 Merlin Luetke-Eversloh
Merlin Luetke-Eversloh Monica Killig
Monica Killig Chiara Romagnani
Chiara Romagnani- Innate Immunity, Deutsches Rheuma-Forschungszentrum, Berlin – A Leibniz Institute, Berlin, Germany
Natural killer (NK) cells are part of the innate lymphoid cell (ILC) family and represent the main cytotoxic population. NK cells develop from bone marrow common lymphoid progenitors and undergo terminal differentiation in the periphery, where they finally gain their cytotoxic competence as well as the ability to produce IFN-γ in response to engagement of activating receptors. This process has been at least partially elucidated and several markers have been identified to discriminate different NK cell stages and other ILC populations. NK cell terminal differentiation is not only associated with progressive phenotypic changes but also with defined effector signatures. In this essay, we will describe the phenotypic and functional characteristics of the main stages of NK cell development and terminal differentiation and discuss them in light of recent discoveries of novel ILC populations.
Natural Killer Cells and the Innate Lymphoid Cell Family
Natural Killer (NK) cells are innate lymphocytes with the capability to rapidly kill virus-infected or transformed target cells and to produce type 1 cytokines, such as IFN-γ. Over the past years, other innate lymphoid effectors have been identified, which share defined developmental requirements with NK cells, such as the dependence on the transcriptional repressor inhibitor of DNA binding 2 (Id2), but clearly differ for phenotype and effector properties. The heterogeneity displayed by the different innate lymphocyte subsets closely resembles the one observed among CD4+ T helper cells. Thus, a new nomenclature was adapted to collectively define the family, which has been named innate lymphoid cells (ILCs). According to this classification, ILCs have been divided in three main groups: Group 1 ILCs, including NK cells, are defined by the expression of the T-box transcription factors Eomesodermin (EOMES) and/or T-bet (TBX21) and produce mainly IFN-γ in response to infected or transformed cells; Group 2 ILCs, expressing GATA3, produce IL-13 and IL-5 for the defense against helminthic infections; Group 3 ILCs are characterized by the expression of the orphan nuclear receptor transcription factor RORγt (RORC) and include fetal lymphoid tissue-inducer (LTi) cells and adult LTi-like cells, also named ILC3 (1). NK cells likely do not represent the only member of Group 1 ILCs. Indeed, NK cells residing in different organs, such as liver, lung, uterus, or intestine are quite dissimilar among each other. It is still unclear whether this peripheral heterogeneity originates from tissue-specific signals influencing either NK cell in situ development or terminal differentiation. Alternatively, tissue resident NK cells might also represent other Group 1 ILC members emerging independently of NK cell precursors. In line with this hypothesis, other potential Group 1 ILC members have been identified recently in the intestine (2, 3). Hence, in the present review we will give an overview on classical human NK cell development and terminal differentiation, and discuss the current knowledge in the frame of this emerging diversity of ILCs.
Human NK Cell Development
Natural killer cells develop during fetal life as well as after birth from hematopoietic stem cells (HSCs) through a common lymphoid progenitor (CLP). Although most markers used in the mouse and human systems are different, we will revise the major findings of human NK cell development also in light of murine data. Fetal as well as bone marrow (BM) CLP still have the potential to give rise to B, T, NK, and dendritic cells (DCs). As the development of all mouse ILC group members relies on Id2, it was proposed that a common Id2+ ILC progenitor might exist, but still needs to be identified (4). Interestingly, fetal as well as BM HSC can give rise to both, Group 3 ILCs and NK cells in mouse. In particular, expression of the integrin α4β7 defines a mouse fetal CLP subset with a developmental potential restricted to T, NK, and LTi (Group 3 ILC) cells (5, 6). In line with these data, mouse fetal liver α4β7+ CLP can up-regulate RORγt and differentiate toward LTi cells (7–9). Similarly, after birth, mouse BM precursors expressing integrin α4β7 and CXCR6 can give rise to Group 3 ILCs as well as to NK cells, although with low efficiency, but have lost B cell as well as T cell differentiation ability, thus potentially represent a common or a mixture of progenitors committed toward Group 3 ILC and NK cell lineage (8). However, it would be relevant to test the differentiation potential of these cells toward Group 2 ILC, which can also be generated from BM CLP (10–12). In humans, a pioneer study from the group of Verneris has clearly shown that total umbilical cord blood-CD34+ cells can give rise to both NK cells and Group 3 ILCs (13); however, these populations might originate from fetal CLP, and the identification of a common human ILC progenitor is not yet identified, similar to mouse. In addition to the CLP, mouse NK cell lineage committed progenitors have been initially identified among Lin− CD122+ NK1.1− DX5− NK cells (14) and more recently redefined according to the expression of CD27 and CD244 or Id2 (15, 16). Nevertheless, as to date it has not been tested whether these cell populations are also able to differentiate toward Groups 2 or 3 ILCs, it still remains to be formally proven whether they represent or not the earliest NK cell committed precursor. Although the exact phenotype is not yet completely established in humans, CLP are enriched among CD34+ CD45RA+ CD38lo CD10± CD7± cells. In particular CD34+ CD7+ CD45RA+ and CD34+ CD10+ CD45RA+ cells are preferentially biased to develop into T/NK and B cells, respectively (17–20). CLP-like cells with NK cell commitment potential and expressing β7 integrin, similar to mice, have also been described in human peripheral blood (PB) and shown to be enriched in extramedullary sites, such as secondary lymphoid organs (SLOs) (21–23), which were therefore proposed as putative sites of human NK cell development. According to this concept, human CLP-like CD34+ β7 integrin+ CD45RA+ (c-Kit− CD94−) cells identified in SLO still display T cell and DC potential and have been termed “stage I” NK cells (23). Human NK cell differentiation in SLO would then proceed through “stage II” (CD34+ CD45RA+ c-Kit+ CD94−) and “stage III” (CD34− c-Kit+ CD94−) NK cells, which would finally give rise to “stage IV” NK cells, also defined as CD56bright CD16neg/lo NK cells (23). More recently, it has been shown that most putative “stage III” human NK cells (CD34− c-Kit+ CD94−) isolated ex vivo in SLO are actually mature ILC3 (24, 25). As mature human ILC3 mostly coexpress c-Kit and CD127, it was proposed that among CD34− c-Kit+ cells only a minor fraction, characterized by the lack of CD127 might represent “stage III” human NK cells (25). Thus, due to the complex overlapping of markers between human NK cells and ILC3, a more detailed analysis of in vitro differentiation of “stage III” and “stage II” NK cell precursors toward different ILC subsets would be of great help. Nevertheless, these studies have contributed crucially to support the idea that extramedullary compartments might indeed represent important sites of NK cell development (22, 23). The unique milieu available in different tissues might influence in situ differentiation of NK cells and could actually explain the large phenotypic and functional heterogeneity found among NK cells derived from different tissues, such as liver, lung, or uterus. NK cell precursors might migrate very early to the tissues and develop in situ influenced by tissue-specific signals, such as cytokines, stromal or epithelial cells, and environmental cues. However, we cannot exclude that this large heterogeneity might actually rather reflect potentially novel Group 1 ILC subsets, displaying NK-like phenotype. Along this line, extramedullary compartments might represent preferential developmental sites not only for most Group 1 but also for Group 3 ILCs. It has been shown that precursors of murine Group 3 ILCs do not up-regulate RORγt expression in the BM and might migrate very early to the periphery, especially to the intestinal lamina propria (LP), for their development/terminal differentiation (8, 26). However, it still remains an open question at which developmental stage (CLP? common ILC, Group 1 ILC, or NK cell precursor?) NK cells migrate to peripheral tissues to undergo further differentiation. Figure 1 illustrates markers associated with early NK cell developmental stages derived from human CLP.
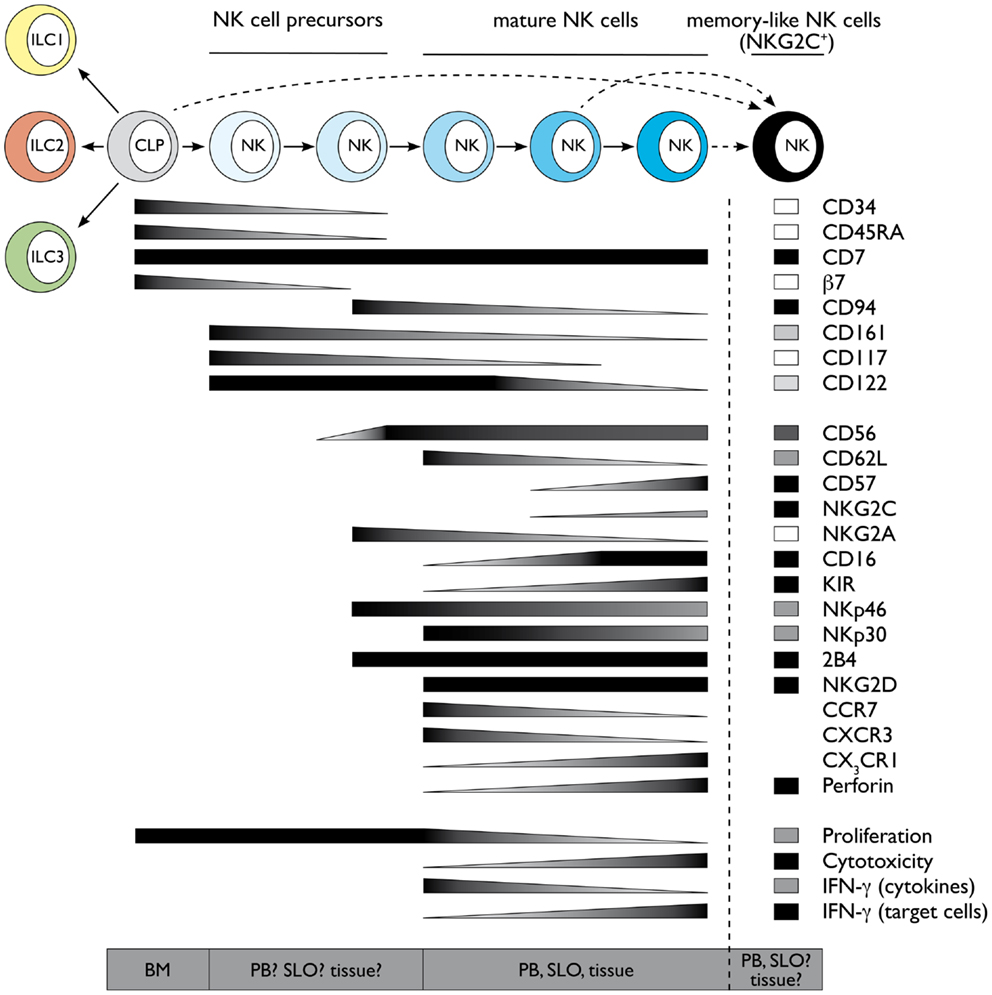
Figure 1. Schematic representation of human NK cell development and terminal differentiation. The development of human NK cells from a common lymphoid progenitor over NK cell precursors to terminally differentiated NK cells is depicted from left to right. The acquisition and loss of indicated surface markers and functional properties propose a linear model of human NK cell development and terminal differentiation. Protein expression levels are depicted as black for high expression and white for no expression, gray indicates intermediate levels. Functional properties are indicated correspondingly. Abbreviations: common lymphoid progenitor (CLP), bone marrow (BM), peripheral blood (PB), secondary lymphoid organs (SLO). See also Ref. 59–61.
Human NK Cell Terminal Differentiation and Related Effector Functions
Despite the presence of NK cells or NK cell-like populations in different organs, most of our knowledge on NK cell biology originates from mouse splenic and human PB-derived NK cells. Even within the same compartment, NK cells are heterogeneous, and one possible interpretation of this observation is that this diversity might reflect a process of terminal differentiation occurring in the periphery. Along this line, different markers have been used in humans and mouse to describe the stages of final NK cell maturation. In mouse, several authors have contributed to delineate a model in which splenic NK1.1+ NK cells can be mainly dissected according to CD27 and CD11b, which enable to foresee the following maturation progression: CD27− CD11b− → CD27+ CD11b− → CD27+ CD11b+ → CD27−CD11b+ NK cells (27–29). Conversely, most of the data concerning this process in humans relies on the analysis of circulating PB-derived NK cells. In this compartment, two main NK subsets were originally described on the basis of the differential expression of CD56, which has no homolog in mice and were named CD56bright and CD56dim NK cells (30, 31). CD56bright and CD56dim NK cells differ in phenotype, functional capabilities, and preferential locations. CD56bright NK cells are only a minority of PB-NK cells, whereas they represent the majority of NK cells in SLO. CD56bright NK cells have been described as preferentially CD62L−/lo CD94/NKG2A+ CD62Lhi CD57− KIR− and commonly termed as the immunomodulatory subset, because of their potential to produce high levels of cytokines like IFN-γ and TNF. In contrast, they express only very low amounts of cytolytic granules and display poor cytotoxic capability. This feature is a characteristic of CD56dim NK cells, which express high amounts of perforin and granzymes and are able to kill target cells like virus-infected and transformed cells. However, the idea that CD56bright are the cytokine producers whereas CD56dim NK cells represent the cytotoxic subset, might be actually misleading as both NK cell subsets can produce large amounts of IFN-γ. CD56bright NK cells produce high amounts of IFN-γ and extensively proliferate in response to DC-derived cytokines like IL-2, IL-15, IL-12, and IL-18 (31), but are unable to produce IFN-γ in response to target cell recognition (32, 33). Conversely, CD56dim NK cells are less efficient in proliferating and producing IFN-γ in response to cytokines (31), but become the main IFN-γ producers after target cell encounter (32, 33). A detailed analysis of CD56dim functional properties and phenotype has actually revealed a consistent heterogeneity also among CD56dim NK cells. When CD56dim NK cells were dissected according to the expression of either CD62L, CD94/NKG2A, or lack of CD57, a subset of NK cells with intermediate phenotype and properties between CD56bright (CD94/NKG2Ahi CD62Lhi CD57−) and CD56dim CD94/NKG2A–, CD62L–, or CD57+ cells could be identified (33–39). These data were also confirmed at global transcriptome level (33). CD56dim NK cells displaying this intermediate signature (CD94/NKG2A+, CD62L+, or CD57−) combine the ability to produce IFN-γ and proliferate in response to cytokines, characteristic for CD56bright NK cells, with the capacity to kill and produce IFN-γ upon engagement of activating receptors (actRs), typical of CD56dim NK cells. The developmental relationship between CD56bright and the different CD56dim NK cell subsets has been controversially discussed, but several studies support the concept that CD56bright cells might represent the progenitors of CD56dim NK cells. CD56bright NK cells display longer telomeres compared to CD56dim NK cells and can acquire the expression of CD16 and killer Ig-like receptors (KIR), as well as of other markers, in vitro as well as in vivo after transfer into immunodeficient mice (33, 36). Further observations support a model of linear maturation from more immature CD56bright toward terminally differentiated CD56dim (CD94/NKG2A–, CD62L–, or CD57+) NK cells passing through CD56dim NK cell subsets displaying intermediate signatures (CD94/NKG2A+, CD62L+, or CD57–). As previously mentioned, this phenotypic sequence is also associated with progressive loss of responsiveness to cytokines and gradual acquisition of responsiveness to actR engagement (33–35) (Luetke-Eversloh and Romagnani, unpublished data). The capability of the more immature CD56bright and CD56dim NK cell subsets to respond to cytokine stimulation strictly correlates to the expression of CD94, CD62L, or CD57 and is reflected by higher expression of cytokine receptors and STAT4 activation (33, 37, 39, 40). Conversely, the ability of the CD56dim subsets to kill and produce IFN-γ in response to actR mainly correlates to the expression of self MHC class I binding inhibitory receptors, in humans belonging to the KIR family or CD94/NKG2A (41–44). This phenomenon, termed education or licensing, was originally described in mice for NK cells expressing self MHC class I specific inhibitory Ly49 receptors or CD94/NKG2A (41–43). According to this concept, during their differentiation or the course of immune responses, NK cells are licensed by self MHC class I molecules through engagement of their inhibitory receptors and this interaction results in NK cell functional competence in response to actR. Thus, licensed NK cells are not only functionally competent but also tolerant because they display at least one inhibitory receptor for self MHC. Conversely, NK cells which do not have a self MHC-specific inhibitory receptor are less functional and therefore still tolerant toward self (41, 45–47). The molecular mechanisms underlying this process have been only partially clarified. In mice, NK cell education seems to require functional immunoreceptor tyrosine-based inhibitory motifs (ITIMs) of self MHC class I binding Ly49 receptors, but to be independent of SHP-1 signaling (41–43). In addition, educated mouse NK cells have an altered membrane distribution of actR, which is instrumental to functional competence in response to actR engagement (41, 45–47). Although further research is needed to clarify the site, the stimuli, and the time of NK cell education, it is now clear that this process is not an on-off switch occurring during BM development, but rather a fine tuning determined by the number and the affinity of MHC-inhibitory receptor interactions (41, 45–48). However, the stage of NK cell terminal differentiation might also contribute to finely tune functional competence in response to actR engagement. Terminally differentiated NK cell subsets are enriched in cells expressing more than one inhibitory receptor (39), and thus in educated cells, suggesting that differentiation and education might not be entirely uncoupled processes. In Figure 1, we summarized several markers defining the phenotype and functions of the different NK cell subsets.
In contrast to individual markers, the combination of expression of CD16, KIR, CD57, CD62L, CD94/NKG2A does not identify three main populations but results in a complex number of intermediate, terminally differentiated, educated, and non-educated NK cell subsets. Although this implies even a larger heterogeneity of PB-NK cells, it does not exclude that NK cell terminal differentiation still proceeds following a linear progression. In support of this model, we have observed that these markers are not acquired or lost synchronously in time during reconstitution from HSC during transplantation and this might contribute to explain NK cell peripheral variety (M.K. and C.R., unpublished data). Moreover, diverse signals, such as homeostatic and pro-inflammatory cytokines or engagement of defined activating or inhibitory receptors at steady state or during infections, might influence NK cell terminal differentiation and/or education and determine the quality and the intensity of this process. One clear example of how environmental stimuli can dramatically poise human NK cell final maturation is cytomegalovirus (CMV) infection, which can globally accelerate NK cell terminal differentiation (49). CMV infection induces the expansion and persistence of a unique NK cell subset, expressing the actR NKG2C and being preferentially positive for CD57 and self MHC class I binding KIR (50–52). Although NKG2C+ NK cells undergoing expansion during CMV infection tend to resemble phenotypically and functionally terminally differentiated NK cells, it has been also suggested that they might represent the human correspondent of the previously described Ly49H+ memory-like murine NK cells expanding and persisting after CMV infection (53, 54) (Figure 1). Interestingly, expansion of NKG2C+ NK cells occurs also in CMV-seropositive patients with chronic hepatitis B or C virus or during hantavirus infection (55, 56). Further experiments are required to understand their origin and developmental relationship with other PB-NK cell subsets.
Interestingly, different stages defined according to CD16, NKG2A, and KIR can also be identified among fetal NK cells, suggesting that similar forces likely govern the process of differentiation before and after birth. However, even in fetal lung where NK cells are strongly enriched in more mature KIR+ cells, CD57 expression is very low and only NKG2A, but not self MHC specific KIR, mediate NK cell education, possibly suggesting that fetal tissue milieu influence NK cell differentiation and functional properties (57).
Despite of all these suggestive evidences, it is however difficult to completely exclude that human NK cell peripheral heterogeneity might not rather represent developmentally unrelated members of the Group 1 ILC family. CD56bright NK cells uniquely express CD127, CD117, and GATA3, which are hallmarks of other ILC subsets and have been also proposed to correspond to mouse thymus-derived NK cells (58). Further investigation employing different experimental approaches might help us to revisit this issue and elicit some surprises.
“It’s a rare occasion when your plans and expectations come down just exactly how you planned them. Who can say, anyway? Time will tell” (“Time will tell,” Tower of Power).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13(2):145–9. doi: 10.1038/nri3365
2. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol (2013) 14(3):221–9. doi:10.1038/ni.2534
3. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity (2013) 38(4):769–81. doi:10.1016/j.immuni.2013.02.010
4. Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol (2012) 30:647–75. doi:10.1146/annurev-immunol-020711-075053
5. Mebius RE, Miyamoto T, Christensen J, Domen J, Cupedo T, Weissman IL, et al. The fetal liver counterpart of adult common lymphoid progenitors gives rise to all lymphoid lineages, CD45+CD4+CD3- cells, as well as macrophages. J Immunol (2001) 166(11):6593–601.
6. Yoshida H, Kawamoto H, Santee SM, Hashi H, Honda K, Nishikawa S, et al. Expression of alpha(4)beta(7) integrin defines a distinct pathway of lymphoid progenitors committed to T cells, fetal intestinal lymphotoxin producer, NK, and dendritic cells. J Immunol (2001) 167(5):2511–21.
7. Cherrier M, Sawa S, Eberl G. Notch, Id2, and RORgammat sequentially orchestrate the fetal development of lymphoid tissue inducer cells. J Exp Med (2012) 209(4):729–40. doi:10.1084/jem.20111594
8. Possot C, Schmutz S, Chea S, Boucontet L, Louise A, Cumano A, et al. Notch signaling is necessary for adult, but not fetal, development of RORgammat(+) innate lymphoid cells. Nat Immunol (2011) 12(10):949–58. doi:10.1038/ni.2105
9. Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, et al. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science (2010) 330(6004):665–9. doi:10.1126/science.1194597
10. Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, et al. Transcription factor RORalpha is critical for nuocyte development. Nat Immunol (2012) 13(3):229–36. doi:10.1038/ni.2208
11. Yang Q, Saenz SA, Zlotoff DA, Artis D, Bhandoola A. Cutting edge: natural helper cells derive from lymphoid progenitors. J Immunol (2011) 187(11):5505–9. doi:10.4049/jimmunol.1102039
12. Halim TY, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity (2012) 37(3):463–74. doi:10.1016/j.immuni.2012.06.012
13. Tang Q, Ahn YO, Southern P, Blazar BR, Miller JS, Verneris MR. Development of IL-22-producing NK lineage cells from umbilical cord blood hematopoietic stem cells in the absence of secondary lymphoid tissue. Blood (2011) 117(15):4052–5. doi:10.1182/blood-2010-09-303081
14. Rosmaraki EE, Douagi I, Roth C, Colucci F, Cumano A, Di Santo JP. Identification of committed NK cell progenitors in adult murine bone marrow. Eur J Immunol (2001) 31(6):1900–9. doi:10.1002/1521-4141(200106)31:6<1900::AID-IMMU1900>3.0.CO;2-M
15. Carotta S, Pang SH, Nutt SL, Belz GT. Identification of the earliest NK-cell precursor in the mouse BM. Blood (2011) 117(20):5449–52. doi:10.1182/blood-2010-11-318956
16. Fathman JW, Bhattacharya D, Inlay MA, Seita J, Karsunky H, Weissman IL. Identification of the earliest natural killer cell-committed progenitor in murine bone marrow. Blood (2011) 118(20):5439–47. doi:10.1182/blood-2011-04-348912
17. Haddad R, Guardiola P, Izac B, Thibault C, Radich J, Delezoide AL, et al. Molecular characterization of early human T/NK and B-lymphoid progenitor cells in umbilical cord blood. Blood (2004) 104(13):3918–26. doi:10.1182/blood-2004-05-1845
18. Barcena A, Muench MO, Galy AH, Cupp J, Roncarolo MG, Phillips JH, et al. Phenotypic and functional analysis of T-cell precursors in the human fetal liver and thymus: CD7 expression in the early stages of T- and myeloid-cell development. Blood (1993) 82(11):3401–14.
19. Miller JS, Alley KA, McGlave P. Differentiation of natural killer (NK) cells from human primitive marrow progenitors in a stroma-based long-term culture system: identification of a CD34+7+ NK progenitor. Blood (1994) 83(9):2594–601.
20. Rossi MI, Yokota T, Medina KL, Garrett KP, Comp PC, Schipul AH Jr, et al. B lymphopoiesis is active throughout human life, but there are developmental age-related changes. Blood (2003) 101(2):576–84. doi:10.1182/blood-2002-03-0896
21. McClory S, Hughes T, Freud AG, Briercheck EL, Martin C, Trimboli AJ, et al. Evidence for a stepwise program of extrathymic T cell development within the human tonsil. J Clin Invest (2012) 122(4):1403–15. doi:10.1172/JCI46125
22. Freud AG, Becknell B, Roychowdhury S, Mao HC, Ferketich AK, Nuovo GJ, et al. A human CD34(+) subset resides in lymph nodes and differentiates into CD56bright natural killer cells. Immunity (2005) 22(3):295–304. doi:10.1016/j.immuni.2005.01.013
23. Freud AG, Yokohama A, Becknell B, Lee MT, Mao HC, Ferketich AK, et al. Evidence for discrete stages of human natural killer cell differentiation in vivo. J Exp Med (2006) 203(4):1033–43. doi:10.1084/jem.20052507
24. Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, et al. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol (2009) 10(1):66–74. doi:10.1038/ni.1668
25. Crellin NK, Trifari S, Kaplan CD, Cupedo T, Spits H. Human NKp44+IL-22+ cells and LTi-like cells constitute a stable RORC+ lineage distinct from conventional natural killer cells. J Exp Med (2010) 207(2):281–90. doi:10.1084/jem.20091509
26. Cherrier M, Eberl G. The development of LTi cells. Curr Opin Immunol (2012) 24(2):178–83. doi:10.1016/j.coi.2012.02.003
27. Kim S, Iizuka K, Kang HS, Dokun A, French AR, Greco S, et al. In vivo developmental stages in murine natural killer cell maturation. Nat Immunol (2002) 3(6):523–8. doi:10.1038/ni796
28. Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood (2009) 113(22):5488–96. doi:10.1182/blood-2008-10-187179
29. Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol (2006) 176(3):1517–24.
30. Lanier LL, Le AM, Civin CI, Loken MR, Phillips JH. The relationship of CD16 (Leu-11) and Leu-19 (NKH-1) antigen expression on human peripheral blood NK cells and cytotoxic T lymphocytes. J Immunol (1986) 136(12):4480–6.
31. Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood (2001) 97(10):3146–51. doi:10.1182/blood.V97.10.3146
32. Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood (2010) 115(11):2167–76. doi:10.1182/blood-2009-08-238469
33. Juelke K, Killig M, Luetke-Eversloh M, Parente E, Gruen J, Morandi B, et al. CD62L expression identifies a unique subset of polyfunctional CD56dim NK cells. Blood (2010) 116(8):1299–307. doi:10.1182/blood-2009-11-253286
34. Romagnani C, Juelke K, Falco M, Morandi B, D’Agostino A, Costa R, et al. CD56brightCD16- killer Ig-like receptor- NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J Immunol (2007) 178(8):4947–55.
35. Chan A, Hong DL, Atzberger A, Kollnberger S, Filer AD, Buckley CD, et al. CD56bright human NK cells differentiate into CD56dim cells: role of contact with peripheral fibroblasts. J Immunol (2007) 179(1):89–94.
36. Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med (2009) 206(1):25–34. doi:10.1084/jem.20082013
37. Yu J, Mao HC, Wei M, Hughes T, Zhang J, Park IK, et al. CD94 surface density identifies a functional intermediary between the CD56bright and CD56dim human NK-cell subsets. Blood (2010) 115(2):274–81. doi:10.1182/blood-2009-04-215491
38. Lopez-Verges S, Milush JM, Pandey S, York VA, Arakawa-Hoyt J, Pircher H, et al. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood (2010) 116(19):3865–74. doi:10.1182/blood-2010-04-282301
39. Bjorkstrom NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood (2010) 116(19):3853–64. doi:10.1182/blood-2010-04-281675
40. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol (2001) 22(11):633–40. doi:10.1016/S1471-4906(01)02060-9
41. Raulet DH, Vance RE. Self-tolerance of natural killer cells. Nat Rev Immunol (2006) 6(7):520–31. doi:10.1038/nri1863
42. Yokoyama WM, Kim S. Licensing of natural killer cells by self-major histocompatibility complex class I. Immunol Rev (2006) 214:143–54. doi:10.1111/j.1600-065X.2006.00458.x
43. Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity (2006) 25(2):331–42. doi:10.1016/j.immuni.2006.06.013
44. Yu J, Heller G, Chewning J, Kim S, Yokoyama WM, Hsu KC. Hierarchy of the human natural killer cell response is determined by class and quantity of inhibitory receptors for self-HLA-B and HLA-C ligands. J Immunol (2007) 179(9):5977–89.
45. Elliott JM, Yokoyama WM. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol (2011) 32(8):364–72. doi:10.1016/j.it.2011.06.001
46. Chalifour A, Scarpellino L, Back J, Brodin P, Devevre E, Gros F, et al. A role for cis interaction between the inhibitory Ly49A receptor and MHC class I for natural killer cell education. Immunity (2009) 30(3):337–47. doi:10.1016/j.immuni.2008.12.019
47. Guia S, Jaeger BN, Piatek S, Mailfert S, Trombik T, Fenis A, et al. Confinement of activating receptors at the plasma membrane controls natural killer cell tolerance. Sci Signal (2011) 4(167):ra21. doi:10.1126/scisignal.2001608
48. Brodin P, Karre K, Hoglund P. NK cell education: not an on-off switch but a tunable rheostat. Trends Immunol (2009) 30(4):143–9. doi:10.1016/j.it.2009.01.006
49. Della Chiesa M, Falco M, Podesta M, Locatelli F, Moretta L, Frassoni F, et al. Phenotypic and functional heterogeneity of human NK cells developing after umbilical cord blood transplantation: a role for human cytomegalovirus? Blood (2012) 119(2):399–410. doi:10.1182/blood-2011-08-372003
50. Kuijpers TW, Baars PA, Dantin C, van den Burg M, van Lier RA, Roosnek E. Human NK cells can control CMV infection in the absence of T cells. Blood (2008) 112(3):914–5. doi:10.1182/blood-2008-05-157354
51. Foley B, Cooley S, Verneris MR, Curtsinger J, Luo X, Waller EK, et al. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J Immunol (2012) 189(10):5082–8. doi:10.4049/jimmunol.1201964
52. Lopez-Verges S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, York VA, et al. Expansion of a unique CD57(+)NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci U S A (2011) 108(36):14725–32. doi:10.1073/pnas.1110900108
53. Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature (2009) 457(7229):557–61. doi:10.1038/nature07665
54. Min-Oo G, Kamimura Y, Hendricks DW, Nabekura T, Lanier LL. Natural killer cells: walking three paths down memory lane. Trends Immunol (2013) 34(6):251–8. doi:10.1016/j.it.2013.02.005
55. Beziat V, Dalgard O, Asselah T, Halfon P, Bedossa P, Boudifa A, et al. CMV drives clonal expansion of NKG2C+ NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur J Immunol (2012) 42(2):447–57. doi:10.1002/eji.201141826
56. Bjorkstrom NK, Lindgren T, Stoltz M, Fauriat C, Braun M, Evander M, et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J Exp Med (2011) 208(1):13–21. doi:10.1084/jem.20100762
57. Ivarsson MA, Loh L, Marquardt N, Kekalainen E, Berglin L, Bjorkstrom NK, et al. Differentiation and functional regulation of human fetal NK cells. J Clin Invest (2013) 123(9):3889–901. doi:10.1172/JCI68989
58. Vosshenrich CA, Garcia-Ojeda ME, Samson-Villeger SI, Pasqualetto V, Enault L, Richard-Le Goff O, et al. A thymic pathway of mouse natural killer cell development characterized by expression of GATA-3 and CD127. Nat Immunol (2006) 7(11):1217–24. doi:10.1038/ni1395
59. Cichocki F, Miller JS, Anderson SK, Bryceson YT. Epigenetic regulation of NK cell differentiation and effector functions. Front Immunol (2013) 4:55. doi:10.3389/fimmu.2013.00055
60. Montaldo E, Del Zotto G, Della Chiesa M, Mingari MC, Moretta A, De Maria A, et al. Human NK cell receptors/markers: a tool to analyze NK cell development, subsets and function. Cytometry A (2013) 83(8):702–13. doi:10.1002/cyto.a.22302
Keywords: NK cells, ILC, differentiation, IFN-γ, CD62L, CD57, NKG2A, KIR
Citation: Luetke-Eversloh M, Killig M and Romagnani C (2013) Signatures of human NK cell development and terminal differentiation. Front. Immunol. 4:499. doi: 10.3389/fimmu.2013.00499
Received: 30 September 2013; Accepted: 18 December 2013;
Published online: 30 December 2013.
Edited by:
Yenan Bryceson, Karolinska Institutet, SwedenReviewed by:
Michael R. Verneris, University of Minnesota, USAJenny Mari Mjösberg, Karolinska Institutet, Sweden
Copyright: © 2013 Luetke-Eversloh, Killig and Romagnani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Romagnani, Innate Immunity, Deutsches Rheuma- Forschungszentrum, Berlin – A Leibniz Institute, Charitéplatz 1, Berlin 10117, Germany e-mail:cm9tYWduYW5pQGRyZnouZGU=