 Eiji Yoshihara
Eiji Yoshihara So Masaki
So Masaki Yoshiyuki Matsuo
Yoshiyuki Matsuo Zhe Chen
Zhe Chen Hai Tian
Hai Tian Junji Yodoi
Junji Yodoi- 1Institute for Virus Research, Kyoto University, Kyoto, Japan
- 2Advanced Chemical Technology Center in Kyoto (ACT Kyoto), JBPA Research Institute, Kyoto, Japan
- 3Redox Bio Science Inc., Kyoto, Japan
During the past few decades, it has been widely recognized that Reduction-Oxidation (redox) responses occurring at the intra- and extra-cellular levels are one of most important biological phenomena and dysregulated redox responses are involved in the initiation and progression of multiple diseases. Thioredoxin1 (Trx1) and Thioredoxin2 (Trx2), mainly located in the cytoplasm and mitochondria, respectively, are ubiquitously expressed in variety of cells and control cellular reactive oxygen species by reducing the disulfides into thiol groups. Thioredoxin interacting protein (Txnip/thioredoxin binding protein-2/vitamin D3 upregulated protein) directly binds to Trx1 and Trx2 (Trx) and inhibit the reducing activity of Trx through their disulfide exchange. Recent studies have revealed that Trx1 and Txnip are involved in some critical redox-dependent signal pathways including NLRP-3 inflammasome activation in a redox-dependent manner. Therefore, Trx/Txnip, a redox-sensitive signaling complex is a regulator of cellular redox status and has emerged as a key component in the link between redox regulation and the pathogenesis of diseases. Here, we review the novel functional concept of the redox-related protein complex, named “Redoxisome,” consisting of Trx/Txnip, as a critical regulator for intra- and extra-cellular redox signaling, involved in the pathogenesis of various diseases such as cancer, autoimmune disease, and diabetes.
Introduction
The thioredoxin (Trx) system, composed of NADPH, thioredoxin reductase (TrxR), and Trx, is a key antioxidant system that protects cells from oxidative stress through its disulfide reductase activity. Trx1 and Trx2 are mainly localized in the cytoplasm and the mitochondria respectively, suggestive of their specific roles at different cellular compartments (Box 1). Trx are highly conserved in many organisms ranging from bacterial organisms to plants and mammals, indicating that the Trx system is a cellular system, essential for cellular survival and function. Recent studies have shown that an important Trx binding protein, thioredoxin interacting protein [Txnip/thioredoxin binding protein-2 (TBP-2)/vitamin D3 upregulated protein (VDUP1)] has the reciprocal function with Trx in the pathogenesis of disease such as autoimmune disease, cancer, and diabetes (1–3). Decades of research have implicated Trx/Txnip regulation as an attractive therapeutic target. Here we review the novel protein signaling complex we call “Redoxisome” as a redox-related signal transducer to highlight the novel therapeutic approach by Trx/Txnip.
Box 1. Localization of Trx and Txnip, predicted existence of redoxisome in nucleus, cytosol, mitochondria, cellular membrane, and extra-cellular space.
It is well known that Trx1 is localized in the cytosol, plasma membrane (PM), and nucleus as well as the extra-cellular space. Since Trx2 is localized in only mitochondria, the Trx/Txnip redoxisome system is mainly works as the Trx2/Txnip complex in mitochondria. It was found that this complex only occurs under the oxidative stress since Txnip is shuttled into mitochondria under stress and remains in the nucleus under the normal condition (4). Recently, it was found that Txnip is also located in PM (5). Poly-ADP-ribose polymerase 1 (Parp1) was found to be a binding protein for Txnip in the nucleus and the inhibition of Parp1 increases PM associated Txnip localization in human umbilical vein endothelial cells (HUVECs), suggesting that the translocation of Txnip from the nucleus to the PM is exclusively related to Parp1 inhibition. Since Txnip and Parp1 are both regulated by changes in cellular redox state in HUVECs, this novel PM associated Txnip could be associated with Trx1 and form a redoxisome system in the PM. Nuclear transport protein, importin-α was identified as a binding protein of Txnip, and the binding leads to the translocation of Txnip from the cytosol to the nucleus (6). Although it is still unknown whether Txnip is released into the extra-cellular space like Trx1, further studies about the localization specific Trx/Txnip, redoxisome system could give us a novel insight for a redox-dependent biological function in the cells.
Thioredoxin1 and Thioredoxin2
In mammalian cells there are two isoforms of Trx called as Thioredoxin1 (Trx1) and Thioredoxin2 (Trx2) (7). Trx1 is mainly located in cytosol but also translocate to the nucleus and can be secreted from cells under certain circumstances [circulatory thioredoxin function were reviewed by our recent paper (8)], whereas Trx2 is located only in mitochondria. Trx1 is a 12-kDa ubiquitous protein that has disulfide-reducing activity. Trx are characterized by the presence of three conserved prolines, with one located between the catalytic cysteine residues of the – Cys-Gly-Pro-Cys – motif. Two cysteine residues (Cys-32 and -35) of the active site – Cys-Gly-Pro-Cys – are responsible for this reducing activity. This proline is the key residue that determines the reducing power of Trx and replacing it by a serine or a threonine has a dramatic effect on the redox and stability properties of the protein (9–13). Trx1 was originally identified as a hydrogen donor for ribonucleotide reductase in Escherichia coli (14). We have identified human Trx1 as an adult T cell leukemia-derived factor (ADF) from the supernatant of human T-cell leukemia type-1 (HTLV-1) infected T cell line, which was initially defined as autoimmune lymphokine and IL-2 receptor-inducing factor (15). In line with its function to respond to oxidative stresses, Trx1 expression is induced by variety of physiochemical stimuli, including virus infection, mitogen, UV-irradiation, hydrogen peroxide, ischemia reperfusion, which we have broadly reviewed (1, 3, 8, 16). The crystal structures of Trx1 in both oxidized and reduced states have been resolved and revealed that Trx1 has a basic Trx-fold (consisting of four β2-strands surrounded by three α2-helices) with additional α2-helices and β2-strands at the N-terminus (10, 17). Natural metabolic or endocrine substances including hemin, estrogen, prostaglandins, sulforaphane, and cAMP can also induce the expression and secretion of Trx1. A series of stress-responsive elements in the promoter region have been identified, including the oxidative stress response element (ORE), antioxidant responsive element (ARE), cAMP responsive element (CRE), xenobiotics responsive element (XRE), and Sp-1. Trx1 is also induced by fragrant unsaturated aldehydes from edible plants, through their ARE in the promoter region, meaning that Trx1 may be beneficial for protection against oxidative stress-induced cellular damage (18). Trx1-knockout mice are lethal due to early development and morphogenesis failure of the mouse embryo (19), whereas Trx1-transgenic (Trx1 Tg) mice are more resistant to oxidative stress with longer life span compared with wild type mice (20). These results suggest that Trx1 is an essential molecule for cellular and organismal survival. Decades of research have shown that Trx1 binds and modulates together proteins such as Nuclear Factor-κB (NF-κB), p53, hypoxia inducible factor-1 (HIF-1), Forkhead box class O1 (Foxo1), glucocorticoid receptor (GR), and Estrogen Receptor (ER) by a thiol-disulfide exchange reaction (3, 21). Trx1 binding protein that we identified in 1999 and named TBP-2, now commonly called as Txnip is a unique Trx binding protein that has a role as an endogenous inhibitor of Trx, since Txnip binding to Trx inhibits. Their protein reducing activity and/or Trx expression (22). This finding has implicated that redox-sensitive proteins and related cellular processes such as metabolism, proliferation, and inflammation could be regulated by Trx/Txnip signaling.
Thioredoxin Interacting Protein (TBP-2/VDUP1)
Thioredoxin interacting protein (TBP-2/VDUP1) was originally cloned as a vitamin D3 target gene in HL-60 cells (23) (named as VDUP1). This molecule has emerged as a key component of cellular redox regulation since it was identified as binding partner of Trx and suggested to be an endogenous Trx inhibitor (22) (named as TBP-2). It has been known that the two Txnip cysteins are important for thioredoxin binding through a disulfide exchange reaction between oxidized Txnip and reduced Trx (24). This clear evidence suggests that the Trx-Txnip complex is important for redox-dependent cell function. Interestingly, Txnip is a member of the α-arrestin protein family (Arrdc1–5 and Txnip) containing two characteristic arrestin-like domains with PxxP sequence, which is a known binding motif for SH3-domains containing proteins, and PPxY sequence that is know binding motifs for WW-domain (2, 24–27). Since Txnip has a specific arrestin-like domain, which is responsible for protein–protein binding, many interacting protein for Txnip have been identified such as importin-α, transcriptional co-repressors SMRT-mSin3-HDAC (histone deacetylase), Jab1, E3 ubiquitinligase itch, Mybbp1a, and NOD-like receptor Protein-3 (NLRP3) as well as Trx (6, 28–32). These finding raises the possibility that Txnip may have a role for scaffolding in a signaling complex. Interestingly, genetic mapping identified a nonsense mutation in the Txnip gene as the cause for the phenotype in the Hcb-19 mutant mouse, which resembles familial combined hyperlipidemia (FCHL) (named as Txnip) (33). The mutation causes the truncation of Txnip in a critical region, which was reported to mediate Txnip’s binding to Trx1. Moreover Hcb-19 mice have decreased CO2 production but increased ketone body synthesis, suggesting that altered redox status may have a role in lipid metabolism such as citric-acid cycle and fatty acid utilization (34).
We reported that disruption of Txnip (Txnip KO) by gene targeting in mice causes predisposition to death with severe bleeding, hypoglycemia, hyperinsulinemia, and liver steatosis during fasting (35). Txnip gene expression is induced in fasting, and the key transcription regulator peroxisome proliferator activated receptor-alpha (PPAR-α) and sterol response element binging protein (SREBP) signaling are dysregulated in the liver of Txnip KO during the feeding-fasting nutritional transition (36). Txnip expression is widely regulated by nutritional status, signal, and enzyme such as feeding-fasting, obesity, high glucose, amino acids, nuclear receptor signal (PPARs, VDR, GR), and AMPK (23, 26, 28, 36–41). These results clearly suggest that Txnip is an important molecule that regulates glucose and lipid homeostasis.
A critical role of Trx and Txnip in inflammation, cancer progression, and diabetes is mentioned in a later part of this review.
Thioredoxin1/Txnip, Redoxisome, a Redox-Related Signal Complex
The Trx system plays an important role in maintaining a reduced environment in the cell. We first identified Txnip/TBP-2/VDUP1 as an endogenous Trx1 binding and inhibiting protein (22). Interestingly, Txnip binds to reduced Trx1 but not to oxidized Trx1 nor to mutant Trx1, in which two redox active cysteine residues are substituted by serine (22). Since the disulfide exchange reaction between oxidized Txnip and reduced Trx1 [Txnip and Trxs form a stable disulfide-linked complex (24)] is known as the essential event for the interaction between Txnip and Trx1, two Txnip cysteines are important for Trx1 binding (Figure 1). These cysteines are not conserved in the broader family of arrestin domain-containing proteins, therefore, the Trx1 binding property of Txnip is unique (24). Thus, the catalytic center of Trx1 seems to be important for the interaction. This interaction is important for cellular redox regulation since the protein reducing activity of Trx1 is actually inhibited by Txnip interaction (22, 24). In COS-7 and HEK293 cells transiently transfected with Txnip expression vector, decreased of insulin reducing activity of Trx1 and diminished expression of Trx1 was observed (22). In addition, treatment of HL-60 cells with 1α, 25-dihydroxyvitamin D3 caused an increase in Txnip expression and down-regulation of the expression and the reducing activity of Trx1. These results suggest that Txnip serves as a negative regulator of the biological function and expression of Trx1 by direct interaction and provides insight into the redox-dependent signal complex (Figure 1). We would like to propose the concept of “Redoxisome” that the signaling complex, composed with Trx and Txnip as a redox-dependent signal complexes, “Redoxisome” since it seems this signal complex could be key regulatory mechanism in multiple condition and diseases (Figure 2).
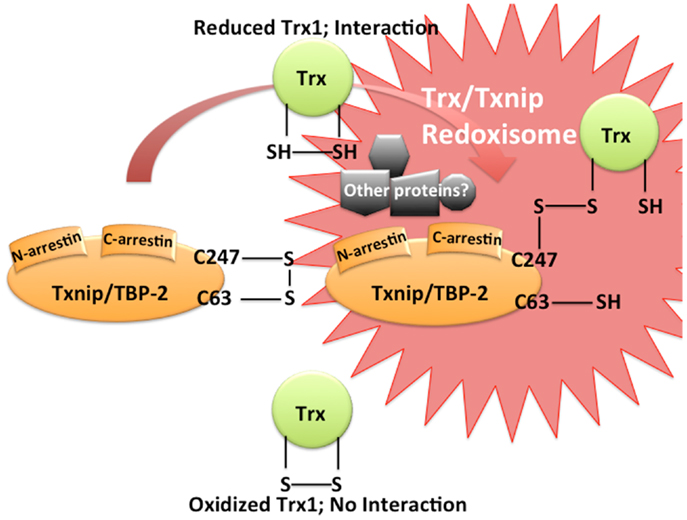
Figure 1. Trx/Txnip signaling complex as redoxisome. Txnip contains an intramolecular disulfide band between Cys-63 and -247 that allows efficient interaction with Trx. Txnip forms disulfide bond with reduced TRX by disulfide exchange, making a stable Trx mixed disulfide.
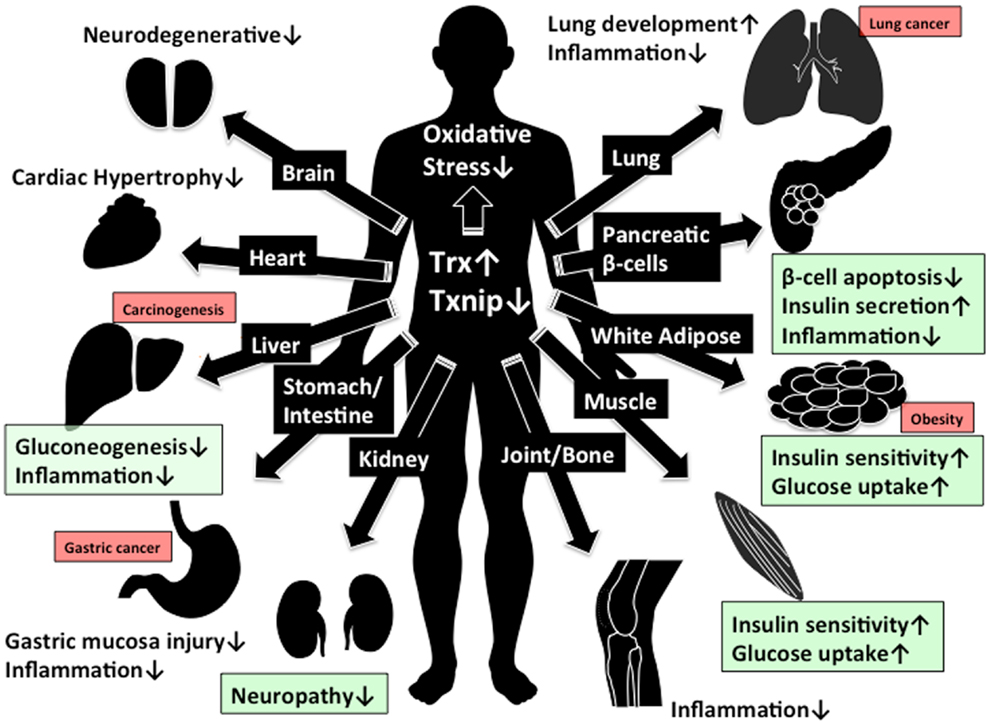
Figure 2. Beneficial effect of Trx1/Txnip signaling for clinical aspect. The beneficial effect by increasing of Trx1 and decreasing of Txnip expression are shown. Green box indicate the beneficial effect in diabetes while red box indicate the concern about adverse effect in cancer development by reduced Txnip expression.
Thioredoxin2/Txnip, Redoxisome in Mitochondria
The Trx/Txnip, redoxisome system also exists in mitochondria (42–44). The major difference between the cytosolic Trx system and the mitochondrial Trx system is that cytosolic Trx system consists of Trx1, TrxR, and Peroxiredoxin (Prx) while that of mitochondria consists of Trx2, TrxR2, and Prx3 (45, 46). Interestingly, Txnip interacts with both cytosolic Trx1 and mitochondrial Trx2 (4, 47), meaning that the Trx/Txnip, redoxisome system works in both cytosol and mitochondria. Txnip can translocate to the mitochondria, where it binds to oxidized Trx2 leading to mitochondrial dysfunction (4). Since Trx2 bind to Apoptosis signal regulating kinase 1 (Ask1), a major mitogen-activated protein kinase kinase kinase (MAPKKK) and inhibits Ask1 phosphorylation and activation, increased binding of Txnip to Trx2 reduces the interaction between Trx2 and Ask1 and induces Ask1 activation for apoptosis (4). It is also reported that Txnip translocation increase reactive oxygen species (ROS) accumulation in mitochondria and leads to activation of the NLRP3 inflammasome (47). However the function of Trx2 in regulating the inflammasome has not been studied to determine whether it is an essential component of NLRP3 activation in mitochondria.
Thioredoxin1/Txnip, Redoxisome, Critical for Pathogenesis of Type 1 and 2 Diabetes
Diabetes is characterized by high blood glucose levels, as results of insufficient insulin due to reduced insulin secretion and/or insulin sensitivity for the body’s required. Type 1 Diabetes (T1DM) is an autoimmune disease that results in β-cells destruction. It usually presents in childhood, accounts for 5–10% of all diabetes, and is associated with the presence of islet-cell antibodies, and patients require lifelong insulin treatment. While, Type 2 Diabetes (T2DM), the most common form of the disease, is characterized by defects in both insulin secretion from pancreatic β-cells and insulin sensitivity in peripheral tissues such as skeletal muscle, liver, and fat. T2DM is influenced by life style factors such as age, pregnancy, and obesity, but also has a strong genetic component (48). Despite the differential mechanism for the pathogenesis of T1DM and T2DM, oxidative stress is commonly related to the pathogenesis of the disease (49). It has been known that low levels of cellular ROS are required for cellular signaling, such as optimal tyrosine phosphorylation-dependent signaling in vitro (49–51), while chronic excessive generation of ROS aggravate insulin sensitivity in skeletal muscle and disrupt β-cell function and survival (52), suggesting that an optimal balance of cellular redox regulation is critical for the pathogenesis of both T1DM and T2DM.
Thioredoxin1-transgenic mice on a C57BL/6J background, in which human Trx1 is systemically overexpressed under control of the β-actin promoter, are more resistant to various oxidative stresses than control mice (20, 53–57). Trx1 Tg mice have more resistance to develop both T1DM and T2DM in mice (55–58). In non-obese diabetic (NOD) mice (T1DM model) or streptozocin (STZ) induced T1DM model mice, β-cell-specific overexpression of Trx1 markedly reduces the incidence of diabetes without improving insulin secretory capacity, insulin content, or the development of insulitis compared with those of littermate controls (55).
Recently it was reported that in an animal model of T2DM, obese diabetic db/db mice, β-cell-specific Trx1 overexpression suppresses progressive β-cell failure (56). Trx1 Tg db/db mice exhibit significantly lower blood glucose levels and higher plasma insulin levels compared with littermate controls (56). These results suggest that Trx1 has a protective effect on reducing oxidative stress inducing failure in T1DM and T2DM.
The endogenous Trx1 inhibitor, Txnip, was implicated as a redox rheostat to control Trx1 activity and expression. Recent studies suggested that Txnip expression is increased in skeletal muscle of human T2DM and impaired glucose tolerance (IGT) patients (59, 60). It was also reported that a genetic variation of the Txnip gene is associated with hypertriglyceridemia and increased diastolic blood pressure (61). Interestingly, Txnip expression is induced by high glucose conditions, while it is reduced by insulin (59). We reported that Txnip is more abundantly expressed in pancreatic islets, skeletal muscle, adipose, and kidney of leptin deficient ob/ob mice (T2DM model) compared with WT control mice and Txnip deletion in ob/ob mice (T2DM model) dramatically improves insulin resistance in skeletal muscle and β-cell insulin secretion function and survival (28). Piled evidence suggested that Txnip induced β-cell apoptosis under various kinds of stresses such as STZ treatment (T1DM model) (62, 63), high glucose (28, 29, 64, 65), ER-stress (66–68), dexamethasone/glucocorticoid (69), and inflammation/cytokine (29, 62), while it has been reported that Txnip aggravates hepatic glucose production (70) and insulin sensitivity in skeletal muscle (28, 71) and adipose tissues (72, 73). These evidences suggest that Trx1 and Txnip have an antagonistic function in progression of both T1DM and T2DM.
Thioredoxin1/Txnip, Redoxisome, a Novel Regulator of NLRP3 Inflammasome and Inflammation
Human Trx1 is originally identified as an ADF (15, 74). Numerous evidences suggest that Trx1 is an anti-inflammatory molecule in both an intracellular and extra-cellular (8, 75–79) environment. Interestingly Trx is highly expressed in human T cell leukemia virus type-1 (HTLV-1)-transformed cell lines (ATL model cell line), whereas the Txnip expression is lost in HTLV-I-positive, interleukin-2-independent T cell lines but not in HTLV-I-negative T cell lines (37, 80, 81), suggesting a role for Trx1/Txnip in virus infection, prevention, and inflammation. Indeed, recent works revealed that Txnip has important role in inflammation in response to excessive nutrition, oxidative stress, and lipopolysaccharide (LPS) stimulation (29, 66–68, 82–85).
Recent publications suggest a physical interaction between Txnip and NLRP3, a key component of NLRP3 inflammasome (29, 66, 67, 82, 83). The inflammasome is the multiprotein complex that controls the activation of caspase-1 in the innate immune system that result in maturation of IL-1β. ROS is the major activator of the NLRP3 inflammasome. The physical interaction between Txnip and NLRP3 may explain the inflammasome activation in a ROS-sensitive manner (29) especially in macrophages in pancreatic islets (29, 67). The study, performed by Zhou et al. suggested that under the unstressed condition, Txnip is bound to Trx1 and NLRP-3 inflammasome is in active because of an absence of Txnip interaction with NLRP3, while under the oxidative stress condition, ROS generation facilitates Trx1-Txnip dissociation, thus increasing NLRP3-Txnip interaction. However there are some conflicting reports. Although Koenen et al. demonstrate that hyperglycemia activates caspase-1 and Txnip, glucose-induced activation of Txnip mediates an increase of IL-1β mRNA and intracellular pro-IL-1β rather than IL-1β processing (73). Furthermore, our collaborator Masters et al. and we found no significant difference in IL-1β secretion in response to inflammasome activators in bone marrow derived macrophages of Txnip KO mice (86). These evidences imply that the redox-dependent NLRP3 inflammasome activation is performed not only by Txnip-NLRP3 direct interaction but also by other mechanisms regulated by Trx1/Txnip with other participants for redoxisome signaling. Thioredoxin reductase (TrxR) might be involved in redox-sensitive inflammasome activation through Trx/Txnip, redoxisome signal since it has reported that under the absence of TrxR condition, Trx1 is able to recycle substrates at the expense of an alternative electron donor that is required to be oxidized form of Trx1 (87). It is possible there is an unknown potential redoxisome signal including TrxR, Trx1, and Txnip that lead to the activation of NLRP3 inflammasome.
Although increased production of ROS is often though to be implicated in the activation of inflammation, redox-related signal molecules are involved in NLRP3 inflammasome activation in a ROS-independent manner. It has reported that cells from patients with chronic granulomatous disease (CGD), characterized by an incapacity of phagocytosis to produce ROS due to a deficiency in the NADPH oxidase system produce more inflammatory cytokines including IL-1β (88). Since TrxR is the mediator of the NADPH oxidase system, TrxR may link redox-related molecules such as Trx1 and Txnip to NLRP3 inflammasome activation without ROS signaling. Further studies addressing ROS-independent activation of NLRP3 inflammasome will be required to understand the contribution of Trx1/Txnip to NLRP3 inflammasome activation.
Cigarette smoke-driven inflammatory airway disease such as chronic obstructive pulmonary disease (COPD) is aggravated by P2 × 7/inflammasome pathway (89). We reported that Trx1 Tg mice are resistant to COPD induced by cigarette smoke (76, 90, 91). It would be another interesting model for the redoxisome to investigate whether the cigarette smoke-induced inflammation is also regulated by the ratio of Trx1-Txnip interaction and NLRP3-Txnip interaction. The association of Txnip with Trx1 and NLRP3 indicates that Txnip plays a major role in the convergence of multiple signaling pathways that contribute to oxidative stress-related disorders.
Collectively, although Trx1/Txnip is likely involved in the activation of NLRP3 inflammasome, the mechanistic insight is still unclear. Therefore the novel concept of redoxisome consisting of Trx1/Txnip and other redox-related molecules could give us novel insight for the currently unknown mechanism of NLRP3 inflammasome activation in ROS-dependent and-independent manner. Future experiments will have to be performed to establish if the Trx1/Txnip redoxisome system contributes as redox regulatory system to NLRP3 inflammasome activation.
Thioredoxin1/Txnip, Redoxisome, Cancer Progression, and Other Disease
Txnip overexpression arrests the growth of cells at G0/G1 phase (92), and induces apoptosis in response to various stresses (30, 38, 41, 64, 93). Txnip is reported to inhibit cell cycles by stabilizing p27 via binding to Jab1 (31), and by repressing transcriptional activities via interaction with HDAC1 (94). Moreover Txnip is down-regulated in various human cancer cells and many reports have shown that the down-regulation of Txnip contributes the malignancy of cancer (2). In HTLV-1-infected T cells, the expression level of Txnip is associated with responsiveness to IL-2-dependent growth, and epigenetic silencing of Txnip results in loss of responsiveness to IL-2 (81). In IL-2-independent stage, Txnip silencing loses the sensitivity against glucocorticoid-induced cell death (38). In in vivo studies, Txnip has been shown to be a suppressor of the incidence and progression of cancer. The Hcb-19 mice strain with a spontaneous mutation of Txnip gene and Txnip KO mice show the higher incidence of hepatocellular carcinoma (95, 96). Also the Txnip KO mice show earlier onset of N-butyl-N-(4-hydroxybutyl) nitrosamine (BBN)-induced bladder carcinoma (97). Moreover, in clinical studies, Txnip expression levels decrease following the progression of cancer stages or malignancy in gastric cancer, melanoma, pheochromocytoma, and bladder cancer (97, 98). These results collectively support that Txnip contributes to controlling the malignancy of cancer. It has been also reported that the deficiency of Txnip promotes TNF-α-induced NF-κB activity, that Txnip inhibits mTOR activity by binding Redd1 (99), and that Txnip deficiency enhances the phosphorylation of Akt in response to insulin (28, 72).
Txnip deficiency also enhances phosphorylation of signal transducers. Regarding the relationship between Txnip and cell signaling, phosphorylation of ERK is enhanced in Txnip KO mice bladders during BBN-induced bladder carcinogenesis (97), and TGF-β signaling is enhanced via Smad2 phosphorylation under Txnip-KO or -knockdown condition (100). The loss of Txnip up-regulates a variety of transcriptional activities for several stimuli or ligands, so that deficiency of Txnip could contribute to integrate excessive biological responses and signals for cell survival, malignancy, and the tumorigenesis of cancer. Here, at the mention of the correlation between Txnip and Trx1, many reports show that Txnip interacts with Trx1. Overexpression of Txnip attenuates the reducing activity, and inhibits transcription of Trx1 resulting in increased ROS, which triggers cell cycle arrest or apoptosis (as referred above). The gene expression of Txnip vs. Trx1 shows a reciprocal pattern under the stimulation of vitamin D3 (22), PPAR-γ ligands (26), and suberoylanilide hydroxamic acid (HDAC inhibitor) (92) (Table 1). In the EGF-induced ERK1/2 phosphorylation, Txnip, and Trx1 apparently show reciprocal functions. As shown in several reports, Trx1 overexpression increases activation of ERK1/2 (101, 102) as well as Txnip deficiency. Interestingly, Txnip overexpression strongly reduced Trx1 expression and activity (22, 24, 103), while Txnip deletion has a minor effect of Trx1 expression and activity (104). In regards to TGF-β signaling, TGF-β-induced transcriptional activation is independent of the protein levels of Trx1 (100). These results give us the idea that the event of redox-dependent interactions between Trx1 and Txnip, resulting in the timely attenuation or enhancement of the functions in response to oxidative stresses, may be more biologically important. Further investigation is required to determine whether chronic oxidative stresses cause the inconsistent expression of Trx1 and Txnip by some feedback pathways. Future studies must unveil the physiological significance of the formation of the Trx1/Txnip complex.
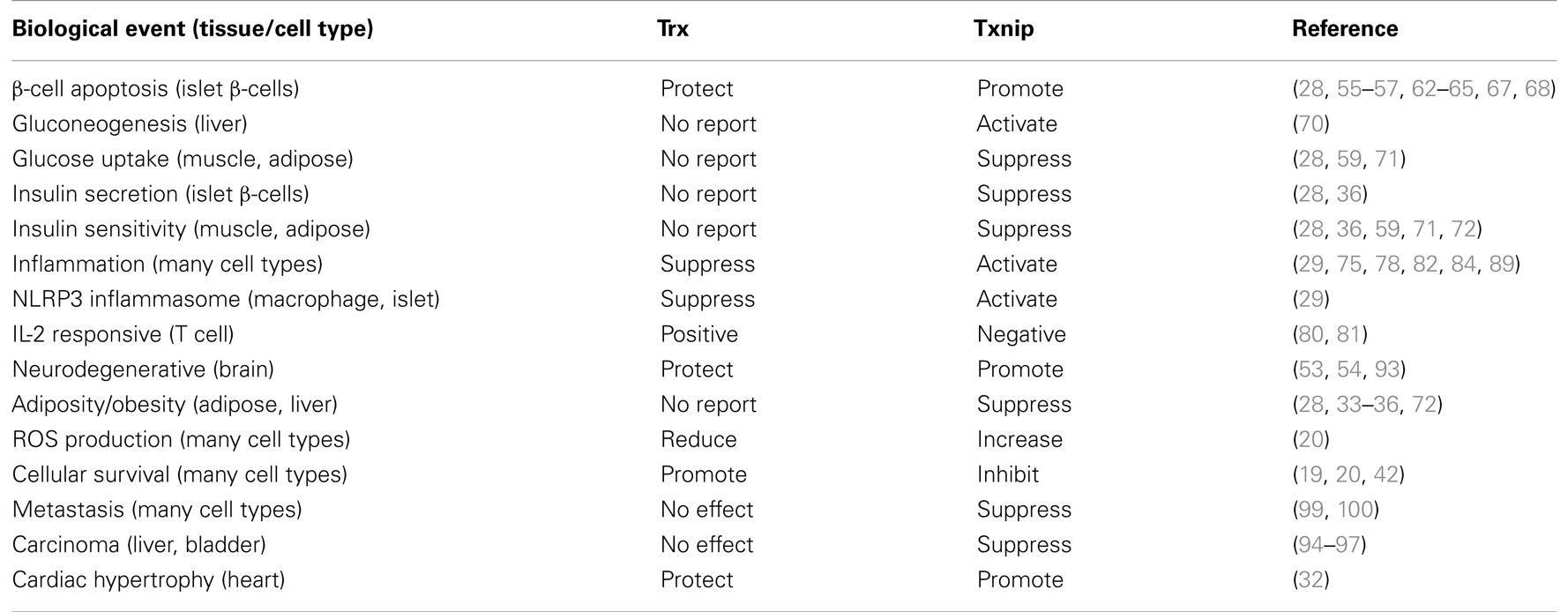
Table 1. Reciprocal function between Trx1 and Txnip.
A Perspective of Trx/Txnip of Clinical Work
The modulation of cellular redox regulation has emerged as a potential clinical approach for cancer, autoimmune disease, and diabetes. In this review, we proposed the Trx/Txnip signal complex is important for redox regulation and related disorders. The function of Trx is to reduce cellular ROS and related stresses, while Txnip has reciprocal function for Trx. This evidence gives us a novel therapeutic approach to control Trx/Txnip. Since the Trx/Txnip complex acts in various kinds of stresses, Trx/Txnip may form a protein complex with other proteins. In this review, we defined this redox-related signal complex as “Redoxisome” and suggest it may be a promising therapeutic target. Many reports suggest that increasing of Trx1 and decreasing of Txnip expression is beneficial for preventing hyper inflammation, neurodegeneration, and progression of diabetes, while there is the risk to increase the chance of incidents for gastric (105–107), lung (108), and liver (30, 95, 96) cancer, especially with reduction of Txnip expression (Figure 2). To eliminate unwanted side effect such as cancer development, the regulation of Trx1 and Txnip interaction might be a more effective therapeutic approach than the regulating their expression. Further elucidation of the mechanism for redox regulation and pathogenesis of diseases by the Trx/Txnip, redoxisome is required to understand how cells and the whole body integrate the various redox signals under the multiple diseases states and the future findings may give us promising redox regulated therapeutic approach by Trx/Txnip, redoxisome signaling (Figure 2).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Maryam Ahmadian for kind English editing. This study was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by the Program for Promotion of Fundamental Studies in Health Sciences of National Institute of Biomedical Innovation (NIBIO). This work was supported in part by the World Class University Grant R31-10010 through the Ewha Womans University. Eiji Yoshihara is supported by the Uehara memorial research foundation and the Japan Society for the Promotion of Science (JSPS) as a research fellow in abroad. We apologize for those references we could not include due to space limitations.
References
1. Watanabe R, Nakamura H, Masutani H, Yodoi J. Anti-oxidative, anti-cancer and anti-inflammatory actions by thioredoxin 1 and thioredoxin-binding protein-2. Pharmacol Ther (2010) 127:261–70. doi:10.1016/j.pharmthera.2010.04.004
2. Masutani H, Yoshihara E, Masaki S, Chen Z, Yodoi J. Thioredoxin binding protein (TBP)-2/Txnip and alpha-arrestin proteins in cancer and diabetes mellitus. J Clin Biochem Nutr (2012) 50:23–34. doi:10.3164/jcbn.11-36SR
3. Yoshihara E, Chen Z, Matsuo Y, Masutani H, Yodoi J. Thiol redox transitions by thioredoxin and thioredoxin-binding protein-2 in cell signaling. Methods Enzymol (2010) 474:67–82. doi:10.1016/S0076-6879(10)74005-2
4. Saxena G, Chen J, Shalev A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J Biol Chem (2010) 285:3997–4005. doi:10.1074/jbc.M109.034421
5. Spindel ON, Yan C, Berk BC. Thioredoxin-interacting protein mediates nuclear-to-plasma membrane communication: role in vascular endothelial growth factor 2 signaling. Arterioscler Thromb Vasc Biol (2012) 32:1264–70. doi:10.1161/ATVBAHA.111.244681
6. Nishinaka Y, Masutani H, Oka S, Matsuo Y, Yamaguchi Y, Nishio K, et al. Importin alpha1 (Rch1) mediates nuclear translocation of thioredoxin-binding protein-2/vitamin D(3)-up-regulated protein 1. J Biol Chem (2004) 279:37559–65. doi:10.1074/jbc.M405473200
7. Tanaka T, Nakamura H, Nishiyama A, Hosoi F, Masutani H, Wada H, et al. Redox regulation by thioredoxin superfamily; protection against oxidative stress and aging. Free Radic Res (2000) 33:851–5. doi:10.1080/10715760000301361
8. Matsuo Y, Yodoi J. Extracellular thioredoxin: a therapeutic tool to combat inflammation. Cytokine Growth Factor Rev (2013) 24(4):345–53. doi:10.1016/j.cytogfr.2013.01.001
9. Schultz LW, Chivers PT, Raines RT. The CXXC motif: crystal structure of an active-site variant of Escherichia coli thioredoxin. Acta Crystallogr D Biol Crystallogr (1999) 55:1533–8. doi:10.1107/S0907444999008756
10. Collet JF, Messens J. Structure, function, and mechanism of thioredoxin proteins. Antioxid Redox Signal (2010) 13:1205–16. doi:10.1089/ars.2010.3114
11. Chivers PT, Raines RT. General acid/base catalysis in the active site of Escherichia coli thioredoxin. Biochemistry (1997) 36:15810–6. doi:10.1021/bi971504l
12. Krause G, Lundstrom J, Barea JL, Pueyo de la Cuesta C, Holmgren A. Mimicking the active site of protein disulfide-isomerase by substitution of proline 34 in Escherichia coli thioredoxin. J Biol Chem (1991) 266:9494–500.
13. Mossner E, Huber-Wunderlich M, Glockshuber R. Characterization of Escherichia coli thioredoxin variants mimicking the active-sites of other thiol/disulfide oxidoreductases. Protein Sci (1998) 7:1233–44. doi:10.1002/pro.5560070519
14. Holmgren A. Thioredoxin. Annu Rev Biochem (1985) 54:237–71. doi:10.1146/annurev.bi.54.070185.001321
15. Tagaya Y, Maeda Y, Mitsui A, Kondo N, Matsui H, Hamuro J, et al. ATL-derived factor (ADF), an IL-2 receptor/Tac inducer homologous to thioredoxin; possible involvement of dithiol-reduction in the IL-2 receptor induction. EMBO J (1989) 8:757–64.
16. Masutani H, Ueda S, Yodoi J. The thioredoxin system in retroviral infection and apoptosis. Cell Death Differ (2005) 12(Suppl 1):991–8. doi:10.1038/sj.cdd.4401625
17. Fritz-Wolf K, Kehr S, Stumpf M, Rahlfs S, Becker K. Crystal structure of the human thioredoxin reductase-thioredoxin complex. Nat Commun (2011) 2:383. doi:10.1038/ncomms1382
18. Masutani H, Otsuki R, Yamaguchi Y, Takenaka M, Kanoh N, Takatera K, et al. Fragrant unsaturated aldehydes elicit activation of the Keap1/Nrf2 system leading to the upregulation of thioredoxin expression and protection against oxidative stress. Antioxid Redox Signal (2009) 11:949–62. doi:10.1089/ARS.2008.2292
19. Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, et al. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol (1996) 178:179–85. doi:10.1006/dbio.1996.0208
20. Mitsui A, Hamuro J, Nakamura H, Kondo N, Hirabayashi Y, Ishizaki-Koizumi S, et al. Overexpression of human thioredoxin in transgenic mice controls oxidative stress and life span. Antioxid Redox Signal (2002) 4:693–6. doi:10.1089/15230860260220201
21. Lillig CH, Holmgren A. Thioredoxin and related molecules – from biology to health and disease. Antioxid Redox Signal (2007) 9:25–47. doi:10.1089/ars.2007.9.25
22. Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, et al. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem (1999) 274:21645–50. doi:10.1074/jbc.274.31.21645
23. Chen KS, DeLuca HF. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim Biophys Acta (1994) 1219:26–32. doi:10.1016/0167-4781(94)90242-9
24. Patwari P, Higgins LJ, Chutkow WA, Yoshioka J, Lee RT. The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J Biol Chem (2006) 281:21884–91. doi:10.1074/jbc.M600427200
25. Patwari P, Chutkow WA, Cummings K, Verstraeten VL, Lammerding J, Schreiter ER, et al. Thioredoxin-independent regulation of metabolism by the alpha-arrestin proteins. J Biol Chem (2009) 284:24996–5003. doi:10.1074/jbc.M109.018093
26. Oka S, Masutani H, Liu W, Horita H, Wang D, Kizaka-Kondoh S, et al. Thioredoxin-binding protein-2-like inducible membrane protein is a novel vitamin D3 and peroxisome proliferator-activated receptor (PPAR)gamma ligand target protein that regulates PPARgamma signaling. Endocrinology (2006) 147:733–43. doi:10.1210/en.2005-0679
27. Alvarez CE. On the origins of arrestin and rhodopsin. BMC Evol Biol (2008) 8:222. doi:10.1186/1471-2148-8-222
28. Yoshihara E, Fujimoto S, Inagaki N, Okawa K, Masaki S, Yodoi J, et al. Disruption of TBP-2 ameliorates insulin sensitivity and secretion without affecting obesity. Nat Commun (2010) 1:127. doi:10.1038/ncomms1127
29. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11:136–40. doi:10.1038/ni.1831
30. Yamaguchi F, Takata M, Kamitori K, Nonaka M, Dong Y, Sui L, et al. Rare sugar D-allose induces specific up-regulation of TXNIP and subsequent G1 cell cycle arrest in hepatocellular carcinoma cells by stabilization of p27kip1. Int J Oncol (2008) 32:377–85.
31. Jeon JH, Lee KN, Hwang CY, Kwon KS, You KH, Choi I. Tumor suppressor VDUP1 increases p27(kip1) stability by inhibiting JAB1. Cancer Res (2005) 65:4485–9. doi:10.1158/0008-5472.CAN-04-2271
32. Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, et al. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell (2008) 133:978–93. doi:10.1016/j.cell.2008.04.041
33. Bodnar JS, Chatterjee A, Castellani LW, Ross DA, Ohmen J, Cavalcoli J, et al. Positional cloning of the combined hyperlipidemia gene Hyplip1. Nat Genet (2002) 30:110–6. doi:10.1038/ng811
34. Donnelly KL, Margosian MR, Sheth SS, Lusis AJ, Parks EJ. Increased lipogenesis and fatty acid reesterification contribute to hepatic triacylglycerol stores in hyperlipidemic Txnip-/- mice. J Nutr (2004) 134:1475–80.
35. Oka S, Liu W, Masutani H, Hirata H, Shinkai Y, Yamada S, et al. Impaired fatty acid utilization in thioredoxin binding protein-2 (TBP-2)-deficient mice: a unique animal model of Reye syndrome. FASEB J (2006) 20:121–3. doi:10.1096/fj.05-4439fje
36. Oka S, Yoshihara E, Bizen-Abe A, Liu W, Watanabe M, Yodoi J, et al. Thioredoxin binding protein-2/thioredoxin-interacting protein is a critical regulator of insulin secretion and peroxisome proliferator-activated receptor function. Endocrinology (2009) 150:1225–34. doi:10.1210/en.2008-0646
37. Chen Z, Yoshihara E, Son A, Matsuo Y, Masutani H, Sugie K, et al. Differential roles of annexin A1 (ANXA1/lipocortin-1/lipomodulin) and thioredoxin binding protein-2 (TBP-2/VDUP1/TXNIP) in glucocorticoid signaling of HTLV-I-transformed T cells. Immunol Lett (2010) 131:11–8. doi:10.1016/j.imlet.2010.04.003
38. Chen Z, Lopez-Ramos DA, Yoshihara E, Maeda Y, Masutani H, Sugie K, et al. Thioredoxin-binding protein-2 (TBP-2/VDUP1/TXNIP) regulates T-cell sensitivity to glucocorticoid during HTLV-I-induced transformation. Leukemia (2011) 25:440–8. doi:10.1038/leu.2010.286
39. Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell (2013) 49:1167–75. doi:10.1016/j.molcel.2013.01.035
40. Liu M, Ling X, Xiong Y, Xu D. Molecular characterization of differentially expressed TXNIP gene and its association with porcine carcass traits. Mol Biol Rep (2012) 39:10439–46. doi:10.1007/s11033-012-1923-2
41. Wang Z, Rong YP, Malone MH, Davis MC, Zhong F, Distelhorst CW. Thioredoxin-interacting protein (txnip) is a glucocorticoid-regulated primary response gene involved in mediating glucocorticoid-induced apoptosis. Oncogene (2006) 25:1903–13. doi:10.1038/sj.onc.1209218
42. Tanaka T, Hosoi F, Yamaguchi-Iwai Y, Nakamura H, Masutani H, Ueda S, et al. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J (2002) 21:1695–703. doi:10.1093/emboj/21.7.1695
43. Wang D, Masutani H, Yodoi J. Are the properties of mitochondrial membranes redox regulated? IUBMB Life (2006) 58:670–3. doi:10.1080/15216540600854957
44. Wang D, Masutani H, Oka S, Tanaka T, Yamaguchi-Iwai Y, Nakamura H, et al. Control of mitochondrial outer membrane permeabilization and Bcl-xL levels by thioredoxin 2 in DT40 cells. J Biol Chem (2006) 281:7384–91. doi:10.1074/jbc.M509876200
45. Spyrou G, Enmark E, Miranda-Vizuete A, Gustafsson J. Cloning and expression of a novel mammalian thioredoxin. J Biol Chem (1997) 272:2936–41. doi:10.1074/jbc.272.5.2936
46. Nonn L, Berggren M, Powis G. Increased expression of mitochondrial peroxiredoxin-3 (thioredoxin peroxidase-2) protects cancer cells against hypoxia and drug-induced hydrogen peroxide-dependent apoptosis. Mol Cancer Res (2003) 1:682–9.
47. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi:10.1038/nature09663
48. Ashcroft FM, Rorsman P. Diabetes mellitus and the beta cell: the last ten years. Cell (2012) 148:1160–71. doi:10.1016/j.cell.2012.02.010
49. Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab (2009) 10:260–72. doi:10.1016/j.cmet.2009.08.009
50. Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science (2006) 312:1882–3. doi:10.1126/science.1130481
51. Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol (2006) 7:833–46. doi:10.1038/nrm2039
52. Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med (2011) 50:567–75. doi:10.1016/j.freeradbiomed.2010.12.006
53. Takagi Y, Mitsui A, Nishiyama A, Nozaki K, Sono H, Gon Y, et al. Overexpression of thioredoxin in transgenic mice attenuates focal ischemic brain damage. Proc Natl Acad Sci U S A (1999) 96:4131–6. doi:10.1073/pnas.96.7.4131
54. Takagi Y, Hattori I, Nozaki K, Mitsui A, Ishikawa M, Hashimoto N, et al. Excitotoxic hippocampal injury is attenuated in thioredoxin transgenic mice. J Cereb Blood Flow Metab (2000) 20:829–33. doi:10.1097/00004647-200005000-00009
55. Hotta M, Tashiro F, Ikegami H, Niwa H, Ogihara T, Yodoi J, et al. Pancreatic beta cell-specific expression of thioredoxin, an antioxidative and antiapoptotic protein, prevents autoimmune and streptozotocin-induced diabetes. J Exp Med (1998) 188:1445–51. doi:10.1084/jem.188.8.1445
56. Yamamoto M, Yamato E, Toyoda S, Tashiro F, Ikegami H, Yodoi J, et al. Transgenic expression of antioxidant protein thioredoxin in pancreatic beta cells prevents progression of type 2 diabetes mellitus. Antioxid Redox Signal (2008) 10:43–9. doi:10.1089/ars.2007.1586
57. Hamada Y, Fujii H, Kitazawa R, Yodoi J, Kitazawa S, Fukagawa M. Thioredoxin-1 overexpression in transgenic mice attenuates streptozotocin-induced diabetic osteopenia: a novel role of oxidative stress and therapeutic implications. Bone (2009) 44:936–41. doi:10.1016/j.bone.2008.12.011
58. Samuel SM, Thirunavukkarasu M, Penumathsa SV, Koneru S, Zhan L, Maulik G, et al. Thioredoxin-1 gene therapy enhances angiogenic signaling and reduces ventricular remodeling in infarcted myocardium of diabetic rats. Circulation (2010) 121:1244–55. doi:10.1161/CIRCULATIONAHA.109.872481
59. Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med (2007) 4:e158. doi:10.1371/journal.pmed.0040158
60. Muoio DM. TXNIP links redox circuitry to glucose control. Cell Metab (2007) 5:412–4. doi:10.1016/j.cmet.2007.05.011
61. van Greevenbroek MM, Vermeulen VM, Feskens EJ, Evelo CT, Kruijshoop M, Hoebee B, et al. Genetic variation in thioredoxin interacting protein (TXNIP) is associated with hypertriglyceridaemia and blood pressure in diabetes mellitus. Diabet Med (2007) 24:498–504. doi:10.1111/j.1464-5491.2007.02109.x
62. Masson E, Koren S, Razik F, Goldberg H, Kwan EP, Sheu L, et al. High beta-cell mass prevents streptozotocin-induced diabetes in thioredoxin-interacting protein-deficient mice. Am J Physiol Endocrinol Metabol (2009) 296:E1251–61. doi:10.1152/ajpendo.90619.2008
63. Xu G, Chen J, Jing G, Shalev A. Preventing beta-cell loss and diabetes with calcium channel blockers. Diabetes (2012) 61:848–56. doi:10.2337/db11-0955
64. Shalev A. Lack of TXNIP protects beta-cells against glucotoxicity. Biochem Soc Trans (2008) 36:963–5. doi:10.1042/BST0360963
65. Chen J, Fontes G, Saxena G, Poitout V, Shalev A. Lack of TXNIP protects against mitochondria-mediated apoptosis but not against fatty acid-induced ER stress-mediated beta-cell death. Diabetes (2010) 59:440–7. doi:10.2337/db09-0949
66. Anthony TG, Wek RC. TXNIP switches tracks toward a terminal UPR. Cell Metab (2012) 16:135–7. doi:10.1016/j.cmet.2012.07.012
67. Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab (2012) 16:265–73. doi:10.1016/j.cmet.2012.07.005
68. Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab (2012) 16:250–64. doi:10.1016/j.cmet.2012.07.007
69. Reich E, Tamary A, Sionov RV, Melloul D. Involvement of thioredoxin-interacting protein (TXNIP) in glucocorticoid-mediated beta cell death. Diabetologia (2012) 55:1048–57. doi:10.1007/s00125-011-2422-z
70. Chutkow WA, Patwari P, Yoshioka J, Lee RT. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J Biol Chem (2008) 283:2397–406. doi:10.1074/jbc.M708169200
71. Hui ST, Andres AM, Miller AK, Spann NJ, Potter DW, Post NM, et al. Txnip balances metabolic and growth signaling via PTEN disulfide reduction. Proc Natl Acad Sci U S A (2008) 105:3921–6. doi:10.1073/pnas.0800293105
72. Chutkow WA, Birkenfeld AL, Brown JD, Lee HY, Frederick DW, Yoshioka J, et al. Deletion of the alpha-arrestin protein Txnip in mice promotes adiposity and adipogenesis while preserving insulin sensitivity. Diabetes (2010) 59:1424–34. doi:10.2337/db09-1212
73. Koenen TB, Stienstra R, van Tits LJ, de Graaf J, Stalenhoef AF, Joosten LA, et al. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1beta transcription in human adipose tissue. Diabetes (2011) 60:517–24. doi:10.2337/db10-0266
74. Yodoi J, Takatsuki K, Masuda T. Letter: two cases of T-cell chronic lymphocytic leukemia in Japan. N Engl J Med (1974) 290:572–3. doi:10.1056/NEJM197403072901019
75. Nakamura T, Nakamura H, Hoshino T, Ueda S, Wada H, Yodoi J. Redox regulation of lung inflammation by thioredoxin. Antioxid Redox Signal (2005) 7:60–71. doi:10.1089/ars.2005.7.60
76. Sato A, Hara T, Nakamura H, Kato N, Hoshino Y, Kondo N, et al. Thioredoxin-1 suppresses systemic inflammatory responses against cigarette smoking. Antioxid Redox Signal (2006) 8:1891–6. doi:10.1089/ars.2006.8.1891
77. Ohashi S, Nishio A, Nakamura H, Kido M, Ueno S, Uza N, et al. Protective roles of redox-active protein thioredoxin-1 for severe acute pancreatitis. Am J Physiol Gastrointest Liver Physiol (2006) 290:G772–81. doi:10.1152/ajpgi.00425.2005
78. Imaoka H, Hoshino T, Takei S, Sakazaki Y, Kinoshita T, Okamoto M, et al. Effects of thioredoxin on established airway remodeling in a chronic antigen exposure asthma model. Biochem Biophys Res Commun (2007) 360:525–30. doi:10.1016/j.bbrc.2007.06.019
79. Ono R, Masaki T, Dien S, Yu X, Fukunaga A, Yodoi J, et al. Suppressive effect of administration of human recombinant thioredoxin on ultraviolet light-induced inflammation and apoptosis of murine skin. Bioengineered (2013) 4(4):254–7. doi:10.1111/j.1346-8138.2012.01566.x
80. Nishinaka Y, Nishiyama A, Masutani H, Oka S, Ahsan KM, Nakayama Y, et al. Loss of thioredoxin-binding protein-2/vitamin D3 up-regulated protein 1 in human T-cell leukemia virus type I-dependent T-cell transformation: implications for adult T-cell leukemia leukemogenesis. Cancer Res (2004) 64:1287–92. doi:10.1158/0008-5472.CAN-03-0908
81. Ahsan MK, Masutani H, Yamaguchi Y, Kim YC, Nosaka K, Matsuoka M, et al. Loss of interleukin-2-dependency in HTLV-I-infected T cells on gene silencing of thioredoxin-binding protein-2. Oncogene (2006) 25:2181–91. doi:10.1038/sj.onc.1209256
82. Wang W, Wang C, Ding XQ, Pan Y, Gu TT, Wang MX, et al. Quercetin and allopurinol reduce liver thioredoxin-interacting protein to improve inflammation and lipid accumulation in diabetic rats. Br J Pharmacol (2013) 169(6):1352–71. doi:10.1111/bph.12226
83. Devi TS, Lee I, Hüttemann M, Kumar A, Nantwi KD, Singh LP. TXNIP links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: implications for diabetic retinopathy. Exp Diabetes Res (2012) 2012:438238. doi:10.1155/2012/438238
84. Perrone L, Devi TS, Hosoya K, Terasaki T, Singh LP. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J Cell Physiol (2009) 221:262–72. doi:10.1002/jcp.21852
85. Oka S, Liu W, Yoshihara E, Ahsan MK, Ramos DA, Son A, et al. Thioredoxin binding protein-2 mediates metabolic adaptation in response to lipopolysaccharide in vivo. Crit Care Med (2010) 38:2345–51. doi:10.1097/CCM.0b013e3181f85b2a
86. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol (2010) 11:897–904. doi:10.1038/ni.1935
87. García-Santamarina S, Boronat S, Calvo IA, Rodríguez-Gabriel M, Ayté J, Molina H, et al. Is oxidized thioredoxin a major trigger for cysteine oxidation? Clues from a redox proteomics approach. Antioxid Redox Signal (2013) 18:1549–56. doi:10.1089/ars.2012.5037
88. Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood (2010) 116:1570–3. doi:10.1182/blood-2010-01-264218
89. Eltom S, Stevenson CS, Rastrick J, Dale N, Raemdonck K, Wong S, et al. P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLoS One (2011) 6:e24097. doi:10.1371/journal.pone.0024097
90. Sato A, Hoshino Y, Hara T, Muro S, Nakamura H, Mishima M, et al. Thioredoxin-1 ameliorates cigarette smoke-induced lung inflammation and emphysema in mice. J Pharmacol Exp Ther (2008) 325:380–8. doi:10.1124/jpet.107.134007
91. Huang YL, Chuang CY, Sung FC, Chen CY. Thioredoxin overexpression modulates remodeling factors in stress responses to cigarette smoke. J Toxicol Environ Health A (2008) 71:1490–8. doi:10.1080/15287390802350030
92. Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA, et al. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci U S A (2002) 99:11700–5. doi:10.1073/pnas.182372299
93. Saitoh T, Tanaka S, Koike T. Rapid induction and Ca(2+) influx-mediated suppression of vitamin D3 up-regulated protein 1 (VDUP1) mRNA in cerebellar granule neurons undergoing apoptosis. J Neurochem (2001) 78:1267–76. doi:10.1046/j.1471-4159.2001.00505.x
94. Kato T, Shimono Y, Hasegawa M, Jijiwa M, Enomoto A, Asai N, et al. Characterization of the HDAC1 complex that regulates the sensitivity of cancer cells to oxidative stress. Cancer Res (2009) 69:3597–604. doi:10.1158/0008-5472.CAN-08-4368
95. Kwon HJ, Won YS, Suh HW, Jeon JH, Shao Y, Yoon SR, et al. Vitamin D3 upregulated protein 1 suppresses TNF-alpha-induced NF-kappaB activation in hepatocarcinogenesis. J Immunol (2010) 185:3980–9. doi:10.4049/jimmunol.1000990
96. Sheth SS, Bodnar JS, Ghazalpour A, Thipphavong CK, Tsutsumi S, Tward AD, et al. Hepatocellular carcinoma in Txnip-deficient mice. Oncogene (2006) 25:3528–36. doi:10.1038/sj.onc.1209394
97. Nishizawa K, Nishiyama H, Matsui Y, Kobayashi T, Saito R, Kotani H, et al. Thioredoxin-interacting protein suppresses bladder carcinogenesis. Carcinogenesis (2011) 32:1459–66. doi:10.1093/carcin/bgr137
98. Ohta S, Lai EW, Pang AL, Brouwers FM, Chan WY, Eisenhofer G, et al. Downregulation of metastasis suppressor genes in malignant pheochromocytoma. Int J Cancer (2005) 114:139–43. doi:10.1002/ijc.20670
99. Jin HO, Seo SK, Kim YS, Woo SH, Lee KH, Yi JY, et al. TXNIP potentiates Redd1-induced mTOR suppression through stabilization of Redd1. Oncogene (2011) 30:3792–801. doi:10.1038/onc.2011.102
100. Masaki S, Masutani H, Yoshihara E, Yodoi J. Deficiency of thioredoxin binding protein-2 (TBP-2) enhances TGF-beta signaling and promotes epithelial to mesenchymal transition. PLoS One (2012) 7:e39900. doi:10.1371/journal.pone.0039900
101. Arai RJ, Ogata FT, Batista WL, Masutani H, Yodoi J, Debbas V, et al. Thioredoxin-1 promotes survival in cells exposed to S-nitrosoglutathione: correlation with reduction of intracellular levels of nitrosothiols and up-regulation of the ERK1/2 MAP kinases. Toxicol Appl Pharmacol (2008) 233:227–37. doi:10.1016/j.taap.2008.07.023
102. Mochizuki M, Kwon YW, Yodoi J, Masutani H. Thioredoxin regulates cell cycle via the ERK1/2-cyclin D1 pathway. Antioxid Redox Signal (2009) 11:2957–71. doi:10.1089/ARS.2009.2623
103. Yoshioka J, Schulze PC, Cupesi M, Sylvan JD, MacGillivray C, Gannon J, et al. Thioredoxin-interacting protein controls cardiac hypertrophy through regulation of thioredoxin activity. Circulation (2004) 109:2581–6. doi:10.1161/01.CIR.0000129771.32215.44
104. Son A, Nakamura H, Okuyama H, Oka S, Yoshihara E, Liu W, et al. Dendritic cells derived from TBP-2-deficient mice are defective in inducing T cell responses. Eur J Immunol (2008) 38:1358–67. doi:10.1002/eji.200737939
105. Lim JY, Yoon SO, Hong SW, Kim JW, Choi SH, Cho JY. Thioredoxin and thioredoxin-interacting protein as prognostic markers for gastric cancer recurrence. World J Gastroenterol (2012) 18:5581–8. doi:10.3748/wjg.v18.i39.5581
106. Takahashi Y, Ishii Y, Murata A, Nagata T, Asai S. Localization of thioredoxin-interacting protein (TXNIP) mRNA in epithelium of human gastrointestinal tract. J Histochem Cytochem (2003) 51:973–6. doi:10.1177/002215540305100713
107. Lee JH, Jeong EG, Choi MC, Kim SH, Park JH, Song SH, et al. Inhibition of histone deacetylase 10 induces thioredoxin-interacting protein and causes accumulation of reactive oxygen species in SNU-620 human gastric cancer cells. Mol Cells (2010) 30:107–12. doi:10.1007/s10059-010-0094-z
Keywords: thioredoxin, Txnip, redoxisome, inflammation, diabetes mellitus, redox regulation
Citation: Yoshihara E, Masaki S, Matsuo Y, Chen Z, Tian H and Yodoi J (2014) Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 4:514. doi: 10.3389/fimmu.2013.00514
Received: 28 November 2013; Accepted: 27 December 2013;
Published online: 09 January 2014.
Edited by:
Pietro Ghezzi, Brighton and Sussex Medical School, UKReviewed by:
Pietro Ghezzi, Brighton and Sussex Medical School, UKAnna Rubartelli, IRCCS AOU San Martino IST, Italy
Copyright: © 2014 Yoshihara, Masaki, Matsuo, Chen, Tian and Yodoi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junji Yodoi, Institute for Virus Research, Kyoto University, Kyoto 606-8397, Japan e-mail:eW9kb2lAdmlydXMua3lvdG8tdS5hYy5qcA==
†Present address: Eiji Yoshihara, Gene Expression Laboratory, Salk Institute for Biological Study, La Jolla, CA, USA; So Masaki, Laboratory for Malignancy Control Research, Medical Innovation Center, Graduate School of Medicine, Kyoto University, Kyoto, Japan; Zhe Chen, Center for Genetics of Host Defense, University of Texas Southwestern Medical Center, Dallas, TX, USA