 Gongxian Liao1*
Gongxian Liao1* Michael S. O’Keeffe1
Michael S. O’Keeffe1 Guoxing Wang1
Guoxing Wang1 Boaz van Driel1
Boaz van Driel1 Rene de Waal Malefyt2
Rene de Waal Malefyt2 Hans-Christian Reinecker3
Hans-Christian Reinecker3 Roland W. Herzog4
Roland W. Herzog4 Cox Terhorst1*
Cox Terhorst1*- 1Division of Immunology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA
- 2Biologics Discovery, Merck Research Laboratories, Palo Alto, CA, USA
- 3Gastrointestinal Unit and Center for the Study of Inflammatory Bowel Disease, Department of Medicine, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
- 4Department of Pediatrics, University of Florida, Gainesville, FL, USA
Glucocorticoid-induced tumor necrosis factor receptor family-related protein (TNFRSF18, CD357) is constitutively expressed on regulatory T cells (Tregs) and is inducible on effector T cells. In this report, we examine the role of glucocorticoid-induced TNF receptor family-related protein ligand (GITR-L), which is expressed by antigen presenting cells, on the development and expansion of Tregs. We found that GITR-L is dispensable for the development of naturally occurring FoxP3+ Treg cells in the thymus. However, the expansion of Treg in GITR-L−/− mice is impaired after injection of the dendritic cells (DCs) inducing factor Flt3 ligand. Furthermore, DCs from the liver of GITR-L−/− mice were less efficient in inducing proliferation of antigen-specific Treg cells in vitro than the same cells from WT littermates. Upon gene transfer of ovalbumin into hepatocytes of GITR-L−/−FoxP3(GFP) reporter mice using adeno-associated virus (AAV8-OVA) the number of antigen-specific Treg in liver and spleen is reduced. The reduced number of Tregs resulted in an increase in the number of ovalbumin specific CD8+ T effector cells. This is highly significant because proliferation of antigen-specific CD8+ cells itself is dependent on the presence of GITR-L, as shown by in vitro experiments and by adoptive transfers into GITR-L−/−Rag−/− and Rag−/− mice that had received AAV8-OVA. Surprisingly, administering αCD3 significantly reduced the numbers of FoxP3+ Treg cells in the liver and spleen of GITR-L−/− but not WT mice. Because soluble Fc-GITR-L partially rescues αCD3 induced in vitro depletion of the CD103+ subset of FoxP3+CD4+ Treg cells, we conclude that expression of GITR-L by antigen presenting cells is requisite for optimal Treg-mediated regulation of immune responses including those in response during gene transfer.
Introduction
CD4+CD25+FoxP3+ regulatory T cells (Treg), which develop in the thymus or can be induced in peripheral organs, control many aspects of the immune response (1–4). Tregs constitutively express glucocorticoid-induced tumor necrosis factor receptor family-related protein (GITR, TNFRSF18, CD357), which is inducible on effector T cells (Teffs) (2, 5–8). Using Fc-GITR-L, a soluble form of the natural ligand of GITR, we found recently that glucocorticoid-induced TNF receptor family-related protein ligand (GITR-L) preferentially induces the in vivo and in vitro expansion of functionally competent Tregs (9). Furthermore, a significantly higher proportion of FoxP3+ Tregs is also found in GITR-L transgenic mouse strains, in which the expression of GITR-L is under control of the CD19- and MHC-II-promoter respectively (10, 11). GITR-L is not expressed by T cells (8), but is found on plasmacytoid dendritic cells (pDCs), Langerhans cells, macrophage subpopulations, and endothelial cells (12–15). Here we use GITR-L−/− mice to examine the role of GITR-L in the induction of Tregs and Treg-mediated suppression in response to hepatic gene transfer with the adeno-associated viral vector AAV8.
Tolerance induction to specific foreign protein by hepatic gene transfer may be established in two steps. First, antigen-specific Tregs are de novo induced in the hepatic microenvironment. Second, antigen-specific Tregs are expanded systemically. Indeed, we previously found that transgene product-specific Treg actively suppresses antibody and T cell responses thereby ensuring long-term gene expression (16). Recently, studies in hemophilic mouse models have shown that AAV-mediated hepatic gene transfer can not only prevent but also reverse pathogenic antibody responses and desensitize from severe allergic reactions to the therapeutic coagulation factor IX protein (17–20). We have recently shown that the immune suppressive cytokine TGF-β is required for Treg induction in hepatic AAV gene transfer and thus necessary for suppression of antibody and CD8+ T cell responses against the transgene product (21). TGF-β, a cytokine highly expressed in mucosal tissues and sites of inflammation, plays a role in conversion of conventional peripheral CD4+ T cells into Treg, and TGF-β up-regulates expression of CD103 (Integrin αEβ7) (22), which is the primary ligand of E-cadherin, an epithelial adhesion molecule. Expression of CD103 marks a subset of peripheral inducible Tregs (about 20–30% of the CD4+FoxP3+ Tregs in the spleen), which inhibit graft-versus-host disease more potently than the CD4+CD25+ Tregs (23, 24).
In this study, we provide evidence in support of the concept that the interactions between GITR and GITR-L are requisite for optimal functioning of Tregs. To this end, we analyze GITR-L−/−FoxP3(GFP) and GITR-L−/−CX3CR1(GFP) mice after gene transfer of ovalbumin into hepatocytes with adeno-associated virus (AAV8-OVA). Coordinate expansion of Treg and dendritic cells (DCs) was assessed after injection of Flt3 ligand in GITR-L−/− mice. The interactions between antigen presenting cells and Tregs are also evaluated after administering αCD3 in GITR-L−/− mice or by co-activation with αCD3 and soluble Fc-GITR-L.
Materials and Methods
Mice
B6, OT-II Tg, and CX3CR1(GFP) reporter mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). OT-I × Rag-/- mice were purchased from Taconic Labs (Germantown, NY, USA). GITR-L-/- and FoxP3-IRES-EGFP-SV40 knock-in [FoxP3(GFP)] B6 mice were described previously (8, 25). GITR-L-/- mice were crossed with FoxP3(GFP) and CX3CR1(GFP) mice to generate GITR-L-/-FoxP3(GFP) and GITR-L-/-CX3CR1(GFP) B6 mice. All animals were housed in the Center for Life Science animal facility of BIDMC. The Guide for the Care and Use of Laboratory Animals was followed in the conduct of the animal studies of the Institutional Animal Care and Use Committee at BIDMC. Veterinary care was given to any animals requiring medical attention.
Antibodies
Anti-CD11b-PacBlu, αCD11b-FITC, αCD4-PE, αCD4-APC, αCD11c-APC, αCD11c-PE, αTCRvα2-PE, and αCD3ε(145-2C11) were purchased from BioLegend (San Diego, CA, USA). Anti-Ly6C-PerCP and αFoxP3-APC were products of eBioscience (San Jose, CA, USA). Anti-Ly6G-PE, αNK1.1-PE, αCD8α-PacBlu, αCD25-PE, and αCD103-Alexa Fluor 647 were products from BD Biosciences (San Jose, CA, USA). Flt3L-Fc fusion protein was purchased from BioXCell (West Lebanon, NH, USA). Anti-IL-2 was purchased from R&D Systems (Minneapolis, MN, USA). Fc-GITR-L fusion protein was produced as described previously (9).
AAV8-OVA Mediated Expression of Foreign Protein in Hepatocytes
AAV8-OVA vector (containing an ovalbumin expression cassette driven by AAV-EF1α) was packaged into serotype 8 capsid as described previously (16). Vector was injected i.v. into FoxP3(GFP) and GITR-L-/-FoxP3(GFP) mice at a dose of 1010 vector genome/mouse. Five weeks later, leukocytes from liver, spleen, and thymus were stained with TCRvα2.
Also, Ly6G-NK1.1-GFP+ cells FACS sorted from the liver of CX3CR1(GFP) mice 7 days after AAV8-OVA injection were incubated with OT-II CD4+ or CFSE-labeled OT-I CD8+ T cells for 3 days. OT-II CD4+ T cell cultures were stained with TCRvα2 and FoxP3. OT-I CD8+ T cell culture was stained with TCRvα2 and proliferating CD8+ cells were evaluated by CFSE dilution.
Induction of Dendritic Cells and Treg with Flt3L
Flt3L-Fc fusion protein (10 ng/mouse/injection) was i.p. injected into FoxP3(GFP) and GITR-L-/-FoxP3(GFP) mice for nine consecutive days as described previously (26). Leukocytes from the spleen and liver were analyzed at day 10.
Cellularity in Mice after αCD3-Mediated Activation of T Cells by In vivo
Anti-CD3ε was i.p. injected into CX3CR1(GFP) and GITR-L−/−CX3CR1(GFP) mice (20 μg/mouse, one injection). After 72 h, leukocytes of the spleen and liver were stained with CD4 and FoxP3. CX3CR1+ cells were evaluated by expression of the reporter gene GFP.
In vitro Activation of CD4+ T Cells
CD4+ T cells from the spleen of FoxP3(GFP) mice were negatively selected using a CD4+ T cells isolation kit (Miltenyi, Auburn, CA, USA) and were activated with αCD3-coupled microbeads in a round bottom 96-well plate in the presence or absence of Fc-GITR-L (1 μg/ml) for 2 days as described previously (9). Cells were stained with CD4 and CD103. Expression of FoxP3 was judged by the reporter protein EGFP. Cell numbers were counted with a Countess Automated Cell Counter (Invitrogen, Grand Island, NY, USA).
Isolation of Liver Leukocytes
Liver leukocytes were isolated as described previously (27). Briefly, liver was mashed and filtered through a 70 μM cell strainer. Hepatocytes and cell debris were removed by spinning at 300 rpm for 10 min. Supernatant was centrifuged at 1500 rpm for 10 min to collect cells. Leukocytes were isolated from the interface of a 40 and 70% Percoll gradient.
Statistical analysis used Prism 4.0c software (GraphPad, San Diego, CA, USA). Statistical comparisons were performed using the two-tailed Student’s t-test. Values of P < 0.05 were considered to be statistically significant.
Results
Flt3L-Induced Expansion of Treg was Impaired in GITR-L Deficient Mice Due to a Partially Reduced Number of Dendritic Cell Subpopulations
We previously found that after administering a Fc-GITR-L fusion protein to WT mice the number of Treg cells increased, which was confirmed by studies with GITR-L transgenic mice (9–11, 28). Surprisingly, we found that GITR-L was dispensable for the development of naturally occurring Treg, as the number of FoxP3+ Treg cells was normal in the thymus and spleen of GITR-L−/−FoxP3(GFP) mice under resting conditions (Figure 1A; Figure S1 in Supplementary Material).
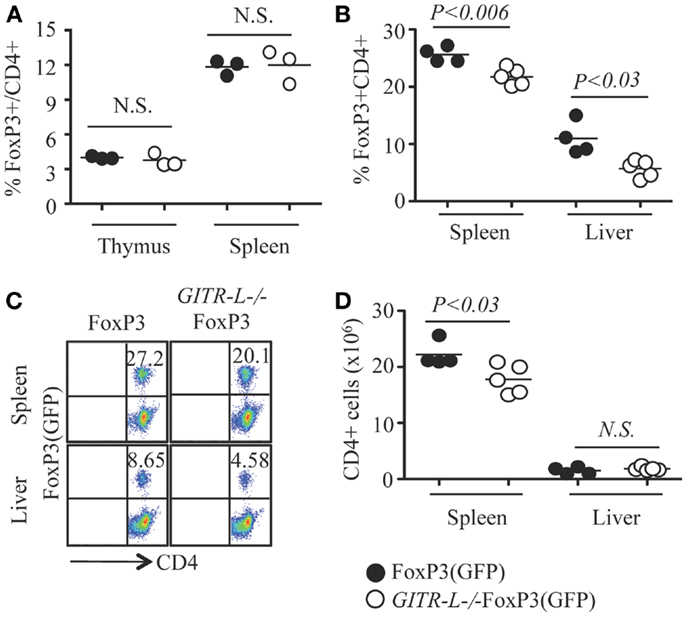
Figure 1. Flt3L-induced expansion of Treg. Flt3L-Fc fusion protein was injected into FoxP3(GFP) and GITR-L−/−FoxP3(GFP) mice (10 ng/mouse/injection, nine injections). CD4+CD8− T cells from the thymus, spleen, and/or liver were analyzed by FACS for FoxP3 expression based on the expression of reporter protein EGFP. (A) Percentages of CD4+FoxP3+ Treg in the thymus and spleen of FoxP3(GFP) and GITR-L−/−FoxP3(GFP) mice without stimulation. Percentages (B) and representative staining (C) of Treg in the spleen and liver after administering Flt3L. (D) Number of CD4+ T cells in the spleen and liver of FoxP3(GFP) and GITR-L−/−FoxP3(GFP) mice after administering Flt3L. Filled circle represents FoxP3(GFP) mouse. Open circle represents GITR-L−/−FoxP3(GFP) mouse. Each circle represents one mouse.
To further investigate the role of GITR-L in controlling Treg development, we assessed the consequences of injecting Fms-related tyrosine kinase 3 ligand (Flt3L) into FoxP3(GFP) and GITR-L−/−FoxP3(GFP) mice for nine consecutive days. Not only is Flt3L a potent inducer of DC and macrophage proliferation (26, 29), several phagocyte subpopulations express GITR-L (12, 30). After the injection of Fc-Flt3L fusion protein, both the numbers and the frequency of FoxP3+ Treg were significantly increased in the spleen and liver. This Fc-Flt3L-induced expansion was, however, significantly reduced in GITR-L−/−FoxP3(GFP) mice (Figures 1B,C). The total number of CD4+ T cells in the spleen was also lower in GITR-L−/−FoxP3(GFP) mice than the WT counterparts (Figure 1D). Thus, GITR-L plays a significant role in the expansion of Treg in the peripheral tissues.
We next evaluated whether the impaired Flt3L-induced expansion of Treg cells in GITR-L−/−FoxP3(GFP) mice correlated with reduced numbers of DCs and macrophages (MØ) (31, 32). As shown in Figure 2A and Figure S2A in Supplementary Material, the percentage of CD11c+CD11b+ and CD11c+CD11b− DCs was reduced in the spleen of GITR-L−/−FoxP3(GFP) mice as compared to FoxP3(GFP) mice. Although the number of conventional CD11c+ DCs in the liver was normal (Figure 2A), the percentage of pDCs in GITR-L−/−FoxP3(GFP) mice was higher than that of their WT counterparts (Figure 2B; Figure S2B in Supplementary Material and Data not shown). The frequency of CD11c−CD11b+ MØ was comparable between these two mice (Figure 2C). Taken together, these data indicate that after Flt3L induction, GITR-L affects the expansion and differentiation of subpopulations of DCs, which in turn leads to expansion of Tregs.
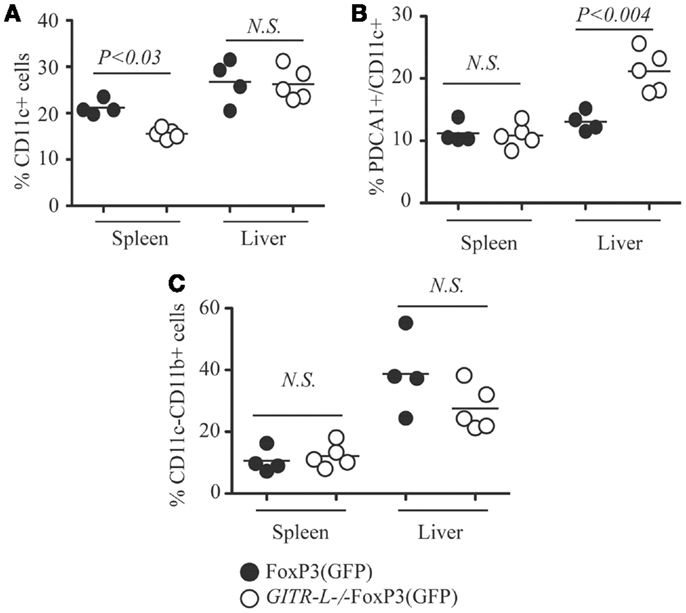
Figure 2. CD11c+ DCs after Flt3L induction. Flt3L-Fc fusion protein was injected into FoxP3(GFP) and GITR-L−/−FoxP3(GFP) mice as described in Figure 1. Different subsets of myeloid cells in the spleen and liver were analyzed. (A) Percentages of CD11c+ cells in the spleen and liver. (B) Percentages of CD11c+PDCA1+ cells in the spleen and liver. (C) Percentages of CD11c−CD11b+ cells. Filled circle represents FoxP3(GFP) mouse. Open circle represents GITR-L−/−FoxP3(GFP) mouse. Each circle represents one mouse.
GITR-L−/− CX3CR1+ DCs Isolated from the Liver are Less Efficient than WT CX3CR1+ DCs in the In vitro Induction of OVA-Specific Treg and CD8+ T Cells
To directly test whether the absence of GITR-L in DC subpopulations affects proliferation of antigen-specific GITR+ Treg and CD8+ cells, we immunized GITR-L−/−CX3CR1(GFP) and WT CX3CR1(GFP) mice by gene transfer with AAV8-OVA (Figure 3A). One week after injection of AAV8-OVA, liver CX3CR1(GFP)+ cells purified by FACS were incubated with OVA-specific OT-II CD4+ T cells or OT-I CD8+ cells for 3 days. GITR-L−/− CX3CR1+ cells were less efficient in inducing Treg as compared to the same cells isolated from WT mice (Figures 3B,C). Since activated CD8+ cells carry GITR on their surface, we also evaluated whether in vitro proliferation of CD8+ T cells would be affected by the absence of GITR-L from the surface of these DCs. Indeed, the proliferation of CD8+ OT-I cells was reduced when cocultured with liver CX3CR1+ cells from AAV8-OVA-primed GITR-L−/−CX3CR1(GFP) mice compared to OT-I cells cultured with WT CX3CR1+ DCs (Figures 3D,E).
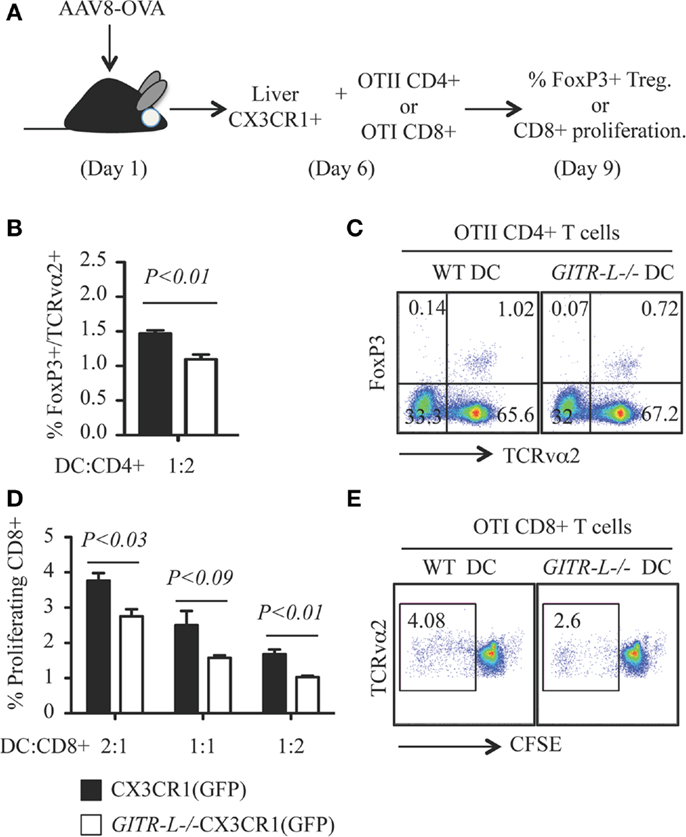
Figure 3. In vitro induction of OVA-specific Treg and CD8+ T cells with hepatic CX3CR1+ DCs. (A) Schematic for in vitro priming of CD8+ OT-I and CD4+ OT-II T cells. Briefly, AAV8-OVA(1010 vector genome/mouse) was i.v. injected into CX3CR1(GFP) and GITR-L−/−CX3CR1(GFP) mice. After 7 days, Ly6G-NK1.1-CX3CR1(GFP)+ cells were purified from the liver and incubated with CD8+ OT-I T and CD4+ OT-II T cells at different ratios for 3 days. Divisions of CD8+ OT-I T cells were evaluated by CFSE dilution. (B) Percentages of in vitro induced of TCRvα2+FoxP3+ Treg. (C) Representative stainings of (B). (D) Ratios of proliferating TCRvα2+CD8+ OT-I T cells. (E) Representative staining of (D). Results represent one of three independent experiments. Error bars indicate mean ± SEM of triplicates.
We conclude that GITR-L on the surface of antigen presenting cells can drive proliferation of both FoxP3+CD4+ Treg cells and activated CD8+ T cells in an antigen-specific manner.
After AAV8-OVA Gene Transfer, the Number of Antigen-Specific Treg in GITR-L−/−FoxP3 Mice is Reduced, Which Results in an Increased Number of OVA-Specific CD8+ T Cells
Because targeted expression of exogenous protein in hepatocytes by AAV8-mediated gene transfer induces a Treg-mediated tolerance (16), we assessed whether this process involves GITR-L. To assess this, we injected an AAV8-OVA vector into in FoxP3(GFP) and GITR-L−/−FoxP3(GFP) mice and determined the number of OVA-specific Treg and CD8+ T cells. Consistent with the results when administering Flt3L, there was a reduced percentage of OVA-specific FoxP3+TCRvα2+ T cells in the spleen and liver of GITR-L−/−FoxP3(GFP) mice as compared to that of WT mice 5 weeks after vector administration (Figure 4A). Conversely, AAV-mediated OVA expression in the hepatocytes induced an increased percentage of OVA-specific CD8+TCRvα2+ T cells in the spleen and liver of GITR-L−/−FoxP3(GFP) mice (Figure 4B). By contrast, the total cell numbers were comparable between these two mouse strains (Figure 4C). The data suggest that GITR-L deficiency may impair the induction of antigen-specific Tregs (16–18, 21, 33), which may at least partially compromise their immunosuppressive capability.
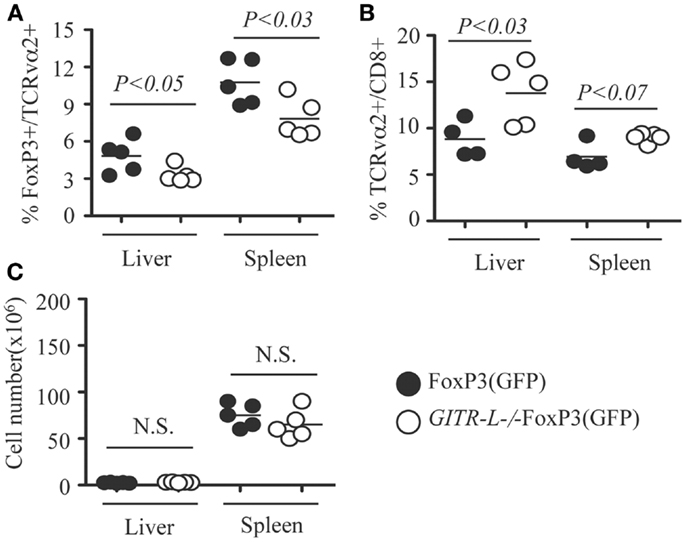
Figure 4. In vivo induction of OVA-specific Treg and CD8+ T cells in GITR-L−/−FoxP3(GFP) mice. AAV8-OVA (1010 vector genome/mouse) was i.v. injected into FoxP3(GFP) and GITR-L−/−FoxP3(GFP) mice. Five weeks later, leukocytes from the liver and spleen were stained with TCRvα2. (A) Percentages of FoxP3+/TCRvα2+ cells. (B) Percentages of TCRvα2+/CD8+ cells. (C) Number of leukocytes. Filled circle represents FoxP3(GFP) mouse. Filled circle represents FoxP3(GFP) mouse. Open circle represents GITR-L−/−FoxP3(GFP) mouse. Each circle represents one mouse.
As the in vitro data suggest that GITR-L expression on DCs causes the expansion of CD8+ cells, this in vivo result might underestimate the consequences of the reduced number of the Tregs in the GITR-L−/− mice. To test whether GITR-L is implicated in the in vivo expansion of antigen-specific CD8+ cells, we used a system in which the Treg-mediated suppression is absent. To this end, we injected AAV8-OVA into Rag−/− and GITR-L−/−Rag−/− mice followed by the adoptive transfer of OT-I CD8+ T cells after 1 week (Figure 5A). Eight weeks after transfer of OT-I CD8+ T cells, the number of CD8+ T cells in the blood of the GITR-L−/−Rag−/− recipients was significantly lower than that of the Rag−/− recipients (Figure 5B). This was not due to an inadequate amount of OVA antigen production in the GITR-L−/−Rag−/− recipients (Figure 5C). Taken together, the data indicate that GITR-L is required for optimal induction and/or expansion of antigen-specific Treg in the context of hepatic AAV8 gene transfer.
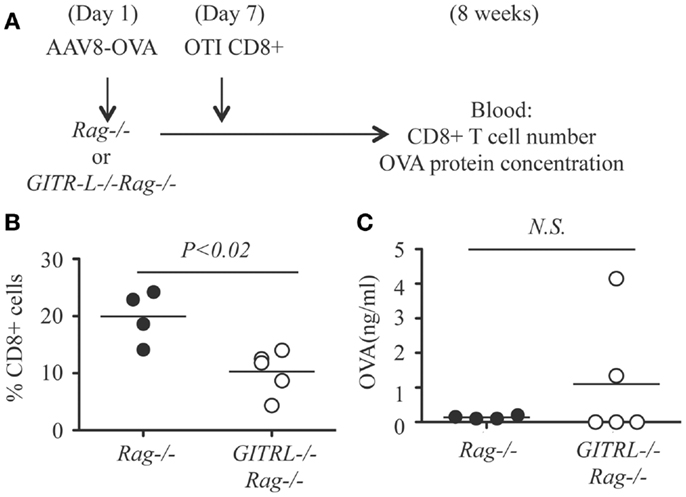
Figure 5. In vivo induction of OVA-specific CD8+ T cells in GITR-L−/−Rag−/− mice. (A) Schematic for in vivo expansion of CD8+ OT-I T cells. Briefly, AAV8-OVA (1010 vector genome/mouse) vector was i.v. injected to Rag−/− or GITR-L−/−Rag−/− mice. Seven days later, CD8+ OT-I T cells (106 cells/mouse) were i.p. injected to mice with AAV8-OVA gene transfer. Mice were euthanized after 8 weeks. (B) Percentages of CD8+ T cells in the blood were evaluated by FACS. (C) Concentrations of chicken ovabulmin protein in the plasma were measured by ELISA. Filled circle represents Rag−/− mouse. Open circle represents GITR-L−/−Rag−/− mouse. Each circle represents one mouse.
Depletion of CX3CR1+ (GFP) Cells by αCD3 in GITR-L−/− Mice Correlates with a Reduced Number of FoxP3+ Treg Cells
In vitro expansion of FoxP3+ Treg cells can be achieved by stimulation with a combination of αCD3 and soluble GITR-L (Fc-GITR-L) (9). We then assessed whether injection of αCD3 into WT and GITR-L−/− mice would affect the Treg population. As shown in Figures 6A,B, αCD3 induced a significant reduction of the percentage of FoxP3+ Treg in the spleen and liver of GITR-L−/−CX3CR1(GFP) mice, but not in WT CX3CR1(GFP) mice. In support of our observations in this paper, the reduced number of Tregs coincided with a reduction of CX3CR1+ DCs in the spleen and liver of GITR-L−/−CX3CR1(GFP) mice (Figures 6C,D). In contrast, the numbers of CX3CR1+ cells in the spleen and liver were comparable in the two mouse strains under homeostasis (Figure S3 in Supplementary Material).
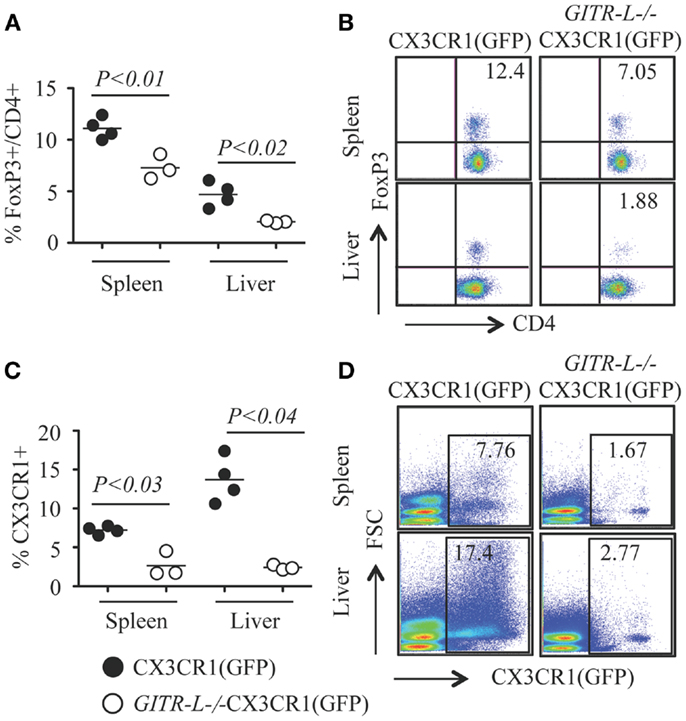
Figure 6. Numbers of Treg and CX3CR1+ DCs after administering αCD3. Anti-CD3ε (20 μg/mouse, one injection) was i.p. injected into CX3CR1(GFP) and GITR-L−/−CX3CR1(GFP) mice. Mice were euthanized at day 3. Splenocytes and liver leukocytes were stained for the expression of CD4 and FoxP3 by FACS. CX3CR1+ cells were assessed by the expression of reporter protein EGFP. Percentages (A) and representative staining (B) of FoxP3+CD4+ Treg cells in the spleen and liver. Percentages (C) and representative staining (D) of CX3CR1+ phagocytes in splenocytes and liver leukocytes. Filled circle represents CX3CR1(GFP) mouse. Open circle represents GITR-L−/−CX3CR1(GFP) mouse. Each circle represents one mouse.
To further investigate the role of GITR-L in the expansion of FoxP3+ Treg, CD4+ T cells were purified from the spleen of FoxP3(GFP) mice and stimulated in vitro with αCD3 with either Fc-GITR-L or IgG. Forty-eight hours after exposure to αCD3, the number of total CD4+ and FoxP3+CD4+ Treg was significantly higher in the presence of Fc-GITR-L than that of IgG (Figures 7A,B). Interestingly, a subset of CD103+ Treg cells, which is induced in epithelium and in sites of inflammation (23, 34) and comprises approximately 20% of all FoxP3+ Treg cells in the spleen, was also expanded by Fc-GITR-L (Figures 7C,D).
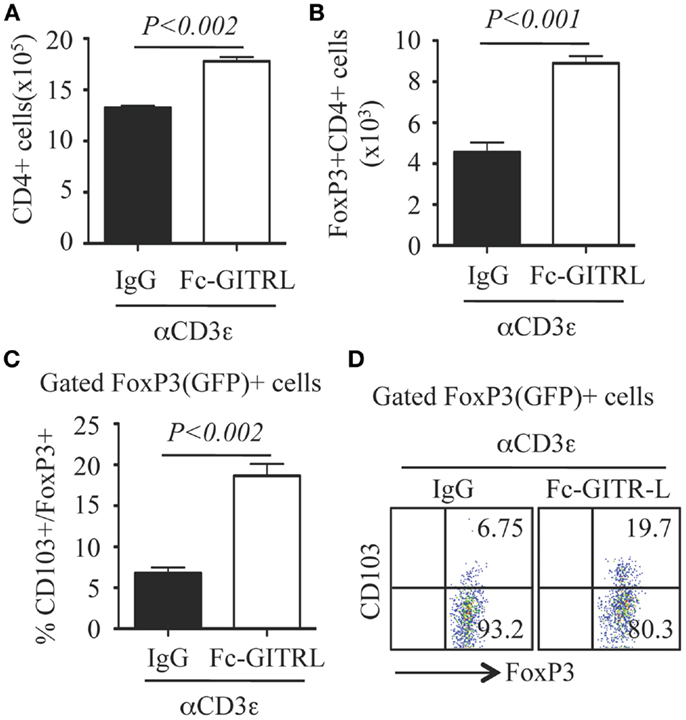
Figure 7. Expansion of FoxP3+ and CD103+ Tregs by Fc-GITR-L in vitro. CD4+ T cells (105 cells/well) from the spleen of FoxP3(GFP) mice were stimulated with αCD3-beads in the presence of Fc-GITR-L or control IgG for 2 days and then stained with CD4 and CD103. FoxP3 expression was judged according to the expression of reporter protein EGFP. (A) Number of total CD4+ cells. (B) Number of FoxP3+CD4+ Tregs. (C) Percentages of CD103+ cells among gated FoxP3+ Tregs. (D) Representative staining of (C). Results were representative of at least three independent experiments. Error bars indicate mean ± SEM of triplicates.
We conclude that while the induction or expansion of Treg is impaired in the absence of GITR-L, Fc-GITR-L provides a positive signal to GITR+ Treg.
Discussion
The receptor-ligand pair GITR/GITR-L (TNFRSF18/TNFSF18) appears to be involved in the development of a variety of inflammation-related diseases in murine models (6, 8, 12, 35, 36). It was originally thought that the suppressor function of Treg cells, which constitutively express GITR, would be abrogated by anti-GITR thus breaking immune self-tolerance (2). More recent additional evidence shows that GITR engagement by its natural ligand GITR-L causes an extensive expansion of functionally competent Tregs (9–11), although the relative role of GITR on Treg and Teff cells remains only partly understood. In this study we find that in the absence of GITR-L the expansion of FoxP3+ Treg cells is impaired in an antigen-specific manner, which can be mimicked by in vivo and in vitro activation of CD4+ Treg cells with αCD3. Our results are consistent with the findings of the Chatila group that expansion and contraction of Teff and Treg dynamically control primary immune responses to foreign antigen (25).
Glucocorticoid-induced TNF receptor family-related protein ligand impacts immune regulation in gene replacement therapy at least at three levels. First, the induction/expansion of antigen-specific Treg cells in the liver after AAV-mediated gene therapy is impaired directly by the absence of GITR-L. Second, the expansion of antigen-specific CD8+ T cells is reduced by GITR-L deficiency. However, impaired expansion of Treg cells can on the other hand up-regulate CD8+ T cell expansion indirectly. Third, GITR-L deficiency affects the infiltration of monocyte-derived MØ to the sites where exogenous protein is expressed and/or the sites of inflammation (30), which changes the local function of different immune cells. These GITR-L-expressing, monocyte-derived MØ may provide a microenvironment for the expression of CD103 in Treg cells, an integrin that facilitates the retention of Treg cells in the sites of inflammation or infection.
Surprisingly, we found that administering αCD3 causes the depletion of CX3CR1+ DCs in the spleen and liver of GITR-L−/− mice, which correlates with a reduced number of FoxP3+ Tregs. It is reported that IL10-secreting GITR+ Tr1 cells may suppress immune responses by granzyme B-mediated killing of myeloid APCs (37, 38). Granzyme B is also important for the ability of Treg, NK cells, and CD8+ T cells to kill their targets (39). It is possible that Tr1, Treg, and CD8+ T cells play a role in the depletion of CX3CR1+ DCs in GITR-L−/− mice. In the presence of GITR-L, an increased expansion of Treg may inhibit this self-destructive cytotoxicity. Depletion of CX3CR1+ DCs, which includes the GITR-L-expressing pDCs and MØ (12, 30), may feedback to cause the reduction of Treg number during immune responses.
Ly6Chi monocytes give rise to CX3CR1+ DCs under both steady state and inflammation. Under resting conditions, CX3CR1+ DCs in the intestine is reported to induce a immunosuppressive CD8+ T cells (40). CX3CR1+ DCs isolated from the liver are able to induce Treg in vitro. However, during inflammation CX3CR1+ DCs give rise to proinflammatory effector cells (41). The mechanism how this Ly6Chi monocyte-derived DC subpopulation is educated to be either protagonist or antagonist is still not well understood. Anti-CD3-mediated depletion of CX3CR1+ DCs in the liver may provide an important tool for the study of migration, colonization, and education of this special DC subset (30).
In conclusion, our data show that GITR and GITR-L have important implications for gene therapy. Optimal induction of an immune regulatory response, which is crucial for tolerance to the transgene product and for immune modulatory gene therapy, requires co-stimulation by GITR-L, which enhances Treg induction and function. Expression of GITR-L on hepatic APCs may in part explain the tolerogenic/Treg inducing capacity of hepatic gene transfer.
Author Contributions
Gongxian Liao performed all the experiments; Michael S. O’Keeffe helped in processing the samples and editing the manuscript; Guoxing Wang and Boaz van Driel helped in processing the samples and discussing the results. Rene de Waal Malefyt generated GITR-L deficient mice; Hans-Christian Reinecker brought deeper insight into αCD3-inducing murine model. Roland W. Herzog helped in discussing and writing the manuscript; Cox Terhorst is the major organizer of this work and designed the experiments with Gongxian Liao.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Talal Chatila for providing the FoxP3EGFP knock-in reporter mice, all other members of the Terhorst Lab for helpful discussions. We thank Dr. Shangzhen Zhou and the AAV research vector core at The Children’s Hospital of Philadelphia for the help with production of AAV8-OVA vector. Grant Support: this work was sponsored by National Institutes of Health (P01 HL078810 to Roland W. Herzog and Cox Terhorst, and R01 DK-52510 and P30 DK-43351 to Cox Terhorst).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fimmu.2014.00035/abstract
Figure S1. Representative staining of Figure 1A.
Figure S2. (A,B) Representative staining of Figures 2A,B.
Figure S3. CX3CR1(GFP)+ phagocytes in spleen and liver leukocytes of CX3CR1(GFP) and GITR-L−/−CX3CR1(GFP) mice under resting condition.
References
1. McHugh RS, Shevach EM. Cutting edge: depletion of CD4+CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ-specific autoimmune disease. J Immunol (2002) 168:5979–83.
2. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol (2002) 3:135–42. doi: 10.1038/ni759
3. Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol (2003) 170:3939–43.
4. Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity (2013) 38:414–23. doi:10.1016/j.immuni.2013.03.002
5. Stephens GL, Mchugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, et al. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol (2004) 173:5008–20.
6. Santucci L, Agostini M, Bruscoli S, Mencarelli A, Ronchetti S, Ayroldi E, et al. GITR modulates innate and adaptive mucosal immunity during the development of experimental colitis in mice. Gut (2007) 56:52–60. doi:10.1136/gut.2006.091181
7. Nocentini G, Cuzzocrea S, Genovese T, Bianchini R, Mazzon E, Ronchetti S, et al. Glucocorticoid-induced tumor necrosis factor receptor-related (GITR)-Fc fusion protein inhibits GITR triggering and protects from the inflammatory response after spinal cord injury. Mol Pharmacol (2008) 73:1610–21. doi:10.1124/mol.107.044354
8. Liao G, Detre C, Berger SB, Engel P, De Waal Malefyt R, Herzog RW, et al. Glucocorticoid-induced tumor necrosis factor receptor family-related protein regulates CD4(+)T cell-mediated colitis in mice. Gastroenterology (2012) 142:582.e–91.e. doi:10.1053/j.gastro.2011.11.031
9. Liao G, Nayak S, Regueiro JR, Berger SB, Detre C, Romero X, et al. GITR engagement preferentially enhances proliferation of functionally competent CD4+CD25+FoxP3+ regulatory T cells. Int Immunol (2010) 22:259–70. doi:10.1093/intimm/dxq001
10. van Olffen RW, Koning N, Van Gisbergen KP, Wensveen FM, Hoek RM, Boon L, et al. GITR triggering induces expansion of both effector and regulatory CD4+ T cells in vivo. J Immunol (2009) 182:7490–500. doi:10.4049/jimmunol.0802751
11. Carrier Y, Whitters MJ, Miyashiro JS, Labranche TP, Ramon HE, Benoit SE, et al. Enhanced GITR/GITRL interactions augment IL-27 expression and induce IL-10-producing Tr-1 like cells. Eur J Immunol (2012) 42:1393–404. doi:10.1002/eji.201142162
12. Grohmann U, Volpi C, Fallarino F, Bozza S, Bianchi R, Vacca C, et al. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med (2007) 13:579–86. doi:10.1038/nm1563
13. Heng TS, Painter MW. The immunological genome project: networks of gene expression in immune cells. Nat Immunol (2008) 9:1091–4. doi:10.1038/ni1008-1091
14. Azuma M. Role of the glucocorticoid-induced TNFR-related protein (GITR)-GITR ligand pathway in innate and adaptive immunity. Crit Rev Immunol (2010) 30:547–57. doi:10.1615/CritRevImmunol.v30.i6.40
15. Lacal PM, Petrillo MG, Ruffini F, Muzi A, Bianchini R, Ronchetti S, et al. Glucocorticoid-induced TNF receptor family-related ligand triggering up-regulates VCAM-1 and ICAM-1 and promotes leukocyte adhesion. J Pharmacol Exp Ther (2013) 347(1):164–72. doi:10.1124/jpet.113.207605
16. Cao O, Dobrzynski E, Wang L, Nayak S, Mingle B, Terhorst C, et al. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood (2007) 110:1132–40. doi:10.1182/blood-2007-02-073304
17. Mingozzi F, Liu YL, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest (2003) 111:1347–56. doi:10.1172/JCI16887
18. Dobrzynski E, Fitzgerald JC, Cao O, Mingozzi F, Wang L, Herzog RW. Prevention of cytotoxic T lymphocyte responses to factor IX-expressing hepatocytes by gene transfer-induced regulatory T cells. Proc Natl Acad Sci U S A (2006) 103:4592–7. doi:10.1073/pnas.0508685103
19. Markusic DM, Hoffman BE, Perrin GQ, Nayak S, Wang X, Loduca PA, et al. Effective gene therapy for haemophilic mice with pathogenic factor IX antibodies. EMBO Mol Med (2013) 5:1698–709. doi:10.1002/emmm.201302859
20. Wang X, Sherman A, Liao G, Leong KW, Daniell H, Terhorst C, et al. Mechanism of oral tolerance induction to therapeutic proteins. Adv Drug Deliv Rev (2013) 65:759–73. doi:10.1016/j.addr.2012.10.013
21. Hoffman BE, Martino AT, Sack BK, Cao O, Liao G, Terhorst C, et al. Nonredundant roles of IL-10 and TGF-beta in suppression of immune responses to hepatic AAV-factor IX gene transfer. Mol Ther (2011) 19:1263–72. doi:10.1038/mt.2011.33
22. Robinson PW, Green SJ, Carter C, Coadwell J, Kilshaw PJ. Studies on transcriptional regulation of the mucosal T-cell integrin alphaEbeta7 (CD103). Immunology (2001) 103:146–54. doi:10.1046/j.1365-2567.2001.01232.x
23. McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity (2002) 16:311–23. doi:10.1016/S1074-7613(02)00280-7
24. Zhao D, Zhang C, Yi T, Lin CL, Todorov I, Kandeel F, et al. In vivo-activated CD103+CD4+ regulatory T cells ameliorate ongoing chronic graft-versus-host disease. Blood (2008) 112:2129–38. doi:10.1182/blood-2008-02-140277
25. Haribhai D, Lin W, Relland LM, Truong N, Williams CB, Chatila TA. Regulatory T cells dynamically control the primary immune response to foreign antigen. J Immunol (2007) 178:2961–72.
26. Laouar Y, Welte T, Fu XY, Flavell RA. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity (2003) 19:903–12. doi:10.1016/S1074-7613(03)00332-7
27. Crispe IN. Isolation of mouse intrahepatic lymphocytes. Curr Protoc Immunol (2001) Chapter 3:Unit3.21. doi:10.1002/0471142735.im0321s22
28. Ephrem A, Epstein AL, Stephens GL, Thornton AM, Glass D, Shevach EM. Modulation of Treg cells/T effector function by GITR signaling is context-dependent. Eur J Immunol (2013) 43:2421–9. doi:10.1002/eji.201343451
29. Lean JM, Fuller K, Chambers TJ. FLT3 ligand can substitute for macrophage colony-stimulating factor in support of osteoclast differentiation and function. Blood (2001) 98:2707–13. doi:10.1182/blood.V98.9.2707
30. Liao G, Van Driel B, Magelky E, O’keeffe MS, De Waal Malefyt R, Engel P, et al. Glucocorticoid-induced TNF receptor family-related protein ligand regulates the migration of monocytes to the inflamed intestine. FASEB J (2013) 28(1):474–84. doi:10.1096/fj.13-236505
31. Darrasse-Jeze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med (2009) 206:1853–62. doi:10.1084/jem.20090746
32. Swee LK, Bosco N, Malissen B, Ceredig R, Rolink A. Expansion of peripheral naturally occurring T regulatory cells by Fms-like tyrosine kinase 3 ligand treatment. Blood (2009) 113:6277–87. doi:10.1182/blood-2008-06-161026
33. Nayak S, Cao O, Hoffman BE, Cooper M, Zhou S, Atkinson MA, et al. Prophylactic immune tolerance induced by changing the ratio of antigen-specific effector to regulatory T cells. J Thromb Haemost (2009) 7:1523–32. doi:10.1111/j.1538-7836.2009.03548.x
34. Suffia I, Reckling SK, Salay G, Belkaid Y. A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. J Immunol (2005) 174:5444–55.
35. Cuzzocrea S, Nocentini G, Di Paola R, Mazzon E, Ronchetti S, Genovese T, et al. Glucocorticoid-induced TNF receptor family gene (GITR) knockout mice exhibit a resistance to splanchnic artery occlusion (SAO) shock. J Leukoc Biol (2004) 76:933–40. doi:10.1189/jlb.0204110
36. Cuzzocrea S, Ayroldi E, Di Paola R, Agostini M, Mazzon E, Bruscoli S, et al. Role of glucocorticoid-induced TNF receptor family gene (GITR) in collagen-induced arthritis. FASEB J (2005) 19:1253–65. doi:10.1096/fj.04-3556com
37. Magnani CF, Alberigo G, Bacchetta R, Serafini G, Andreani M, Roncarolo MG, et al. Killing of myeloid APCs via HLA class I, CD2 and CD226 defines a novel mechanism of suppression by human Tr1 cells. Eur J Immunol (2011) 41:1652–62. doi:10.1002/eji.201041120
38. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med (2013) 19:739–46. doi:10.1038/nm.3179
39. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity (2007) 27:635–46. doi:10.1016/j.immuni.2007.08.014
40. Chang SY, Song JH, Guleng B, Cotoner CA, Arihiro S, Zhao Y, et al. Circulatory antigen processing by mucosal dendritic cells controls CD8+ T cell activation. Immunity (2013) 38:153–65. doi:10.1016/j.immuni.2012.09.018
Keywords: GITR-L, TNFSF18, Flt3L, Treg, CX3CR1
Citation: Liao G, O’Keeffe MS, Wang G, van Driel B, de Waal Malefyt R, Reinecker H-C, Herzog RW and Terhorst C (2014) Glucocorticoid-induced TNF receptor family-related protein ligand is requisite for optimal functioning of regulatory CD4+ T cells. Front. Immunol. 5:35. doi: 10.3389/fimmu.2014.00035
Received: 13 November 2013; Accepted: 21 January 2014;
Published online: 03 February 2014.
Edited by:
Federico Mingozzi, UPMC Paris 6, France; Genethon, FranceReviewed by:
Federico Mingozzi, UPMC Paris 6, France; Genethon, FranceDavid William Scott, Uniformed Services University of the Health Sciences, USA
Copyright: © 2014 Liao, O’Keeffe, Wang, van Driel, de Waal Malefyt, Reinecker, Herzog and Terhorst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gongxian Liao, Division of Immunology, Beth Israel Deaconess Medical Center, Harvard Medical School, 3 Blackfan Circle, CLS-928, Boston, MA 02115, USA e-mail:Z2xpYW9AYmlkbWMuaGFydmFyZC5lZHU=;
Cox Terhorst, Division of Immunology, Beth Israel Deaconess Medical Center, Harvard Medical School, 3 Blackfan Circle, CLS-938, Boston, MA 02115, USA e-mail:Y3RlcmhvcnNAYmlkbWMuaGFydmFyZC5lZHU=