 Ariel D. Stock
Ariel D. Stock Jing Wen
Jing Wen Chaim Putterman
Chaim Putterman- 1Department of Microbiology and Immunology, Albert Einstein College of Medicine, Bronx, NY, USA
- 2Division of Rheumatology, Albert Einstein College of Medicine, Bronx, NY, USA
Patients with systemic lupus erythematosus (SLE) can experience acute neurological events such as seizures, cerebrovascular accidents, and delirium, psychiatric conditions including depression, anxiety, and psychosis, as well as memory loss and general cognitive decline. Neuropsychiatric SLE (NPSLE) occurs in between 30 and 40% of SLE patients, can constitute the initial patient presentation, and may occur outside the greater context of an SLE flare. Current efforts to elucidate the mechanistic underpinnings of NPSLE are focused on several different and potentially complementary pathways, including thrombosis, brain autoreactive antibodies, and complement deposition. Furthermore, significant effort is dedicated to understanding the contribution of neuroinflammation induced by TNF, IL-1, IL-6, and IFN-γ. More recent studies have pointed to a possible role for the TNF family ligand TWEAK in the pathogenesis of neuropsychiatric disease in human lupus patients, and in a murine model of this disease. The blood brain barrier (BBB) consists of tight junctions between endothelial cells (ECs) and astrocytic projections which regulate paracellular and transcellular flow into the central nervous system (CNS), respectively. Given the privileged environment of the CNS, an important question is whether and how the integrity of the BBB is compromised in NPSLE, and its potential pathogenic role. Evidence of BBB violation in NPSLE includes changes in the albumin quotient (Qalb) between plasma and cerebrospinal fluid, activation of brain ECs, and magnetic resonance imaging. This review summarizes the evidence implicating BBB damage as an important component in NPSLE development, occurring via damage to barrier integrity by environmental triggers such as infection and stress; cerebrovascular ischemia as result of a generally prothrombotic state; and immune mediated EC activation, mediated by antibodies and/or inflammatory cytokines. Additionally, new evidence supporting the role of TWEAK/Fn14 signaling in compromising the integrity of the BBB in lupus will be presented.
Introduction
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease characterized by hyper-activation of B and T lymphocytes resulting in the overproduction of autoantibodies, tissue deposition of immune complexes, and high levels of inflammatory cytokines, cumulatively resulting in a systemic pro-inflammatory state (1). SLE patients may suffer from skin, joint, hematologic, and renal disease, the latter being a predominant contributor to morbidity and mortality. Treatments have traditionally consisted of corticosteroids and potent immunosuppressive agents such as cyclophosphamide, though biologic medications targeting particular cytokines may eventually prove to be promising alternatives. Additionally, the course of the disease is highly variable between patients, with certain manifestations more common than others, and the overall impact on quality of life dependent on the individual patient’s circumstances and particular disease manifestations.
Central nervous system (CNS) presentations in SLE patients consist of a broad array of symptoms, which can be generally divided between focal neurological and diffuse psychiatric manifestations. Focal episodes may include seizures and cerebrovascular events, while psychiatric presentations often consist of anxiety and depression (2). A neuropsychiatric SLE (NPSLE) phenotype can be a presenting feature of lupus, and is eventually found in up to 40% of SLE patients (3). Research into the underlying mechanisms of NPSLE has taken several different and potentially complementary directions. Human, murine and in vitro systems have all been utilized to examine the effects of autoantibodies, cytokines, vascular disease, and cellular effectors in the development of NPSLE symptoms. TNF-like weak inducer of apoptosis, better known as TWEAK, is a TNF family member cytokine which together with its sole confirmed receptor Fn14 have recently shown to be instrumental in the pathogenesis of murine NPSLE. Furthermore, it is increasingly evident that blood brain barrier (BBB) disruption is an essential component of NPSLE pathogenesis (4), and that TWEAK may play an important role in this process (5).
NPSLE in Human Lupus and Experimental Models
Studying NPSLE in humans poses some obvious limitations, including the scarcity of CNS tissue samples and the heterogeneity of NPSLE presentations. Most available data from NPSLE patients consist of blood and cerebrospinal fluid (CSF) analysis, and radiologic imaging, including magnetic resonance imaging (MRI). CSF is often remarkable for the presence of increased immunoglobulins, elevated concentrations of cytokines, and evidence of BBB disruption, as measured by increased albumin concentrations. MRI data has additionally proven useful in identifying the brain regions most frequently involved in NPSLE, as well as which CNS tissues are affected (6). Additionally, extensive work has gone into the correlation between certain systemic autoantibody titers and NPSLE phenotypes (7).
There are several spontaneous mouse models of SLE, including the NZB × NZW F1 (NZB/W F1), BXSB, and MRL/Tnfrsf6lpr/lpr (MRL/lpr) strains. These three models all develop some measure of neuropsychiatric disease. BXSB mice, for example, demonstrate problems with both spatial and non-spatial learning tasks (8). One issue with the BXSB model, however, is the sex bias toward males, which is inconsistent with the strong female predominance found in human SLE. Additionally, BXSB and NZB/W F1 mice may have congenital structural abnormalities of the brain (9), potentially confounding structure-function analyses. Both NZB/W F1 and MRL/lpr mice demonstrate neurological deficits (10, 11), though the MRL/lpr model has a greater incidence of neuropsychiatric disease (12). The MRL/lpr has the added benefit of a congenic control (MRL+/+), which does not develop disease.
The MRL/lpr strain develops a disease phenotype consistent with the affective and behavioral pathologies seen in human lupus (13). Gao et al. found that depressive symptoms appear as early as 6 weeks of age in female MRL/lpr mice, preceding onset of renal pathology. Additionally, we found a correlation between depressive symptoms and several autoantibodies, including anti-NMDAR and anti-dsDNA (14). Similarly, depression and other neuropsychiatric symptoms can appear early in the disease course in human disease (15). MRL/lpr mice demonstrate increased immobility on the forced swim test, which is a widely accepted indicator of depression in rodents (if strength and locomotion are otherwise normal). Additionally, MRL/lpr mice display decreased preference for sweetened water (anhedonia), as well as an acquired anosmia, both manifestations of murine depressive-like behavior (16, 17). Finally, cognitive tests in MRL/lpr mice reveal clear deficits in the object placement task indicating deficits in spatial memory, relatable to the cognitive decline found in NPSLE patients.
Another experimental model of NPSLE is induced by treatment with anti-N-methyl-d-aspartate (anti-NMDA; also known as anti-NR2) receptor antibodies coupled with BBB disruption in BALB/c mice (18). Depending on the method of BBB disruption, resulting symptoms may include impaired performance on memory tasks or altered fear responses (19, 20). More recently, Kivity et al. showed that intracerebroventricular transfer of the 16/6 idotype (a human anti-ssDNA antibody) into C3H mice resulted in hippocampal inflammation and decreased performance in memory tasks (21).
Neuropathic Antibodies in NPSLE
As previously mentioned, NPSLE deficits can be defined as either focal or diffuse in nature. Focal findings in NPSLE are most readily associated with the presence of antiphospholipid (aPL) antibodies, including anti-cardiolipin antibodies, anti-β2glycoprotein I antibodies, and lupus anticoagulant (22). These antibodies dramatically increase susceptibility to thrombosis, resulting in an increased rate of cerebrovascular accidents (CVA) and transient ischemic attacks (TIA). It is thought that aPL antibodies may act through increasing oxidative stress, as measured by increased level of oxidized low-density lipoprotein, which itself is associated with atherosclerosis and thrombosis (23). While these patients will present with typical focal findings, such as motor and cranial nerve deficits, seropositivity for aPL antibodies is not typically associated with diffuse psychiatric and cognitive presentations (7).
Other circulating and intrathecal antibodies are also associated with NPSLE manifestations. Anti-ribosomal-P (anti-P) antibodies have long been associated with NPSLE presentations (24–26), and more recently have been found to induce depression in mice when injected intraventricularly (16). Recent work by Matus et al. found that anti-P antibodies from human lupus serum induced calcium influx and subsequent apoptosis in p331 positive neurons in rats, which they characterized as a new P-antigen. Death of these neurons, found in the hippocampus, amygdala, and certain neocortical layers, account for a broad range of potential symptoms, including depression, memory deficits, and cognitive decline (27). Anti-NMDA receptor antibodies are also associated with psychiatric symptoms, such as depression and memory dysfunction in SLE patients, and altered fear responses when transferred to mice, due to excitotoxic glutamatergic effects on neurons (14, 19, 28–30). Anti-U1-RNP antibodies have been reported by Sato et al. as a more specific marker of NPSLE than anti-P or anti-NR2 antibodies (31), and may potently induce expression of interferons (32). Anti-ganglioside (anti-GM1) antibodies were once thought to be associated with NPSLE pathogenesis (4, 33) through disruption of voltage gated Na+ channels found near nodes of Ranvier (34), though it has since been found that they are more likely associated with peripheral neuropathies than central NPSLE presentations (35). Other antibodies associated with NPSLE include anti-dsDNA, anti-Microtubule-associated Protein 2, anti-Triose-phosphate isomerase, and brain reactive autoantibodies (BRAA) (36–38). The reader is referred to a number of excellent reviews for further detail regarding these and other neuropathic antibodies (39–42). Regardless of the antigenic specificity of these neuropathic autoantibodies, the question remains how they gain entry into the central nervous system (CNS) from the systemic circulation, implicating a failure in the BBB as a key component of NPSLE pathogenesis.
The Blood Brain Barrier
The CNS is maintained as a privileged environment due to the combination of tight junctions between endothelial cells (ECs) limiting paracellular transport, and astrocytic processes, which regulate transcellular transport from systemic circulation. The choroid plexus (CP) and arachnoid epithelia similarly maintain this barrier through tight junctions, as well as secrete and reabsorb CSF, respectively. Another element that seems essential to a competent blood brain barrier is the presence of resident microglia. Microglia populations of monocytic origin are found perivascularly within the CNS parenchyma, limit paracellular transport across ECs, and may serve as a bridge between CNS and systemic immune activity (43).
The isolating nature of brain microvascular ECs is attributable to the tight junctions, including members of the zonula occludens (ZO-1) and claudin (claudin-5) families, resulting in impermeability to macromolecules (44). Additionally, these multi-laminated junctional complexes provide very high electrical resistance, dependent on the basal parenchymal presence of astrocytes, yielding remarkably low permeability to smaller and ionic molecules (45, 46). Astrocytes are also required to provide the environmental cues ECs need to develop and maintain their unique CNS phenotype, and are involved in bidirectional cytokine signaling with ECs (47). Microglia are involved as well in modulating EC through secretion of TNF, which upregulates MHC-I presentation on ECs and promotes entry of T-cells into brain parenchyma during pathological states (48).
Beyond serving as a barrier, ECs are directly responsible for regulating the immune response in the brain. ECs help maintain the CNS in its basal state of suppressed immunity through secretion of TGFβ (49) and soluble cellular adhesion molecules (50). Brain ECs can, however, activate an immune response, such as under LPS stimulation, which induces production of IL-6 and GM-CSF. Interestingly, Verma et al. showed that LPS itself does not appear to induce disruption of EC junctional complexes; rather, further signaling by luminal and parenchymal effector cells likely potentiates BBB disruption following LPS stimulation (51).
The CP vasculature is unique within the CNS, as it consists of fenestrated ECs necessary for CSF volume maintenance. This vasculature is isolated from brain parenchyma by a blood-CSF barrier (BCSFB). The BCSFB consists of cuboidal CP epithelial cells, which are interconnected by tight junctions, effectively providing for a barrier similar to the BBB (52, 53). CSF not only provides physical support to CNS tissue by reducing its apparent weight, it is also able to rapidly transmit hormonal signals within the CNS (54) and provide for drainage, much as the lymphatics do systemically (55). Additionally, CSF is enriched with memory T-cells due to one of its primary roles, immune surveillance of the CNS. Normally, non-autoreactive T-cells are allowed to pass through the CP epithelial barrier unhindered (56). It is believed, however, that through increased expression of cellular adhesion molecules, CP ECs are involved in the pathogenesis of certain autoimmune conditions, such as multiple sclerosis (57).
Evidence of BBB Damage in SLE
There are several markers available to monitor the integrity of the BBB. Albumin is a large and extensively charged protein that is not synthesized intrathecally, and whose transport into the CNS is tightly regulated. As such, an elevated albumin concentration gradient (Qalb) between CSF and plasma (normal [Alb]CSF/[Alb]Plasma ≤ 7.6 × 10−3) serves as an indicator of BBB disruption, and has been found repeatedly in NPSLE patients (31, 58, 59) and MRL/lpr mice (60). While Qalb provides a useful measurement of relatively large scale leakage across the BBB, it lacks the finer resolution needed to appreciate small and transient leakage (4). The IgG index [CSF(IgG/Albumin)]/[Serum(IgG/Albumin)] is another useful measure of BBB permeability that can also identify the relative intrathecal vs. systemic origin of IgG within the CNS (61, 62), and is found elevated in both NPSLE patients and experimental models (31, 60). Sato et al. found both an elevated Qalb and an elevated IgG index in 8 and 9 of 14 NPSLE patients, respectively (31). Similarly, Sidor et al. found elevated Qalb and IgG index measurements in MRL/lpr mice when compared with MRL+/+ mice, along with increased neurodegeneration in those mice with a disrupted BBB (60). Jacob et al. further demonstrated that IgG enters CNS parenchyma in MRL/lpr mice (63). Finally, Ma et al. have shown extensive penetration of CD3+ cells into the CP and in brain parenchyma, as well as the presence of CD19+-B-cells in MRL/lpr mice, providing further evidence of BBB failure in NPSLE (64).
The availability of new and improved modalities with finer resolution will likely continue to be an asset in measuring BBB function non-invasively in NPSLE. Since the calcium binding protein S100B is predominately found in astrocytes, its presence in serum serves as valuable indicator of BBB injury (65–67). Schenatto et al., examining a cohort of 89 SLE patients, identified elevated S100B levels in NPSLE vs. non-NPSLE, a finding which was even more prominent during acute episodes (68). Evolution of MRI is also proving to be useful in the clinical characterization of BBB breaches. The increasing availability of 3.0 T MRI magnets and newer gadolinium (Gd) containing contrast agents, which visualizes contrast flow into CNS parenchyma with T1-weighted imaging, are improving the ability to highlight areas of BBB insufficiency (69). Recently, Toledano et al. utilized multiple MRI modalities in imaging NPSLE patient brains, and found vascular damage in a third of patients with the vast majority consisting of small vessel damage (70). One could reasonably speculate that evidence of BBB disruption may be obscured due to transient changes in BBB integrity, or the reparative effects of treatment. Indeed, findings of BBB disruption by Gd-contrast enhancement are likely under-reported, since the frequent use of corticosteroids in SLE treatment likely results in stabilization of BBB damage (71).
Mechanisms of Brain EC Activation and BBB Disruption in SLE
EC Activation in SLE
As described above, brain ECs are not mere bystanders in the regulation of the CNS environment; they play an active role in concert with both CNS and luminal effector cells and molecules. Infection and systemic inflammation are potent activators of ECs, resulting in upregulated expression of cytokines and chemokines, thereby potentiating a local immune response (51, 72). IL-1, TNF, and LPS signaling each result in upregulation of E-selectin, ICAM-1, and VCAM-1 in microvascular ECs in vitro (73). MRL/lpr mice are found to have elevated expression of ICAM-1 and VCAM-1 in predominately CP associated ECs when compared to congenic controls (74). Sun et al. have recently shown that immune complexes in SLE induce production of inflammatory cytokines and cellular adhesion molecules in ECs via NF-κB signaling, due to HMGB1-RAGE axis activation (75).
TREX1 is a major endogenous 3′–5′ DNA exonuclease. Mutations in TREX1 are associated with chilblain lupus erythematosus, a rare form of cutaneous disease, as well as with sporadic SLE (76). In addition, TREX1 variants are found in two other diseases with neurological manifestations, autosomal dominant retinal vasculopathy with cerebral leukodystrophy and Aicardi–Goutieres syndrome (77). TREX1 deficiency or dysfunction may lead to accumulation of cytosolic DNA and enhanced alpha-interferon signaling. Furthermore, TREX1 deficiency in lymphocytes modulates vascular EC angiogenesis (78), suggesting an interesting possible link between the genetic susceptibility for lupus associated with TREX1, ECs, and CNS disease (76). Irrespective of the cause, endothelial activation results in increased vascular permeability and diapedesis, and increased local aggregation of humoral and cellular effectors.
Environmentally Induced BBB Disruption
Diamond et al. have done extensive work on anti-NMDA receptor antibodies as effectors of NPSLE symptomatology. In their early studies, CNS disease was induced by direct injection of anti-NMDA receptor antibodies into the cerebral cortices of healthy mice, thereby bypassing the BBB (79). They then utilized several models of extraneous BBB disruption, including LPS as model of infection and epinephrine as a model of stress. When LPS was used to induce BBB disruption, mice showed poorer performance on the Morris water maze and T-maze tasks, indicative of learning and memory deficits, and consistent with hippocampal damage. Neuron loss in the hippocampus was found histologically and was evident as structural abnormalities on MRI (80). When using epinephrine, neuron loss occurred selectively in the amygdala, inducing alterations in conditioned fear responses in mice, while sparing hippocampal neurons (81). These findings provide a possible explanation for the phenotypic differences between NPSLE patients, and the role environmental mediators may play.
Anti-Endothelial Cell Antibodies
Conti et al. found that 64.7% of NPSLE patients sera are positive for anti-endothelial cell antibodies (AECAs), as compared to only 29.4% of non-NPSLE patients (82). AECAs have been previously characterized as inducers of increased EC cellular adhesion molecules, including E-selectin, ICAM-1, and VCAM-1. Nara et al. stimulated HUVEC with monoclonal antibodies targeted toward thrombomodulin, a proposed antigenic substrate of some AECAs in SLE patients (83), and found increased endothelial production of IL-6 and IL-8 mediated through the NF-κB pathway (84). More recently, Yoshio et al. found that anti-NR2 antibodies recognize antigenic targets on HUVECs, and can induce IL-6 and IL-8 expression with IL-1β co-treatment (85). Collectively, AECAs may induce endothelial activation, which is pivotal in many inflammatory processes, but in the brain is an important component of BBB disruption as well (86).
Complement
One of the hallmarks of increased disease activity in SLE patients is the depletion of complement components from serum, due to consumption by circulating immune complexes, deposition in target tissues and targeting by anti-C1q antibodies (87, 88). Low circulating levels of complement components C3 and C4 have also been suggested as potential biomarkers of human NPSLE activity (89). Alexander et al. first demonstrated that complement inhibition can attenuate NPSLE presentations in MRL/lpr mice (90). Jacob et al. then showed that C5aR activation induces EC cytoskeletal alteration in vitro and laminin disruption in MRL/lpr brain vasculature, both indicators of BBB damage. Additionally, C5aR activation results in increased CCL2 and CXCL2 production by mouse microvascular ECs, when pretreated with IL-6 (91). Finally, in further characterizing the BBB disruption that occurs with C5aR activation, Jacob et al. found increased expression of MAP-kinase, increased nuclear NF-κB translocation and decreased zona occludin (ZO) levels, indicative of EC junctional complex interruption, which would dramatically increase BBB permeability (92).
Cytokines and Chemokines
Stimulation of human brain microvessel ECs (HBMEC) with cytokines such as IL-1β, IL-8, TNF, and IFN-γ are known to induce increased permeability across monolayers (86, 93). CCL2 signaling has been shown to play a role in BBB disruption both in vitro and in CCR2−/− mice (94, 95). Zameer et al. found elevated expression levels of ICAM-1 and VCAM-1 in the CNS of MRL/lpr mice, providing further evidence of an inflammatory process involving the BBB (74, 96). Trysberg et al., analyzing CSF from lupus patients, found elevated levels of IL-6 and IL-8 which were correlated with elevated MMP-9 levels, the latter associated with degradation of BBB extracellular matrix (97).
In most, if not all of the above models of BBB disruption, it is clear that the cytokine and chemokine environment is critical. While complement and AECAs may indeed be effectors of BBB disruption, they cannot do so on their own; cytokines are needed to fully activate and disrupt the BBB. Amongst these cytokines, TWEAK has recently been demonstrated as a potent effector of multiple downstream pathways needed for BBB disruption, including activating intracellular signaling cascades and inducing production of additional cytokines, chemokines, and metalloproteinases.
The causes and consequences of EC activation are summarized in Table 1.
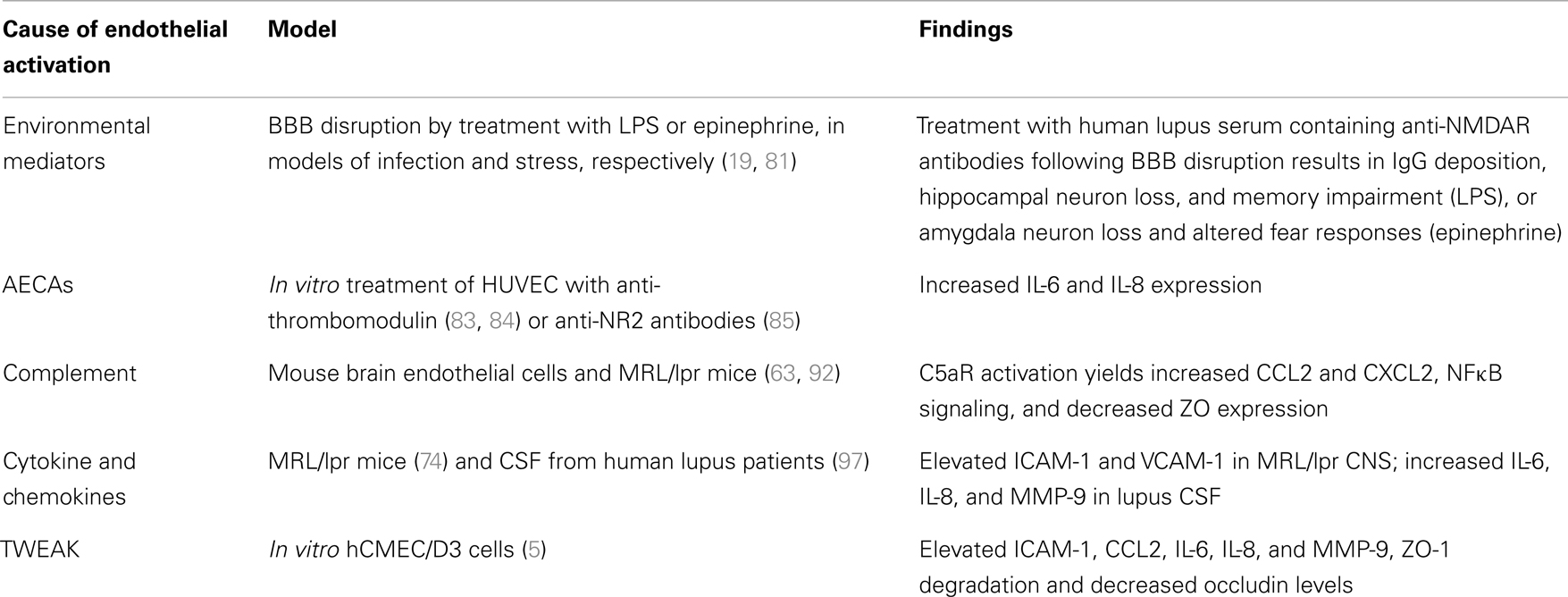
Table 1. Potential routes of endothelial cell activation in SLE.
TWEAK, NPSLE, and BBB Disruption
TWEAK is a pro-inflammatory cytokine member of the TNF superfamily. Through activation of its sole receptor, Fn14, TWEAK variably induces cellular proliferation, angiogenesis, inflammation, and apoptosis (98). As previously mentioned, MRL/lpr mice develop a neuropsychiatric phenotype remarkably similar to human SLE. We recently found increased TWEAK and Fn14 expression in the cerebral cortices of MRL/lpr mice. Furthermore, in MRL/lpr Fn14-knockout (Fn14KO) mice we found significantly improved cognitive function, decreased depression, and less anhedonia, as demonstrated by object placement tasks, the forced swim test, and preference for sweetened water, respectively (17). The attenuated neuropsychiatric phenotype in Fn14 deficient lupus mice may be due to decreases in brain expression of CCL5 and C3, which have been found associated with depression and cognitive decline (99–101). Other mechanisms by which the Fn14KO is protective of NPSLE development in MRL/lpr mice are under active investigation.
Fn14 is expressed both in the human cerebral microvascular EC line (hCMEC/D3) and astrocytes, while TWEAK is secreted by only the latter (5, 102). As described above, we found decreased Qalb ratios in MRL/lpr Fn14KO mice as well as decreased CSF titers of anti-dsDNA antibodies (17). More recently, we characterized the effects of TWEAK on endothelial cytokine expression and barrier disruption using hCMEC/D3 brain ECs, and found TWEAK-induced increases in ICAM-1, CCL2, IL-6, and IL-8 (5). Furthermore, TWEAK-induced activation of the MAPK pathway yielded increased expression of MMP-9, which degraded ZO-1, decreased occludin expression (Figure 1), and increased permeability (5). A similar finding was seen by Polavarapu et al. in their investigation of cerebral ischemic injury, where intracerebral injection of TWEAK in wild type mice increased MMP-9 activity and BBB permeability (103). Furthermore, TWEAK induced expression of ICAM-1, IL-8, and IL-6 in cultured astrocytes, typical of reactive astrocytes, another effector of BBB disruption (104). Together, this data supports the conclusion that TWEAK is instrumental in the development of lupus associated neuropsychiatric disease, with BBB disruption as an important mechanistic contribution of this cytokine. The lack of effect of TWEAK on systemic autoantibodies (17) and our current understanding of the mechanism of action of the TWEAK/Fn14 signaling pathway also suggest that attenuated neuropsychiatric disease in MRL/lpr Fn14 KO mice is not due to a reduced systemic autoantibody response, but rather local (i.e., brain) effects of blocking this pathway such as the effect on the BBB discussed here. Finally, it is important to note that not only is TWEAK/Fn14 signaling involved in NPSLE, but this cytokine/receptor pair has been implicated in the pathogenesis of injury in other major lupus target organs, including the kidney [for which there is evidence both in murine models (105, 106), and in human disease (107)], and in cutaneous lupus as well (108).
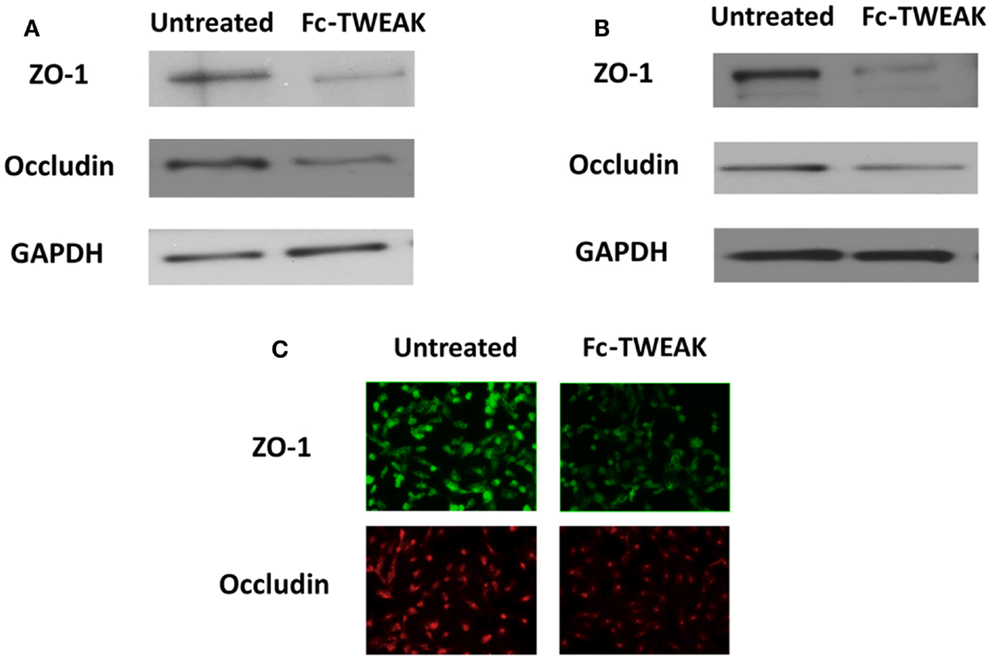
Figure 1. ZO-1 and occludin expression after treatment with Fc-TWEAK. ZO-1 and occludin expression was measured by western blot. Treatment with Fc-TWEAK (100 ng/mL, 48 h) decreased ZO-1 and occludin expression in hCMEC/D3 cells (A). ZO-1 and occludin expression was decreased in hBMEC cells after treatment with Fc-TWEAK (100 ng/mL, 24 h) as well (B). Similarly, immunofluorescent detection of ZO-1 and occludin in hBMECs was reduced after treatment with Fc-TWEAK (100 ng/mL, 24 h) (C).
The success of mAb treatment targeting pathogenic cytokines such as TNF, IL-1, and BLyS in inflammatory rheumatic diseases, together with the data presented above, strongly suggest the need to examine anti-TWEAK antibody treatment as novel treatment approach in NPSLE. Antibodies given intravenously can have therapeutic effects on the brain, especially if the blood brain barrier is already breached (as in NPSLE) (109). Alternatively, it would be necessary to bypass the BBB, or develop a delivery system to deliver the antibody to the CNS despite the compartmentalization of the brain from the blood (e.g., bispecific antibodies using the transferrin receptor) (110).
Conclusion
Neuropsychiatric SLE is often associated with the presence of neuropathic antibodies within the CNS, making the question of how they gain entry into this anatomically privileged space increasingly important. Evidence points to entry of autoantibodies across the BBB, with entry into different brain regions and specific autoantibody subtypes potentially associated with the variable phenotypes found in both murine experimental models and NPSLE patients. There is strong support for the roles of AECAs, complement components, and environmental mediators in increasing permeability across the BBB, though in each of these cases, cytokines and chemokines have an essential role as well. TWEAK appears to be one such cytokine that is necessary for the development of NPSLE; one mechanism central to the contributions of the TWEAK/Fn14 axis appears to be its role in BBB disruption.
Interestingly, TWEAK/Fn14 signaling has been implicated in other neurologic diseases besides NPSLE, including hypoxic brain damage and autoimmune brain disease. Elsewhere in this special issue, Yepes describes how TWEAK/Fn14 signaling in middle cerebral artery occlusion (a model of ischemic stroke) induces inflammation and MMP-9 mediated basal lamina disruption (111). Desplat-Jego et al. demonstrated the involvement of TWEAK in the development of experimental autoimmune encephalomyelitis (EAE), a murine model of multiple sclerosis, and that TWEAK over-expression in transgenic mice further exacerbates the EAE phenotype (102). In both these disease models, preventing TWEAK signaling via Fn14 deficiency, treatment with a Fn14-Fc decoy receptor, or treatment with anti-TWEAK monoclonal antibodies results in attenuated disease. Ameliorating the disruption of the BBB may be a valuable tool in the control of NPSLE as well other neurologic disorders, and targeting the TWEAK pathway may be one way to do so.
Conflict of Interest Statement
The authors Ariel D. Stock and Jing Wen declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Research presented in this paper was supported in part by a grant from Biogen Idec to Chaim Putterman.
Acknowledgments
Funding: Studies reported in this paper were supported by research grants from Biogen Idec and the National Institutes of Health (NIDDK, RO1 DK090319) to C. Putterman.
References
1. Frieri M. Mechanisms of disease for the clinician: systemic lupus erythematosus. Ann Allergy Asthma Immunol (2013) 110(4):228–32. doi:10.1016/j.anai.2012.12.010
2. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum (1999) 42(4):599–608. doi:10.1002/1529-0131(199904)42:4<599::AID-ANR2>3.0.CO;2-F
3. Bertsias GK, Boumpas DT. Pathogenesis, diagnosis and management of neuropsychiatric SLE manifestations. Nat Rev Rheumatol (2010) 6(6):358–67. doi:10.1038/nrrheum.2010.62
4. Abbott NJ, Mendonça LLF, Dolman DEM. The blood-brain barrier in systemic lupus erythematosus. Lupus (2003) 12(12):908–15. doi:10.1191/0961203303lu501oa
5. Stephan D, Sbai O, Wen J, Couraud PO, Putterman C, Khrestchatisky M, et al. TWEAK/Fn14 pathway modulates properties of a human microvascular endothelial cell model of blood brain barrier. J Neuroinflammation (2013) 10:9. doi:10.1186/1742-2094-10-9
6. Peterson PL, Axford JS, Isenberg D. Imaging in CNS lupus. Best Pract Res Clin Rheumatol (2005) 19(5):727–39. doi:10.1016/j.berh.2005.04.001
7. Borowoy AM, Pope JE, Silverman E, Fortin PR, Pineau C, Smith CD, et al. Neuropsychiatric lupus: the prevalence and autoantibody associations depend on the definition: results from the 1000 faces of lupus cohort. Semin Arthritis Rheum (2012) 42(2):179–85. doi:10.1016/j.semarthrit.2012.03.011
8. Denenberg VH, Sherman GF, Schrott LM, Rosen GD, Galaburda AM. Spatial learning, discrimination learning, paw preference and neocortical ectopias in two autoimmune strains of mice. Brain Res (1991) 562(1):98–104. doi:10.1016/0006-8993(91)91192-4
9. Sherman GF, Galaburda AM, Behan PO, Rosen GD. Neuroanatomical anomalies in autoimmune mice. Acta Neuropathol (1987) 74(3):239–42. doi:10.1007/BF00688187
10. Šakic B, Szechtman H, Keffer M, Talangbayan H, Stead R, Denburg JA. A behavioral profile of autoimmune lupus-prone MRL mice. Brain Behav Immun (1992) 6(3):265–85. doi:10.1016/0889-1591(92)90048-S
11. Forster MJ, Popper MD, Retz KC, Lal H. Age differences in acquisition and retention of one-way avoidance learning in C57BL/6NNia and autoimmune mice. Behav Neural Biol (1988) 49(2):139–51. doi:10.1016/S0163-1047(88)90462-1
12. Vogelweid CM, Wright DC, Johnson JC, Hewett JE, Walker SE. Evaluation of memory, learning ability, and clinical neurologic function in pathogen-free mice with systemic lupus erythematosus. Arthritis Rheum (1994) 37(6):889–97. doi:10.1002/art.1780370617
13. Gulinello M, Putterman C. The MRL/lpr mouse strain as a model for neuropsychiatric systemic lupus erythematosus. J Biomed Biotechnol (2011) 2011:207504. doi:10.1155/2011/207504
14. Gao HX, Sanders E, Tieng AT, Putterman C. Sex and autoantibody titers determine the development of neuropsychiatric manifestations in lupus prone mice. J Neuroimmunol (2010) 229:112–22. doi:10.1016/j.jneuroim.2010.07.020
15. Meszaros ZS, Perl A, Faraone SV. Psychiatric symptoms in systemic lupus erythematosus: a systematic review. J Clin Psychiatry (2012) 73(7):993–1001. doi:10.4088/JCP.11m07043
16. Katzav A, Solodeev I, Brodsky O, Chapman J, Pick CG, Blank M, et al. Induction of autoimmune depression in mice by anti-ribosomal P antibodies via the limbic system. Arthritis Rheum (2007) 56(3):938–48. doi:10.1002/art.22419
17. Wen J, Xia Y, Stock A, Michaelson JS, Burkly LC, Gulinello M, et al. Neuropsychiatric disease in murine lupus is dependent on the TWEAK/Fn14 pathway. J Autoimmun (2013) 43:44–54. doi:10.1016/j.jaut.2013.03.002
18. Kowal C, Diamond B. Aspects of CNS lupus: mouse models of anti-NMDA receptor antibody mediated reactivity. Methods Mol Biol (2012) 900:181–206. doi:10.1007/978-1-60761-720-4_9
19. Kowal C, Degiorgio LA, Lee JY, Edgar MA, Huerta PT, Volpe BT, et al. Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc Natl Acad Sci USA (2006) 103(52):19854–9. doi:10.1073/pnas.0608397104
20. Diamond B, Kowal C, Huerta PT, Aranow C, Mackay M, DeGiorgio LA, et al. Immunity and acquired alterations in cognition and emotion: lessons from SLE. Adv Immunol (2006) 89:289–320. doi:10.1016/S0065-2776(05)89007-8
21. Kivity S, Katzav A, Arango MT, Landau-Rabi M, Zafrir Y, Agmon-Levin N, et al. 16/6-Idiotype expressing antibodies induce brain inflammation and cognitive impairment in mice: the mosaic of central nervous system involvement in lupus. BMC Med (2013) 11:90. doi:10.1186/1741-7015-11-90
22. Devreese KM. Standardization of antiphospholipid antibody assays. Where do we stand? Lupus (2012) 21(7):718–21. doi:10.1177/0961203312439335
23. Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med (2013) 368(11):1033–44. doi:10.1056/NEJMra1112830
24. Bonfa E, Golombek SJ, Kaufman LD, Skelly S, Weissbach H, Brot N, et al. Association between lupus psychosis and anti-ribosomal P protein antibodies. N Engl J Med (1987) 317(5):265–71. doi:10.1056/NEJM198707303170503
25. Watanabe T, Sato T, Uchiumi T, Arakawa M. Neuropsychiatric manifestations in patients with systemic lupus erythematosus: diagnostic and predictive value of longitudinal examination of anti-ribosomal P antibody. Lupus (1996) 5(3):178–83. doi:10.1177/096120339600500303
26. Nojima Y, Minota S, Yamada A, Takaku F, Aotsuka S, Yokohari R. Correlation of antibodies to ribosomal P protein with psychosis in patients with systemic lupus erythematosus. Ann Rheum Dis (1992) 51(9):1053–5. doi:10.1136/ard.51.9.1053
27. Matus S, Burgos PV, Bravo-Zehnder M, Kraft R, Porras OH, Farias P, et al. Antiribosomal-P autoantibodies from psychiatric lupus target a novel neuronal surface protein causing calcium influx and apoptosis. J Exp Med (2007) 204(13):3221–34. doi:10.1084/jem.20071285
28. Fragoso-Loyo H, Cabiedes J, Orozco-Narvaez A, Davila-Maldonado L, Atisha-Fregoso Y, Diamond B, et al. Serum and cerebrospinal fluid autoantibodies in patients with neuropsychiatric lupus erythematosus. Implications for diagnosis and pathogenesis. PLoS One (2008) 3(10):e3347. doi:10.1371/journal.pone.0003347
29. Omdal R, Brokstad K, Waterloo K, Koldingsnes W, Jonsson R, Mellgren SI. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur J Neurol (2005) 12(5):392–8. doi:10.1111/j.1468-1331.2004.00976.x
30. Lapteva L, Nowak M, Yarboro CH, Takada K, Roebuck-Spencer T, Weickert T, et al. Anti-N-methyl-D-aspartate receptor antibodies, cognitive dysfunction, and depression in systemic lupus erythematosus. Arthritis Rheum (2006) 54(8):2505–14. doi:10.1002/art.22031
31. Sato T, Fujii T, Yokoyama T, Fujita Y, Imura Y, Yukawa N, et al. Anti-U1 RNP antibodies in cerebrospinal fluid are associated with central neuropsychiatric manifestations in systemic lupus erythematosus and mixed connective tissue disease. Arthritis Rheum (2010) 62(12):3730–40. doi:10.1002/art.27700
32. Santer DM, Yoshio T, Minota S, Moller T, Elkon KB. Potent induction of IFN-alpha and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus. J Immunol (2009) 182(2):1192–201.
33. Pereira RM, Yoshinari NH, De Oliveira RM, Cossermelli W. Antiganglioside antibodies in patients with neuropsychiatric systemic lupus erythematosus. Lupus (1992) 1(3):175–9. doi:10.1177/096120339200100309
34. Weber F, Rüdel R, Aulkemeyer P, Brinkmeier H. Anti-GM1 antibodies can block neuronal voltage-gated sodium channels. Muscle Nerve (2000) 23(9):1414–20. doi:10.1002/1097-4598(200009)23:9<1414::AID-MUS13>3.0.CO;2-0
35. Labrador-Horrillo M, Martinez-Valle F, Gallardo E, Rojas-Garcia R, Ordi-Ros J, Vilardell M. Anti-ganglioside antibodies in patients with systemic lupus erythematosus and neurological manifestations. Lupus (2012) 21(6):611–5. doi:10.1177/0961203312436856
36. Williams RC Jr., Sugiura K, Tan EM. Antibodies to microtubule-associated protein 2 in patients with neuropsychiatric systemic lupus erythematosus. Arthritis Rheum (2004) 50(4):1239–47. doi:10.1002/art.20156
37. Watanabe H, Seino T, Sato Y. Antibodies to triosephosphate isomerase in patients with neuropsychiatric lupus. Biochem Biophys Res Commun (2004) 321(4):949–53. doi:10.1016/j.bbrc.2004.07.051
38. Tin SK, Xu Q, Thumboo J, Lee LY, Tse C, Fong KY. Novel brain reactive autoantibodies: prevalence in systemic lupus erythematosus and association with psychoses and seizures. J Neuroimmunol (2005) 169(1–2):153–60. doi:10.1016/j.jneuroim.2005.07.015
39. Arnson Y, Shoenfeld Y, Alon E, Amital H. The antiphospholipid syndrome as a neurological disease. Semin Arthritis Rheum (2010) 40(2):97–108. doi:10.1016/j.semarthrit.2009.05.001
40. Zandman-Goddard G, Chapman J, Shoenfeld Y. Autoantibodies involved in neuropsychiatric SLE and antiphospholipid syndrome. Semin Arthritis Rheum (2007) 36(5):297–315. doi:10.1016/j.semarthrit.2006.11.003
41. Eber T, Chapman J, Shoenfeld Y. Anti-ribosomal P-protein and its role in psychiatric manifestations of systemic lupus erythematosus: myth or reality? Lupus (2005) 14(8):571–5. doi:10.1191/0961203305lu2150rr
42. Bosch X, Ramos-Casals M, Khamashta MA. The DWEYS peptide in systemic lupus erythematosus. Trends Mol Med (2012) 18(4):215–23. doi:10.1016/j.molmed.2012.01.008
43. Correale J, Villa A. Cellular elements of the blood-brain barrier. Neurochem Res (2009) 34(12):2067–77. doi:10.1007/s11064-009-0081-y
44. Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annu Rev Neurosci (1999) 22:11–28. doi:10.1146/annurev.neuro.22.1.11
45. Crone C, Olesen SP. Electrical resistance of brain microvascular endothelium. Brain Res (1982) 241(1):49–55. doi:10.1016/0006-8993(82)91227-6
46. Nag S. Morphology and properties of brain endothelial cells. Methods Mol Biol (2011) 686:3–47. doi:10.1007/978-1-60761-938-3_1
47. Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci (2006) 7(1):41–53. doi:10.1038/nrn1824
48. Yang YM, Shang DS, Zhao WD, Fang WG, Chen YH. Microglial TNF-alpha-dependent elevation of MHC class I expression on brain endothelium induced by amyloid-beta promotes T cell transendothelial migration. Neurochem Res (2013) 38(11):2295–304. doi:10.1007/s11064-013-1138-5
49. Antonelli-Orlidge A, Saunders KB, Smith SR, D’Amore PA. An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci USA (1989) 86(12):4544–8. doi:10.1073/pnas.86.12.4544
50. Frigerio S, Gelati M, Ciusani E, Corsini E, Dufour A, Massa G, et al. Immunocompetence of human microvascular brain endothelial cells: cytokine regulation of IL-1beta, MCP-1, IL-10, sICAM-1 and sVCAM-1. J Neurol (1998) 245(11):727–30. doi:10.1007/s004150050275
51. Verma S, Nakaoke R, Dohgu S, Banks WA. Release of cytokines by brain endothelial cells: a polarized response to lipopolysaccharide. Brain Behav Immun (2006) 20(5):449–55. doi:10.1016/j.bbi.2005.10.005
52. Engelhardt B, Sorokin L. The blood-brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol (2009) 31(4):497–511. doi:10.1007/s00281-009-0177-0
53. Johanson CE, Stopa EG, McMillan PN. The blood-cerebrospinal fluid barrier: structure and functional significance. Methods Mol Biol (2011) 686:101–31. doi:10.1007/978-1-60761-938-3_4
54. Brown PD, Davies SL, Speake T, Millar ID. Molecular mechanisms of cerebrospinal fluid production. Neuroscience (2004) 129(4):957–70. doi:10.1016/j.neuroscience.2004.07.003
55. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med (2012) 4(147):147ra11. doi:10.1126/scitranslmed.3003748
56. Wolburg H, Paulus W. Choroid plexus: biology and pathology. Acta Neuropathol (2010) 119(1):75–88. doi:10.1007/s00401-009-0627-8
57. Vercellino M, Votta B, Condello C, Piacentino C, Romagnolo A, Merola A, et al. Involvement of the choroid plexus in multiple sclerosis autoimmune inflammation: a neuropathological study. J Neuroimmunol (2008) 199(1–2):133–41. doi:10.1016/j.jneuroim.2008.04.035
58. McLean BN, Miller D, Thompson EJ. Oligoclonal banding of IgG in CSF, blood-brain barrier function, and MRI findings in patients with sarcoidosis, systemic lupus erythematosus, and Behcet’s disease involving the nervous system. J Neurol Neurosurg Psychiatry (1995) 58(5):548–54. doi:10.1136/jnnp.58.5.548
59. Nishimura K, Harigai M, Omori M, Sato E, Hara M. Blood-brain barrier damage as a risk factor for corticosteroid-induced psychiatric disorders in systemic lupus erythematosus. Psychoneuroendocrinology (2008) 33(3):395–403. doi:10.1016/j.psyneuen.2007.12.007
60. Sidor MM, Sakic B, Malinowski PM, Ballok DA, Oleschuk CJ, Macri J. Elevated immunoglobulin levels in the cerebrospinal fluid from lupus-prone mice. J Neuroimmunol (2005) 165(1–2):104–13. doi:10.1016/j.jneuroim.2005.04.022
61. Mehta PD. Diagnostic usefulness of cerebrospinal fluid in multiple sclerosis. Crit Rev Clin Lab Sci (1991) 28(3):233–51. doi:10.3109/10408369109106864
62. Killingsworth LM. Clinical applications of protein determinations in biological fluids other than blood. Clin Chem (1982) 28(5):1093–102.
63. Jacob A, Hack B, Chiang E, Garcia JG, Quigg RJ, Alexander JJ. C5a alters blood-brain barrier integrity in experimental lupus. FASEB J (2010) 24(6):1682–8. doi:10.1096/fj.09-138834
64. Ma X, Foster J, Sakic B. Distribution and prevalence of leukocyte phenotypes in brains of lupus-prone mice. J Neuroimmunol (2006) 179(1–2):26–36. doi:10.1016/j.jneuroim.2006.06.023
65. Bertsch T, Casarin W, Kretschmar M, Zimmer W, Walter S, Sommer C, et al. Protein S-100B: a serum marker for ischemic and infectious injury of cerebral tissue. Clin Chem Lab Med (2001) 39(4):319–23. doi:10.1515/cclm.2001.050
66. Jaranyi Z, Szekely M, Bobek I, Galfy I, Geller L, Selmeci L. Impairment of blood-brain barrier integrity during carotid surgery as assessed by serum S-100B protein concentrations. Clin Chem Lab Med (2003) 41(10):1320–2. doi:10.1515/cclm.2003.201
67. Kapural M, Krizanac-Bengez L, Barnett G, Perl J, Masaryk T, Apollo D, et al. Serum S-100beta as a possible marker of blood-brain barrier disruption. Brain Res (2002) 940(1–2):102–4. doi:10.1016/S0006-8993(02)02586-6
68. Schenatto CB, Xavier RM, Bredemeier M, Portela LV, Tort AB, Dedavid e Silva TL, et al. Raised serum S100B protein levels in neuropsychiatric lupus. Ann Rheum Dis (2006) 65(6):829–31. doi:10.1136/ard.2005.048330
69. Essig M, Dinkel J, Gutierrez JE. Use of contrast media in neuroimaging. Magn Reson Imaging Clin N Am (2012) 20(4):633–48. doi:10.1016/j.mric.2012.08.001
70. Toledano P, Sarbu N, Espinosa G, Bargallo N, Cervera R. Neuropsychiatric systemic lupus erythematosus: magnetic resonance imaging findings and correlation with clinical and immunological features. Autoimmun Rev (2013) 12(12):1166–70. doi:10.1016/j.autrev.2013.07.004
71. de Boer AG, Gaillard PJ. Blood-brain barrier dysfunction and recovery. J Neural Transm (2006) 113(4):455–62. doi:10.1007/s00702-005-0375-4
72. Eugenin EA, Berman JW. Chemokine-dependent mechanisms of leukocyte trafficking across a model of the blood-brain barrier. Methods (2003) 29(4):351–61. doi:10.1016/S1046-2023(02)00359-6
73. Haraldsen G, Kvale D, Lien B, Farstad IN, Brandtzaeg P. Cytokine-regulated expression of E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) in human microvascular endothelial cells. J Immunol (1996) 156(7):2558–65.
74. Zameer A, Hoffman SA. Increased ICAM-1 and VCAM-1 expression in the brains of autoimmune mice. J Neuroimmunol (2003) 142(1–2):67–74. doi:10.1016/S0165-5728(03)00262-5
75. Sun W, Jiao Y, Cui B, Gao X, Xia Y, Zhao Y. Immune complexes activate human endothelium involving the cell-signaling HMGB1-RAGE axis in the pathogenesis of lupus vasculitis. Lab Invest (2013) 93(6):626–38. doi:10.1038/labinvest.2013.61
76. Crispin JC, Hedrich CM, Tsokos GC. Gene-function studies in systemic lupus erythematosus. Nat Rev Rheumatol (2013) 9(8):476–84. doi:10.1038/nrrheum.2013.78
77. Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, et al. C-terminal truncations in human 3’-5’ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet (2007) 39(9):1068–70. doi:10.1038/ng2082
78. Pulliero A, Marengo B, Domenicotti C, Longobardi MG, Fazzi E, Orcesi S, et al. Inhibition of neuroblastoma cell growth by TREX1-mutated human lymphocytes. Oncol Rep (2012) 27(5):1689–94. doi:10.3892/or.2012.1696
79. DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med (2001) 7(11):1189–93. doi:10.1038/nm1101-1189
80. Kowal C, DeGiorgio LA, Nakaoka T, Hetherington H, Huerta PT, Diamond B, et al. Cognition and immunity; antibody impairs memory. Immunity (2004) 21(2):179–88. doi:10.1016/j.immuni.2004.07.011
81. Huerta PT, Kowal C, DeGiorgio LA, Volpe BT, Diamond B. Immunity and behavior: antibodies alter emotion. Proc Natl Acad Sci USA (2006) 103(3):678–83. doi:10.1073/pnas.0510055103
82. Conti F, Alessandri C, Bompane D, Bombardieri M, Spinelli FR, Rusconi AC, et al. Autoantibody profile in systemic lupus erythematosus with psychiatric manifestations: a role for anti-endothelial-cell antibodies. Arthritis Res Ther (2004) 6(4):R366–72. doi:10.1186/ar1198
83. Oosting JD, Preissner KT, Derksen RH, de Groot PG. Autoantibodies directed against the epidermal growth factor-like domains of thrombomodulin inhibit protein C activation in vitro. Br J Haematol (1993) 85(4):761–8. doi:10.1111/j.1365-2141.1993.tb03220.x
84. Nara H, Okamoto H, Minota S, Yoshio T. Mouse monoclonal anti-human thrombomodulin antibodies bind to and activate endothelial cells through NF-kappaB activation in vitro. Arthritis Rheum (2006) 54(5):1629–37. doi:10.1002/art.21797
85. Yoshio T, Okamoto H, Hirohata S, Minota S. IgG anti-NR2 glutamate receptor autoantibodies from patients with systemic lupus erythematosus activate endothelial cells. Arthritis Rheum (2013) 65(2):457–63. doi:10.1002/art.37745
86. Wong D, Dorovini-Zis K, Vincent SR. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol (2004) 190(2):446–55. doi:10.1016/j.expneurol.2004.08.008
87. Valentijn RM, van Overhagen H, Hazevoet HM, Hermans J, Cats A, Daha MR, et al. The value of complement and immune complex determinations in monitoring disease activity in patients with systemic lupus erythematosus. Arthritis Rheum (1985) 28(8):904–13. doi:10.1002/art.1780280810
88. Coremans IE, Spronk PE, Bootsma H, Daha MR, van der Voort EA, Kater L, et al. Changes in antibodies to C1q predict renal relapses in systemic lupus erythematosus. Am J Kidney Dis (1995) 26(4):595–601. doi:10.1016/0272-6386(95)90595-2
89. Karassa FB, Ioannidis JP, Touloumi G, Boki KA, Moutsopoulos HM. Risk factors for central nervous system involvement in systemic lupus erythematosus. QJM (2000) 93(3):169–74. doi:10.1093/qjmed/93.3.169
90. Alexander JJ, Jacob A, Bao L, Macdonald RL, Quigg RJ. Complement-dependent apoptosis and inflammatory gene changes in murine lupus cerebritis. J Immunol (2005) 175(12):8312–9.
91. Laudes IJ, Chu JC, Huber-Lang M, Guo RF, Riedemann NC, Sarma JV, et al. Expression and function of C5a receptor in mouse microvascular endothelial cells. J Immunol (2002) 169(10):5962–70.
92. Jacob A, Hack B, Chen P, Quigg RJ, Alexander JJ. C5a/CD88 signaling alters blood-brain barrier integrity in lupus through nuclear factor-kappaB. J Neurochem (2011) 119(5):1041–51. doi:10.1111/j.1471-4159.2011.07490.x
93. Matsumoto T, Ikeda K, Mukaida N, Harada A, Matsumoto Y, Yamashita J, et al. Prevention of cerebral edema and infarct in cerebral reperfusion injury by an antibody to interleukin-8. Lab Invest (1997) 77(2):119–25.
94. Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke (2007) 38(4):1345–53. doi:10.1161/01.STR.0000259709.16654.8f
95. Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, et al. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab (2005) 25(5):593–606. doi:10.1038/sj.jcbfm.9600055
96. Stielke S, Keilhoff G, Kirches E, Mertens PR, Neumann K-H, Tsokos GC, et al. Adhesion molecule expression precedes brain damages of lupus-prone mice and correlates with kidney pathology. J Neuroimmunol (2012) 252(1–2):24–32. doi:10.1016/j.jneuroim.2012.07.011
97. Trysberg E, Blennow K, Zachrisson O, Tarkowski A. Intrathecal levels of matrix metalloproteinases in systemic lupus erythematosus with central nervous system engagement. Arthritis Res Ther (2004) 6(6):R551–6. doi:10.1186/ar1228
98. Jakubowski A, Ambrose C, Parr M, Lincecum JM, Wang MZ, Zheng TS, et al. TWEAK induces liver progenitor cell proliferation. J Clin Invest (2005) 115(9):2330–40. doi:10.1172/JCI23486
99. Marksteiner J, Kemmler G, Weiss EM, Knaus G, Ullrich C, Mechtcheriakov S, et al. Five out of 16 plasma signaling proteins are enhanced in plasma of patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging (2011) 32(3):539–40. doi:10.1016/j.neurobiolaging.2009.03.011
100. Loeffler DA, Camp DM, Bennett DA. Plaque complement activation and cognitive loss in Alzheimer’s disease. J Neuroinflammation (2008) 5:9. doi:10.1186/1742-2094-5-9
101. Maes M, Delange J, Ranjan R, Meltzer HY, Desnyder R, Cooremans W, et al. Acute phase proteins in schizophrenia, mania and major depression: modulation by psychotropic drugs. Psychiatry Res (1997) 66(1):1–11. doi:10.1016/S0165-1781(96)02915-0
102. Desplat-Jego S, Varriale S, Creidy R, Terra R, Bernard D, Khrestchatisky M, et al. TWEAK is expressed by glial cells, induces astrocyte proliferation and increases EAE severity. J Neuroimmunol (2002) 133(1–2):116–23. doi:10.1016/S0165-5728(02)00368-5
103. Polavarapu R, Gongora MC, Winkles JA, Yepes M. Tumor necrosis factor-like weak inducer of apoptosis increases the permeability of the neurovascular unit through nuclear factor-kappa B pathway activation. J Neurosci (2005) 25(44):10094–100. doi:10.1523/JNEUROSCI.3382-05.2005
104. Saas P, Boucraut J, Walker PR, Quiquerez AL, Billot M, Desplat-Jego S, et al. TWEAK stimulation of astrocytes and the proinflammatory consequences. Glia (2000) 32(1):102–7. doi:10.1002/1098-1136(200010)32:1<102::AID-GLIA100>3.3.CO;2-L
105. Xia Y, Campbell SR, Broder A, Herlitz L, Abadi M, Wu P, et al. Inhibition of the TWEAK/Fn14 pathway attenuates renal disease in nephrotoxic serum nephritis. Clin Immunol (2012) 145(2):108–21. doi:10.1016/j.clim.2012.08.008
106. Zhao Z, Burkly LC, Campbell S, Schwartz N, Molano A, Choudhury A, et al. TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J Immunol (2007) 179(11):7949–58.
107. Lu J, Kwan BC, Lai FM, Choi PC, Tam LS, Li EK, et al. Gene expression of TWEAK/Fn14 and IP-10/CXCR3 in glomerulus and tubulointerstitium of patients with lupus nephritis. Nephrology (2011) 16(4):426–32. doi:10.1111/j.1440-1797.2011.01449.x
108. Doerner JW, Xia Y, Friedman A, Michaelson JS, Burkly LC, Wu L, et al. Elucidating the role for TWEAK in the pathogenesis of cutaneous lupus [abstract]. Federation of Clinical Immunology Societies Annual Meeting, Boston (2013).
109. Banks WA, Terrell B, Farr SA, Robinson SM, Nonaka N, Morley JE. Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer’s disease. Peptides (2002) 23(12):2223–6. doi:10.1016/S0196-9781(02)00261-9
110. Couch JA, Yu YJ, Zhang Y, Tarrant JM, Fuji RN, Meilandt WJ, et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med (2013) 5(183):183ra57,1–12. doi:10.1126/scitranslmed.3005338
Keywords: TWEAK, Fn14, blood brain barrier, neuropsychiatric lupus, MRL/lpr
Citation: Stock AD, Wen J and Putterman C (2013) Neuropsychiatric lupus, the blood brain barrier, and the TWEAK/Fn14 pathway. Front. Immunol. 4:484. doi: 10.3389/fimmu.2013.00484
Received: 15 October 2013; Accepted: 10 December 2013;
Published online: 25 December 2013.
Edited by:
Linda C. Burkly, Biogen Idec, Inc., USACopyright: © 2013 Stock, Wen and Putterman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaim Putterman, Division of Rheumatology, Albert Einstein College of Medicine, F701N, 1300 Morris Park Avenue, Bronx, NY 10461, USA e-mail:Y2hhaW0ucHV0dGVybWFuQGVpbnN0ZWluLnl1LmVkdQ==