 Joan Vendrell
Joan Vendrell Matilde R. Chacón*
Matilde R. Chacón*- Research Unit, Centro de Investigación Biomédica en Red de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM), Hospital Universitari de Tarragona Joan XXIII, Institut d’Investigació Sanitària Pere Virgili, Universitat Rovira i Virgili, Tarragona, Spain
Obesity and type 2 diabetes (T2D) are associated with chronic low-grade inflammation. Mounting evidence suggests the involvement of an inflammatory switch in adipose tissue, both in mature adipocytes and immune-competent cells from the stromal vascular compartment, in the progression of obesity and insulin resistance. Several inflammatory cytokines secreted by obese adipose tissue, including TNFα and IL-6 have been described as hallmark molecules involved in this process, impairing insulin signaling in insulin-responsive organs. An increasing number of new molecules affecting the local and systemic inflammatory imbalance in obesity and T2D have been identified. In this complex condition, some molecules may exhibit opposing actions, depending on the cell type and on systemic or local influences. Tumor necrosis factor weak inducer of apoptosis (TWEAK), a cytokine of the tumor necrosis (TNF) superfamily, is gaining attention as an important player in chronic inflammatory diseases. TWEAK can exist as a full-length membrane-associated (mTWEAK) form and as a soluble (sTWEAK) form and, by acting through its cognate receptor Fn14, can control many cellular activities including proliferation, migration, differentiation, apoptosis, angiogenesis, and inflammation. Notably, sTWEAK has been proposed as a biomarker of cardiovascular diseases. Here, we will review the recent findings relating to TWEAK and its receptor within the context of obesity and the associated disorder T2D.
Introduction
A characteristic feature of obesity is a low-grade level of inflammation, likely originating in the expanding adipose tissue, which is illustrated by infiltration of immune cells, including macrophages, lymphocytes, and leukocytes (1). Pro-inflammatory cytokines released by activated immune cells and adipocytes can impair insulin signaling in insulin-responsive organs, promoting systemic insulin resistance, which increases the risk of developing type 2 diabetes (T2D) (2).
Tumor necrosis factor alpha (TNFα) was the first described cytokine to have a relevant role in obesity and an associated insulin-resistant state. Studies first conducted by Hotamisligil et al. (3), demonstrated an upregulation of TNFα in adipose tissue from obese patients. Since then, many members of the TNF superfamily have been shown to participate in obesity related diseases, including TNF-related apoptosis inducing ligand (TRAIL) (4), B cell activating factor (BAFF) (5), Lymphotoxin-α (LTα) (6, 7), Lymphotoxin β receptor (LTβR) (8), and Tumor necrosis factor ligand superfamily member 14 (TNFSF14) (9) among others. Recently, Tumor Necrosis Factor Weak Inducer of Apoptosis (TWEAK) has gained attention as a potentially important regulator of the inflammatory/anti-inflammatory equilibrium which takes place in the insulin-resistant milieu. TWEAK is a cytokine belonging to the TNF superfamily and triggers multiple, and often seemingly conflicting, cellular activities in a wide variety of cells, ranging from proliferation to cell death (10, 11). Like most TNF members, TWEAK protein exists as a membrane-bound (mTWEAK) form, and also as a soluble variant (sTWEAK), formed after proteolytic cleavage by a furin endoproteinase. Both forms are biologically active and can bind to Fibroblast growth factor-inducible 14 (Fn14), its only bona fide signal transducing receptor (12). Studies in vitro suggests that mTWEAK, can function as a juxtacrine signaling molecule and sTWEAK can elicit qualitatively different states of activity through Fn14 (12–14). Moreover, some authors have proposed sTWEAK as a potential biomarker in human cardiovascular diseases (CVDs) (15).
The cytoplasmic domain of Fn14 contains a TNF Receptor-Associated Factor (TRAF) binding site allowing recruitment of TRAF adapter proteins. This interaction is shared by most members of the TNF receptor family including TNFα, and plays a pivotal role in activating the nuclear factor κB (NF-κB) or mitogen-activated protein kinase (MAPK) pathway, which can be also activated by TWEAK (16). In particular, TRAF2 is implicated in the activation of TWEAK signaling in several human cell lines (17).
Interestingly, new data from human studies points to the TWEAK/Fn14 axis as a component of the network that contributes to the inflammatory imbalance occurring in insulin resistance-associated diseases (18–22). Here we review the role of TWEAK in adipocyte biology, and the prognostic and diagnostic value of its soluble form sTWEAK, within the context of insulin-resistant associated diseases, obesity, and diabetes.
TWEAK and Adipocyte Biology
Tumor necrosis factor weak inducer of apoptosis mRNA expression was initially described in multiple human tissues (23). However, the first data concerning the expression of this cytokine in adipose tissue revealed mRNA gene expression of both TWEAK and its receptor Fn14 in human fat depots from subcutaneous and visceral origin (18, 19, 24). Furthermore, altered patterns of TWEAK and Fn14 protein expression were observed in subcutaneous adipose samples from severely obese subjects when compared to healthy subjects (21).
In addition to adipocytes, adipose tissue contains a heterogeneous population of cells including preadipocytes, mesenchymal stem cells (MSC), endothelial cells, and macrophages among others cell types (25). Together, this collection of cells is termed the stromal vascular fraction (SVF). From this compartment, TWEAK expression has been detected mainly on the surface of macrophages and lymphoid cells (21, 26), whereas Fn14 expression has been detected in mature adipocytes (18, 21, 27, 28), preadipocytes (28, 29), MSC (29), and endothelial cells (30).
It is well recognized that Fn14 is a highly inducible receptor (11). Inflammation can regulate the expression of Fn14 in adipocytes and TWEAK in macrophages (19). In agreement with this data, isolated adipocytes from severely obese subjects exposed to a striking inflammatory environment, displayed increased levels of Fn14 (18, 21). Independently of inflammation, hypoxic stress is suggested to be a contributing factor in the adipocyte metabolism in the setting of obesity (31). Insufficient oxygen supply can lead to endoplasmic reticulum (ER) stress and mitochondrial dysfunction, and hypoxia alters the balance between pro- and anti-inflammatory activities in adipose tissue (32, 33). Although an up-regulation of the TWEAK/Fn14 axis, in parallel with hypoxia and ER-specific genes has been observed in the adipose tissue of severely obese subjects, studies emulating hypoxia and ER stress in vitro do not report any change in TWEAK/Fn14 gene expression in isolated adipocytes or macrophages (19).
The pro-inflammatory milieu facilitated by hypoxia in adipose tissue may represent an important stimulus for macrophage attraction (34, 35). These cells may have a different polarized state (termed M1 for the pro-inflammatory and M2 for the anti-inflammatory subtypes) (35). Although there is a controversy regarding the balance between M1 and M2 macrophages in human obesity, and in obese mice models (36), many authors agree that the M2 type constitute the most abundant infiltrating macrophages found in human obesity (37, 38). In SVF from obese subjects mTWEAK protein has been found over-expressed. Since macrophages are an important component of the SVF cells, this finding points to macrophages as one of the cells that expresses this cytokine in the adipose tissue of obese subjects. In this regard, in vitro studies describe a higher level of mTWEAK expression in M2 human monocyte derived-macrophages when compared to M1 cells (21).
Tumor necrosis factor weak inducer of apoptosis stimulus may induce a pro-inflammatory activity in several human cell types including endothelial, kidney, synoviocytes, and muscle cells, among others (11). In human adipocytes, TWEAK stimulus alone resulted in a modest pro-inflammatory state, with up-regulation of the cytokines IL-6 and MCP-1, whereas leptin and adiponectin expression were unaltered (19, 28). This inflammatory effect seemed to be mediated through both canonical and non-canonical NF-κB pathway activation. Whereas the canonical pathway was activated in subcutaneous adipocytes, the non-canonical pathway appeared activated only in visceral adipocytes (21, 22). Furthermore, a moderate induction of the MAPKs ERK1/2 and p38 after TWEAK stimulus, has also been observed in both subcutaneous and visceral adipocytes (19, 22).
It is known that TWEAK can interfere with the differentiation ability of several cell types, including myogenic, osteoblast, chondrocyte, and erythroblast lineages (11). In addition, TWEAK can also inhibit adipocyte differentiation at an early stage, as indicated by a rapid reduction of the key adipogenic transcription factors Peroxisome proliferator-activated receptor gamma (PPARγ) and CCAAT/Enhancer Binding Protein α (C/EBPα) (28, 39).
In contrast to its effects on the differentiation capacity of adipocytes, TWEAK does not influence the metabolic function of these cells, such as glucose uptake and lipolysis. Furthermore, distinct from its clear apoptotic effect in neurons, monocytes, and tumor cell lines (40), a TWEAK stimulus does not induce apoptosis in adipocytes (22, 28).
Overall, the main impact of TWEAK on adipocytes appears to be an inhibition of adipocyte differentiation and the induction of a moderate inflammatory response.
Circulating Levels of sTWEAK in Obesity and Diabetes
Diabesity is a new term coined to describe the common clinical associations between obesity and T2D, and highlights the close relationship between both states and their shared pathophysiological mechanisms. Chronic and subtle inflammation is usually documented in both states and can markedly influence cardiovascular (CV) outcomes (41).
Since the observation of lower levels of sTWEAK in patients with atherosclerosis and a negatively correlation between circulating sTWEAK levels and the intima/media thickness in asymptomatic subjects (15), decreased sTWEAK levels have been confirmed in many other CVD indications (40, 42–51). Consistent with these findings, obese individuals also present a similar trend of decreased sTWEAK in peripheral blood (21). Reinforcing the hypothesis that low sTWEAK levels associate with a poor CV profile, circulating levels of sTWEAK have also been found negatively associated with levels of glucose, glycated hemoglobin (HbA1), and also insulin resistance index (HOAM-IR) and central obesity; all of which are well known CV risk factors (20, 21, 46, 52). In contrast, the atherogenic lipid profile does not show a clear inverse association with circulating sTWEAK, and opposing data are reported in the literature. Whereas some studies describe a negative relationship with total cholesterol and triglycerides in severe obesity (21), other authors describe either a positive relationship with triglycerides (53) or no association at all (45). In severe obesity, changes in the levels of Free Fatty Acids were found to negatively influence circulating sTWEAK, indicating that lipotoxicity could be a modulator of sTWEAK levels. The observation of a lower rate of release of sTWEAK in carotid atheroma plaques, compared to normal arteries, supports a link between the lipotoxic effects of abnormal lipid accumulation and TWEAK synthesis (15).
Some of the evidence mentioned may lead to speculation about the potential anti-inflammatory behavior of sTWEAK, at least in high-risk atherogenic conditions. Indeed, the inverse relationship with inflammatory markers or surrogate inflammatory scores lends support to this hypothesis (20, 53). The rise of circulating sTWEAK levels after massive weight loss in severely obese subjects reinforces the parallelism between expression of this cytokine and other well recognized anti-inflammatory molecules such as adiponectin (21). Along similar lines, a trend toward positive correlation between levels of sTWEAK and adiponectin in patients on chronic hemodialysis has been reported (52).
Recently, a new study highlighted the relevance of decreased serum sTWEAK as a predictive marker of T2D. Interestingly this study was conducted in a high CV risk population, in which the incidence of T2D was assessed during a follow up (54). In this large prospective nested case-control study lower sTWEAK serum levels were found in incident cases compared to matched controls. Indeed, previous cross-sectional studies have also proposed a link between sTWEAK concentration and T2D (52).
The rationale that low levels of sTWEAK, in contrast to other cytokines, appears as protective in conditions with high CV risk diseases associated with an increased of chronic inflammatory activity, is incompletely understood. Several conceivable explanations have been proposed. A reduction of sTWEAK in serum, due to uptake by the Fn14 receptor has been postulated. Endothelial dysfunction is the initial pathophysiological step in the progression of vascular damage that precedes and leads to clinically visible CVD (55). Under these conditions, Fn14 expression is increased in the endothelium. Recently, we reported increased Fn14 expression in human adipocytes from severely obese subjects. These cells also showed an increase in Fn14 expression after inflammatory stimulation, thus increasing the availability for sTWEAK ligand, which could lead to a peripheral reduction of serum sTWEAK (19, 21).
An alternative hypothesis proposes the involvement of CD163, a monocyte-macrophage surface receptor which has been suggested to act as a scavenger receptor for sTWEAK (56). Soluble CD163 (sCD163) is a macrophage-specific serum marker which is elevated in inflammatory conditions (57). Circulating levels of sCD163 and sTWEAK are expressed in an opposite trend in human carotid atherosclerotic plaques (58). Moreover, CD163-expressing macrophages can bind and internalize sTWEAK in vitro (58). Thus, the reduction of sTWEAK could be related to the presence of sCD163, which is up-regulated both in patients with chronic kidney disease (CKD), and in obese subjects (51, 59–61). This incremental increase could enable sTWEAK degradation by inflammatory macrophages, leading to decreased sTWEAK levels, represented by the reduction in the sTWEAK/sCD163 ratio observed in some diseases such CKD (51). Thus, low sTWEAK levels may be related to the degree of macrophage activation. However, these observations are not fully coincident in TD2 patients. High serum levels of sCD163 have been reported as a useful predictive biomarker of T2D (62), but a more recent study reveals no association between circulating sCD163 and the incidence of T2D (54).
In contrast to the hypothesis of the potential anti-inflammatory behavior of sTWEAK, animal studies with different approaches to investigate the role of TWEAK/Fn14 axis in the development and progression of atherosclerosis, gain of function, or loss of function, have showed that TWEAK participates in the atherogenic process (63, 64) indicating that the “net” effect of the pathway is damaging rather than protective in this condition.
sTWEAK Modulates TNFα Activity
To date, TNFα has focused the attention as a preponderant inflammatory cytokine with important implications both at local and systemic levels in obesity and related diseases. The action of TNFα on adipose tissue can alter the production of many adipokines, and this is relevant for the systemic effects of TNFα on insulin sensitivity and whole body energy homeostasis (65). sTWEAK and TNFα co-exist within the obese adipose tissue milieu, and both cytokines have a pro-inflammatory potential, although at the same concentrations TNFα is a much more potent and rapid inflammatory mediator than TWEAK (66).
Examination of the biological mechanisms through which sTWEAK improves insulin sensitivity has demonstrated that, in visceral adipocytes, treatment with sTWEAK ameliorates TNFα-induced insulin resistance on glucose uptake. This occurs by abolishing the stimulatory effect of TNFα on JNK1/2 kinase, which is directly involved in the development of insulin resistance (67). This effect is produced, at least in part, through a reduction in the cellular concentration of TRAF2, leading to a curbing of TNFα intracellular signaling events. Furthermore, this modulation of TNFα-induced changes in insulin sensitivity was found to be associated with an increase in the activity of PP2A, a Ser/Thr protein phosphatase known to negatively regulate cytokine signaling (22). Additionally, in human subcutaneous adipocytes, sTWEAK exerts a modulator effect over TNFα-induced cytokine production by inhibiting the MAPK and NF-κB signaling cascades commonly used by TNFα (21).
This protective/modulatory effect of sTWEAK on TNFα activity has been observed in different cell types such cultured fibroblast like synoviocytes obtained from synovial tissues of rheumatoid arthritis patients (68), in mouse cerebral cortical neurons (69), and also in several tumor human epithelial cell lines (70), suggesting a broader and general competitive behavior between sTWEAK and TNFα.
Concluding Remarks
The duality between inflammatory and anti-inflammatory activity seems to be one of the major elements in the evolution of high CV risk diseases, such as obesity and T2D. In this scenario, some molecules may display contradictory actions, depending on the cell type and the location, and on the systemic, or local influence.
Here we have summarized emerging data on the role of TWEAK within the context of metabolic inflammation. Despite the moderate inflammatory activity of the sTWEAK cytokine in adipocytes, a competitive interference ability of sTWEAK on TNFα signaling in the adipocyte has been revealed. Contrary to that observed with TNFα in obese and T2D patients, circulating sTWEAK appears as a protective element under these conditions. Interestingly, mTWEAK and sTWEAK have been shown to have different effects on signal transduction pathways. Since mTWEAK appears to be mainly expressed in macrophages, the metabolic effects of TWEAK may therefore differ in cells having contact with macrophages (e.g., adipocytes in an obesity context) and in more distant cells living in a macrophage-free environment. Thus, it is tempting to speculate that the decrease in sTWEAK levels, together with an increase of mTWEAK, may help to maintain the local pro-inflammatory effect of the TNF-α-driven response in obese and T2D conditions (Figure 1). Hence, the use of recombinant sTWEAK or Fn14 agonists to manipulate the TWEAK/Fn14 axis offers an exciting perspective for the treatment of insulin resistance and should be explored further.
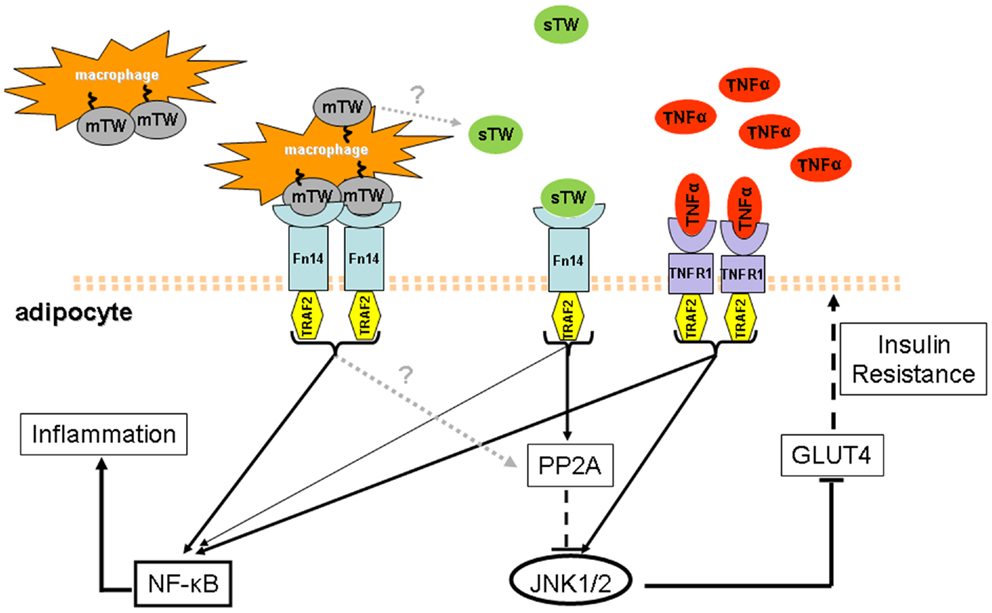
Figure 1. Model of the state of the art of TWEAK and TNFα in obesity and T2D conditions. In obesity and T2D states, higher circulating levels of TNFα and lower levels of sTWEAK have been observed. Additionally, obese adipose tissue is highly infiltrated with macrophages that could be a source of sTWEAK production and also express mTWEAK on the surface. The latter may establish a strong pro-inflammatory response in adipocytes where the Fn14 receptor is over-expressed and activates the NF-κB pathway. In contrast, sTWEAK has been identified as a negative regulator of TNFα signaling since both signal via similar TNFR-associated factors, including TRAF2. Amelioration of TNFα-induced insulin resistance by sTWEAK in obesity and T2D could be a consequence, at least in part, of its direct regulation of JNK1/2 phosphorylation, controlled by PP2A phosphatase which is known to negatively regulate cytokine signaling. PPA2 activation could thereby be linked to the protective role of sTWEAK during the development of insulin resistance. Thus it is tempting to speculate that in obesity and T2D, the increase of mTWEAK and the decrease of sTWEAK may help to maintain the pro-inflammatory effect of TNFα-driven response.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author’s studies cited here were supported by grants from Fondo de Investigación Sanitaria: CP06/00119, PI08/0733 and PI 11/00049. All projects from FIS are co-financed by the European Regional Development Fund (ERDF). Dr. Matilde R. Chacón is supported by the Research Stabilization Programme of the Instituto de Salud Carlos III (ISCIII) co-financed by Institut Català de Salut (ICS) in Catalonia.
Abbreviations
BMI, body mass index; CD163, cluster of differentiation 163; CVDs, cardiovascular disease; ERK, extracellular signal-regulated kinases; Fn14, fibroblast growth factor-inducible 14; HOMA-IR, homeostasis model assessment of insulin resistance; IL-6, interleukin 6; MAPKs, mitogen-activated protein kinases; mTWEAK, full-length TWEAK isoform; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; p38, mitogen-activated protein kinase; PP2A, protein phosphatase 2A; sTWEAK, soluble TWEAK isoform; SVF, stromal vascular fraction; T2D, type 2 diabetes; TNFα, tumor necrosis factor alpha; TRAF2, TNF receptor-associated factor 2; TWEAK, tumor necrosis factor weak inducer of apoptosis.
References
1. Sethi JK, Vidal-Puig AJ. Thematic review series: adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. J Lipid Res (2007) 48:1253–62. doi: 10.1194/jlr.R700005-JLR200
2. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol (2011) 11:98–107. doi:10.1038/nri2925
3. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science (1993) 259:87–91. doi:10.1126/science.7678183
4. Keuper M, Wernstedt Asterholm I, Scherer PE, Westhoff MA, Möller P, Debatin KM, et al. TRAIL (TNF-related apoptosis-inducing ligand) regulates adipocyte metabolism by caspase-mediated cleavage of PPARgamma. Cell Death Dis (2013) 4:e474. doi:10.1038/cddis.2012.212
5. Tada F, Abe M, Kawasaki K, Miyake T, Shiyi C, Hiasa Y, et al. B cell activating factor in obesity is regulated by oxidative stress in adipocytes. J Clin Biochem Nutr (2013) 52:120–7. doi:10.3164/jcbn.12-115
6. Naoum JJ, Chai H, Lin PH, Lumsden AB, Yao Q, Chen C. Lymphotoxin-alpha and cardiovascular disease: clinical association and pathogenic mechanisms. Med Sci Monit (2006) 7:RA121–4.
7. Lau WB, Zhang Y, Zhao J, Liu B, Wang X, Yuan Y, et al. Lymphotoxin-α is a novel adiponectin expression suppressor following myocardial ischemia/reperfusion. Am J Physiol Endocrinol Metab (2013) 304(6):E661–7. doi:10.1152/ajpendo.00012.2013
8. Owens AW, Matulevicius S, Rohatgi A, Ayers CR, Das SR, Khera A, et al. Circulating lymphotoxin β receptor and atherosclerosis: observations from the Dallas Heart Study. Atherosclerosis (2010) 212:601–6. doi:10.1016/j.atherosclerosis.2010.06.003
9. Tiller G, Laumen H, Fischer-Posovszky P, Finck A, Skurk T, Keuper M, et al. LIGHT (TNFSF14) inhibits adipose differentiation without affecting adipocyte metabolism. Int J Obes (Lond) (2011) 35:208–16. doi:10.1038/ijo.2010.126
10. Burkly LC, Michaelson JS, Zheng TS. TWEAK/Fn14 pathway: an immunological switch for shaping tissue responses. Immunol Rev (2011) 244:99–114. doi:10.1111/j.1600-065X.2011.01054.x
11. Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov (2008) 5:411–25. doi:10.1038/nrd2488
12. Brown SA, Ghosh A, Winkles JA. Full-length, membrane-anchored TWEAK can function as a juxtacrine signaling molecule and activate the NF-kappaB pathway. J Biol Chem (2010) 285:17432–41. doi:10.1074/jbc.M110.131979
13. Roos C, Wicovsky A, Müller N, Salzmann S, Rosenthal T, Kalthoff H, et al. Soluble and transmembrane TNF-like weak inducer of apoptosis differentially activate the classical and noncanonical NF-kappa B pathway. J Immunol (2010) 185:1593–605. doi:10.4049/jimmunol.0903555
14. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell (2001) 104:487–501. doi:10.1016/S0092-8674(01)00237-9
15. Blanco-Colio LM, Martín-Ventura JL, Muñóz-García B, Orbe J, Páramo JA, Michel JB, et al. Identification of soluble tumor necrosis factor-like weak inducer of apoptosis (sTWEAK) as a possible biomarker of subclinical atherosclerosis. Arterioscler Thromb Vasc Biol (2007) 27:916–22. doi:10.1161/01.ATV.0000258972.10109.ff
16. Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ (2003) 10:45–65. doi:10.1038/sj.cdd.4401189
17. Vince JE, Chau D, Callus B, Wong WW, Hawkins CJ, Schneider P, et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J Cell Biol (2008) 182:171–84. doi:10.1083/jcb.200801010
18. Chacón MR, Richart C, Gómez JM, Megía A, Vilarrasa N, Fernández-Real JM, et al. Expression of TWEAK and its receptor Fn14 in human subcutaneous adipose tissue. Relationship with other inflammatory cytokines in obesity. Cytokine (2006) 33:129–37. doi:10.1016/j.cyto.2005.12.005
19. Vendrell J, Maymó-Masip E, Tinahones F, García-España A, Megia A, Caubet E, et al. Tumor necrosis-like weak inducer of apoptosis as a pro-inflammatory cytokine in human adipocyte cells: up-regulation in severe obesity is mediated by inflammation but not hypoxia. J Clin Endocrinol Metab (2010) 95:2983–92. doi:10.1210/jc.2009-2481
20. Llauradó G, González-Clemente JM, Maymó-Masip E, Subías D, Vendrell J, Chacón MR. Serum levels of TWEAK and scavenger receptor CD163 in type 1 diabetes mellitus: relationship with cardiovascular risk factors. a case-control study. PLoS One (2012) 7:e43919. doi:10.1371/journal.pone.0043919
21. Maymó-Masip E, Fernández-Veledo S, García-España A, Vázquez-Carballo A, Tinahones FJ, García-Fuentes E, et al. The rise of soluble TWEAK levels in severely obese subjects after bariatric surgery may affect adipocyte-cytokine production induced by TNFα. J Clin Endocrinol Metab (2013) 98(8):E1323–33. doi:10.1210/jc.2012-4177
22. Vázquez-Carballo A, Ceperuelo-Mallafré V, Chacon MR, Maymo-Masip E, Lorenzo M, Porras A, et al. TWEAK prevent TNF-α-induced insulin resistance through PP2a activation in human adipocytes. Am J Physiol Endocrinol Metab (2013) 305(1):E101–12. doi:10.1152/ajpendo.00589.2012
23. Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, et al. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem (1997) 272:32401–10. doi:10.1074/jbc.272.51.32401
24. Díaz M, Chacón MR, López-Bermejo A, Maymó-Masip E, Salvador C, Vendrell J, et al. Ethinyl estradiol-cyproterone acetate versus low-dose pioglitazone-flutamide-metformin for adolescent girls with androgen excess: divergent effects on CD163, TWEAK receptor, ANGPTL4, and LEPTIN expression in subcutaneous adipose tissue. J Clin Endocrinol Metab (2012) 97:3630–8. doi:10.1210/jc.2012-1754
25. Alexander RW. Understanding adipose-derived stromal vascular fraction (AD-SVF) cell biology and use on the basis of cellular, chemical, structural and paracrine components: a concise review. J Prolother (2012) 4:e855–69.
26. Maecker H, Varfolomeev E, Kischkel F, Lawrence D, LeBlanc H, Lee W, et al. TWEAK attenuates the transition from innate to adaptive immunity. Cell (2005) 123:931–44. doi:10.1016/j.cell.2005.09.022
27. Alexaki VI, Notas G, Pelekanou V, Kampa M, Valkanou M, Theodoropoulos P, et al. Adipocytes as immune cells: differential expression of TWEAK, BAFF, and APRIL and their receptors (Fn14, BAFF-R, TACI, and BCMA) at different stages of normal and pathological adipose tissue development. J Immunol (2009) 183:5948–56. doi:10.4049/jimmunol.0901186
28. Tiller G, Fischer-Posovszky P, Laumen H, Finck A, Skurk T, Keuper M, et al. Effects of TWEAK (TNF superfamily member 12) on differentiation, metabolism, and secretory function of human primary preadipocytes and adipocytes. Endocrinology (2009) 150:5373–83. doi:10.1210/en.2009-0488
29. Girgenrath M, Weng S, Kostek CA, Browning B, Wang M, Brown SA, et al. TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J (2006) 25:5826–39. doi:10.1038/sj.emboj.7601441
30. Harada N, Nakayama M, Nakano H, Fukuchi Y, Yagita H, Okumura K. Pro-inflammatory effect of TWEAK/Fn14 interaction on human umbilical vein endothelial cells. Biochem Biophys Res Commun (2002) 299:488–93. doi:10.1016/S0006-291X(02)02670-0
31. Trayhurn T, Wang B, Wood I. Hypoxia in adipose tissue: a basis for the dysregulation of tissue function in obesity? Br J Nutr (2008) 100:227–35. doi:10.1017/S0007114508971282
32. Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes (2007) 56:901–11. doi:10.2337/db06-0911
33. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell (2010) 140:900–17. doi:10.1016/j.cell.2010.02.034
34. Cancello R, Henegar C, Viguerie N, Taleb S, Poitou C, Rouault C, et al. Reduction of macrophage infiltration and chemo-attractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes (2005) 54:2277–86. doi:10.2337/diabetes.54.8.2277
35. Gordon S. Alternative activation of macrophages. Nat Rev Immunol (2003) 3:23–35. doi:10.1038/nri978
36. Dalmas E, Clément K, Guerre-Millo M. Defining macrophage phenotype and function in adipose tissue. Trends Immunol (2011) 32:307–14. doi:10.1016/j.it.2011.04.008
37. Zeyda M, Stulnig TM. Adipose tissue macrophages. Immunol Lett (2007) 112:61–7. doi:10.1016/j.imlet.2007.07.003
38. Fischer-Posovszky P, Wang QA, Asterholm IW, Rutkowski JM, Scherer PE. Targeted deletion of adipocytes by apoptosis leads to adipose tissue recruitment of alternatively activated M2 macrophages. Endocrinology (2011) 152:3074–81. doi:10.1210/en.2011-1031
39. Burkly LC, Michaelson JS, Hahm K, Jakubowski A, Zheng TS. TWEAKing tissue remodeling by a multifunctional cytokine: role of TWEAK/Fn14 pathway in health and disease. Cytokine (2007) 40:1–16. doi:10.1016/j.cyto.2007.09.007
40. Sanz AB, Sanchez-Niño MD, Ortiz A. TWEAK, a multifunctional cytokine in kidney injury. Kidney Int (2011) 80:708–18. doi:10.1038/ki.2011.180
41. Musso G, Paschetta E, Gambino R, Cassader M, Molinaro F. Interactions among bone, liver, and adipose tissue predisposing to diabesity and fatty liver. Trends Mol Med (2013) 19(9):522–35. doi:10.1016/j.molmed.2013.05.006
42. Chorianopoulos E, Rosenberg M, Zugck C, Wolf J, Katus HA, Frey N. Decreased soluble TWEAK levels predict an adverse prognosis in patients with chronic stable heart failure. Eur J Heart Fail (2009) 11:1050–6. doi:10.1093/eurjhf/hfp139
43. Chorianopoulos E, Jarr K, Steen H, Giannitsis E, Frey N, Katus HA. Soluble TWEAK is markedly upregulated in patients with ST-elevation myocardial infarction and related to an adverse short-term outcome. Atherosclerosis (2010) 211:322–6. doi:10.1016/j.atherosclerosis.2010.02.016
44. Jelic-Ivanovic Z, Bujisic N, Spasic S, Bogavac-Stanojevic N, Spasojevic-Kalimanovska V, Kotur-Stevuljevic J. Circulating sTWEAK improves the prediction of coronary artery disease. Clin Biochem (2009) 42:1381–6. doi:10.1016/j.clinbiochem.2009.06.001
45. Yilmaz MI, Carrero JJ, Ortiz A, Martín-Ventura JL, Sonmez A, Saglam M, et al. Soluble TWEAK plasma levels as a novel biomarker of endothelial function in patients with chronic kidney disease. Clin J Am Soc Nephrol (2009) 4:1716–23. doi:10.2215/CJN.02760409
46. Yilmaz MI, Sonmez A, Ortiz A, Saglam M, Kilic S, Eyileten T, et al. Soluble TWEAK and PTX3 in nondialysis CKD patients: impact on endothelial dysfunction and cardiovascular outcomes. Clin J Am Soc Nephrol (2011) 6:785–92. doi:10.2215/CJN.09231010
47. Moreno JA, Dejouvencel T, Labreuche J, Smadja DM, Dussiot M, Martin-Ventura JL, et al. Peripheral artery disease is associated with a high CD163/TWEAK plasma ratio. Arterioscler Thromb Vasc Biol (2010) 30:1253–62. doi:10.1161/ATVBAHA.110.203364
48. Filusch A, Zelniker T, Baumgärtner C, Eschricht S, Frey N, Katus HA, et al. Soluble TWEAK predicts hemodynamic impairment and functional capacity in patients with pulmonary arterial hypertension. Clin Res Cardiol (2011) 100:879–85. doi:10.1007/s00392-011-0318-z
49. Richter B, Rychli K, Hohensinner PJ, Berger R, Mörtl D, Neuhold S, et al. Differences in the predictive value of tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in advanced ischemic and non-ischemic heart failure. Atherosclerosis (2010) 213:545–8. doi:10.1016/j.atherosclerosis.2010.08.061
50. Martín-Ventura JL, Lindholt JS, Moreno JA, Vega de Céniga M, Meilhac O, et al. Soluble TWEAK plasma levels predict expansion of human abdominal aortic aneurysms. Atherosclerosis (2011) 214:486–9. doi:10.1016/j.atherosclerosis.2010.11.009
51. Valdivielso JM, Coll B, Martín-Ventura JL, Moreno JA, Egido J, Fernández E, et al. Soluble TWEAK is associated with atherosclerotic burden in patients with chronic kidney disease. J Nephrol (2013) 26(6):1105–13. doi:10.5301/jn.5000245
52. Kralisch S, Ziegelmeier M, Bachmann A, Seeger J, Lossner U, Bluher M, et al. Serum levels of the atherosclerosis biomarker sTWEAK are decreased in type 2 diabetes and end-stage renal disease. Atherosclerosis (2008) 199:440–4. doi:10.1016/j.atherosclerosis.2007.10.022
53. Carrero JJ, Ortiz A, Qureshi AR, Martín-Ventura JL, Bárány P, Heimbürger O, et al. Additive effects of soluble TWEAK and inflammation on mortality in hemodialysis patients. Clin J Am Soc Nephrol (2009) 1:110–8. doi:10.2215/CJN.02790608
54. Díaz-López A, Chacón MR, Bulló M, Maymó-Masip E, Martínez-González MA, Estruch R, et al. Serum sTWEAK concentrations and risk of developing type 2 diabetes in a high cardiovascular risk population: a nested case- control study. J Clin Endocrinol Metab (2013) 98(8):3482–9. doi:10.1210/jc.2013-1848
55. Vita JA, Keaney JF Jr. Endothelial function: a barometer for cardiovascular risk? Circulation (2002) 106:640–2. doi:10.1161/01.CIR.0000028581.07992.56
56. Bover LC, Cardó-Vila M, Kuniyasu A, Sun J, Rangel R, Takeya M, et al. A previously unrecognized protein-protein interaction between TWEAK and CD163: potential biological implications. J Immunol (2007) 178:8183–94.
57. Møller HJ. Soluble CD163. Scand J Clin Lab Invest (2012) 72:1–13. doi:10.3109/00365513.2011.626868
58. Moreno JA, Muñoz-García B, Martín-Ventura JL, Madrigal-Matute J, Orbe J, Páramo JA, et al. The CD163-expressing macrophages recognize and internalize TWEAK: potential consequences in atherosclerosis. Atherosclerosis (2009) 207:103–10. doi:10.1016/j.atherosclerosis.2009.04.033
59. Sporrer D, Weber M, Wanninger J, Weigert J, Neumeier M, Stögbauer F, et al. Adiponectin downregulates CD163 whose cellular and soluble forms are elevated in obesity. Eur J Clin Invest (2009) 39:671–9. doi:10.1111/j.1365-2362.2009.02170.x
60. Fjeldborg K, Christiansen T, Bennetzen M, Møller HJ, Pedersen SB, Richelsen B. The macrophage specific serum marker, soluble CD163, is increased in obesity and reduced after dietary induced weight loss. Obesity (Silver Spring) (2013) 21(12):2437–43. doi:10.1002/oby.20376
61. Al-Daghri NM, Al-Attas OS, Bindahman LS, Alokail MS, Alkharfy KM, Draz HM, et al. Soluble CD163 is associated with body mass index and blood pressure in hypertensive obese Saudi patients. Eur J Clin Invest (2012) 42:1221–6. doi:10.1111/j.1365-2362.2012.02714.x
62. Møller HJ, Frikke-Schmidt R, Moestrup SK, Nordestgaard BG, Tybjaerg-Hansen A. Serum soluble CD163 predicts risk of type 2 diabetes in the general population. Clin Chem (2011) 57:291–7. doi:10.1373/clinchem.2010.154724
63. Muñoz-García B, Moreno JA, Lopez-Franco O, Sanz AB, Martin-Ventura JL, Blanco J, et al. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) enhances vascular and renal damage induced by hyperlipidemic diet in ApoE-knockout mice. Arterioscler Thromb Vasc Biol (2009) 29:2061–8. doi:10.1161/ATVBAHA.109.194852
64. Schapira K, Burkly LC, Zheng TS, Wu P, Groeneweg M, Rousch M, et al. Fn14-Fc fusion protein regulates atherosclerosis in ApoE-/- mice and inhibits macrophage lipid uptake in vitro. Arterioscler Thromb Vasc Biol (2009) 29:2021–7. doi:10.1161/ATVBAHA.109.195040
65. Cawthorn WP, Sethi JK. TNF-alpha and adipocyte biology. FEBS Lett (2008) 582:117–31. doi:10.1016/j.febslet.2007.11.051
66. Fick A, Lang I, Schäfer V, Seher A, Trebing J, Weisenberger D, et al. Studies of binding of tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) to fibroblast growth factor inducible 14 (Fn14). J Biol Chem (2012) 287:484–95. doi:10.1074/jbc.M111.287656
67. Fernández-Veledo S, Vila-Bedmar R, Nieto-Vazquez I, Lorenzo M. c-Jun N-terminal kinase 1/2 activation by tumor necrosis factor-alpha induces insulin resistance in human visceral but not subcutaneous adipocytes: reversal by liver X receptor agonists. J Clin Endocrinol Metab (2009) 94:3583–93. doi:10.1210/jc.2009-0558
68. Yamana J, Morand EF, Manabu T, Sunahori K, Takasugi K, Makino H, et al. Inhibition of TNF-induced IL-6 by the TWEAK-Fn14 interaction in rheumatoid arthritis fibroblast like synoviocytes. Cell Immunol (2012) 272:293–8. doi:10.1016/j.cellimm.2011.09.004
69. Echeverry R, Wu F, Haile WB, Wu J, Yepes M. The cytokine tumor necrosis factor-like weak inducer of apoptosis and its receptor fibroblast growth factor-inducible 14 have a neuroprotective effect in the central nervous system. J Neuroinflammation (2012) 9:45. doi:10.1186/1742-2094-9-45
Keywords: TWEAK, obesity, type 2 diabetes, adipose tissue, TNFα, insulin resistance, inflammation
Citation: Vendrell J and Chacón MR (2013) TWEAK: a new player in obesity and diabetes. Front. Immunol. 4:488. doi: 10.3389/fimmu.2013.00488
Received: 17 September 2013; Accepted: 12 December 2013;
Published online: 30 December 2013.
Edited by:
Linda C. Burkly, Biogen Idec, Inc., USAReviewed by:
Martin S. Obin, Tufts University, USAAndreas Tsapis, U976 Institut national de la santé et de la recherche médicale, France
Copyright: © 2013 Vendrell and Chacón. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matilde R. Chacón, Research Unit, Centro de Investigación Biomédica en Red de Diabetes y Enfermedades Metabólicas Asociadas CIBERDEM, Hospital Universitari de Tarragona Joan XXIII, Institut d’Investigació Sanitària Pere Virgili, Universitat Rovira i Virgili, C/Dr. Mallafré Guasch 4, Tarragona 43007, Spain e-mail:bXJvZHJpZ3Vlei5oajIzLmljc0BnZW5jYXQuY2F0