 Luis M. Blanco-Colio
Luis M. Blanco-Colio- Vascular Research Laboratory, IIS-Fundación Jiménez Díaz, Madrid, Spain
Cardiovascular diseases (CVD) are the first cause of mortality in Western countries. CVD include several pathologies such as coronary heart disease, stroke or cerebrovascular accident, congestive heart failure, peripheral arterial disease, and aortic aneurysm, among others. Interaction between members of the tumor necrosis factor (TNF) superfamily and their receptors elicits several biological actions that could participate in CVD. TNF-like weak inducer of apoptosis (TWEAK) and its functional receptor and fibroblast growth factor-inducible molecule 14 (Fn14) are two proteins belonging to the TNF superfamily that activate NF-κB by both canonical and non-canonical pathways and regulate several cell functions such as proliferation, migration, differentiation, cell death, inflammation, and angiogenesis. TWEAK/Fn14 axis plays a beneficial role in tissue repair after acute injury. However, persistent TWEAK/Fn14 activation mediated by blocking experiments or overexpression experiments in animal models has shown an important role of this axis in the pathological remodeling underlying CVD. In this review, we summarize the role of TWEAK/Fn14 pathway in the development of CVD, focusing on atherosclerosis and stroke and the molecular mechanisms by which TWEAK/Fn14 interaction participates in these pathologies. We also review the role of the soluble form of TWEAK as a biomarker for the diagnosis and prognosis of CVD. Finally, we highlight the results obtained with other members of the TNF superfamily that also activate canonical and non-canonical NF-κB pathway.
Introduction
Cardiovascular diseases (CVD) claim more lives worldwide than any other, causing an estimated 17 million deaths worldwide each year due to heart attacks and strokes. CVD included several pathologies such as coronary heart disease, stroke or cerebrovascular accident, congestive heart failure, peripheral arterial disease, and aortic aneurysm, among others.
Cumulative evidence supports the important role of the tumor necrosis factor (TNF) superfamily of proteins in the development of CVD. The majority of ligands included in this family are synthesized as type II transmembrane proteins with a common structural motif (THD) that mediates self-trimerization and receptor binding (1). The extracellular domain of ligands can be cleaved to generate soluble cytokines. Receptors are usually type I transmembrane glycoproteins characterized by the presence of extracellular cysteine-rich domains (1). As their ligands, functional receptors are also usually trimerics. Many TNF members, including TNF-like weak inducer of apoptosis (TWEAK), activate the nuclear factor kappaB (NF-κB) family of transcription factors (2). NF-κB DNA-binding complex are homo- or heterodimers of five Rel proteins: NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB, and c-Rel. NF-κB dimers translocate to the nucleus and bind to DNA by two different pathways, canonical or non-canonical NF-κB activation. Binding of NF-κB to DNA activates the transcription of several target genes that are implicated in the inflammatory response as well as in cell proliferation, migration, and differentiation. All these processes are closely related to pathological vascular remodeling.
TWEAK and Fn14: Structure, Expression, and Function
The human TWEAK gene is located at chromosomal position 17p13.1 and encodes a 249-amino acid (aa) type II transmembrane protein (3). TWEAK is expressed as a full-length, membrane-bound protein (mTWEAK) and then is proteolytically processed by furin, leading to the release of a 156-aa, 18 kDa soluble form (sTWEAK) (3). The extracellular domain contains the receptor-binding site and the intracellular domain contains a putative serine phosphorylation site. In 2001, Fn14 was identified as the functional TWEAK receptor using a cDNA expression library screening approach (4). The human Fn14 gene is located at the chromosomal position 16p13.3 (5), and encodes a 129-aa type I transmembrane protein of 14 kDa that is processed into a mature form of 102-aa (4). The extracellular domain contains the ligand-binding site and the intracellular domain contains a TNFR-associated factor (TRAF)-binding site (6) implicated in signal transduction induced by TWEAK (7). A second receptor for TWEAK, CD163, has recently been identified (8, 9). CD163 is a hemoglobin scavenger receptor that is exclusively expressed by monocytes/macrophages (10). It has been proposed that CD163 acts as a scavenger receptor for TWEAK, thus preventing TWEAK from exerting its biological actions by sequestering it from the environment. However, it has been reported that recombinant CD163 failed to decrease cell death induced by TWEAK in macrophages (11). The relevance of TWEAK/CD163 interaction needs to be confirmed and more studies are needed in order to determine whether this interaction takes place either in vitro or in vivo. In addition, the existence of a third alternative receptor for TWEAK has been proposed, since murine RAW264.7 cells differentiation induced by TWEAK occurs in an Fn14-independent manner (12). However, no reports describing this alternative receptor have been published and we have observed that this monocytic/macrophage cell line expresses functional Fn14 (Blanco-Colio, unpublished observation).
TNF-like weak inducer of apoptosis is expressed in several cell types and tissues including the intestine, pancreas, lung, brain, ovary, skeletal muscle, and vasculature, and to a lesser degree in kidney and liver (3). Although TWEAK can be upregulated after injury (13), changes in TWEAK gene expression are usually moderated. By contrast, Fn14 expression in healthy tissues, including the vasculature and heart, is usually low or undetectable, although it is rapidly and highly upregulated under pathological conditions as demonstrated in experimental models of chronic liver injury (14), myocardial infarction (15), colitis (16), denervation-induced skeletal muscle atrophy (17), restenosis after balloon injury (4), atherosclerosis (18), autoimmune encephalomyelitis (19), acute kidney injury (20), and cardiac dysfunction (21). Once Fn14 is upregulated, TWEAK binds and causes Fn14 trimerization and signal transduction (7). Although soluble TWEAK is responsible for the responses associated with Fn14, it has been recently reported that full-length, membrane-anchored TWEAK can, in a juxtacrine manner, bind to Fn14 on neighboring cells and activate the NF-κB signaling pathway, thus initiating the cellular response (22).
Fn14 is upregulated by several growth factors, cytokines, and interleukins in cells present in the injured vascular wall such as endothelial cells, vascular smooth muscle cells (SMCs), and monocyte/macrophages, but not in T and B lymphocytes (4, 18, 23, 24). However, little is known of the regulatory mechanisms of Fn14 expression, and only the RhoA/ROCK pathway has been related to Fn14 upregulation in cardiomyocytes (25). TWEAK protein can be upregulated by PMA and IFN-γ in cultured peripheral mononuclear cells and natural killer cells (24, 26). Fn14 trimerization induces the recruitment of TRAF2 and TRAF5 through its TRAF-binding motif (PIEET). This motif is responsible for activating different signaling pathways such as NF-κB and mitogen-activated protein kinases (MAPK) (Figure 1) (7, 27). Activation of NF-κB by TWEAK participates in the upregulation of several cytokines implicated in the recruitment of inflammatory cells within the injured vessel wall. Thus, TWEAK increases MCP-1 and RANTES in SMCs (28). TWEAK also activates NF-κB in cultured Thp-1 monocytic cell line (29). In addition, TWEAK induces the expression of CCL19 and CCL21 in murine tubular cells (30), and both cytokines are also expressed in atherosclerotic plaques of ApoE-deficient mice, a model of hyperlipidemic-induced atherosclerosis (31). TWEAK also activates MAPK, although activation of ERK, c-Jun N-terminal kinase (JNK), or p38 pathways is context-dependent. MAPK activation has been reported in several cell lines, including Thp-1 monocytic cell line, endothelial cells, cardiomyocytes, fibroblast, and others (23, 29, 32–34). There are also different reports indicating that TWEAK activates PI3K/AKT in different cell types. Thus, TWEAK increases HMGB1 secretion by cultured monocytes through PI3K activation (29). In addition, TWEAK also activates transforming growth factor-β activated kinase 1 (TAK1), implicated in NF-κB activation (35, 36) and JNK, related with AP-1 activation (37). Overall, TWEAK activates several signaling pathways that participate in the inflammatory response of the injured tissues.
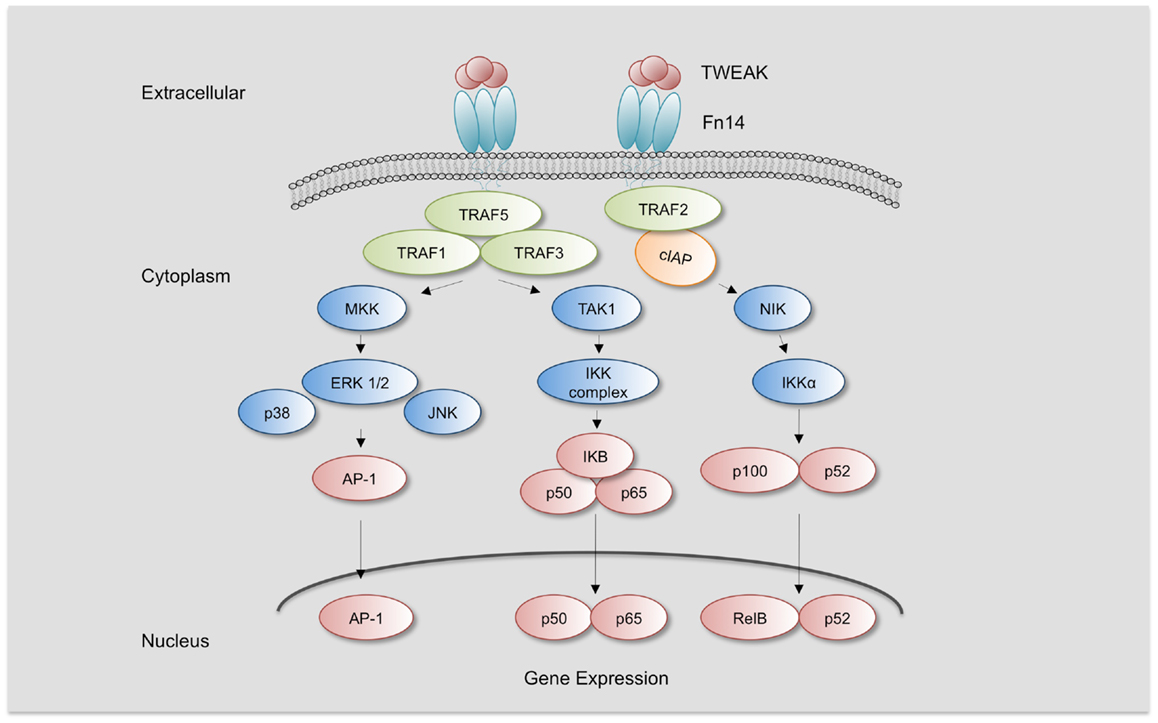
Figure 1. TWEAK/Fn14 Signaling. TWEAK/Fn14 binding induces the recruitment of cIAP and TRAF-1, 2, 3, and 5 and leads to activation of different kinases such as mitogen-activated protein kinase kinases (MKK), transforming growth factor β-activated kinase 1 (TAK1), and NF-κB inducing kinase (NIK). TAK1 activates IKKβ and NIK activates IKKα, leading to the activation of canonical or non-canonical NF-κB pathway, respectively. MMK are responsible for the activation of c-Jun N-terminal kinase 1 (JNK1) and p38 MAPK, which activate transcription factor activator protein-1 (AP-1). Increased activation of NF-κB and AP-1 leads to the expression of specific target genes responsible for TWEAK-mediated responses.
Several functions with potential pathological significance have been related to TWEAK/Fn14 interaction and are dependent on the cell type, microenvironment, and cell activation. However, the basis for these differential responses is poorly understood. TWEAK can regulate cell proliferation, migration, differentiation, and death as well as tissue inflammation, angiogenesis, and regeneration (Figure 2) (3, 5, 38–40). The precise role of TWEAK in different pathological situations needs to be characterized, since TWEAK has beneficial or deleterious effects depending on the stage of the disease (13, 41).
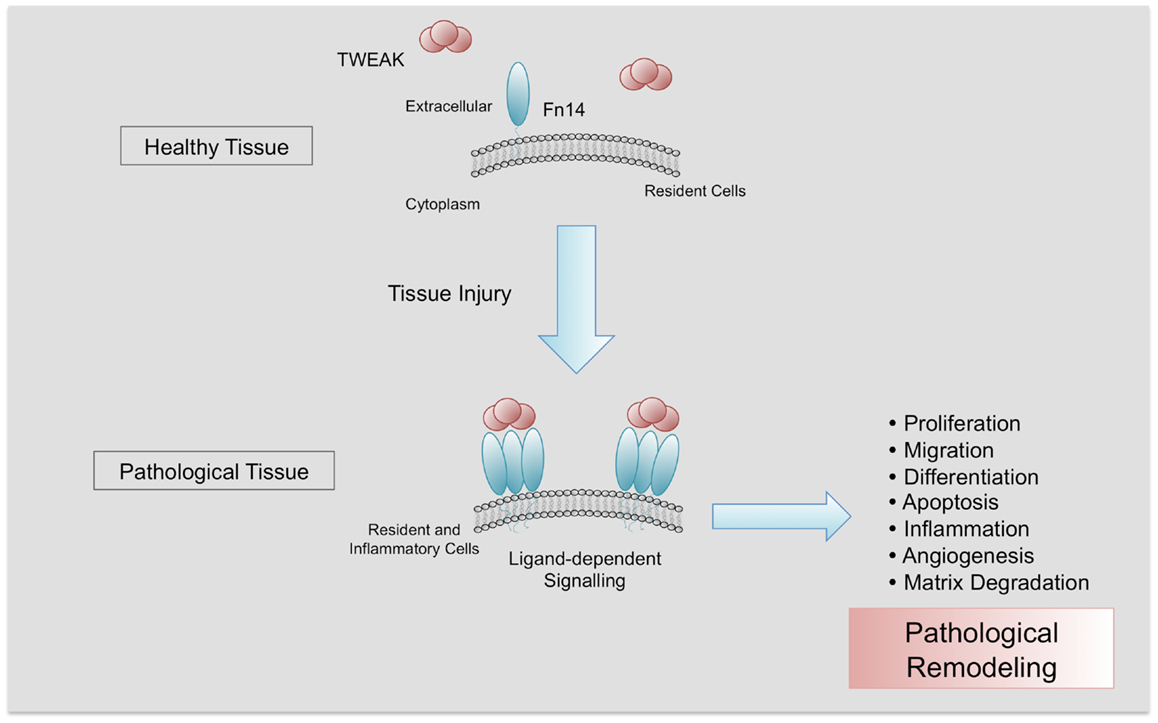
Figure 2. Pathological actions of TWEAK/Fn14 interaction. Fn14 is almost absent in healthy tissues. After injury, Fn14 is upregulated, facilitating the interaction with its ligand TWEAK and its trimerization. In a context of chronic injury, TWEAK/Fn14 interaction participates in pathological tissue remodeling, promoting proliferation, migration, differentiation, apoptosis, inflammation, angiogenesis, and matrix degradation.
TWEAK and Atherosclerosis
Atherosclerosis is a multifactorial disease characterized by chronic inflammation and excessive cell proliferation (Figure 3). Vascular lesions begin as a fatty streak in the subendothelial space of large arteries. Endothelial cells acquire an activated phenotype and express adhesion molecules such as intercellular adhesion molecules (ICAMs), selectins, and vascular adhesion molecules (VCAMs) that act as receptors for proteins expressed by leukocytes (monocytes and lymphocytes and neutrophils). Recruitment of monocytes to the subendothelial space causes their differentiation to macrophages that uptake oxidized low density lipoproteins (ox-LDL). In addition, chemokines and cytokines are secreted by inflammatory cells and induce proliferation and migration of SMCs from the media forming the neointima (42). The transition of relatively early lesions to more advanced lesions is characterized by the proliferation of SMCs and continuous uptake of ox-LDL by macrophages, forming foam cells. In addition, SMCs synthesize extracellular matrix proteins that lead to the development of the fibrous cap. This cap confers resistance to rupture by the accumulation of collagen synthesized by SMCs. The continuous ingestion of ox-LDL by foam cells induces death of these cells, releasing of insoluble lipids and contributing to the formation of the necrotic core characteristic of advanced lesions. Expression of different proteases by macrophages and SMCs leads to degradation of the fibrous cap, promoting plaque instability and subsequent plaque rupture. Rupture of an atherosclerotic plaque may result in the occlusion of an artery by the formation of a thrombus over an atherosclerotic lesion, causing myocardial infarction, stroke, or peripheral vascular disease (42).
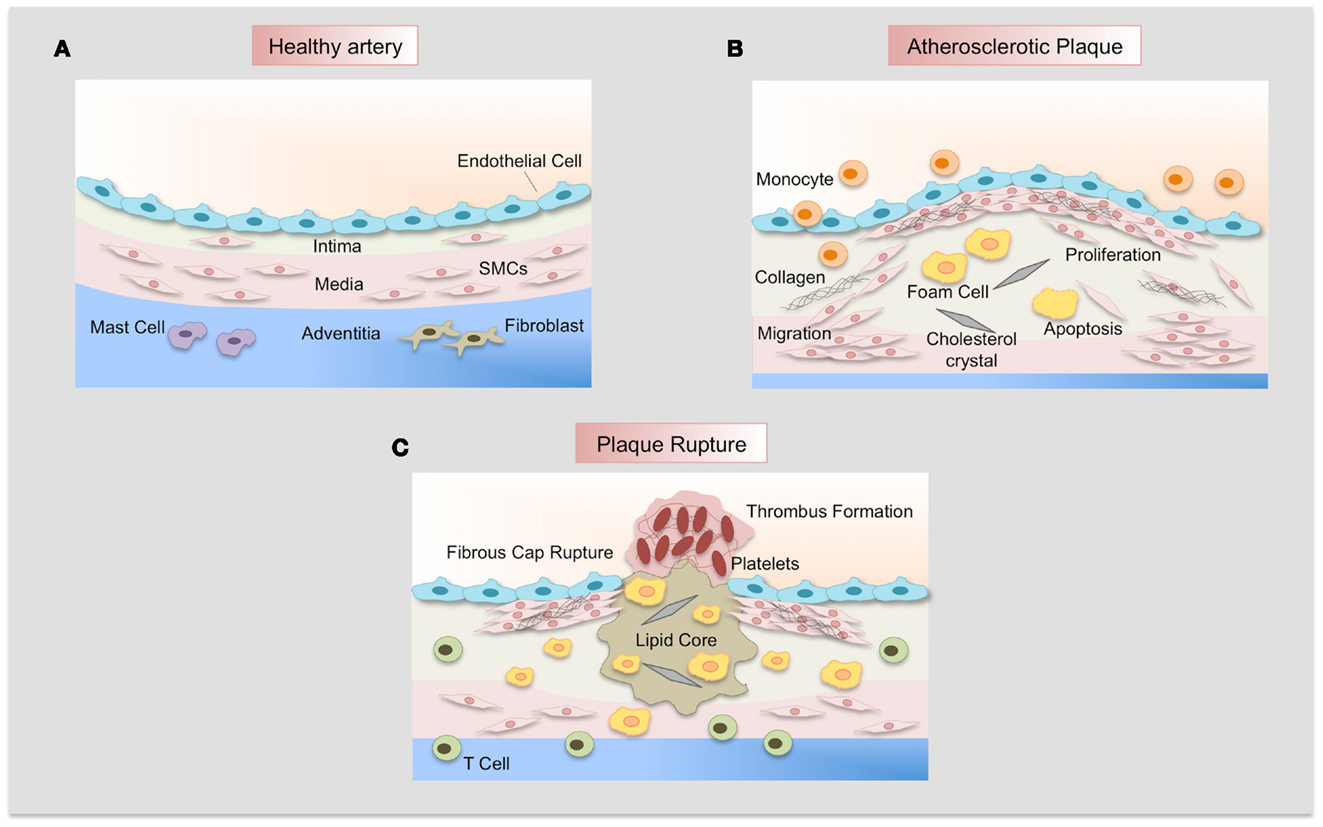
Figure 3. Atherosclerotic plaque development. (A) Normal artery is formed by a monolayer of endothelial cells, the tunica media of resident smooth muscle cells embedded in an extracellular matrix, and the adventitia that contains mast cells, fibroblasts, and microvessels. (B) Adhesion of leukocytes to dysfunctional endothelium leads to their migration into the vascular wall, forming the neointima. Monocytes differentiate to macrophages and uptake lipids yielding foam cells. SMCs migrate from the media, proliferate, and synthesize extracellular matrix proteins such as collagen and elastin. SMCs and macrophages can die and lipid derived from dead cells accumulates in the central region of the plaque, the necrotic core that is covered by a fibrous cap. (C) Finally, when the fibrous cap is broken, blood coagulant components trigger thrombus formation, which occludes the lumen and interrupts the blood flow.
The TWEAK/Fn14 axis plays an important role in several steps of atherosclerotic plaque development including initiation, progression, destabilization/rupture, and subsequent thrombosis. As commented, TWEAK is expressed in both the normal and pathological arterial wall (18), but Fn14 is almost absent in healthy arteries and its expression is highly upregulated in the carotid artery (18), femoral atherosclerotic plaques (43), and in abdominal aortic aneurysms (44). Different stimuli induce Fn14 expression in resident and inflammatory cells present in the vascular wall. Thus, pro-inflammatory cytokines (IL-1β and INF-γ), growth factors (PDGF-BB, EGF, FGF-2), Angiotensin II, or α-thrombin increase Fn14 expression in human and rat aortic SMCs (4, 18). In addition, VEGF-A and FGF-2 increase Fn14 expression in human umbilical endothelial cells (23) and human CD14+ monocytes express Fn14 in response to INF-γ or PMA stimulation (24).
In the first steps of atherosclerosis development, adhesion molecules and chemokines are responsible for the recruitment of the inflammatory cells to the injured tissue. In vitro studies have demonstrated that TWEAK/Fn14 interaction induces expression of adhesion molecules such as ICAM-1 and E-selectin in human umbilical endothelial cells (45). Furthermore, TWEAK increases interleukin-8 and MCP-1 secretion by endothelial cells (45). Infiltrating cells secrete many cytokines that contribute to SMCs migration and proliferation, favoring plaque progression. In this sense, TWEAK/Fn14 interaction directly induces proliferation and migration of human and rat aortic SMCs (39, 45, 46) and human endothelial cells (4, 23, 39, 45). These suggest that the TWEAK/Fn14 axis could participate in neointimal thickening of the pathological arterial wall. In fact, an in vivo study has reported that Fn14 is upregulated in SMCs after balloon injury in mice (4).
The presence of a chronic inflammatory response is an important phenomenon implicated in the development and progression of atherosclerotic plaque. A key transcription factor implicated in vascular inflammation is NF-κB. Activation of signal transduction mediated by NF-κB has been demonstrated at different stages of atherosclerotic lesion development, from plaque formation to plaque rupture (47). NF-κB is activated in SMCs, macrophages, and endothelial cells in human atherosclerotic plaques (48–50). Several molecules can activate this transcription factor in the context of atherogenesis. Inflammatory stimuli such as members of the TNF-α superfamily, IL-1, and ox-LDL induce NF-κB activation, and in consequence amplifying and maintaining a vascular inflammatory response that facilitates atherosclerosis progression. Activation of this transcription factor in endothelial cells enhances the expression of adhesion molecules, chemokines, and metalloproteinases (MMP). These molecules coordinate the invasion of inflammatory cells into the vascular wall, and enhance migration and proliferation of SMCs as well as the remodeling of the extracellular matrix. Inflammatory cells and SMCs also increase cytokine and MMP expression through NF-κB activation, perpetuating the inflammatory response. In particular, TWEAK activates NF-κB in several cell types and increases the expression of pro-inflammatory proteins such as IL-6, IL-8, MCP-1, and RANTES (7, 18, 27, 28, 46, 51, 52); these pro-inflammatory proteins are implicated in atherogenesis. In addition, recombinant TWEAK injection increases atherosclerotic lesion size and inflammatory cell content as well as NF-κB activation in the aortic root of hyperlipidemic ApoE-knockout mice (28). Moreover, anti-TWEAK monoclonal antibody (mAb) therapy diminishes NF-κB activation as well as inflammatory response in ApoE-null mice, indicating that endogenous TWEAK participates in atherogenesis (28). In addition, genetic deletion of TWEAK or treatment with anti-TWEAK mAb diminished NF-κB activation, chemokines secretion and inflammatory response in ApoE-deficient mice (53). The activation of NF-κB by TWEAK observed in this experimental model was related to the canonical pathway, since p50/p65 dimers were detected in the nuclei of cells within atherosclerotic plaques. Until now, non-canonical NF-κB activation induced by TWEAK has not been reported in atherosclerotic plaques.
TWEAK also increases the secretion of HMGB1 through NF-κB activation in human M1 macrophages (29). HMGB1 is a DNA-binding cytokine that activates endothelial cells and monocytes/macrophages to express pro-inflammatory cytokines, chemokines, and adhesion molecules functioning as a critical mediator of inflammation (54). HMGB1 colocalizes with Fn14 in the shoulder region of human atherosclerotic plaques, a macrophage-rich area (29). In addition, systemic injection of recombinant TWEAK augmented HMGB1 expression in atherosclerotic plaques of hyperlipidemic ApoE-null mice (29). The importance of the finding that NF-κB can regulate HMGB1 release induced by TWEAK is because secreted HMGB1 may in turn induce NF-κB activation, forming a loop between NF-κB and HMGB1 that perpetuates vascular pro-inflammatory effects related to TWEAK. These data support the notion that TWEAK/Fn14 interaction has deleterious consequences in the injured vascular wall.
Interestingly, it has been reported that TWEAK can modulate macrophage size within atherosclerotic plaques (55). This finding is related to the capacity of TWEAK to modulate lipid uptake by macrophages. In fact, ApoE-deficient mice treated with Fn14–Fc protein present smaller macrophages in their atherosclerotic plaques, and treatment with anti-Fn14 or anti-TWEAK antibodies or Fn14–Fc protein diminished macrophage uptake of modified lipids in vitro (55).
The stability of the advanced atherosclerotic plaque depends on the integrity of the fibrous cap that encloses its lipid core. Established atherosclerotic lesions usually have a dense fibrous cap. However, areas with sustained inflammation, macrophage accumulation, and apoptosis are prone to rupture due to a weakening of the fibrous cap. Deterioration of the fibrous cup is dependent on the activity of MMP, which are collagen-degrading endopeptidases that are secreted by SMCs and macrophages (56). As commented, TWEAK and Fn14 are expressed in macrophages/foam cells rich regions of atheroma plaques and colocalized with MMP (57). Moreover, an activating anti-Fn14 antibody increases the expression of MMP-9 and MMP-1/13 in cultured monocytes (57). In addition, anti-TWEAK mAb treatment diminishes MMP activity in atherosclerotic plaques present in the aortic root of ApoE-deficient mice (53). In addition, features of greater plaque stability included augmented collagen/lipid ratio, reduced macrophages content, and less presence of lateral xanthomas, buried caps, medial erosion, intraplaque hemorrhage, and calcium content have been observed in TWEAK/ApoE-double knockout mice or in anti-TWEAK mAb treated ApoE-deficient mice (53). These data indicate a potential role of TWEAK in extracellular matrix degradation, which favors plaque instability.
Plaque rupture or erosion, and subsequent thrombosis, represent the main complications of atherosclerosis and could lead to an acute cardiovascular event. Different molecules, such as plasminogen activator inhibitor 1 (PAI-1) and tissue factor (TF), are responsible for hemostasis and thrombosis (58). TF is the principal initiator of the clotting cascade, while PAI-1 plays a critical role in inhibiting fibrinolysis, and thereby the activity of both molecules promotes thrombotic states and plays a crucial role in vascular diseases (59). Fn14 colocalizes with PAI-1 and TF in human carotid atherosclerotic plaques (60). In addition, TWEAK increases TF and PAI-1 mRNA and protein expression as well as activity in cultured human aortic SMCs (60). Furthermore, systemic injection of recombinant TWEAK augmented TF and PAI-1 expression in atherosclerotic plaques of ApoE-deficient mice and, conversely, anti-TWEAK treatment diminished the expression of both prothrombotic proteins (60). These data indicate that TWEAK favors thrombus formation after plaque rupture.
Overall, data obtained from in vitro and in vivo studies indicate that TWEAK participates in different stages of atherosclerotic plaque development from early stages to progression and subsequent plaque rupture that lead to an acute cardiovascular event, such as myocardial infarction or stroke. Anti-TWEAK treatment has the capacity to diminish pro-inflammatory response associated with atherosclerotic plaque progression and to alter plaque morphology toward a stable phenotype.
TWEAK and Stroke Outcome
Stroke is the third most common cause of death in the world (61). A stroke, or cerebrovascular accident, causes rapid loss of brain function due to a lack of oxygen; sudden death of brain cells also takes place. The two main types of stroke include ischemic (when a blood clot or thrombus forms) and hemorrhagic stroke. The outcome after a stroke depends on where it occurs and how much of the brain is affected. Smaller strokes may result in minor problems, such as weakness in an arm or leg. Larger strokes may lead to paralysis or death. Ischemic stroke triggers a cascade of pathophysiological events such as energy depletion, excitotoxicity, peri-infarct depolarization, inflammation, and apoptotic cell death (62). The onset of the ischemic insult is followed by an increase in the expression of pro-inflammatory molecules in the ischemic tissue, which has been associated with neuronal death and poor outcome.
Recent reports have shown the role of TWEAK/Fn14 axis after an ischemic stroke. In fact, it has been reported that ischemic stroke in humans (63) and experimental middle cerebral artery occlusion (MCAO) in mice (64, 65) increase the expression of both TWEAK and Fn14 in the ischemic tissue. In the central nervous system (CNS), TWEAK and Fn14 are expressed mainly in endothelial cells, perivascular astrocytes, microglia, and neurons. There are two principal mechanisms by which TWEAK participates in stroke pathogenesis: neuronal apoptosis and breakdown of the blood–brain barrier (BBB) (64, 66). It has been demonstrated that in response to hypoxia/ischemia, TWEAK induces cell death in neurons via NF-κB activation and PARP-1 and caspase-3 cleavage (64, 67). However, oxygen–glucose deprivation conditions did not affect cell survival in neurons from Fn14- or TWEAK-deficient mice, indicating that cell death is mediated by the TWEAK/Fn14 interaction (67).
On the other hand, during cerebral ischemia, disruption of the architecture of the neurovascular unit (NVU) results in an increase in the permeability of the BBB with the development of cerebral edema, which is a major cause of mortality among patients with acute stroke. NVU is a dynamic structure consisting of endothelial cells, the basal lamina, astrocytic end-feet processes, pericytes, and neurons (68). The permeability of BBB is increased by pro-inflammatory cytokines that act on the NVU under ischemic conditions (69). It has been demonstrated that TWEAK has a detrimental effect on the structure of the NVU and the permeability of the BBB in the early stages of cerebral ischemia. Recombinant TWEAK injection directly into the brain induces activation of NF-κB and MMP-9 expression, resulting in the disruption of the structure of NVU and an increase in the permeability of BBB (70). Furthermore, inhibition of TWEAK actions by Fn14–Fc decoy receptor or Fn14 deficiency diminished cerebral ischemia-induced increase in the permeability of the NVU. This protection was associated with a faster recovery of locomotor activity (66).
Finally, intraperitoneal administration of anti-TWEAK monoclonal antibodies (64) or intracerebroventricular administration of Fn14–Fc decoy receptor (65) diminished the infarct size by around 30–40% after 48 h of MCAO. In addition, Fn14-deficient mice exhibited a 60% reduction in the volume of the ischemic lesion following MCAO compared to wild-type animals (66). Overall, TWEAK may play an important role during ischemia-induced brain injury and its inhibition in the brain could be a novel neuroprotective strategy for the treatment of ischemic stroke.
Diagnostic and Prognostic Value of Soluble TWEAK for Cardiovascular Diseases
As commented above, TWEAK is expressed as a full-length, membrane-bound protein and then is proteolytically processed by furin, leading to the release of a 156-amino acid, 18 kDa soluble form (sTWEAK) (3). Among the potential biomarkers that could be differentially secreted by the pathological arterial wall, sTWEAK was identified as a protein that is highly released by normal arteries in comparison with carotid atherosclerotic plaques (71). Plasma levels of sTWEAK were also found to be diminished in patients with atherosclerosis compared to control subjects. The association of sTWEAK with the presence of CVD or CVD-related diseases has been extensively validated in other cohorts of individuals (Figure 4). Thus, sTWEAK concentrations were significantly reduced in patients with chronic kidney disease (CKD) and/or type II diabetes (72). In addition, sTWEAK plasma concentrations were diminished in patients undergoing hemodialysis compared to healthy subjects (73). In addition, a gradual decrease in sTWEAK along with a reduction in estimated glomerular filtration rate in CKD patients has been observed (74–76). Reduced levels of sTWEAK have also been associated with the presence of coronary artery disease (CAD) (25, 77), systolic heart failure (78), peripheral artery disease (PAD) (43), and aortic abdominal aneurysm (AAA) (44). Finally, elevated circulating sTWEAK levels have been described in patients after myocardial infarction (25) and stroke (63). However, although the study of circulating proteins in subjects suffering an acute event could unveil novel proteins implicated in atherothrombosis, some of these proteins could be released by the tissue necrosis that takes place during an acute event.
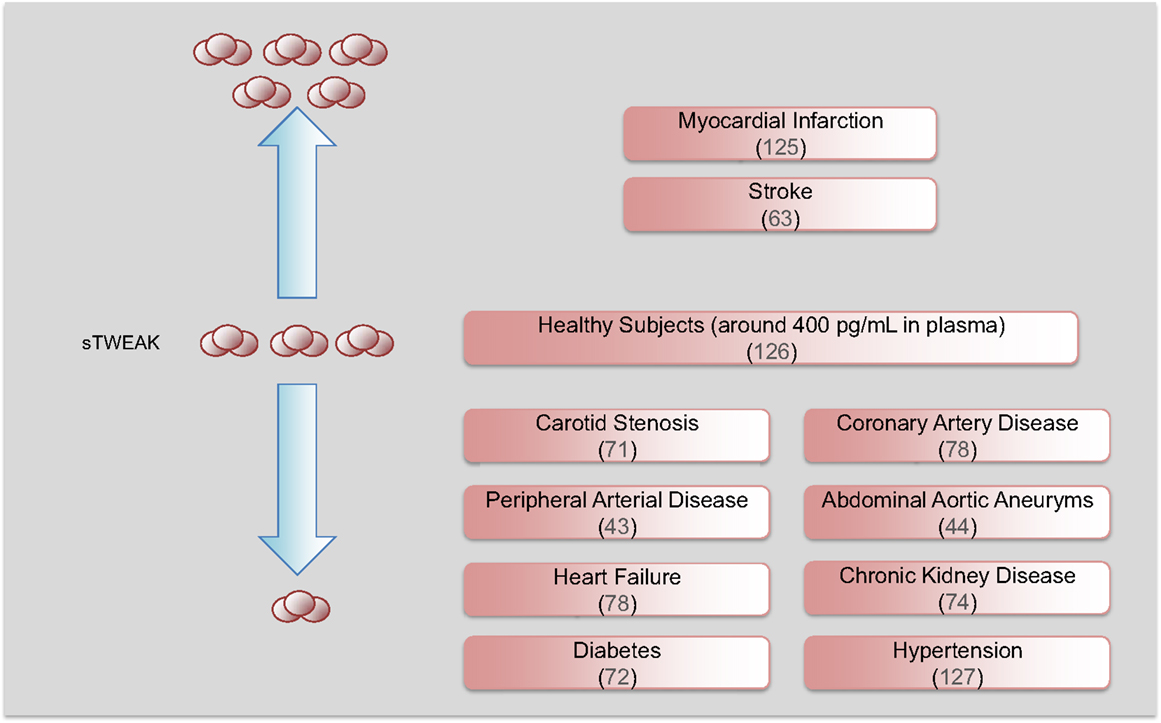
Figure 4. Soluble TWEAK as a biomarker of cardiovascular diseases. Circulating sTWEAK concentrations are diminished in subjects with chronic vascular damage including carotid stenosis, coronary artery disease, peripheral artery disease, abdominal aortic aneurysm, heart failure, and chronic kidney disease. Soluble TWEAK levels are also diminished in subjects with cardiovascular risk factors such as diabetes or hypertension. However, circulating sTWEAK levels are increased in patients suffering an acute cardiovascular event (myocardial infarction or stroke).
Two different surrogate markers of atherosclerosis such as flow-mediated dilation (FMD) and intima/media thickness (IMT) have been negatively associated with sTWEAK concentrations (71, 74, 75). As mentioned previously, endothelial dysfunction participates in the formation of vascular lesion (79). FMD described the vasodilation of a conduit artery following an augmentation in shear stress induced by an ischemia period. FMD provides us information about the integrity of the endothelium (80). It has been reported that sTWEAK plasma concentrations are negatively associated with FMD in patients with CKD (74), persisting after adjustment for factors related to FMD in CKD subjects such as blood pressure, C-reactive protein, and estimated glomerular filtration rate. This result was later confirmed in hypertensive CKD patients (75). In addition, IMT can be measured non-invasively by means of B-mode ultrasound, and increases in IMT have been associated with an augmentation for future cardiovascular outcomes (81). Thus, IMT has been negatively associated with sTWEAK concentrations in asymptomatic subjects (71) and in patients with CKD (76, 82, 83), even after adjustment for traditional risk factors and inflammatory biomarkers. Moreover, sTWEAK is associated with atherosclerotic burden in CKD patients (83). However, IMT was positively correlated with sTWEAK in renal transplant patients (84, 85). Overall, the association of sTWEAK levels with different surrogate markers of atherosclerosis indicates that this protein could be a novel and independent biomarker of CVD.
Finally, different reports have indicated the potential use of sTWEAK as a prognostic biomarker of CVD or CVD-related diseases as well as its impact on survival. Thus, individuals in the upper two tertiles of sTWEAK concentrations presented a lower incidence of PAD (43). In addition, a decreased sTWEAK concentration was significantly and independently associated with long-term cardiovascular mortality in patients with lower-extremity PAD (86). sTWEAK levels were also negatively related with AAA size and AAA expansion rate after a 5-year follow-up, and sTWEAK concentrations were predictive for subjects expanding more than 2 mm/year in AAA size (44). As for CKD, decreasing sTWEAK concentration was associated with increased risk of cardiovascular events independently of basic confounders (age, gender, estimated glomerular filtration rate, C-reactive protein, diabetes, and cardiovascular comorbidity) (76). However, high levels of sTWEAK were associated with atherosclerosis in patients with systemic lupus erythematosus (SLE), but not in control subjects (87). In addition, although sTWEAK plasma levels were diminished in HD patients compared with controls, subjects belonging to the upper tertile of sTWEAK presented a higher risk of all-cause and cardiovascular mortality (73). This discrepancy could be due to the existence of the known reverse epidemiology observed in HD patients. Finally, sTWEAK also provides prognostic information on subjects with heart failure. Subjects with chronic stable heart failure with reduced sTWEAK plasma concentrations (78) present a higher mortality rate than those with elevated sTWEAK levels. In addition, the increase of sTWEAK concentrations diminishes the risk of mortality in subjects with non-ischemic heart failure (88).
The mechanism/s by which sTWEAK is diminished in subjects with vascular damage should be related to the expression of their receptors. As commented, Fn14 expression is undetectable in the vasculature in normal conditions (18). However, under pathological conditions including systemic inflammatory states, Fn14 is highly upregulated in the vasculature, favoring sTWEAK binding and retention in the pathological tissues (18, 57). In addition, the expression of CD163 by M2 macrophages in pathological tissues could be responsible for the decrease in sTWEAK, since CD163 can bind and internalize sTWEAK in vitro (8). On the basis of this preceding literature, we speculate that the reduction in sTWEAK concentrations in cardiovascular-related diseases could potentially reflect either Fn14 binding or CD163 degradation (Figure 5). However, this hypothesis needs to be tested in future studies. Overall, all these data reveal that sTWEAK could be a novel biomarker of CVD. More large-scale studies to consolidate its usefulness are required.
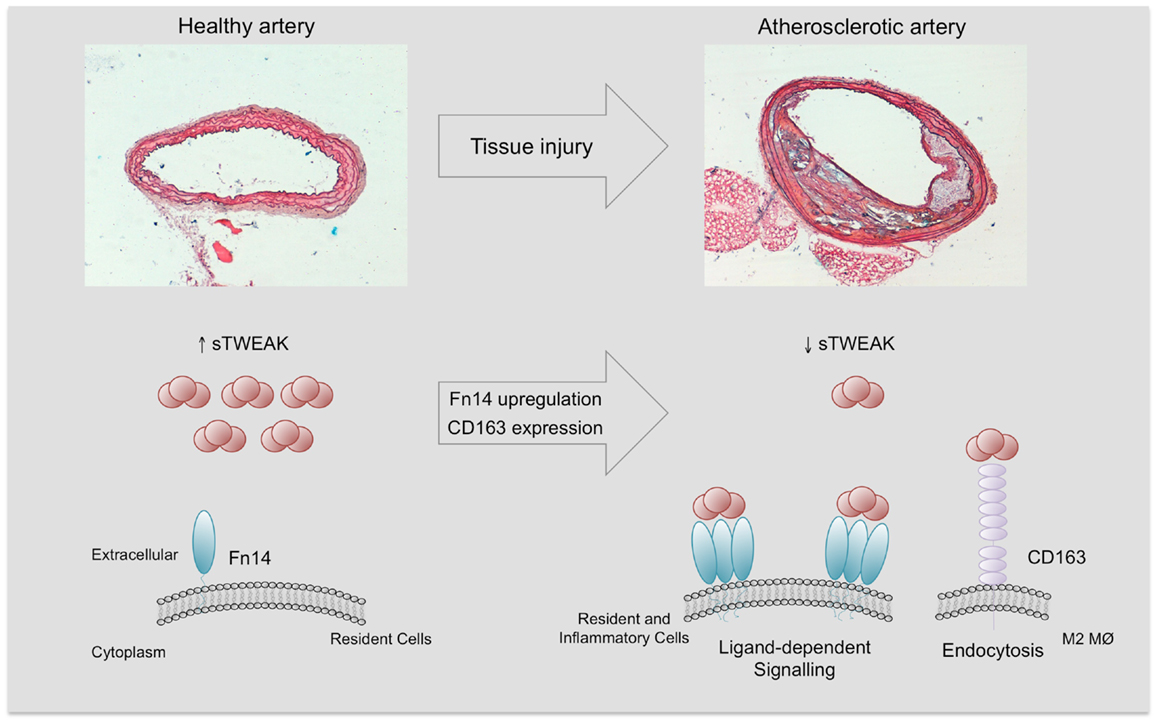
Figure 5. Soluble TWEAK in health and disease. Fn14 expression is almost absent and minimal Fn14 activation is expected in the healthy arterial wall. However, under pathological conditions, Fn14 expression is highly upregulated in resident (smooth muscle cells) and inflammatory cells (e.g., M1 macrophages). This increase facilitates the interaction with sTWEAK and would trigger sTWEAK-dependent Fn14 signaling. On the other hand, pathological tissues are infiltrated by anti- inflammatory macrophages (M2) that express CD163, a scavenger receptor of sTWEAK. The tissue consumption of sTWEAK by Fn14 interaction and CD163 degradation could be responsible for the decrease in circulating sTWEAK levels observed in subjects with atherosclerostic complications.
Other TNF Superfamily Members Implicated in Atherothrombosis That Activate Both Canonical and Non-Canonical NF-κB Pathway
In recent years, the number of TNF receptors that are known to potentially activate the non-canonical NF-κB pathway has increased. These include, in addition to Fn14, CD40, B-cell activating factor receptor (BAFFR), lymphotoxin β receptor (LTβR), receptor activator of NF-κB (RANK), and CD27 (89–93). Some of these receptors have been implicated in different functions related to the pathogenesis of atherosclerosis (Table 1).
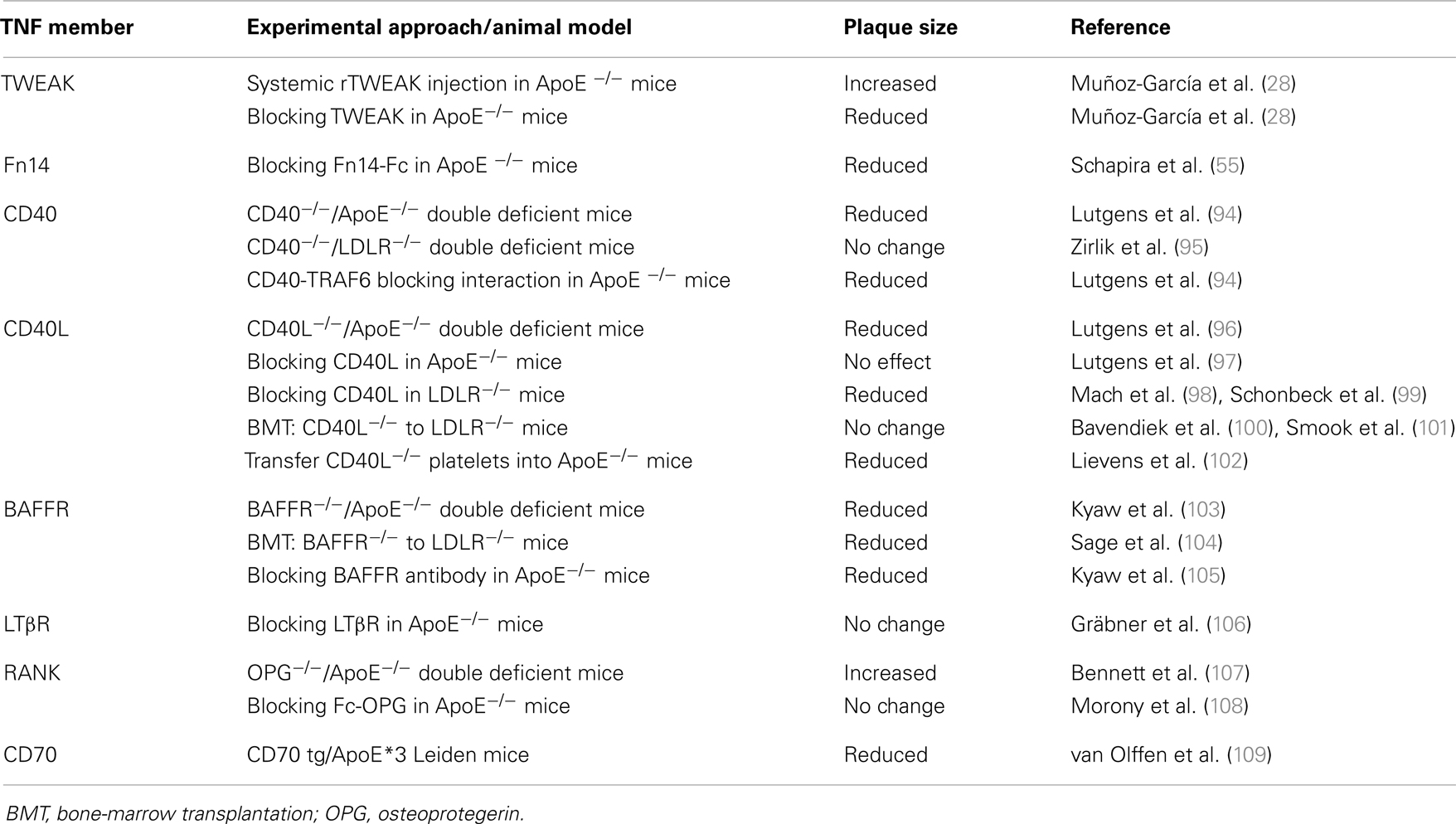
Table 1. Effect of TNF superfamily members that activate non-canonical NF-κB in experimental models of murine atherosclerosis.
The role of CD40 and its ligand CD40L in atherosclerosis has been extensively studied. Both proteins are expressed in various cell types implicated in atherogenesis such as platelets, endothelial cells, monocytes/macrophages, and SMCs (110). CD40L induces a broad inflammatory response in these cell types, including increased expression of adhesion molecules, pro-inflammatory cytokines, matrix degrading enzymes, and pro-coagulants (111, 112). Different groups have analyzed the role of CD40 and CD40L in athero-prone mice by using diverse strategies such as gene modification, blocking antibody treatment, or bone-marrow transplantation (BMT). Thus, CD40L and ApoE double deficient mice develop markedly reduced atherosclerosis (96, 98, 100). In addition, treatment with neutralizing anti-CD40L antibodies diminished atherosclerotic lesion size (98, 99) in LDLR-deficient mice but failed to modify plaque size in ApoE-null mice (97). The decrease of atherosclerotic plaques was associated with features of higher plaque stability such as reduced macrophage and lipid content as well as increased collagen deposition. The effect of CD40L on atherosclerotic plaque progression seems to be related with resident cells (endothelial and SMCs), since BMT failed to modify atherosclerosis plaque size in LDLR−/− mice (100, 101). However, a recent study describes that transfer of CD40L−/− platelets into ApoE-null mice diminished atherosclerotic burden, an effect that results from the capacity of platelets to synthesize higher amounts of CD40L (102). The role of CD40 in atherosclerosis remains controversial. It has been demonstrated that CD40 and ApoE double deficient mice develop reduced levels of atherosclerosis when given a normal chow diet compared with control animals (94). However, a similar atherosclerotic burden was observed in CD40−/−LDLR−/− mice on a high-cholesterol diet (95). These data could suggest that CD40L mediates atherosclerosis development independently of CD40 (95). However, specific interruption of CD40/TRAF6 interaction in ApoE-deficient mice diminished atherosclerotic plaque size, indicating that CD40L/CD40 interaction participates in atherosclerotic plaque development (94).
It has been demonstrated that the depletion of B cells diminished atherosclerosis in mice (113). The proatherogenic effect of B cells is mainly driven by the B2 subset, which responds to T-cell-dependent antigens and is part of the adaptive immune response (114), while the atheroprotective effect is attributed to the B1 subset, which responds to T-cell-independent antigens (115). The survival and maturation of B2 lymphocytes depends on the interaction of BAFF with its receptor, BAFFR (116). Genetic disruption of BAFFR induces a significant reduction in mature B2 cells without affecting B1a cells (117) and BAFFR/ApoE double deficient mice present a reduced atherosclerotic plaque size and macrophage content in their aortic root; this effect is also related to a decrease in the number of B2 cells (103). In addition, BMT of BAFFR deficient cells to LDLR−/− mice also leads to a reduction in plaque size and inflammation (104). These data could suggest that BAFFR is an interesting therapeutic target to limit the development of atherosclerosis. Indeed, atherosclerosis development is diminished in ApoE−/− treated with a BAFFR blocking mAB (105).
Until now, the role of LTβR in atherosclerotic plaque development is unclear. As commented above, cells present in atherosclerotic lesions elicit persistent inflammation and trigger immune adaptive response toward arterial wall-derived autoantigens, such as ox-LDL or heat shock proteins. Coronary patients with atherosclerosis present infiltrates of leukocytes in the adventitia, and the presence of adventitial B-cell follicle-like aggregates in human aorta has been demonstrated (118). Moreover, adventitia of ApoE-deficient mice also contains T- and B-cell aggregates. These aggregates are the precursors of fully structured aorta tertiary lymphoid organs (ATLOs) that contain a high number of germinal centers, endothelial venules, regulatory T cells, and LN-like conduits that connect ATLOs to medial SMCs (106). The recruitment of T- and B-cell aggregates is dependent on CXCL13 and CCL21 secretion by SMCs through an LTβR-dependent signaling pathway. The formation of ATLOs in ApoE-deficient mice is restricted to abdominal aorta, and ATLOs are communicated with medial SMC by conducts that serve as channels for both transport of molecules (e.g., cytokines and chemokines) and soluble antigens (119). Although it is conceivable that ATLOs play a role in atherosclerotic plaque progression, ApoE-null mice treated with anti-LTβR have not modified their atherosclerotic plaques. Future gene deletion studies would help to understand the role of LTβR and ATLOs in atherogenesis.
RANKL, which is expressed in human atherosclerotic plaques (120), is capable of modulating different cell-type activities through its receptor, RANK. RANK/RANKL interaction activates several intracellular signal transduction pathways such as MAPKs and NF-κB (121). Several proatherogenic actions of RANKL have been described. For example, RANKL induces MCP-1 expression and secretion and matrix MMP activity in SMCs (122). In addition, RANKL induces TF expression in macrophages mainly through the cooperative action of NF-κB, AP-1, and Egr-1, supporting a role of RANKL in the thrombogenicity of atherosclerotic plaques (123). Furthermore, RANK, RANKL, and the decoy receptor for RANKL osteoprotegerin (OPG) have been related with vascular calcification, a crucial step for plaque destabilization and rupture. In the absence of OPG, mice display vascular calcification and present increased atherosclerotic plaque size (107). However, treatment of LDLR-deficient mice with Fc-OPG diminished vascular calcification but failed to reduce atherosclerotic plaque size (108).
Finally, CD27 is mainly found in T cells, and its ligand CD70 is expressed in B cells, activated T cells, and dendritic cells. Interaction of both proteins is necessary for the generation and long-term maintenance of T-cell immune responses (124). Overexpression of CD70 in B cells leads to an expansion of Th1 T cells in mice. This indicates that these mice should have a proatherogenic phenotype. However, mice overexpressing CD70 are protective against atherosclerosis development, possibly due to a reduced viability of circulating monocytes (109). After these results, further research is clearly needed to clarify the relevance of CD27 in atherosclerosis.
Concluding Remarks
The evidence gathered to date supports a role of the TWEAK/Fn14 axis in the development and outcome of atherosclerosis and ischemic stroke. Data from experimental models make TWEAK and its functional receptor Fn14 a promising target for the treatment of patients with different CVD. Treatment with the TWEAK neutralizing antibody or Fn14–Fc decoy protein has demonstrated a beneficial effect on the development and progression of atherosclerotic plaques in mice. Furthermore, Fn14 deletion or anti-TWEAK administration diminished the volume of the ischemic lesion after stroke, a related complication of atherosclerotic plaque rupture. Although the use of monoclonal antibodies offers – unlike small-molecule drugs – high target specificity and allows less frequent, albeit parenteral, administration, there is a need for further drug development in this area, including Fn14-specific antagonists.
The role of the TWEAK/Fn14 axis on pathological vascular remodeling is not completely understood, and many questions need to be answered. Could TWEAK induce vascular remodeling in contexts other than atherosclerosis? As in stroke, could TWEAK participate in the outcome of myocardial infarction? Could anti-TWEAK therapy prevent atherosclerotic plaque development and progression in the presence of different cardiovascular risk factors such as diabetes or hypertension? Since TWEAK is also able to interact with CD163 (8), could overexpression of CD163 diminish the proatherogenic effect of TWEAK? Statins can diminish Fn14 expression in cultured SMCs (18), so could statins inhibit atherosclerotic plaque progression induced by TWEAK? In the near future, these and other potential questions could help us to understand the role of this axis in different cardiovascular pathologies.
Finally, the evidence accumulated indicates that sTWEAK could be a biomarker for the diagnosis and prognosis of CVD. The reduction of sTWEAK plasma levels has been demonstrated in subjects with different vascular affectations and its association with total and cardiovascular morbidity and mortality has been reported in different cohorts. However, after an acute cardiovascular event, sTWEAK concentrations are increased. Current and future studies in large-scale populations will help us to determine the relevance of sTWEAK as a CVD biomarker and its potential implementation in clinical practice.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author would like to thank C. Gómez-Guerrero and J. L. Martín-Ventura (Vascular Research Lab. IIS-Fundación Jiménez Díaz) for their critical review of the manuscript and Oliver Shaw (IDC, Spain) for editing this work. The author’s studies cited here were supported by grants from Fondo de Investigaciones Sanitarias (Programa Estabilización Investigadores to Luis M. Blanco-Colio), Instituto de Salud Carlos III (PI10/00234; PI13/00395; RETICS RD12/0042/0038), Fundación Mutua Madrileña, Sociedad Española de Arteriosclerosis, and a research grant from Biogen Idec (to Luis M. Blanco-Colio).
References
1. Bodmer JL, Schneider P, Tschopp J. The molecular architecture of the TNF superfamily. Trends Biochem Sci (2002) 27:19–26. doi:10.1016/S0968-0004(01)01995-8
2. Napetschnig J, Wu H. Molecular basis of NF-κB signaling. Annu Rev Biophys (2013) 42:443–68. doi:10.1146/annurev-biophys-083012-130338
3. Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, et al. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem (1997) 272:32401–10. doi:10.1074/jbc.272.51.32401
4. Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, et al. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity (2001) 15:837–46. doi:10.1016/S1074-7613(01)00232-1
5. Feng SL, Guo Y, Factor VM, Thorgeirsson SS, Bell DW, Testa JR, et al. The Fn14 immediate-early response gene is induced during liver regeneration and highly expressed in both human and murine hepatocellular carcinomas. Am J Pathol (2000) 156:1253–61. doi:10.1016/S0002-9440(10)64996-6
6. Brown SA, Hanscom HN, Vu H, Brew SA, Winkles JA. TWEAK binding to the Fn14 cysteine-rich domain depends on charged residues located in both the A1 and D2 modules. Biochem J (2006) 397:297–304. doi:10.1042/BJ20051362
7. Brown SA, Richards CM, Hanscom HN, Feng SL, Winkles JA. The Fn14 cytoplasmic tail binds tumour-necrosis-factor-receptor-associated factors 1, 2, 3 and 5 and mediates nuclear factor-kappaB activation. Biochem J (2003) 371:395–403. doi:10.1042/BJ20021730
8. Bover LC, Cardó-Vila M, Kuniyasu A, Sun J, Rangel R, Takeya M, et al. A previously unrecognized protein-protein interaction between TWEAK and CD163: potential biological implications. J Immunol (2007) 178:8183–94.
9. Moreno JA, Muñoz-García B, Martín-Ventura JL, Madrigal-Matute J, Orbe J, Páramo JA, et al. The CD163-expressing macrophages recognize and internalize TWEAK: potential consequences in atherosclerosis. Atherosclerosis (2009) 207:103–10. doi:10.1016/j.atherosclerosis.2009.04.033
10. Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature (2001) 409:198–201. doi:10.1038/35051594
11. Fick A, Lang I, Schäfer V, Seher A, Trebing J, Weisenberger D, et al. Studies of binding of tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) to fibroblast growth factor 14 (Fn14). J Biol Chem (2012) 287:484–95. doi:10.1074/jbc.M111.287656
12. Polek TC, Talpaz M, Darnay BG, Spivak-Kroizman T. TWEAK mediates signal transduction and differentiation of RAW264.7 cells in the absence of Fn14/TweakR. Evidence for a second TWEAK receptor. J Biol Chem (2003) 278:32317–23. doi:10.1074/jbc.M302518200
13. Burkly LC, Michaelson JS, Hahm K, Jakubowski A, Zheng TS. TWEAKing tissue remodeling by a multifunctional cytokine: role of TWEAK/Fn14 pathway in health and disease. Cytokine (2007) 40:1–16. doi:10.1016/j.cyto.2007.09.007
14. Tirnitz-Parker JE, Viebahn CS, Jakubowski A, Klopcic BR, Olynyk JK, Yeoh GC, et al. Tumor necrosis factor like weak inducer of apoptosis is a mitogen for liver progenitor cells. Hepatology (2010) 52:291–302. doi:10.1002/hep.23663
15. Mustonen E, Sakkinen H, Tokola H, Isopoussu E, Aro J, Leskinen H, et al. Tumour necrosis factor-like weak inducer of apoptosis (TWEAK) and its receptor Fn14 during cardiac remodelling in rats. Acta Physiol (Oxf) (2010) 199:11–22. doi:10.1111/j.1748-1716.2010.02080.x
16. Dohi T, Borodovsky A, Wu P, Shearstone JR, Kawashima R, Runkel L, et al. TWEAK/Fn14 pathway: a nonredundant role in intestinal damage in mice through a TWEAK/intestinal epithelial cell axis. Gastroenterology (2009) 136:912–23. doi:10.1053/j.gastro.2008.11.017
17. Mittal A, Bhatnagar S, Kumar A, Lach-Trifilieff E, Wauters S, Li H, et al. The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J Cell Biol (2010) 188:833–49. doi:10.1083/jcb.200909117
18. Muñoz-García B, Martín-Ventura JL, Martínez E, Sánchez S, Hernández G, Ortega L, et al. Fn14 is upregulated in cytokine-stimulated vascular smooth muscle cells and is expressed in human carotid atherosclerotic plaques: modulation by atorvastatin. Stroke (2006) 37:2044–53. doi:10.1161/01.STR.0000230648.00027.00
19. Desplat-Jégo S, Creidy R, Varriale S, Allaire N, Luo Y, Bernard D, et al. Anti-TWEAK monoclonal antibodies reduce immune cell infiltration in the central nervous system and severity of experimental autoimmune encephalomyelitis. Clin Immunol (2005) 117:15–23. doi:10.1016/j.clim.2005.06.005
20. Justo P, Sanz AB, Sanchez-Nino MD, Winkles JA, Lorz C, Egido J, et al. Cytokine cooperation in renal tubular cell injury: the role of TWEAK. Kidney Int (2006) 70:1750–8. doi:10.1038/sj.ki.5001866
21. Jain M, Jakubowski A, Cui L, Shi J, Su L, Bauer M, et al. A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure. Circulation (2009) 119:2058–68. doi:10.1161/CIRCULATIONAHA.108.837286
22. Brown SA, Ghosh A, Winkles JA. Full-length, membrane-anchored TWEAK can function as a juxtacrine signaling molecule and activate the NF-kappaB pathway. J Biol Chem (2010) 285:17432–41. doi:10.1074/jbc.M110.131979
23. Donohue PJ, Richards CM, Brown SA, Hanscom HN, Buschman J, Thangada S, et al. TWEAK is an endothelial cell growth and chemotactic factor that also potentiates FGF-2 and VEGF-A mitogenic activity. Arterioscler Thromb Vasc Biol (2003) 23:594–600. doi:10.1161/01.ATV.0000062883.93715.37
24. Maecker H, Varfolomeev E, Kischkel F, Lawrence D, Leblanc H, Lee W, et al. TWEAK attenuates the transition from innate to adaptive immunity. Cell (2005) 123:931–44. doi:10.1016/j.cell.2005.09.022
25. Chorianopoulos E, Heger T, Lutz M, Frank D, Bea F, Katus HA, et al. FGF inducible 14-kDa protein (Fn14) is regulated via the RhoA/ROCK kinase pathway in cardiomyocytes and mediates nuclear factor-kappaB activation by TWEAK. Basic Res Cardiol (2010) 105:301–13. doi:10.1007/s00395-009-0046-y
26. Nakayama M, Kayagaki N, Yamaguchi N, Okumura K, Yagita H. Involvement of TWEAK in interferon γ-stimulated monocyte cytotoxicity. J Exp Med (2000) 192:1373–80. doi:10.1084/jem.192.9.1373
27. Saitoh T, Nakayama M, Nakano H, Yagita H, Yamamoto N, Yamaoka S. TWEAK induces NF-kappaB2 p100 processing and long lasting NF-kappaB activation. J Biol Chem (2003) 278:36005–12. doi:10.1074/jbc.M304266200
28. Muñoz-García B, Moreno JA, López-Franco O, Sanz AB, Martín-Ventura JL, Blanco J, et al. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) enhances vascular and renal damage induced by hyperlipidemic diet in ApoE-knockout mice. Arterioscler Thromb Vasc Biol (2009) 29:2061–8. doi:10.1161/ATVBAHA.109.194852
29. Moreno JA, Sastre C, Madrigal-Matute J, Muñoz-García B, Ortega L, Burkly LC, et al. HMGB1 expression and secretion are increased via TWEAK-Fn14 interaction in atherosclerotic plaques and cultured monocytes. Arterioscler Thromb Vasc Biol (2013) 33:612–20. doi:10.1161/ATVBAHA.112.300874
30. Sanz AB, Sanchez-Niño MD, Izquierdo MC, Jakubowski A, Justo P, Blanco-Colio LM, et al. TWEAK activates the non-canonical NFkappaB pathway in murine renal tubular cells: modulation of CCL21. PLoS One (2010) 5:e8955. doi:10.1371/journal.pone.0008955
31. Damås JK, Smith C, Øie E, Fevang B, Halvorsen B, Waehre T, et al. Enhanced expression of the homeostatic chemokines CCL19 and CCL21 in clinical and experimental atherosclerosis: possible pathogenic role in plaque destabilization. Arterioscler Thromb Vasc Biol (2007) 27:614–20. doi:10.1161/01.ATV.0000255581.38523.7c
32. Burkly LC, Michaelson JS, Zheng TS. TWEAK/Fn14 pathway: an immunological switch for shaping tissue responses. Immunol Rev (2011) 244:99–114. doi:10.1111/j.1600-065X.2011.01054.x
33. Hosokawa Y, Hosokawa I, Ozaki K, Nakae H, Matsuo T. Proinflammatory effects of tumour necrosis factor-like weak inducer of apoptosis (TWEAK) on human gingival fibroblasts. Clin Exp Immunol (2006) 146:540–9. doi:10.1111/j.1365-2249.2006.03233.x
34. Li H, Mittal A, Paul PK, Kumar M, Srivastava DS, Tyagi SC, et al. Tumor necrosis factor-related weak inducer of apoptosis augments matrix metalloproteinase 9 (MMP-9) production in skeletal muscle through the activation of nuclear factor-kappaB-inducing kinase and p38 mitogen-activated protein kinase: a potential role of MMP-9 in myopathy. J Biol Chem (2009) 284:4439–50. doi:10.1074/jbc.M805546200
35. Srivastava AK, Qin X, Wedhas N, Arnush M, Linkhart TA, Chadwick RB, et al. Tumor necrosis factor-alpha augments matrix metalloproteinase-9 production in skeletal muscle cells through the activation of transforming growth factor-beta-activated kinase 1 (TAK1)-dependent signaling pathway. J Biol Chem (2007) 282:35113–24. doi:10.1074/jbc.M705329200
36. Kumar M, Makonchuk DY, Li H, Mittal A, Kumar A. TNF-like weak inducer of apoptosis (TWEAK) activates proinflammatory signaling pathways and gene expression through the activation of TGF-beta-activated kinase 1. J Immunol (2009) 182:2439–48. doi:10.4049/jimmunol.0803357
37. Dogra C, Hall SL, Wedhas N, Linkhart TA, Kumar A. Fibroblast growth factor inducible 14 (Fn14) is required for the expression of myogenic regulatory factors and differentiation of myoblasts into myotubes. Evidence for TWEAK-independent functions of Fn14 during myogenesis. J Biol Chem (2007) 282:15000–10. doi:10.1074/jbc.M608668200
38. Jakubowski A, Browning B, Lukashev M, Sizing I, Thompson JS, Benjamin CD, et al. Dual role for TWEAK in angiogenic regulation. J Cell Sci (2002) 115:267–74.
39. Lynch CN, Wang YC, Lund JK, Chen YW, Leal JA, Wiley SR. TWEAK induces angiogenesis and proliferation of endothelial cells. J Biol Chem (1999) 274:8455–9. doi:10.1074/jbc.274.13.8455
40. Meighan-Mantha RL, Hsu DK, Guo Y, Brown SA, Feng SL, Peifley KA, et al. The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J Biol Chem (1999) 274:33166–76. doi:10.1074/jbc.274.46.33166
41. Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov (2008) 7:411–25. doi:10.1038/nrd2488
42. Glass CK, Witzum JL. Atherosclerosis: the road ahead. Cell (2001) 104:503–16. doi:10.1016/S0092-8674(01)00238-0
43. Moreno JA, Dejouvencel T, Labreuche J, Smadja DM, Dussiot M, Martin-Ventura JL, et al. Peripheral artery disease is associated with a high CD163/TWEAK plasma ratio. Arterioscler Thromb Vasc Biol (2010) 30:1253–62. doi:10.1161/ATVBAHA.110.203364
44. Martín-Ventura JL, Lindholt JS, Moreno JA, Vega de Céniga M, Meilhac O, Michel JB, et al. Soluble TWEAK plasma levels predict expansion of human abdominal aortic aneurysms. Atherosclerosis (2011) 214:486–9. doi:10.1016/j.atherosclerosis.2010.11.009
45. Harada N, Nakayama M, Nakano H, Fukuchi Y, Yagita H, Okumura K. Pro-inflammatory effect of TWEAK/Fn14 interaction on human umbilical vein endothelial cells. Biochem Biophys Res Commun (2002) 299:488–93. doi:10.1016/S0006-291X(02)02670-0
46. Han S, Yoon K, Lee K, Kim K, Jang H, Lee NK, et al. TNF-related weak inducer of apoptosis receptor, a TNF receptor superfamily member, activates NF-kappa B through TNF receptor-associated factors. Biochem Biophys Res Commun (2003) 305:789–96. doi:10.1016/S0006-291X(03)00852-0
47. Pamukcu B, Lip GYH, Shantsila E. The nuclear factor – kappa B pathway in atherosclerosis: a potential therapeutic target for atherothrombotic vascular disease. Thromb Res (2011) 128:117–23. doi:10.1016/j.thromres.2011.03.025
48. Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, et al. Activated transcription factor nuclear factor kappa B is present in the atherosclerotic lesion. J Clin Invest (1996) 97:1715–22. doi:10.1172/JCI118598
49. Bourcier T, Sukhova G, Libby P. The nuclear factor kappa-B signaling pathway participates in dysregulation of vascular smooth muscle cells in vitro and in human atherosclerosis. J Biol Chem (1997) 272:15817–24. doi:10.1074/jbc.272.25.15817
50. Martin-Ventura JL, Blanco-Colio LM, Munoz-Garcia B, Gomez-Hernandez A, Arribas A, Ortega L, et al. NF-kappaB activation and Fas ligand overexpression in blood and plaques of patients with carotid atherosclerosis: potential implication in plaque instability. Stroke (2004) 35:458–63. doi:10.1161/01.STR.0000114876.51656.7A
51. Droga C, Changotra H, Mohan S, Kumar A. Tumor necrosis factor-like inducer of apoptosis inhibits skeletal myogenesis through sustained activation of nuclear factor kappa B and degradation of MyoD protein. J Biol Chem (2006) 281:10327–33. doi:10.1074/jbc.M511131200
52. Ho DH, Vu H, Brown SA, Donohue PJ, Hanscom HN, Winkles JA. Soluble tumor necrosis factor-like weak inducer of apoptosis overexpression in HEK293 cells promotes tumor growth and angiogenesis in athymic nude mice. Cancer Res (2004) 64:8968–72. doi:10.1158/0008-5472.CAN-04-1879
53. Sastre C, Fernández-Laso V, Madrigal-Matute J, Muñoz-García B, Moreno JA, Pastor-Vargas C, et al. Genetic deletion or TWEAK blocking antibody administration reduce atherosclerosis and enhance plaque stability in mice. J Cell Mol Med (2013). doi:10.1111/jcmm.12221
54. Andersson U, Erlandsson-Harris H, Yang H, Tracey KJ. HMGB1 as a DNA-binding cytokine. J Leukoc Biol (2002) 72:1084–91.
55. Schapira K, Burkly LC, Zheng TS, Wu P, Groeneweg M, Rousch M, et al. Fn14-Fc fusion protein regulates atherosclerosis in ApoE−/− mice and inhibits macrophage lipid uptake in vitro. Arterioscler Thromb Vasc Biol (2009) 29:2021–7. doi:10.1161/ATVBAHA.109.195040
56. Ketelhuth DF, Bäck M. The role of matrix metalloproteinases in atherothrombosis. Curr Atheroscler Rep (2011) 13:162–9. doi:10.1007/s11883-010-0159-7
57. Kim SH, Lee WH, Kwon BS, Oh GT, Choi YH, Park JE. Tumor necrosis factor receptor superfamily 12 may destabilize atherosclerotic plaques by inducing matrix metalloproteinases. Circ J (2001) 65:136–8. doi:10.1253/jcj.65.136
58. Mackman N. Role of tissue factor in hemostasis and thrombosis. Blood Cells Mol Dis (2006) 36:104–7. doi:10.1016/j.bmcd.2005.12.008
59. Agirbasli M. Pivotal role of plasminogen-activator inhibitor 1 in vascular disease. Int J Clin Pract (2005) 59:102–6. doi:10.1111/j.1742-1241.2005.00379.x
60. Muñoz-García B, Madrigal-Matute J, Moreno JA, Martin-Ventura JL, López-Franco O, Sastre C, et al. TWEAK-Fn14 interaction enhances plasminogen activator inhibitor 1 and tissue factor expression in atherosclerotic plaques and in cultured vascular smooth muscle cells. Cardiovasc Res (2011) 89:225–33. doi:10.1093/cvr/cvq278
61. Lloyd-Jones D, Adams R, Carnethon M, De SG, Ferguson TB, Flegal K, et al. Heart disease and stroke statistics – 2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation (2009) 119:480–6. doi:10.1161/CIRCULATIONAHA.108.191259
62. Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci (1999) 22:391–7. doi:10.1016/S0166-2236(99)01401-0
63. Inta I, Frauenknecht K, Dörr H, Kohlhof P, Rabsilber T, Auffarth GU, et al. Induction of the cytokine TWEAK and its receptor Fn14 in ischemic stroke. J Neurol Sci (2008) 275:117–20. doi:10.1016/j.jns.2008.08.005
64. Potrovita I, Zhang W, Burkly L, Hahm K, Lincecum J, Wang MZ, et al. Tumor necrosis factor-like weak inducer of apoptosis-induced neurodegeneration. J Neurosci (2004) 24:8237–44. doi:10.1523/JNEUROSCI.1089-04.2004
65. Yepes M, Brown SA, Moore EG, Smith EP, Lawrence DA, Winkles JA. A soluble Fn14-Fc decoy receptor reduces infarct volume in a murine model of cerebral ischemia. Am J Pathol (2005) 166:511–20. doi:10.1016/S0002-9440(10)62273-0
66. Zhang X, Winkles JA, Gongora MC, Polavarapu R, Michaelson JS, Hahm K, et al. TWEAK-Fn14 pathway inhibition protects the integrity of the neurovascular unit during cerebral ischemia. J Cereb Blood Flow Metab (2007) 27:534–44. doi:10.1038/sj.jcbfm.9600368
67. Haile WB, Echeverry R, Wu F, Guzman J, An J, Wu J, et al. Tumor necrosis factor-like weak inducer of apoptosis and fibroblast growth factor-inducible 14 mediate cerebral ischemia-induced poly (ADP-ribose) polymerase-1 activation and neuronal death. Neuroscience (2010) 171:1256–64. doi:10.1016/j.neuroscience.2010.10.029
68. del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab (2003) 23:879–94. doi:10.1097/01.WCB.0000078322.96027.78
69. Rieckmann P, Engelhardt B. Building up the blood-brain barrier. Nat Med (2003) 9:828–9. doi:10.1038/nm0703-828
70. Polavarapu R, Gongora MC, Winkles JA, Yepes M. Tumor necrosis factor-like weak inducer of apoptosis increases the permeability of the neurovascular unit through nuclear factor-kappa B pathway activation. J Neurosci (2005) 25:10094–100.
71. Blanco-Colio LM, Martín-Ventura JL, Muñóz-García B, Orbe J, Páramo JA, Michel JB, et al. Identification of soluble tumor necrosis factor-like weak inducer of apoptosis (sTWEAK) as a possible biomarker of subclinical atherosclerosis. Arterioscler Thromb Vasc Biol (2007) 27:916–22. doi:10.1161/01.ATV.0000258972.10109.ff
72. Kralisch S, Ziegelmeier M, Bachmann A, Seeger J, Lössner U, Blüher M, et al. Serum levels of the atherosclerosis biomarker sTWEAK are decreased in type 2 diabetes and end-stage renal disease. Atherosclerosis (2008) 199:440–4. doi:10.1016/j.atherosclerosis.2007.10.022
73. Carrero JJ, Ortiz A, Qureshi AR, Martín-Ventura JL, Heimbürger O, Marrón B, et al. Additive effects of soluble TWEAK and inflammation on mortality in hemodialysis patients. Clin J Am Soc Nephrol (2009) 4:110–8. doi:10.2215/CJN.02790608
74. Yilmaz MI, Carrero JJ, Ortiz A, Martín-Ventura JL, Sonmez A, Saglam M, et al. Soluble TWEAK plasma levels as a novel biomarker of endothelial function in patients with chronic kidney disease. Clin J Am Soc Nephrol (2009) 4:1716–23. doi:10.2215/CJN.02760409
75. Yilmaz MI, Carrero JJ, Martín-Ventura JL, Sonmez A, Saglam M, Celik T, et al. Combined therapy with renin-angiotensin system and calcium channel blockers in type 2 diabetic hypertensive patients with proteinuria: effects on soluble TWEAK, PTX3, and flow-mediated dilation. Clin J Am Soc Nephrol (2010) 5:1174–81. doi:10.2215/CJN.01110210
76. Yilmaz MI, Sonmez A, Ortiz A, Saglam M, Kilic S, Eyileten T, et al. Soluble TWEAK and PTX3 in nondialysis CKD patients: impact on endothelial dysfunction and cardiovascular outcomes. Clin J Am Soc Nephrol (2011) 6:785–92. doi:10.2215/CJN.09231010
77. Jelic-Ivanovic Z, Bujisic N, Spasic S, Bogavac-Stanojevic N, Spasojevic-Kalimanovska V, Kotur-Stevuljevic J. Circulating sTWEAK improves the prediction of coronary artery disease. Clin Biochem (2009) 42:1381–6. doi:10.1016/j.clinbiochem.2009.06.001
78. Chorianopoulos E, Rosenberg M, Zugck C, Wolf J, Katus HA, Frey N. Decreased soluble TWEAK levels predict an adverse prognosis in patients with chronic stable heart failure. Eur J Heart Fail (2009) 11:1050–6. doi:10.1093/eurjhf/hfp139
79. Widlansky ME, Gokce N, Keaney JF Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol (2003) 42:1149–60. doi:10.1016/S0735-1097(03)00994-X
80. Vita JA, Keaney JF Jr. Endothelial function: a barometer for cardiovascular risk? Circulation (2002) 106:640–2. doi:10.1161/01.CIR.0000028581.07992.56
81. Bots ML, Hoes AW, Koudstaal PJ, Hofman A, Grobbee DE. Common carotid intima-media thickness and risk of stroke and myocardial infarction: the Rotterdam study. Circulation (1997) 128:262–9. doi:10.1161/01.CIR.96.5.1432
82. Hassan SB, El-demery AB, Ahmed AI, Abukhalil RE. Soluble TWEAK and cardiovascular morbidity and mortality in chronic kidney disease patients. Arab J Nephrol Transplant (2012) 5:27–32.
83. Valdivielso JM, Coll B, Martín-Ventura JL, Moreno JA, Egido J, Fernández E, et al. Soluble TWEAK is associated with atherosclerotic burden in patients with chronic kidney disease. J Nephrol (2013) 26:1105–13. doi:10.5301/jn.5000245
84. Turkmen K, Tonbul HZ, Erdur FM, Toker A, Biyik Z, Ozbiner H, et al. Soluble TWEAK independently predicts atherosclerosis in renal transplant patients. BMC Nephrol (2013) 14:144. doi:10.1186/1471-2369-14-144
85. Gungor O, Kismali E, Sisman AR, Kircelli F, Carrero JJ, Tatar E, et al. The relationships between serum sTWEAK, FGF-23 levels, and carotid atherosclerosis in renal transplant patients. Ren Fail (2013) 35:77–81. doi:10.3109/0886022X.2012.734890
86. Urbonaviciene G, Martin-Ventura JL, Lindholt JS, Urbonavicius S, Moreno JA, Egido J, et al. Impact of soluble TWEAK and CD163/TWEAK ratio on long-term cardiovascular mortality in patients with peripheral arterial disease. Atherosclerosis (2011) 219:892–9. doi:10.1016/j.atherosclerosis.2011.09.016
87. McMahon M, Skaggs BJ, Grossman J, Sahakian L, Fitzgerald J, Wong WK, et al. A panel of biomarkers is associated with increased risk for the presence and progression of atherosclerosis in women with systemic lupus erythematosus. Arthritis Rheum (2013) 66(1):130–9. doi:10.1002/art.38204
88. Richter B, Rychli K, Hohensinner PJ, Berger R, Mörtl D, Neuhold S, et al. Differences in the predictive value of tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in advanced ischemic and non-ischemic heart failure. Atherosclerosis (2010) 213:545–8. doi:10.1016/j.atherosclerosis.2010.08.061
89. Coope HJ, Atkinson PG, Huhse B, Belich M, Janzen J, Holman MJ, et al. CD40 regulates the processing of NF-[kappa]B2 p100 to p52. EMBO J (2000) 21:5375–85. doi:10.1093/emboj/cdf542
90. Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol (2002) 3:958–65. doi:10.1038/ni842
91. Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, et al. The lymphotoxin-b receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity (2002) 17:525–35. doi:10.1016/S1074-7613(02)00423-5
92. Novack DV, Yin L, Hagen-Stapleton A, Schreiber RD, Goeddel DV, Ross FP, et al. The IkappaB function of NF-kappaB2 p100 controls stimulated osteoclastogenesis. J Exp Med (2003) 198:771–81. doi:10.1084/jem.20030116
93. Ramakrishnan P, Wang W, Wallach D. Receptor-specific signaling for both the alternative and the canonical NF-kappaB activation pathways by NF-kappaB-inducing kinase. Immunity (2004) 21:477–89. doi:10.1016/j.immuni.2004.08.009
94. Lutgens E, Lievens D, Beckers L, Wijnands E, Soehnlein O, Zernecke A, et al. Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med (2010) 207:391–404. doi:10.1084/jem.20091293
95. Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, Bavendiek U, et al. CD40 ligand mediates inflammation independently of CD40 by interaction with Mac-1. Circulation (2007) 115:1571–80. doi:10.1161/CIRCULATIONAHA.106.683201
96. Lutgens E, Gorelik L, Daemen MJ, de Muinck ED, Grewal IS, Koteliansky VE, et al. Requirement for CD154 in the progression of atherosclerosis. Nat Med (1999) 5:1313–6. doi:10.1038/15271
97. Lutgens E, Cleutjens KB, Heeneman S, Koteliansky VE, Burkly LC, Daemen MJ. Both early and delayed anti-CD40L antibody treatment induces a stable plaque phenotype. Proc Natl Acad Sci U S A (2000) 97:7464–9. doi:10.1073/pnas.97.13.7464
98. Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature (1998) 394:200–3. doi:10.1038/28204
99. Schonbeck U, Sukhova GK, Shimizu K, Match F, Libby P. Inhibition of CD40 signaling limits evolution of established atherosclerosis in mice. Proc Natl Acad Sci U S A (2000) 97:7458–63. doi:10.1073/pnas.97.13.7458
100. Bavendiek U, Zirlik A, LaClair S, MacFarlane L, Libby P, Schönbeck U. Atherogenesis in mice does not require CD40 ligand from bone marrow-derived cells. Arterioscler Thromb Vasc Biol (2005) 25:1244–9. doi:10.1161/01.ATV.0000161420.55482.ef
101. Smook ML, Heeringa P, Damoiseaux JG, Daemen MJ, de Winther MP, Gijbels MJ, et al. Leukocyte CD40L deficiency affects the CD25(+) CD4 T cell population but does not affect atherosclerosis. Atherosclerosis (2005) 183:275–82. doi:10.1016/j.atherosclerosis.2005.03.051
102. Lievens D, Zernecke A, Seijkens T, Soehnlein O, Beckers L, Munnix IC, et al. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood (2010) 116:4317–27. doi:10.1182/blood-2010-01-261206
103. Kyaw T, Tay C, Hosseini H, Kanellakis P, Gadowski T, MacKay F, et al. Depletion of B2 but not B1a B cells in BAFF receptor-deficient ApoE mice attenuates atherosclerosis by potently ameliorating arterial inflammation. PLoS One (2012) 7:e29371. doi:10.1371/journal.pone.0029371
104. Sage AP, Tsiantoulas D, Baker L, Harrison J, Masters L, Murphy D, et al. BAFF receptor deficiency reduces the development of atherosclerosis in mice – brief report. Arterioscler Thromb Vasc Biol (2012) 32:1573–6. doi:10.1161/ATVBAHA.111.244731
105. Kyaw T, Cui P, Tay C, Kanellakis P, Hosseini H, Liu E, et al. BAFF receptor mAb treatment ameliorates development and progression of atherosclerosis in hyperlipidemic ApoE(−/−) mice. PLoS One (2013) 8:e60430. doi:10.1371/journal.pone.0060430
106. Gräbner R, Lötzer K, Döpping S, Hildner M, Radke D, Beer M, et al. Lymphotoxin β receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE −/− mice. J Exp Med (2009) 206:233–48. doi:10.1084/jem.20080752
107. Bennett BJ, Scatena M, Kirk EA, Rattazzi M, Varon RM, Averill M, et al. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE-/- mice. Arterioscler Thromb Vasc Biol (2006) 26:2117–24. doi:10.1161/01.ATV.0000236428.91125.e6
108. Morony S, Tintut Y, Zhang Z, Cattley RC, Van G, Dwyer D, et al. Osteoprotegerin inhibits vascular calcification without affecting atherosclerosis in LDLR(−/−) mice. Circulation (2008) 117:411–20. doi:10.1161/CIRCULATIONAHA.107.707380
109. van Olffen RW, de Bruin AM, Vos M, Staniszewska AD, Hamann J, van Lier RA, et al. CD70-driven chronic immune activation is protective against atherosclerosis. J Innate Immun (2010) 2:344–52. doi:10.1159/000314772
110. Mach F, Schonbeck U, Sukhova GK, Bourcier T, Bonnefoy JY, Pober JS, et al. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci USA (1997) 94:1931–6.
111. Schonbeck U, Libby P. CD40 signaling and plaque instability. Circ Res (2001) 89:1092–103. doi:10.1161/hh2401.101272
112. Lievens D, Eijgelaar WJ, Biessen EA, Daemen MJ, Lutgens E. The multi-functionality of CD40L and its receptor CD40 in atherosclerosis. Thromb Haemost (2009) 102:206–14. doi:10.1160/TH09-01-0029
113. Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med (2010) 207:1579–87. doi:10.1084/jem.20100155
114. Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K, et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol (2010) 185:4410–9. doi:10.4049/jimmunol.1000033
115. Kyaw T, Tay C, Krishnamurthi S, Kanellakis P, Agrotis A, Tipping P, et al. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ Res (2011) 109:830–40. doi:10.1161/CIRCRESAHA.111.248542
116. Mackay F, Figgett WA, Saulep D, Lepage M, Hibbs ML. B-cell stage and context-dependent requirements for survival signals from BAFF and the B-cell receptor. Immunol Rev (2010) 237:205–25. doi:10.1111/j.1600-065X.2010.00944.x
117. Miller DJ, Hayes CE. Phenotypic and genetic characterization of a unique B lymphocyte deficiency in strain A/WySnJ mice. Eur J Immunol (1991) 21:1123–30. doi:10.1002/eji.1830210506
118. Houtkamp MA, de Boer OJ, van der Loos CM, van der Wal AC, Becker AE. Adventitial infiltrates associated with advanced atherosclerotic plaques: structural organization suggests generation of local humoral immune responses. J Pathol (2001) 193:263–9. doi:10.1002/1096-9896(2000)9999:9999<::AID-PATH774>3.0.CO;2-N
119. Nolte MA, Belien JA, Schadee-Eestermans I, Jansen W, Unger WW, van Rooijen N, et al. A conduit system distributes chemokines and small blood-borne molecules through the splenic white pulp. J Exp Med (2003) 198:505–12. doi:10.1084/jem.20021801
120. Tintut Y, Demer L. Role of osteoprotegerin and its ligands and competing receptors in atherosclerotic calcification. J Investig Med (2006) 54:395–401. doi:10.2310/6650.2006.06019
121. Mosheimer BA, Kaneider NC, Feistritzer C, Sturn DH, Wiedermann CJ. Expression and function of RANK in human monocyte chemotaxis. Arthritis Rheum (2004) 50:2309–16. doi:10.1002/art.20352
122. Sandberg WJ, Yndestad A, Øie E, Smith C, Ueland T, Ovchinnikova O, et al. Enhanced T-cell expression of RANK ligand in acute coronary syndrome: possible role in plaque destabilization. Arterioscler Thromb Vasc Biol (2006) 26:857–63. doi:10.1161/01.ATV.0000204334.48195.6A
123. Kim J, Min JK, Park JA, Doh HJ, Choi YS, Rho J, et al. Receptor activator of nuclear factor kappaB ligand is a novel inducer of tissue factor in macrophages. Circ Res (2010) 107:871–6. doi:10.1161/CIRCRESAHA.110.221168
124. Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol (2005) 23:23–68. doi:10.1146/annurev.immunol.23.021704.115839
125. Chorianopoulos E, Jarr K, Steen H, Giannitsis E, Frey N, Katus HA. Soluble TWEAK is markedly upregulated in patients with ST-elevation myocardial infarction and related to an adverse short-term outcome. Atherosclerosis (2010) 211:322–6. doi:10.1016/j.atherosclerosis.2010.02.016
126. Blanco-Colio LM, Martín-Ventura JL, Carrero JJ, Yilmaz MI, Moreno JA, Gómez-Guerrero C, et al. Vascular proteomics and the discovery process of clinical biomarkers: the case of TWEAK. Proteomics Clin Appl (2011) 5:281–8. doi:10.1002/prca.201000102
Keywords: atherosclerosis, stroke, biomarker, TWEAK, TNF superfamily
Citation: Blanco-Colio LM (2014) TWEAK/Fn14 axis: a promising target for the treatment of cardiovascular diseases. Front. Immunol. 5:3. doi: 10.3389/fimmu.2014.00003
Received: 13 September 2013; Accepted: 03 January 2014;
Published online: 20 January 2014.
Edited by:
Linda C. Burkly, Biogen Idec, Inc., USAReviewed by:
Maureen McMahon, University of California Los Angeles, USARonglih Liao, Brigham and Women’s Hospital and Harvard Medical School, USA
Copyright: © 2014 Blanco-Colio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis M. Blanco-Colio, Vascular Reseach Laboratory, IIS-Fundación Jiménez Díaz, Avenue Reyes Católicos 2, Madrid 28040, Spain e-mail:bGJsYW5jb0BmamQuZXM=