated in a stepwise manner to avoid possible adverse side effects, such as autoimmunity (113). However, combination therapies allow clinicians to take advantage of mechanistic synergies that can effectively boost NK cell function using agents that have limited impacts when used alone. An appropriate starting platform combines strategies that boost the immune system and block immune suppression (157), and the hard wiring of NK cells makes them particularly receptive to this strategy. Of particular interest, several phase I clinical trials are currently in progress that combine antibodies designed to block multiple inhibitory immune receptors on NK cells and other leukocytes simultaneously. These include combining the anti-CTLA antibody, ipilimumab with the anti-KIR antibody, IPH2102 (lirilumab) to treat advanced tumors (NCT01750580) and an anti-PD-1 antibody (nivolumab) in combination with either ipilimumab to treat melanoma (NCT01024231) or with lirilumab to treat solid tumors (NCT01714739). Combination therapies may also allow the use of more toxic conventional anti-tumor therapies at lower doses when new therapies are added. Also, patients who have weakened immune function, due to prior radiotherapy or chemotherapy, may particularly benefit from NK cell boosting therapies.
In using these immune therapies, it is important to consider the phenotype of individual patients (158, 159). It is possible that most of these agents can be used together in various combinations or with conventional therapies, depending on the biomarkers present in a particular tumor environment. Altogether, there are several new tools in the medicine cabinet that offer the possibility to improve patient outcomes through boosting NK cell functions. While no one-size-fits-all solution is available to universally improving anti-tumor therapy, proper patient screening should allow the application of personalized combination therapies that harness the beneficial attributes of NK cell-mediated anti-tumor responses to systematically improve overall patient survival.
Conflict of Interest Statement
Kerry S. Campbell and Adam D. Cohen have consulted for and received funding to study elotuzumab from Bristol-Myers Squibb, which provides partial salary support for Ashley Mentlik James.
Acknowledgments
We thank Drs. Hossein Borghaei, Amanda Purdy, and Jun Hasegawa for critical reading of the manuscript. This work was supported by NIH grants CA083859 (Kerry S. Campbell) and CA06927 (FCCC), Collaborative Science Center of Excellence grants from Bristol-Myers Squibb (Kerry S. Campbell and Adam D. Cohen), the Lawrence H. Greenwald Postdoctoral Fellowship award (Ashley Mentlik James), and a Health Research Formula Fund (CURE) grant from the Pennsylvania Department of Health (Kerry S. Campbell). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions.
References
1. Moretta A, Vitale M, Bottino C, Orengo AM, Morelli L, Augugliaro R, et al. P58 molecules as putative receptors for major histocompatibility complex (MHC) class I molecules in human natural killer (NK) cells. Anti-p58 antibodies reconstitute lysis of MHC class I-protected cells in NK clones displaying different specificities. J Exp Med (1993) 178:597–604. doi: 10.1084/jem.178.2.597
2. D’Andrea A, Chang C, Phillips JH, Lanier LL. Regulation of T cell lymphokine production by killer cell inhibitory receptor recognition of self HLA class I alleles. J Exp Med (1996) 184:789–94. doi:10.1084/jem.184.2.789
3. Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, Long EO. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol Cell Biol (2003) 23:6291–9. doi:10.1128/MCB.23.17.6291-6299.2003
4. Purdy AK, Campbell KS. SHP-2 expression negatively regulates NK cell function. J Immunol (2009) 183:7234–43. doi:10.4049/jimmunol.0900088
5. MacFarlane AW IV, Campbell KS. Signal transduction in natural killer cells. Curr Top Microbiol Immunol (2006) 298:23–57.
6. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol (2008) 9:495–502. doi:10.1038/ni1581
7. Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol (2001) 19:197–223. doi:10.1146/annurev.immunol.19.1.197
8. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285:727–9. doi:10.1126/science.285.5428.727
9. Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity (2001) 14:123–33. doi:10.1016/S1074-7613(01)00095-4
10. Deguine J, Breart B, Lemaitre F, Bousso P. Cutting edge: tumor-targeting antibodies enhance NKG2D-mediated NK cell cytotoxicity by stabilizing NK cell-tumor cell interactions. J Immunol (2012) 189:5493–7. doi:10.4049/jimmunol.1202065
11. Abdool K, Cretney E, Brooks AD, Kelly JM, Swann J, Shanker A, et al. NK cells use NKG2D to recognize a mouse renal cancer (Renca), yet require intercellular adhesion molecule-1 expression on the tumor cells for optimal perforin-dependent effector function. J Immunol (2006) 177:2575–83.
12. Peruzzi G, Femnou L, Gil-Krzewska A, Borrego F, Weck J, Krzewski K, et al. Membrane-type 6 matrix metalloproteinase regulates the activation-induced downmodulation of CD16 in human primary NK cells. J Immunol (2013) 191:1883–94. doi:10.4049/jimmunol.1300313
13. Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood (2013) 121:3599–608. doi:10.1182/blood-2012-04-425397
14. Zhou Q, Gil-Krzewska A, Peruzzi G, Borrego F. Matrix metalloproteinases inhibition promotes the polyfunctionality of human natural killer cells in therapeutic antibody-based anti-tumour immunotherapy. Clin Exp Immunol (2013) 173:131–9. doi:10.1111/cei.12095
15. Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Gleason MK, et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res (2013) 19:3844–55. doi:10.1158/1078-0432.CCR-13-0505
16. Renner C, Pfreundschuh M. Tumor therapy by immune recruitment with bispecific antibodies. Immunol Rev (1995) 145:179–209. doi:10.1111/j.1600-065X.1995.tb00082.x
17. Hombach A, Jung W, Pohl C, Renner C, Sahin U, Schmits R, et al. A CD16/CD30 bispecific monoclonal antibody induces lysis of Hodgkin’s cells by unstimulated natural killer cells in vitro and in vivo. Int J Cancer (1993) 55:830–6. doi:10.1002/ijc.2910550523
18. Weiner LM, Clark JI, Davey M, Li WS, Garcia De Palazzo I, Ring DB, et al. Phase I trial of 2B1, a bispecific monoclonal antibody targeting c-erbB-2 and Fc gamma RIII. Cancer Res (1995) 55:4586–93.
19. Xie Z, Shi M, Feng J, Yu M, Sun Y, Shen B, et al. A trivalent anti-erbB2/anti-CD16 bispecific antibody retargeting NK cells against human breast cancer cells. Biochem Biophys Res Commun (2003) 311:307–12. doi:10.1016/j.bbrc.2003.09.211
20. Lu H, Shi M, Wang M, Xie Z, Hu M, Yu M, et al. In vitro and in vivo antitumor effect of a trivalent bispecific antibody targeting ErbB2 and CD16. Cancer Biol Ther (2008) 7:1744–50. doi:10.4161/cbt.7.11.6725
21. Kellner C, Bruenke J, Horner H, Schubert J, Schwenkert M, Mentz K, et al. Heterodimeric bispecific antibody-derivatives against CD19 and CD16 induce effective antibody-dependent cellular cytotoxicity against B-lymphoid tumor cells. Cancer Lett (2011) 303:128–39. doi:10.1016/j.canlet.2011.01.020
22. Gleason MK, Verneris MR, Todhunter DA, Zhang B, Mccullar V, Zhou SX, et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol Cancer Ther (2012) 11:2674–84. doi:10.1158/1535-7163.MCT-12-0692
23. von Strandmann EP, Hansen HP, Reiners KS, Schnell R, Borchmann P, Merkert S, et al. A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood (2006) 107:1955–62. doi:10.1182/blood-2005-05-2177
24. Shahied LS, Tang Y, Alpaugh RK, Somer R, Greenspon D, Weiner LM. Bispecific minibodies targeting HER2/neu and CD16 exhibit improved tumor lysis when placed in a divalent tumor antigen binding format. J Biol Chem (2004) 279:53907–14. doi:10.1074/jbc.M407888200
25. Helfrich MH, Livingston E, Franklin IM, Soutar RL. Expression of adhesion molecules in malignant plasma cells in multiple myeloma: comparison with normal plasma cells and functional significance. Blood Rev (1997) 11:28–38. doi:10.1016/S0268-960X(97)90004-7
26. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin (2010) 60:277–300. doi:10.3322/caac.20073
27. Ludwig H, Durie BG, Mccarthy P, Palumbo A, San Miguel J, Barlogie B, et al. IMWG consensus on maintenance therapy in multiple myeloma. Blood (2012) 119:3003–15. doi:10.1182/blood-2011-11-374249
28. Koehne G, Giralt S. Allogeneic hematopoietic stem cell transplantation for multiple myeloma: curative but not the standard of care. Curr Opin Oncol (2012) 24:720–6. doi:10.1097/CCO.0b013e328358f619
29. Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood (2008) 111:2516–20. doi:10.1182/blood-2007-10-116129
30. Shi J, Tricot G, Szmania S, Rosen N, Garg TK, Malaviarachchi PA, et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br J Haematol (2008) 143:641–53. doi:10.1111/j.1365-2141.2008.07340.x
31. Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A, Aloisi T, et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood (2007) 110:433–40. doi:10.1182/blood-2006-07-038687
32. Shortt J, Hsu AK, Johnstone RW. Thalidomide-analogue biology: immunological, molecular and epigenetic targets in cancer therapy. Oncogene (2013) 32:4191–202. doi:10.1038/onc.2012.599
33. Sampaio EP, Sarno EN, Galilly R, Cohn ZA, Kaplan G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J Exp Med (1991) 173:699–703. doi:10.1084/jem.173.3.699
34. D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A (1994) 91:4082–5. doi:10.1073/pnas.91.9.4082
35. Haslett PA, Corral LG, Albert M, Kaplan G. Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J Exp Med (1998) 187:1885–92. doi:10.1084/jem.187.11.1885
36. Quach H, Ritchie D, Stewart AK, Neeson P, Harrison S, Smyth MJ, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia (2010) 24:22–32. doi:10.1038/leu.2009.236
37. Schafer PH, Gandhi AK, Loveland MA, Chen RS, Man HW, Schnetkamp PP, et al. Enhancement of cytokine production and AP-1 transcriptional activity in T cells by thalidomide-related immunomodulatory drugs. J Pharmacol Exp Ther (2003) 305:1222–32. doi:10.1124/jpet.102.048496
38. Bartlett JB, Michael A, Clarke IA, Dredge K, Nicholson S, Kristeleit H, et al. Phase I study to determine the safety, tolerability and immunostimulatory activity of thalidomide analogue CC-5013 in patients with metastatic malignant melanoma and other advanced cancers. Br J Cancer (2004) 90:955–61. doi:10.1038/sj.bjc.6601579
39. Lioznov M, El-Cheikh J Jr, Hoffmann F, Hildebrandt Y, Ayuk F, Wolschke C, et al. Lenalidomide as salvage therapy after allo-SCT for multiple myeloma is effective and leads to an increase of activated NK (NKp44(+)) and T (HLA-DR(+)) cells. Bone Marrow Transplant (2010) 45:349–53. doi:10.1038/bmt.2009.155
40. Hsu AK, Quach H, Tai T, Prince HM, Harrison SJ, Trapani JA, et al. The immunostimulatory effect of lenalidomide on NK-cell function is profoundly inhibited by concurrent dexamethasone therapy. Blood (2011) 117:1605–13. doi:10.1182/blood-2010-04-278432
41. Corral LG, Kaplan G. Immunomodulation by thalidomide and thalidomide analogues. Ann Rheum Dis (1999) 58(Suppl 1):I107–13. doi:10.1136/ard.58.2008.i107
42. Dredge K, Marriott JB, Todryk SM, Muller GW, Chen R, Stirling DI, et al. Protective antitumor immunity induced by a costimulatory thalidomide analog in conjunction with whole tumor cell vaccination is mediated by increased Th1-type immunity. J Immunol (2002) 168:4914–9.
43. Davies FE, Raje N, Hideshima T, Lentzsch S, Young G, Tai YT, et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood (2001) 98:210–6. doi:10.1182/blood.V98.1.210
44. Hayashi T, Hideshima T, Akiyama M, Podar K, Yasui H, Raje N, et al. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: clinical application. Br J Haematol (2005) 128:192–203. doi:10.1111/j.1365-2141.2004.05286.x
45. Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, et al. Lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res (2008) 14:4650–7. doi:10.1158/1078-0432.CCR-07-4405
46. Tai YT, Li XF, Catley L, Coffey R, Breitkreutz I, Bae J, et al. Immunomodulatory drug lenalidomide (CC-5013, IMiD3) augments anti-CD40 SGN-40-induced cytotoxicity in human multiple myeloma: clinical implications. Cancer Res (2005) 65:11712–20. doi:10.1158/0008-5472.CAN-05-1657
47. Wang M, Fayad L, Wagner-Bartak N, Zhang L, Hagemeister F, Neelapu SS, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol (2012) 13:716–23. doi:10.1016/S1470-2045(12)70200-0
48. Badoux XC, Keating MJ, Wen S, Wierda WG, O’Brien SM, Faderl S, et al. Phase II study of lenalidomide and rituximab as salvage therapy for patients with relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol (2013) 31:584–91. doi:10.1200/JCO.2012.42.8623
49. Weber DM, Chen C, Niesvizky R, Wang M, Belch A, Stadtmauer EA, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med (2007) 357:2133–42. doi:10.1056/NEJMoa070596
50. Rajkumar SV, Jacobus S, Callander NS, Fonseca R, Vesole DH, Williams ME, et al. Lenalidomide plus high-dose dexamethasone versus lenalidomide plus low-dose dexamethasone as initial therapy for newly diagnosed multiple myeloma: an open-label randomised controlled trial. Lancet Oncol (2010) 11:29–37. doi:10.1016/S1470-2045(09)70284-0
51. Benson DM Jr, Bakan CE, Zhang S, Collins SM, Liang J, Srivastava S, et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood (2011) 118:6387–91. doi:10.1182/blood-2011-06-360255
52. Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, et al. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol (2008) 180:163–70.
53. Shi J, Tricot GJ, Garg TK, Malaviarachchi PA, Szmania SM, Kellum RE, et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood (2008) 111:1309–17. doi:10.1182/blood-2007-03-078535
54. van Rhee F, Szmania SM, Dillon M, Van Abbema AM, Li X, Stone MK, et al. Combinatorial efficacy of anti-CS1 monoclonal antibody elotuzumab (HuLuc63) and bortezomib against multiple myeloma. Mol Cancer Ther (2009) 8:2616–24. doi:10.1158/1535-7163.MCT-09-0483
55. Lundqvist A, Su S, Rao S, Childs R. Cutting edge: bortezomib-treated tumors sensitized to NK cell apoptosis paradoxically acquire resistance to antigen-specific T cells. J Immunol (2010) 184:1139–42. doi:10.4049/jimmunol.0902856
56. Lundqvist A, Yokoyama H, Smith A, Berg M, Childs R. Bortezomib treatment and regulatory T-cell depletion enhance the antitumor effects of adoptively infused NK cells. Blood (2009) 113:6120–7. doi:10.1182/blood-2008-11-190421
57. Lundqvist A, Berg M, Smith A, Childs RW. Bortezomib treatment to potentiate the anti-tumor immunity of ex-vivo expanded adoptively infused autologous natural killer cells. J Cancer (2011) 2:383–5. doi:10.7150/jca.2.383
58. Boles KS, Mathew PA. Molecular cloning of CS1, a novel human natural killer cell receptor belonging to the CD2 subset of the immunoglobulin superfamily. Immunogenetics (2001) 52:302–7. doi:10.1007/s002510000274
59. Bouchon A, Cella M, Grierson HL, Cohen JI, Colonna M. Activation of NK cell-mediated cytotoxicity by a SAP-independent receptor of the CD2 family. J Immunol (2001) 167:5517–21.
60. Veillette A, Guo H. CS1, a SLAM family receptor involved in immune regulation, is a therapeutic target in multiple myeloma. Crit Rev Oncol Hematol (2013) 88:168–77. doi:10.1016/j.critrevonc.2013.04.003
61. Jakubowiak AJ, Benson DM, Bensinger W, Siegel DS, Zimmerman TM, Mohrbacher A, et al. Phase I trial of anti-CS1 monoclonal antibody elotuzumab in combination with bortezomib in the treatment of relapsed/refractory multiple myeloma. J Clin Oncol (2012) 30:1960–5. doi:10.1200/JCO.2011.37.7069
62. Lonial S, Vij R, Harousseau JL, Facon T, Moreau P, Mazumder A, et al. Elotuzumab in combination with lenalidomide and low-dose dexamethasone in relapsed or refractory multiple myeloma. J Clin Oncol (2012) 30:1953–9. doi:10.1200/JCO.2011.37.2649
63. Zonder JA, Mohrbacher AF, Singhal S, Van Rhee F, Bensinger WI, Ding H, et al. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood (2012) 120:552–9. doi:10.1182/blood-2011-06-360552
64. Kumaresan PR, Lai WC, Chuang SS, Bennett M, Mathew PA. CS1, a novel member of the CD2 family, is homophilic and regulates NK cell function. Mol Immunol (2002) 39:1–8. doi:10.1016/S0161-5890(02)00094-9
65. Hsi ED, Steinle R, Balasa B, Szmania S, Draksharapu A, Shum BP, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res (2008) 14:2775–84. doi:10.1158/1078-0432.CCR-07-4246
66. Tai YT, Dillon M, Song W, Leiba M, Li XF, Burger P, et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood (2008) 112:1329–37. doi:10.1182/blood-2007-08-107292
67. Benson DM Jr, Byrd JC. CS1-directed monoclonal antibody therapy for multiple myeloma. J Clin Oncol (2012) 30:2013–5. doi:10.1200/JCO.2011.40.4061
68. Cruz-Munoz ME, Dong Z, Shi X, Zhang S, Veillette A. Influence of CRACC, a SLAM family receptor coupled to the adaptor EAT-2, on natural killer cell function. Nat Immunol (2009) 10:297–305. doi:10.1038/ni.1693
69. Collins SM, Bakan CE, Swartzel GD, Hofmeister CC, Efebera YA, Kwon H, et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother (2013) 62:1841–49. doi:10.1007/s00262-013-1493-8
70. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med (2011) 364:1046–60. doi:10.1056/NEJMra1011442
71. Purdy AK, Campbell KS. Natural killer cells and cancer: regulation by the killer cell Ig-like receptors (KIR). Cancer Biol Ther (2009) 8:2211–20. doi:10.4161/cbt.8.23.10455
72. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295:2097–100. doi:10.1126/science.1068440
73. Hsu KC, Keever-Taylor CA, Wilton A, Pinto C, Heller G, Arkun K, et al. Improved outcome in HLA-identical sibling hematopoietic stem-cell transplantation for acute myelogenous leukemia predicted by KIR and HLA genotypes. Blood (2005) 105:4878–84. doi:10.1182/blood-2004-12-4825
74. Miller JS, Soignier Y, Panoskaltsis-Mortari A, Mcnearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105:3051–7. doi:10.1182/blood-2004-07-2974
75. Ljunggren HG, Malmberg KJ. Prospects for the use of NK cells in immunotherapy of human cancer. Nat Rev Immunol (2007) 7:329–39. doi:10.1038/nri2073
76. Kroger N, Zabelina T, Berger J, Duske H, Klyuchnikov E, Binder T, et al. Donor KIR haplotype B improves progression-free and overall survival after allogeneic hematopoietic stem cell transplantation for multiple myeloma. Leukemia (2011) 25:1657–61. doi:10.1038/leu.2011.138
77. Romagne F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood (2009) 114:2667–77. doi:10.1182/blood-2009-02-206532
78. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood (2010) 116:2411–9. doi:10.1182/blood-2010-05-283051
79. Venstrom JM, Pittari G, Gooley TA, Chewning JH, Spellman S, Haagenson M, et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N Engl J Med (2012) 367:805–16. doi:10.1056/NEJMoa1200503
80. Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol (2008) 180:6392–401.
81. Sola C, Andre P, Lemmers C, Fuseri N, Bonnafous C, Blery M, et al. Genetic and antibody-mediated reprogramming of natural killer cell missing-self recognition in vivo. Proc Natl Acad Sci U S A (2009) 106:12879–84. doi:10.1073/pnas.0901653106
82. Benson DM Jr, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood (2012) 120:4324–33. doi:10.1182/blood-2012-06-438028
83. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood (2012) 120:4317–23. doi:10.1182/blood-2012-06-437558
84. Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol (2002) 169:4098–102.
85. Salih HR, Antropius H, Gieseke F, Lutz SZ, Kanz L, Rammensee HG, et al. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood (2003) 102:1389–96. doi:10.1182/blood-2003-01-0019
86. Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia (2004) 6:558–68. doi:10.1593/neo.04316
87. Song H, Kim J, Cosman D, Choi I. Soluble ULBP suppresses natural killer cell activity via down-regulating NKG2D expression. Cell Immunol (2006) 239:22–30. doi:10.1016/j.cellimm.2006.03.002
88. Hilpert J, Grosse-Hovest L, Grünebach F, Buechele C, Nuebling T, Raum T, et al. Comprehensive analysis of NKG2D ligand expression and release in leukemia: implications for NKG2D-mediated NK cell responses. J Immunol (2012) 189:1360–71. doi:10.4049/jimmunol.1200796
89. Huergo-Zapico L, Gonzalez-Rodriguez AP, Contesti J, Gonzalez E, Lopez-Soto A, Fernandez-Guizan A, et al. Expression of ERp5 and GRP78 on the membrane of chronic lymphocytic leukemia cells: association with soluble MICA shedding. Cancer Immunol Immunother (2012) 61:1201–10. doi:10.1007/s00262-011-1195-z
90. Chitadze G, Lettau M, Bhat J, Wesch D, Steinle A, Furst D, et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int J Cancer (2013) 133:1557–66. doi:10.1002/ijc.28174
91. Ashiru O, Boutet P, Fernandez-Messina L, Aguera-Gonzalez S, Skepper JN, Vales-Gomez M, et al. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res (2010) 70:481–9. doi:10.1158/0008-5472.CAN-09-1688
92. Chen Z, Chen L, Baker K, Olszak T, Zeissig S, Huang YH, et al. CEACAM1 dampens antitumor immunity by down-regulating NKG2D ligand expression on tumor cells. J Exp Med (2011) 208:2633–40. doi:10.1084/jem.20102575
93. Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res (2005) 65:6321–9. doi:10.1158/0008-5472.CAN-04-4252
94. Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res (2005) 65:11136–45. doi:10.1158/0008-5472.CAN-05-0599
95. Diermayr S, Himmelreich H, Durovic B, Mathys-Schneeberger A, Siegler U, Langenkamp U, et al. NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood (2008) 111:1428–36. doi:10.1182/blood-2007-07-101311
96. Zhang C, Wang Y, Zhou Z, Zhang J, Tian Z. Sodium butyrate upregulates expression of NKG2D ligand MICA/B in HeLa and HepG2 cell lines and increases their susceptibility to NK lysis. Cancer Immunol Immunother (2009) 58:1275–85. doi:10.1007/s00262-008-0645-8
97. Jinushi M, Takehara T, Tatsumi T, Kanto T, Groh V, Spies T, et al. Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int J Cancer (2003) 104:354–61. doi:10.1002/ijc.10966
98. Rohner A, Langenkamp U, Siegler U, Kalberer CP, Wodnar-Filipowicz A. Differentiation-promoting drugs up-regulate NKG2D ligand expression and enhance the susceptibility of acute myeloid leukemia cells to natural killer cell-mediated lysis. Leuk Res (2007) 31:1393–402. doi:10.1016/j.leukres.2007.02.020
99. Poggi A, Catellani S, Garuti A, Pierri I, Gobbi M, Zocchi MR. Effective in vivo induction of NKG2D ligands in acute myeloid leukaemias by all-trans- retinoic acid or sodium valproate. Leukemia (2009) 23:641–8. doi:10.1038/leu.2008.354
100. Fionda C, Soriani A, Malgarini G, Iannitto ML, Santoni A, Cippitelli M. Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation. J Immunol (2009) 183:4385–94. doi:10.4049/jimmunol.0901797
101. Lu X, Ohata K, Kondo Y, Espinoza JL, Qi Z, Nakao S. Hydroxyurea upregulates NKG2D ligand expression in myeloid leukemia cells synergistically with valproic acid and potentially enhances susceptibility of leukemic cells to natural killer cell-mediated cytolysis. Cancer Sci (2010) 101:609–15. doi:10.1111/j.1349-7006.2009.01439.x
102. Hervieu A, Rebe C, Vegran F, Chalmin F, Bruchard M, Vabres P, et al. Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth. J Invest Dermatol (2013) 133:499–508. doi:10.1038/jid.2012.273
103. Sauer M, Reiners KS, Hansen HP, Engert A, Gasser S, Strandmann EP. Induction of the DNA damage response by IAP inhibition triggers natural immunity via upregulation of NKG2D ligands in Hodgkin lymphoma in vitro. Biol Chem (2013) 394:1325–31. doi:10.1515/hsz-2013-0161
104. Yamanegi K, Yamane J, Kobayashi K, Kato-Kogoe N, Ohyama H, Nakasho K, et al. Sodium valproate, a histone deacetylase inhibitor, augments the expression of cell-surface NKG2D ligands, MICA/B, without increasing their soluble forms to enhance susceptibility of human osteosarcoma cells to NK cell-mediated cytotoxicity. Oncol Rep (2010) 24:1621–7. doi:10.3892/or_00001026
105. Yang F, Shao Y, Liu M, Huang J, Zhu K, Guo C, et al. Valproic acid upregulates NKG2D ligand expression and enhances susceptibility of human renal carcinoma cells to NK cell-mediated cytotoxicity. Arch Med Sci (2013) 9:323–31. doi:10.5114/aoms.2013.34413
106. Chavez-Blanco A, De La Cruz-Hernandez E, Dominguez GI, Rodriguez-Cortez O, Alatorre B, Perez-Cardenas E, et al. Upregulation of NKG2D ligands and enhanced natural killer cell cytotoxicity by hydralazine and valproate. Int J Oncol (2011) 39:1491–9. doi:10.3892/ijo.2011.1144
107. Yamanegi K, Yamane J, Kobayashi K, Kato-Kogoe N, Ohyama H, Nakasho K, et al. Valproic acid cooperates with hydralazine to augment the susceptibility of human osteosarcoma cells to Fas- and NK cell-mediated cell death. Int J Oncol (2012) 41:83–91. doi:10.3892/ijo.2012.1438
108. Huang B, Sikorski R, Sampath P, Thorne SH. Modulation of NKG2D-ligand cell surface expression enhances immune cell therapy of cancer. J Immunother (2011) 34:289–96. doi:10.1097/CJI.0b013e31820e1b0d
109. Leung WH, Vong QP, Lin W, Janke L, Chen T, Leung W. Modulation of NKG2D ligand expression and metastasis in tumors by spironolactone via RXRgamma activation. J Exp Med (2013) 210:2675–92. doi:10.1084/jem.20122292
110. Fionda C, Malgarini G, Soriani A, Zingoni A, Cecere F, Iannitto ML, et al. Inhibition of glycogen synthase kinase-3 increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: role of STAT3. J Immunol (2013) 190:6662–72. doi:10.4049/jimmunol.1201426
111. Wilcox RA, Flies DB, Zhu G, Johnson AJ, Tamada K, Chapoval AI, et al. Provision of antigen and CD137 signaling breaks immunological ignorance, promoting regression of poorly immunogenic tumors. J Clin Invest (2002) 109:651–9. doi:10.1172/JCI14184
112. Wang C, Lin GH, Mcpherson AJ, Watts TH. Immune regulation by 4-1BB and 4-1BBL: complexities and challenges. Immunol Rev (2009) 229:192–215. doi:10.1111/j.1600-065X.2009.00765.x
113. Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer (2007) 7:95–106. doi:10.1038/nrc2051
114. Murillo O, Arina A, Hervas-Stubbs S, Gupta A, Mccluskey B, Dubrot J, et al. Therapeutic antitumor efficacy of anti-CD137 agonistic monoclonal antibody in mouse models of myeloma. Clin Cancer Res (2008) 14:6895–906. doi:10.1158/1078-0432.CCR-08-0285
115. Houot R, Goldstein MJ, Kohrt HE, Myklebust JH, Alizadeh AA, Lin JT, et al. Therapeutic effect of CD137 immunomodulation in lymphoma and its enhancement by Treg depletion. Blood (2009) 114:3431–8. doi:10.1182/blood-2009-05-223958
116. Melero I, Johnston JV, Shufford WW, Mittler RS, Chen L. NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell Immunol (1998) 190:167–72. doi:10.1006/cimm.1998.1396
117. Ito F, Li Q, Shreiner AB, Okuyama R, Jure-Kunkel MN, Teitz-Tennenbaum S, et al. Anti-CD137 monoclonal antibody administration augments the antitumor efficacy of dendritic cell-based vaccines. Cancer Res (2004) 64:8411–9. doi:10.1158/0008-5472.CAN-04-0590
118. Wang Q, Zhang P, Zhang Q, Wang X, Li J, Ma C, et al. Analysis of CD137 and CD137L expression in human primary tumor tissues. Croat Med J (2008) 49:192–200. doi:10.3325/cmj.2008.2.192
119. Baessler T, Charton JE, Schmiedel BJ, Grunebach F, Krusch M, Wacker A, et al. CD137 ligand mediates opposite effects in human and mouse NK cells and impairs NK-cell reactivity against human acute myeloid leukemia cells. Blood (2010) 115:3058–69. doi:10.1182/blood-2009-06-227934
120. Barao I. The TNF receptor-ligands 4-1BB-4-1BBL and GITR-GITRL in NK cell responses. Front Immunol (2013) 3:402. doi:10.3389/fimmu.2012.00402
121. Buechele C, Baessler T, Schmiedel BJ, Schumacher CE, Grosse-Hovest L, Rittig K, et al. 4-1BB ligand modulates direct and Rituximab-induced NK-cell reactivity in chronic lymphocytic leukemia. Eur J Immunol (2012) 42:737–48. doi:10.1002/eji.201141920
122. Wilcox RA, Tamada K, Strome SE, Chen L. Signaling through NK cell-associated CD137 promotes both helper function for CD8+ cytolytic T cells and responsiveness to IL-2 but not cytolytic activity. J Immunol (2002) 169:4230–6.
123. Lin W, Voskens CJ, Zhang X, Schindler DG, Wood A, Burch E, et al. Fc-dependent expression of CD137 on human NK cells: insights into “agonistic” effects of anti-CD137 monoclonal antibodies. Blood (2008) 112:699–707. doi:10.1182/blood-2007-11-122465
124. Kohrt HE, Houot R, Goldstein MJ, Weiskopf K, Alizadeh AA, Brody J, et al. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood (2011) 117:2423–32. doi:10.1182/blood-2010-08-301945
125. Kohrt HE, Houot R, Weiskopf K, Goldstein MJ, Scheeren F, Czerwinski D, et al. Stimulation of natural killer cells with a CD137-specific antibody enhances trastuzumab efficacy in xenotransplant models of breast cancer. J Clin Invest (2012) 122:1066–75. doi:10.1172/JCI61226
126. Melero I, Hirschhorn-Cymerman D, Morales-Kastresana A, Sanmamed MF, Wolchok JD. Agonist antibodies to TNFR molecules that costimulate T and NK cells. Clin Cancer Res (2013) 19:1044–53. doi:10.1158/1078-0432.CCR-12-2065
127. Houot R, Kohrt H, Levy R. Boosting antibody-dependant cellular cytotoxicity against tumor cells with a CD137 stimulatory antibody. Oncoimmunology (2012) 1:957–8. doi:10.4161/onci.19974
128. Zhou P, L’Italien L, Hodges D, Schebye XM. Pivotal roles of CD4+ effector T cells in mediating agonistic anti-GITR mAb-induced-immune activation and tumor immunity in CT26 tumors. J Immunol (2007) 179:7365–75.
129. Schaer DA, Cohen AD, Wolchok JD. Anti-GITR antibodies – potential clinical applications for tumor immunotherapy. Curr Opin Investig Drugs (2010) 11:1378–86.
130. Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med (2005) 202:885–91. doi:10.1084/jem.20050940
131. Cohen AD, Diab A, Perales MA, Wolchok JD, Rizzuto G, Merghoub T, et al. Agonist anti-GITR antibody enhances vaccine-induced CD8(+) T-cell responses and tumor immunity. Cancer Res (2006) 66:4904–12. doi:10.1158/0008-5472.CAN-05-2813
132. Coe D, Begom S, Addey C, White M, Dyson J, Chai JG. Depletion of regulatory T cells by anti-GITR mAb as a novel mechanism for cancer immunotherapy. Cancer Immunol Immunother (2010) 59:1367–77. doi:10.1007/s00262-010-0866-5
133. Cohen AD, Schaer DA, Liu C, Li Y, Hirschhorn-Cymmerman D, Kim SC, et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS One (2010) 5:e10436. doi:10.1371/journal.pone.0010436
134. Liu B, Li Z, Mahesh SP, Pantanelli S, Hwang FS, Siu WO, et al. Glucocorticoid-induced tumor necrosis factor receptor negatively regulates activation of human primary natural killer (NK) cells by blocking proliferative signals and increasing NK cell apoptosis. J Biol Chem (2008) 283:8202–10. doi:10.1074/jbc.M708944200
135. Baltz KM, Krusch M, Baessler T, Schmiedel BJ, Bringmann A, Brossart P, et al. Neutralization of tumor-derived soluble glucocorticoid-induced TNFR-related protein ligand increases NK cell anti-tumor reactivity. Blood (2008) 112:3735–43. doi:10.1182/blood-2008-03-143016
136. Buechele C, Baessler T, Wirths S, Schmohl JU, Schmiedel BJ, Salih HR. Glucocorticoid-induced TNFR-related protein (GITR) ligand modulates cytokine release and NK cell reactivity in chronic lymphocytic leukemia (CLL). Leukemia (2012) 26:991–1000. doi:10.1038/leu.2011.313
137. Schmiedel BJ, Werner A, Steinbacher J, Nuebling T, Buechele C, Grosse-Hovest L, et al. Generation and preclinical characterization of a Fc-optimized GITR-Ig fusion protein for induction of NK cell reactivity against leukemia. Mol Ther (2013) 21:877–86. doi:10.1038/mt.2013.11
138. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev (2010) 236:219–42. doi:10.1111/j.1600-065X.2010.00923.x
139. Nirschl CJ, Drake CG. Molecular pathways: co-expression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res (2013) 19:4917–24. doi:10.1158/1078-0432.CCR-12-1972
140. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med (1999) 5:1365–9. doi:10.1038/70932
141. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192:1027–34. doi:10.1084/jem.192.7.1027
142. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol (2001) 2:261–8. doi:10.1038/85330
143. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi:10.1038/nm730
144. Hallett WH, Jing W, Drobyski WR, Johnson BD. Immunosuppressive effects of multiple myeloma are overcome by PD-L1 blockade. Biol Blood Marrow Transplant (2011) 17:1133–45. doi:10.1016/j.bbmt.2011.03.011
145. Norris S, Coleman A, Kuri-Cervantes L, Bower M, Nelson M, Goodier MR. PD-1 expression on natural killer cells and CD8(+) T cells during chronic HIV-1 infection. Viral Immunol (2012) 25:329–32. doi:10.1089/vim.2011.0096
146. Zitvogel L, Kroemer G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology (2012) 1:1223–5. doi:10.4161/onci.21335
147. Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood (2010) 116:2286–94. doi:10.1182/blood-2010-02-271874
148. Dolina JS, Sung SS, Novobrantseva TI, Nguyen TM, Hahn YS. Lipidoid nanoparticles containing PD-L1 siRNA delivered in vivo enter Kupffer cells and enhance NK and CD8(+) T cell-mediated hepatic antiviral immunity. Mol Ther Nucleic Acids (2013) 2:e72. doi:10.1038/mtna.2012.63
149. Sznol M, Chen L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin Cancer Res (2013) 19:1021–34. doi:10.1158/1078-0432.CCR-12-2063
150. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol (2008) 8:467–77. doi:10.1038/nri2326
151. Thompson RH, Kuntz SM, Leibovich BC, Dong H, Lohse CM, Webster WS, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res (2006) 66:3381–5. doi:10.1158/0008-5472.CAN-05-4303
152. Thompson RH, Dong H, Lohse CM, Leibovich BC, Blute ML, Cheville JC, et al. PD-1 is expressed by tumor-infiltrating immune cells and is associated with poor outcome for patients with renal cell carcinoma. Clin Cancer Res (2007) 13:1757–61. doi:10.1158/1078-0432.CCR-06-2599
153. MacFarlane AW IV, Jillab M, Plimack ER, Hudes GR, Uzzo RG, Litwin S, et al. PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Clin Cancer Res (In press). doi:10.1158/2326-6066.CIR-13-0133
154. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol (2010) 28:3167–75. doi:10.1200/JCO.2009.26.7609
155. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, Mcdermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi:10.1056/NEJMoa1200690
156. Moreau P. The future of therapy for relapsed/refractory multiple myeloma: emerging agents and novel treatment strategies. Semin Hematol (2012) 49(Suppl 1):S33–46. doi:10.1053/j.seminhematol.2012.05.004
157. Takeda K, Okumura K, Smyth MJ. Combination antibody-based cancer immunotherapy. Cancer Sci (2007) 98:1297–302. doi:10.1111/j.1349-7006.2007.00529.x
158. Ascierto PA, Kalos M, Schaer DA, Callahan MK, Wolchok JD. Biomarkers for immunostimulatory monoclonal antibodies in combination strategies for melanoma and other tumor types. Clin Cancer Res (2013) 19:1009–20. doi:10.1158/1078-0432.CCR-12-2982
159. Melero I, Grimaldi AM, Perez-Gracia JL, Ascierto PA. Clinical development of immunostimulatory monoclonal antibodies and opportunities for combination. Clin Cancer Res (2013) 19:997–1008. doi:10.1158/1078-0432.CCR-12-2214
160. Armeanu S, Krusch M, Baltz KM, Weiss TS, Smirnow I, Steinle A, et al. Direct and natural killer cell-mediated antitumor effects of low-dose bortezomib in hepatocellular carcinoma. Clin Cancer Res (2008) 14:3520–8. doi:10.1158/1078-0432.CCR-07-4744
161. Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J Immunother (2011) 34:409–18. doi:10.1097/CJI.0b013e31821ca6ce
162. Gardiner D, Lalezari J, Lawitz E, Dimicco M, Ghalib R, Reddy KR, et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS One (2013) 8:e63818. doi:10.1371/journal.pone.0063818
Keywords: NK cells, immunotherapy of cancer, antibodies, monoclonal, ADCC, multiple myeloma
Citation: Mentlik James A, Cohen AD and Campbell KS (2013) Combination immune therapies to enhance anti-tumor responses by NK cells. Front. Immunol. 4:481. doi: 10.3389/fimmu.2013.00481
Received: 10 October 2013; Paper pending published: 25 October 2013;
Accepted: 09 December 2013; Published online: 23 December 2013.
Edited by:
Konrad Krzewski, National Institutes of Health, USAReviewed by:
Francisco Borrego, Food and Drug Administration, USAFrank Momburg, Deutsches Krebsforschungszentrum, Germany
Copyright: © 2013 Mentlik James, Cohen and Campbell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kerry S. Campbell, The Research Institute at Fox Chase Cancer Center, 333 Cottman Avenue, Philadelphia, PA 19111, USA e-mail:a2VycnkuY2FtcGJlbGxAZmNjYy5lZHU=