 Bilal Osta
Bilal Osta Pierre Miossec
Pierre Miossec- Immunogenomics and Inflammation Research Unit EA 4130, Department of Immunology and Rheumatology, Hospital Edouard Herriot, University of Lyon 1, Lyon, France
Tumor necrosis factor-α (TNF-α) plays an essential role in the regulation of bone homeostasis in several chronic immune and inflammatory joint diseases, where inhibition of TNF has led to significant clinical improvement. However, TNF-activated pathways and mechanisms involved in bone remodeling remain unclear. So far, TNF-α was known as an inhibitor of osteoblast differentiation and an activator of osteoclastogenesis. Recent contradictory findings indicated that TNF-α can also activate osteoblastogenesis. The paradoxical role of TNF-α in bone homeostasis seems to depend on the concentration and the differentiation state of the cell type used as well as on the exposure time. This review aims to summarize the recent contradictory findings on the regulation of bone homeostasis by TNF-α at the isolated cell, whole bone, and whole body levels. In addition, the involvement of TNF-α in the bone remodeling imbalance is observed in inflammatory joint diseases including rheumatoid arthritis and ankylosing spondylitis, which are associated with bone destruction and ectopic calcified matrix formation, respectively. Both diseases are associated with systemic/vertebral osteoporosis.
Introduction
Tumor necrosis factor-α (TNF-α) is one of the several pro-inflammatory cytokines involved in bone remodeling. Classically, TNF-α plays a major role in the regulation of bone homeostasis by stimulating osteoclastogenesis and inhibiting osteoblast (OB) function. It is also involved in the pathogenesis of chronic inflammation in both mouse and human models. Based on these observations, treatments of several chronic inflammatory diseases such as Crohn’s disease, ulcerative colitis, and psoriasis target TNF-α. TNF-α is also associated with the pathogenesis of inflammatory joint diseases including rheumatoid arthritis (RA), which is characterized by massive juxta-articular bone destruction, and ankylosing spondylitis (AS), which is characterized by simultaneous bone destruction and excessive formation, respectively. However, opposite findings suggest that TNF-α may also induce osteogenic differentiation. This review summarizes the classical and paradoxical effects of TNF-α on bone homeostasis by focusing on osteoblastogenesis and osteoclastogenesis.
TNF-α and Its Signaling Pathways
The immune response is modulated by several inflammatory cytokines including TNF-α. TNF-α acts through several pathways including the activation of nuclear factor kappa-B (NF-κB) involved in inflammation and apoptosis (1). TNF-α is produced mainly by monocytes, but can also be secreted by many other cell types (1–3). It is produced as a transmembrane protein (mTNF-α) that is subsequently cleaved by the TNF-α converting enzyme (TACE) to a soluble form (sTNF-α) (4). Both types are biologically active and bind as a trimer to either TNF receptors TNFR1 (also known as TNFRSF1A or p55) or to TNFR2 (also known as TNFRSF1B or p75).
Activation of TNFR1 death domain initiates the apoptotic signaling through the activation of several factors such as caspase and NF-κB resulting in transcription of pro-inflammatory cytokines, chemokines, and anti-apoptotic molecules (5). Unlike TNFR1, the cell expression of TNFR2 is far more limited and does not contain a death domain. Its effects include T cell activation and proliferation via signaling pathways that involve NF-κB, activator protein 1 (AP-1), and the mitogen-activated protein kinases (MAPKs) (6). The pro-inflammatory effects of this cytokine are well documented (7) and are involved in many osteo-immunological diseases including RA and AS (8).
In humans, TNF-α antagonists are clearly effective in a large number of conditions, as first shown in patients with RA (9, 10). However, incidents of lupus-like syndrome and skin lesions have been reported in patients receiving anti-TNF therapies (11). The cellular and molecular mechanisms underlying the TNF-α-mediated pro-inflammatory effects and the occasionally paradoxical effects of TNF-α antagonists in chronic inflammatory diseases remain incompletely understood (10). This also applies to bone biology.
Bone Biology
Bone structure is affected by several paracrine and endocrine factors including mechanical abrasion and serum electrolyte levels such as calcium and phosphorus. This remodeling is regulated by several cells such as osteoclasts (OCs), OBs, and osteocytes (OYs) (12). This continuous remodeling prevents and removes fatigue-related micro-damage and allows adaptation of the bone mass and structure. The number and activity of OCs and OBs are determined by a multitude of factors such as hormones, cytokines, and locally produced signaling molecules under the influence of mechanical stimuli (13–15). Among these factors, TNF-α was found to have great effects on both bone regulator cells, OBs and OCs.
Biology of Osteoclasts
Osteoclasts are hematopoietic cells derived from the monocyte lineage. They undergo a series of differentiation steps to become mature bone-resorbing cells. Intracellular signaling cascade of transcriptional regulation of osteoclast-specific genes is activated by macrophage-colony-stimulating factor (M-CSF) and receptor activator of nuclear factor kappa-B ligand (RANKL) (16) (Figure 1). Upon differentiation, OC precursor cells express strongly RANK, the receptor for RANKL.
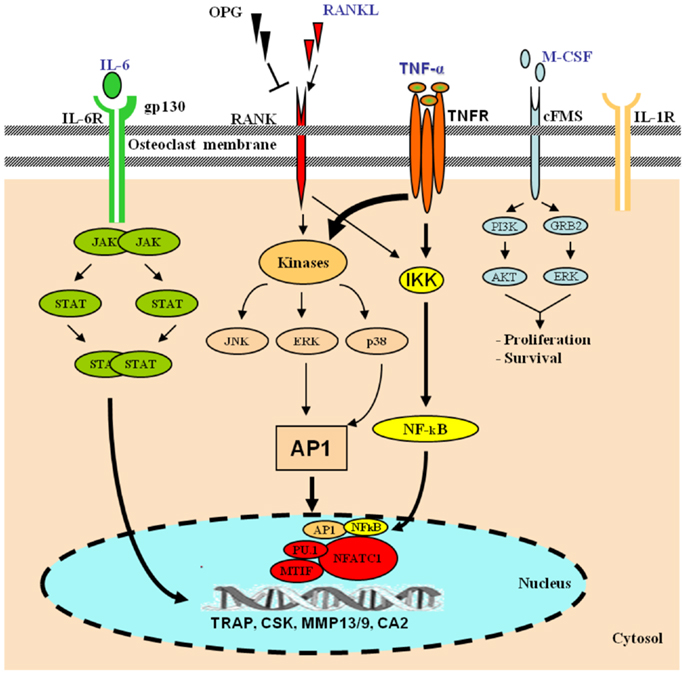
Figure 1. Role of TNF-α in osteoclast differentiation. Hematopoietic stem cells differentiate along the myelo-monocytic pathway with M-CSF stimulation. Differentiating cells continue along a trajectory toward the osteoclast phenotype under the influence of RANKL, which signals through the transcription factors AP-1 and NF-κB. In the presence of RANKL, various pro-inflammatory cytokines, including TNF-α, IL-6, and IL-1, cooperatively orchestrate enhanced osteoclastogenesis. TNF-α signals via NF-κB and the mitogen-activated protein kinases (MAPKs), and IL-6 via the JAK–STAT pathway. The activation of osteoclasts with the inflammatory mechanisms mentioned leads to exaggerated systemic and local bone loss. [AKT, protein kinase B; CA2, carbonic anhydrase 2; CSK, cathepsin K; ERK, extracellular signal-regulated kinase; IKK, IκB kinase; JAK, janus kinase; gp130, glycoprotein 130; GRB2, growth factor receptor-bound protein 2; MITF, microphthalmia-associated transcription factor; MMP, matrix metalloproteinase; NFATc1, nuclear factor of activated T-cells, cytoplasmic 1; PI3K, phosphoinositide 3-kinase; SPI1, spleen focus forming virus (SFFV) proviral integration oncogene spi1; STAT, signal transducer and activator of transcription; TRAP, tartrate-resistant acid phosphatase].
Biology of Osteoblasts
Osteoblasts are derived from pluripotent stem cells called “mesenchymal stem cells (MSCs)” capable of differentiating into adipocytes, chondrocytes, fibroblasts, skeletal muscle cells, and tendon cells (17). A number of factors can stimulate the differentiation toward an osteoblastic direction. The first step includes the proliferation of osteoprogenitors leading to the production of proteins such as histones, fibronectin, and type I collagen. Subsequently, osteogenic matrix is formed through the expression of genes responsible for the production of alkaline phosphatase (ALP), bone sialoprotein, and type I collagen. Finally, extracellular matrix is mineralized by activation of genes involved in the release of osteocalcin (OCN), osteopontin, and collagenase. This complex cascade of gene expression and cellular differentiation is orchestrated by several transcription factors including the runt-related transcription factor-2 (Runx2) and surface expression of RANKL (Figure 2A).
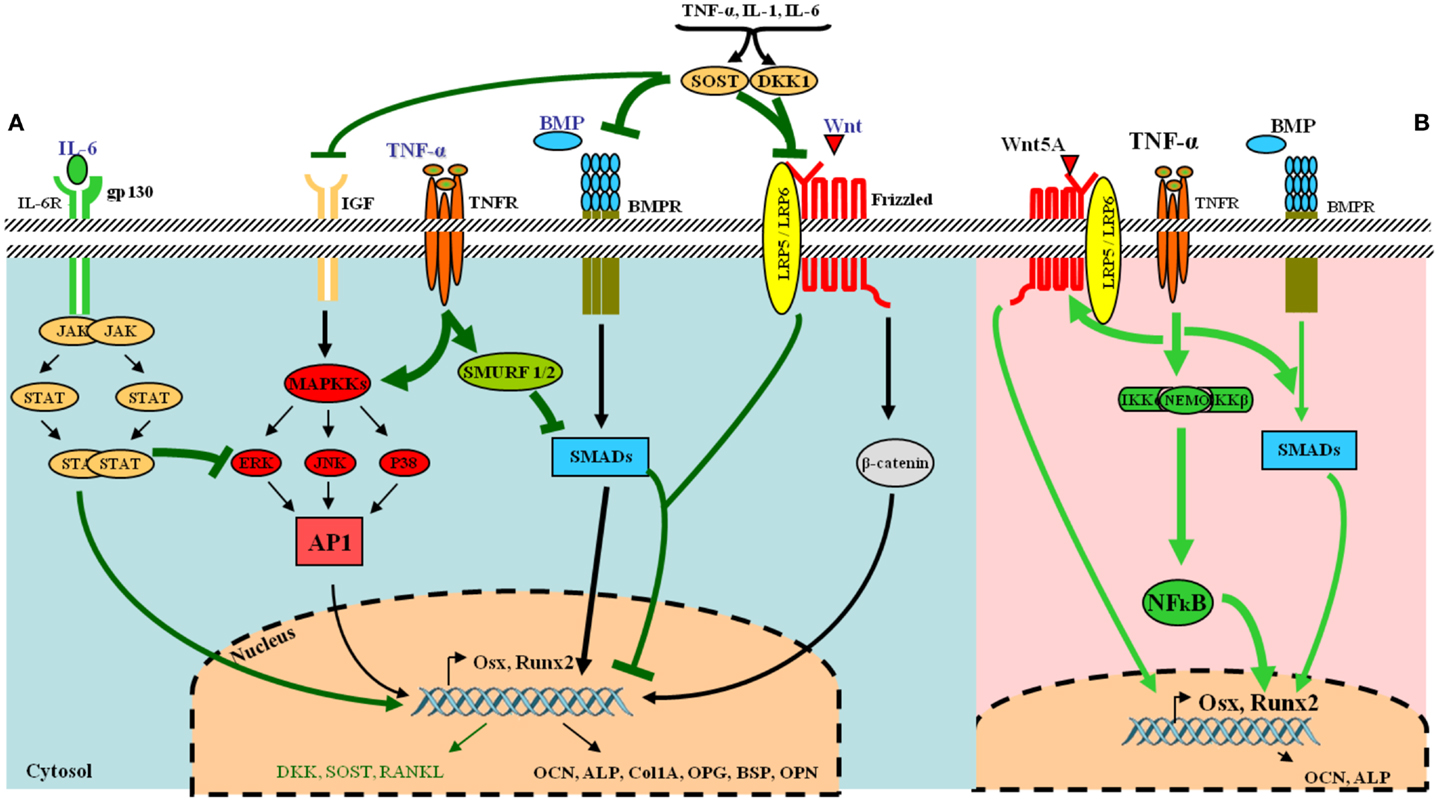
Figure 2. Tumor necrosis factor-α and its osteoblastogenesis paradoxical effects. (A) Under normal condition, various receptors and downstream signaling pathways can be activated in osteoblasts. The most common ligands, receptors, and their signal transduction are represented in black. Essential pathways involve bone morphogenetic proteins (BMPs) and their receptors, acting via SMAD proteins to directly or transcriptionally activate RUNX2 and its subsequent downstream cellular events and the Wnt–frizzled pathway, which utilizes β-catenin for further activities. Under inflammatory conditions, the release of pro-inflammatory cytokines such as TNF-α and IL-6 leads to the inhibition of osteogenic differentiation via several mechanisms (represented in green in the figure). IL-6 inhibits mitogen-activated protein kinase (MAPK) activities by activated signal transducers and activators of transcription (STAT). TNF-α activates SMAD ubiquitination regulatory factor-1 (SMURF1) and SMURF2 leading to the inhibition of SMADs. The pro-inflammatory cytokines also up-regulate dickkopf-related protein 1 (DKK-1) and sclerostin (SOST), which inhibit the Wnt–frizzled pathway, whereas many other osteoblast gene products are down-regulated. (B) TNF-α can also favor osteogenic differentiation via NF-κB by increasing expression of BMP-2, Osx, Runx2, OCN, and Wnt pathway. [AP-1, activator protein 1; BMPR, BMP receptor; ERK, extracellular signal-regulated kinase; FGF, fibroblast growth factor; gp130, glycoprotein 130; IL-6R, IL-6 receptor; JAK, janus kinase; JNK, JUN N-terminal kinase; LRP5-6, low-density lipoprotein receptor-related protein 5 or 6; Osx, osterix (Sp7); OPG, osteoprotegerin; ALP, alkaline phosphatase; BSP, bone sialoprotein; OPN, osteopontin; OCN, osteocalcin; p38, p38 MAPK].
Biology of Osteocytes
Osteocytes are derived from MSCs through OB differentiation. They are located within the bone matrix and represent 90–95% of all bone cells in adults (18). In addition, they have the longest life span among bone cells, up to decades, within their mineralized environment (19). OYs are involved in the dynamics of bone structure through expression of different markers that are released during OB-to-OY ontogeny. In addition, OYs affect the function of other organs such as phosphate transport in kidney (20). Furthermore, OYs are now known to be the principal sensors for mechanical loading of bone. They translate the canalicular flow resulting from bone loading into OCs and OBs recruiting signals (21). Due to the high expression of RANKL by OYs, they have a more significant contribution to osteoclastogenesis as compared to OBs and bone MSCs (22, 23).
Effects of TNF-α on Isolated Cells
Effects of TNF-α on Osteoblasts
Previous in vitro studies have indicated an inhibitory effect of TNF-α on OB differentiation (Table 1). This blockage of osteogenic differentiation resulted from the inhibition of insulin-like growth factor-1 (IGF-1), osterix (Osx also known as SP7), and Runx2 (24–26). The inhibition of Runx2 seemed to be mediated by the TNF-induced up-regulation of Smurf1, a negative regulator of OB differentiation that causes the degradation of Runx2 (27). Moreover, TNF-α inhibited MSC differentiation into OBs via the ubiquitin protein ligase Wwp1 in TNF transgenic mice expressing human TNF-α (28). Another human study demonstrated an inhibitory effect of TNF-α on Runx2 and collagen expression. However, an increase in alkaline phosphatase activity and matrix mineralization was noted (29).
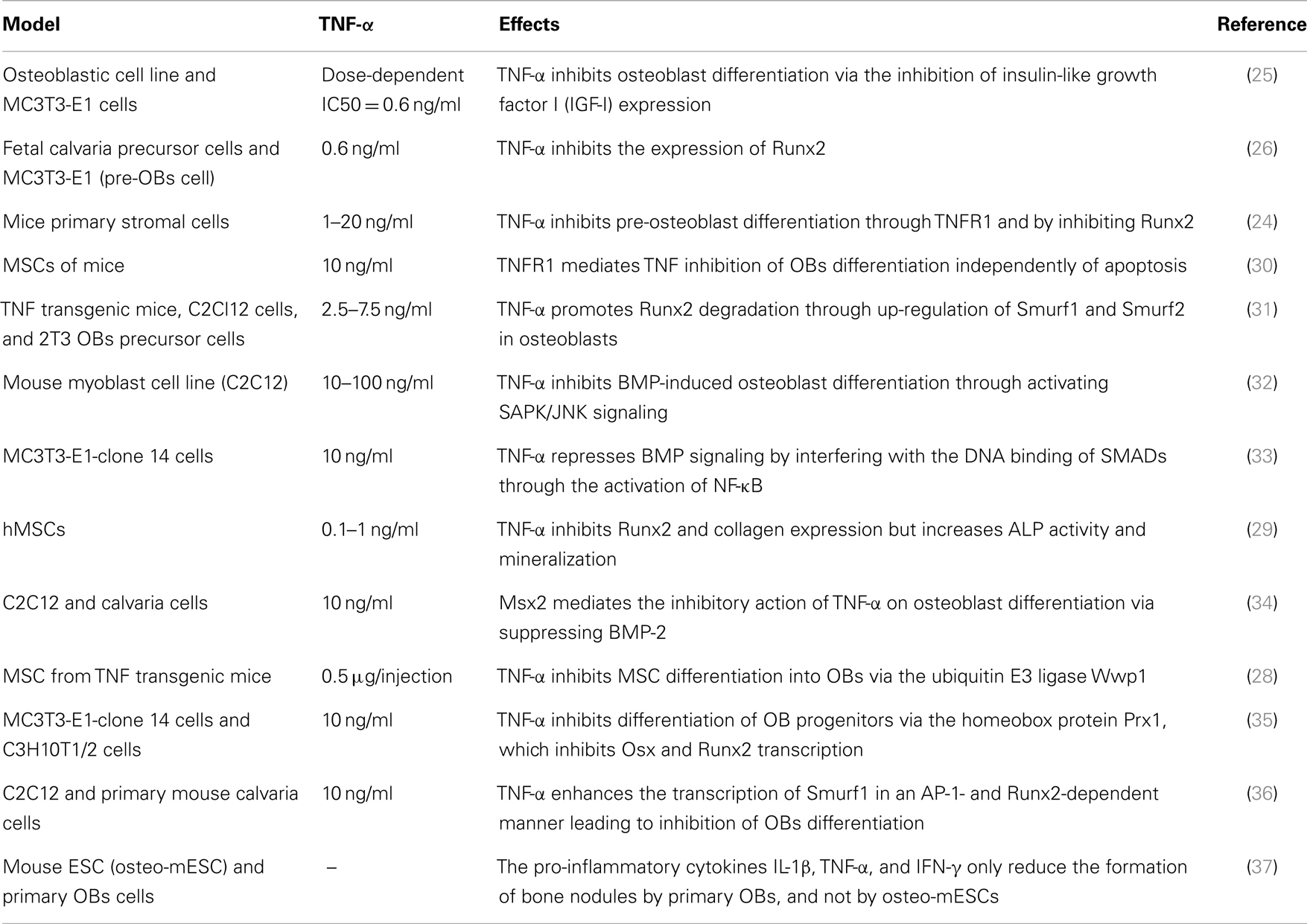
Table 1. Overview of studies indicating a TNF-mediated inhibition of osteogenic differentiation.
In full contrast with these findings, other murine and human MSC studies have reported that TNF-α can also induce osteogenic differentiation (Table 2). In three in vitro rodent models, low concentrations of TNF-α increased osteogenic differentiation via an up-regulation of Runx2, Osx, OCN, and ALP levels (38, 39). Moreover, four human experimental models indicated a similar osteogenic activity for TNF-α. In these models, osteogenic differentiation was promoted via induction of BMP-2, Osx, Runx2, and OCN (40–43). In addition, human models revealed that the dual role of TNF-α on osteogenic differentiation was directly dependent on the concentration of TNF-α, the cell type, and the exposure time (Table 2). Furthermore, a recent study showed that TNF-α stimulated the expression of Wnt5a, which was directly associated with an increase in tissue non-specific alkaline phosphatase (TNAP) levels and mineralization. This suggests an autocrine stimulation of OB activity by Wnt5a in response to TNF in hMSCs (44). This paradoxical effect of TNF-α in inhibiting or activating osteoblastogenesis, lies in the differentiation stage of the responding cells. MSCs can differentiate into several lineages. At this early stage, TNF-α binds its receptors and favors osteogenic differentiation through activation of multiple signaling pathways, notably NF-kB (40) (Figure 2B) (Table 2). On the other hand, TNF-α inhibits osteoblastogenesis in the presence of pre-OB cells, which are already on the differentiation process. This inhibition occurs at different levels, where TNF-α induces DKK-1 expression that inhibits Wnt pathway (45), or activates Smurf that inhibits BMP-2 pathway (31) (Figure 2A) (Table 1).
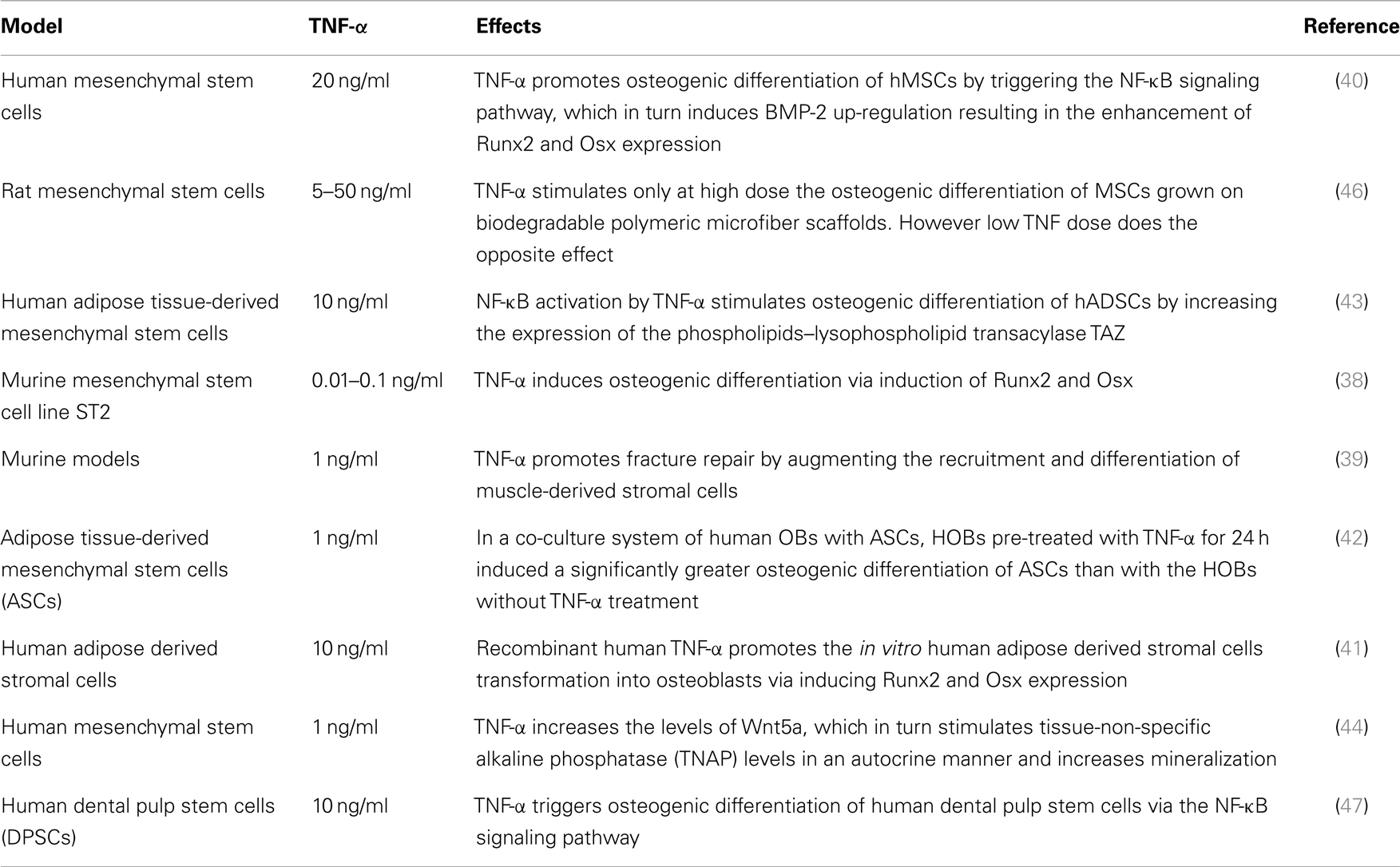
Table 2. Overview of studies indicating a TNF-mediated activation of osteogenic differentiation.
Effects of TNF-α on Osteocytes
Limited results are available on the effect of TNF-α on OYs, which are known to be affected by the surrounding environment. TNF-α and interleukin-1 (IL-1), which increase with estrogen deficiency, can induce OYs apoptosis (48). In a fluid shear stress study, in which the mechanical bone loading was mimicked by applying pulsating fluid flow, the TNF-α-induced apoptosis observed in OYs was inhibited by mechanical loading. However, this effect was not observed in OBs and periosteal fibroblasts. Since apoptotic OYs attract OCs, these results suggest a key role for OYs apoptosis in osteoclastic bone resorption during bone remodeling that is in part modulated by TNF-α (49). Additionally, TNF-α was shown to inhibit the increase in nitric oxide (NO) production and intracellular calcium while strongly reducing F-actin content. This resulted in a reduction of OYs stiffness and provided a possible mechanism to explain the contribution of inflammation to loss of bone mass (50).
Effects of TNF-α on Osteoclasts
The role of TNF-α as a stimulator of osteoclastogenesis is well established (51–55). The expression of a number of transcription factors, including NF-κB, is critical for osteoclastogenesis (56, 57) (Figure 1). Early bone marrow progenitors embark on a path toward pre-OCs or monocytes–macrophages under the influence of M-CSF while TNF-α, IL-1, and RANKL promote a progression toward the functional OCs phenotype. Thus, both RANKL and TNF-α are not only required for, but also synergize to induce osteoclastogenesis. The requirement for both TNF-α and RANKL allows for a more precise control of OCs numbers through a dual level of regulation. First, they affect the selective signaling of the precursor cells via the two different receptors, and subsequently their synergy further increases the activation of NF-κB and AP-1 signaling. Furthermore, the effect of the pro-inflammatory cytokines TNF-α, IL-17, IL-6, IL-1β, and IL-23 on osteoclastogenesis in vitro demonstrated that they have specific characteristic osteoclastogenic properties. However, they exhibited osteoclastogenesis-related activity only in the presence of permissive levels of RANKL. TNF-α appears to have osteoclastogenic properties in the early stage of osteoclastogenesis when bone marrow-derived macrophages (BMMs) are at the stage of osteoclast precursor cells (58).
Effects of TNF-α on Whole Bone
These results on isolated cells need to be expanded to whole bone studies in order to reproduce the cell–cell interactions between bone specific cells and immune cells in the bone marrow. Only a few studies have examined the effects of TNF alone or combined to other cytokines such as IL-1 and IL-17 in ex vivo models or explants where both osteoclast and OB cells are present. In a RA ex vivo model of bone resorption, an IL-4-mediated inhibition of TNF-α levels corroborated with an inhibition in bone resorption, manifested as a 35% increase in the mean total bone area with IL-4 (59). Another study with RA-derived bone explants showed that inhibition of TNF-α decreased inflammation as measured by levels of IL-6 and bone resorption markers. These inhibitory effects were enhanced in combination with IL-1 and IL-17 blockade (60). Histomorphometric analysis of bone samples in patients with different diseases affecting bone such as RA, Crohn’s disease, and bronchial asthma showed that eroded surface over bone surface (ES/BS), a parameter of bone resorption, was significantly increased in the context of high TNF-α levels (61). On the contrary, one study showed that addition of TNF-α to a cultured fetal mouse long bone explant induced NO production resulting in suppression of osteoclast bone resorption (62). These findings suggest a mixed effect of TNF-α on bone structure, which corroborates with the paradoxical role of TNF-α observed in vitro.
Effects of TNF-α in vivo
Tumor necrosis factor-α disrupts bone homeostasis by activating osteoclastic resorption, inhibiting osteoblastic proliferation and matrix synthesis, and activating TNF receptor-associated Factor-2 (TRAF-2). The latter activates NF-κB, AP-1, and MAPKs signaling pathways, which leads to decreased bone formation.
In human TNF transgenic mice, TNF-α induced DKK-1, a regulatory molecule of the Wnt pathway, which led to impaired local bone formation. In addition, blockade of DKK-1 interfered with local bone resorption by reducing OCs numbers in the joints of animal models with inflammatory arthritis (45, 63, 64). A recent study of collagen-induced arthritis (CIA) in rats showed that TNF inhibition did not significantly affect RANKL, TNF-α, and OPG mRNA expression, which are linked to osteoclastogenesis, while it promoted bone formation via suppression of DKK-1 expression. This was confirmed by histomorphometric analysis, which demonstrated a significant increase in bone formation parameters while the changes in bone destruction parameters were not significant (63). Clinical trials in human RA confirmed the association of TNF with arthritic bone destruction as observed in several human TNF-α transgenic mouse models.
However, this observation was not clear in TNF-deficient mouse models, due to insignificant reduction in the clinical parameters of CIA and absence of major histological and morphological abnormalities in the joints. Moreover, some mice exhibited severe disease in the absence of TNF, suggesting the contribution of other cytokines such as IL-1 (65–68).
TNF-α and Inflammatory Joint Diseases
Tumor necrosis factor-α has been associated with an increasing number of chronic inflammatory diseases. Among these diseases, the two most common inflammatory joint diseases RA and AS will be discussed. RA causes massive destruction of juxta-articular and systemic bone while AS leads to the concomitant bone destruction and ectopic bone formation (especially at the vertebral site) because of inflammation of tendon/ligament insertions. Thus, patients with AS experience stiffness of the spine known as ankylosis, and functional disability (69–71). Inhibition of TNF-α has substantially advanced the treatment of these two inflammatory diseases.
Rheumatoid Arthritis
Rheumatoid arthritis is a chronic disease characterized by joint inflammation leading to destruction of cartilage and bone (71). In RA, focal bone loss is due to an excessive bone resorption by OCs (72). Moreover, bone formation by OBs is greatly impaired at the erosion site in RA (73). TNF-α plays an important role in the bone destruction observed in RA patients. TNF inhibitors delay appearance of bone erosion in RA patients with no progression of bone destruction in responders and possibly to some extend, in non-responders (9, 74–76). In a murine CIA model, neutralization of TNF-α reduced joint pathology, especially when the treatment was started early after the onset of disease (77). Moreover, simultaneous inhibition of TNF-α and IL-17A further improved protection against joint damage even at the late stages of CIA (78). Mechanistic models of human TNF transgenic mice confirmed that ongoing TNF-driven inflammation suppresses new bone formation by Wnt signaling through up-regulation of DKK-1, and then the release of this inhibition allows new bone formation (45). Furthermore, several murine autoimmune arthritis models provided evidence that TNF deficiency reduces disease incidence (79–81).
The pro-inflammatory influence of TNF-α in RA acts at several levels. It induces other pro-inflammatory cytokines such as IL-1 and IL-17 and regulates osteoclastogenesis via several mechanisms. TNF-α accelerates cartilage destruction through inducing the production of matrix metalloproteinases (MMPs) and aggrecanases (ADAMTSs), which are enzymes that degrade components of the cartilage extracellular matrix. As an example, in human RA patients and in the human TNF-α transgenic mouse model, TNF-α was shown to regulate MMP3 and 13 by the small ubiquitin-like modifier-1 (SUMO-1) through NF-κB, which led to bone destruction (82). In addition, TNF-α stimulates OCs maturation both directly and indirectly by inducing RANKL on synovial fibroblasts, the synovial lining cells, and T-cells in the synovium (52, 83–85). This increased OCs maturation, and in turn, osteoclastic bone resorption. Moreover, TNF-α possibly promotes OCs activity via enhancing oxidative stress. The production of reactive oxygen species (ROS) by OCs helps accelerate the destruction of calcified tissue (86). This was confirmed in RA patients using an anti-TNF-α antibody therapy. In these patients, reduced levels of oxidative stress markers were associated with the control of joint and bone damage (87). Thus, TNF-α in RA has a destructive function as net effect.
Ankylosing Spondylitis
Ankylosing spondylitis is the second most common form of chronic inflammatory arthritis. The unique hallmark of the disease is pathologic new bone formation (88). AS and related spondyloarthritis are characterized by a paradoxical and simultaneous bone destruction and formation occurring in proximal anatomical sites. The strongest evidence for a key role of TNF-α in AS pathophysiology comes from in vivo inhibition of TNF in AS patients. TNF blockers reduce signs and symptoms of AS, which translates into better physical function and quality of life. However, little or no effect on structural remodeling is achieved (89, 90).
In AS, relevant human samples are not readily available even so TNF expression has been shown at the site of sacroiliitis. Thus, most observations are only made in rodent models. Recently, mice that specifically overexpress the transmembrane but not the soluble form of TNF-α, so called tmTNF transgenic mice, developed clinically spinal abnormalities similar to those in AS (91). These mice developed pronounced local inflammatory infiltration of the axial and peripheral joints (88). In sharp contrast with the other TNF overexpression models, these mice do not develop systemic inflammation, yet display pronounced axial and peripheral new bone formation leading to ankylosis (92). This model strongly argues for a role of tmTNF in the pathogenesis of AS but the exact molecular mechanisms still need to be elucidated. AS patients are particularly prone to develop syndesmophytes, a bony growth originating inside a ligament, and enthesitis, an inflammation at the sites where tendons or ligaments insert to bone. Therefore, in these settings, OCs may not be in direct contact with tendon-derived OBs and TNF-α may exhibit an osteogenic effect there. This is different from the effect found in whole bone where massive bone loss is seen. Thus in AS, even with TNF-α inhibition, ectopic bone formation is still observed, meaning that additional factors are governing this effect.
Osteoporosis
Osteoporosis is characterized by a decrease in bone mass and density leading to bone weakness and prone to fracture. In RA patients, several studies have shown an increased incidence of osteoporosis. This could be due to the highly systemic inflammatory environment observed in these patients and the use of glucocorticoid treatment, which can affect bone remodeling (93, 94). Despite ectopic bone formation, AS patients show massive systemic osteoporosis (95). Among other cytokines, TNF-α seems to be an important player implicating in osteoporosis via TNFR1 (96). In RA patients treated with TNF-α inhibitors, control of bone loss was observed in parallel to that seen on arthritis levels (76, 97).
Conclusion
Tumor necrosis factor-α has an important role in the regulation of bone homeostasis via activation of complex signaling pathways leading to gene transcription of several regulators of bone homeostasis. The classical view remains that TNF-α is an established inducer of osteoclastogenesis and inhibitor of osteoblastogenesis. To add further complexity, it was recently discovered that TNF-α also possessed osteogenic differentiation effects. Most of these paradoxical effects were only described in in vitro models and were concentration- and time-dependent, and cell-type specific. Despite the fact that TNF-α levels are high in both RA and AS, the bone dynamics are different. In RA, the dynamics are shifted toward bone destruction, combined with inhibition of specific osteogenic genes. In AS, systemic bone destruction is accompanied by ectopic bone formation at sites where OCs and OBs are disconnected. Future research should focus on the delineation of the molecular pathways used by this mediator and understand the conditions affecting and the purpose of this dual role in bone remodeling.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Bilal Osta was supported by a fellowship from the Région Rhône-Alpes. Professor Miossec is a senior member of and supported by the Institut Universitaire de France. His laboratory is supported by grants from the IHU prometteur OPERA (Grant ANR-10-IBHU-004) and the EU Marie Curie Network EUTRAIN.
References
1. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol (2003) 3:745–56. doi:10.1038/nri1184
2. Sethi G, Sung B, Aggarwal BB. TNF: a master switch for inflammation to cancer. Front Biosci (2008) 13:5094–107. doi:10.2741/3066
3. Vassalli P. The pathophysiology of tumor necrosis factors. Annu Rev Immunol (1992) 10:411–52. doi:10.1146/annurev.iy.10.040192.002211
4. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature (1997) 385:729–33. doi:10.1038/385729a0
5. MacEwan DJ. TNF ligands and receptors – a matter of life and death. Br J Pharmacol (2002) 135:855–75. doi:10.1038/sj.bjp.0704549
6. Faustman D, Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov (2010) 9:482–93. doi:10.1038/nrd3030
7. Biton J, Boissier MC, Bessis N. TNFalpha: activator or inhibitor of regulatory T cells? Joint Bone Spine (2012) 79:119–23. doi:10.1016/j.jbspin.2011.09.017
9. Feldmann M, Maini SR. Role of cytokines in rheumatoid arthritis: an education in pathophysiology and therapeutics. Immunol Rev (2008) 223:7–19. doi:10.1111/j.1600-065X.2008.00626.x
10. Feldmann M, Williams RO, Paleolog E. What have we learnt from targeted anti-TNF therapy? Ann Rheum Dis (2010) 69(Suppl 1):i97–9. doi:10.1136/ard.2009.117143
11. Sfikakis PP. The first decade of biologic TNF antagonists in clinical practice: lessons learned, unresolved issues and future directions. Curr Dir Autoimmun (2010) 11:180–210. doi:10.1159/000289205
12. Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell (2007) 129:1081–95. doi:10.1016/j.cell.2007.03.055
13. Klein-Nulend J, Bacabac RG, Bakker AD. Mechanical loading and how it affects bone cells: the role of the osteocyte cytoskeleton in maintaining our skeleton. Eur Cell Mater (2012) 24:278–91.
14. Onal M, Xiong J, Chen X, Thostenson JD, Almeida M, Manolagas SC, et al. Receptor activator of nuclear factor kappaB ligand (RANKL) protein expression by B lymphocytes contributes to ovariectomy-induced bone loss. J Biol Chem (2012) 287:29851–60. doi:10.1074/jbc.M112.377945
15. Vezeridis PS, Semeins CM, Chen Q, Klein-Nulend J. Osteocytes subjected to pulsating fluid flow regulate osteoblast proliferation and differentiation. Biochem Biophys Res Commun (2006) 348:1082–8. doi:10.1016/j.bbrc.2006.07.146
16. Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone (2007) 40:251–64. doi:10.1016/j.bone.2006.09.023
17. Kulterer B, Friedl G, Jandrositz A, Sanchez-Cabo F, Prokesch A, Paar C, et al. Gene expression profiling of human mesenchymal stem cells derived from bone marrow during expansion and osteoblast differentiation. BMC Genomics (2007) 8:70. doi:10.1186/1471-2164-8-70
18. Franz-Odendaal TA, Hall BK, Witten PE. Buried alive: how osteoblasts become osteocytes. Dev Dyn (2006) 235:176–90. doi:10.1002/dvdy.20603
19. Schaffler MB, Kennedy OD. Osteocyte signaling in bone. Curr Osteoporos Rep (2012) 10:118–25. doi:10.1007/s11914-012-0105-4
20. Pereira RC, Juppner H, Azucena-Serrano CE, Yadin O, Salusky IB, Wesseling-Perry K. Patterns of FGF-23, DMP1, and MEPE expression in patients with chronic kidney disease. Bone (2009) 45:1161–8. doi:10.1016/j.bone.2009.08.008
21. Price C, Zhou X, Li W, Wang L. Real-time measurement of solute transport within the lacunar-canalicular system of mechanically loaded bone: direct evidence for load-induced fluid flow. J Bone Miner Res (2011) 26:277–85. doi:10.1002/jbmr.211
22. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med (2011) 17:1231–4. doi:10.1038/nm.2452
23. Nakashima T, Hayashi M, Takayanagi H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol Metab (2012) 23:582–90. doi:10.1016/j.tem.2012.05.005
24. Abbas S, Zhang YH, Clohisy JC, Abu-Amer Y. Tumor necrosis factor-alpha inhibits pre-osteoblast differentiation through its type-1 receptor. Cytokine (2003) 22:33–41. doi:10.1016/S1043-4666(03)00106-6
25. Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, et al. Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology (2000) 141:3956–64. doi:10.1210/endo.141.11.7739
26. Gilbert L, He X, Farmer P, Rubin J, Drissi H, van Wijnen AJ, et al. Expression of the osteoblast differentiation factor RUNX2 (Cbfa1/AML3/Pebp2alpha A) is inhibited by tumor necrosis factor-alpha. J Biol Chem (2002) 277:2695–701. doi:10.1074/jbc.M106339200
27. Lee HL, Yi T, Baek K, Kwon A, Hwang HR, Qadir AS, et al. Tumor necrosis factor-alpha enhances the transcription of smad ubiquitination regulatory factor 1 in an activating protein-1- and Runx2-dependent manner. J Cell Physiol (2012) 228(5):1076–86. doi:10.1002/jcp.24256
28. Zhao L, Huang J, Zhang H, Wang Y, Matesic LE, Takahata M, et al. Tumor necrosis factor inhibits mesenchymal stem cell differentiation into osteoblasts via the ubiquitin E3 ligase Wwp1. Stem Cells (2011) 29:1601–10. doi:10.1002/stem.703
29. Ding J, Ghali O, Lencel P, Broux O, Chauveau C, Devedjian JC, et al. TNF-alpha and IL-1beta inhibit RUNX2 and collagen expression but increase alkaline phosphatase activity and mineralization in human mesenchymal stem cells. Life Sci (2009) 84:499–504. doi:10.1016/j.lfs.2009.01.013
30. Gilbert LC, Rubin J, Nanes MS. The p55 TNF receptor mediates TNF inhibition of osteoblast differentiation independently of apoptosis. Am J Physiol Endocrinol Metab (2005) 288:E1011–8. doi:10.1152/ajpendo.00534.2004
31. Kaneki H, Guo R, Chen D, Yao Z, Schwarz EM, Zhang YE. Tumor necrosis factor promotes Runx2 degradation through up-regulation of Smurf1 and Smurf2 in osteoblasts. J Biol Chem (2006) 281:4326–33. doi:10.1074/jbc.M509430200
32. Mukai T, Otsuka F, Otani H, Yamashita M, Takasugi K, Inagaki K. TNF-alpha inhibits BMP-induced osteoblast differentiation through activating SAPK/JNK signaling. Biochem Biophys Res Commun (2007) 356:1004–10. doi:10.1016/j.bbrc.2007.03.099
33. Yamazaki M, Fukushima H, Shin M, Katagiri T, Doi T, Takahashi T. Tumor necrosis factor alpha represses bone morphogenetic protein (BMP) signaling by interfering with the DNA binding of Smads through the activation of NF-kappaB. J Biol Chem (2009) 284:35987–95. doi:10.1074/jbc.M109.070540
34. Lee HL, Yi T, Woo KM, Ryoo HM, Kim GS, Baek JH. Msx2 mediates the inhibitory action of TNF-alpha on osteoblast differentiation. Exp Mol Med (2010) 42:437–45. doi:10.3858/emm.2010.42.6.045
35. Lu X, Beck GR Jr, Gilbert LC, Camalier CE, Bateman NW, Hood BL. Identification of the homeobox protein Prx1 (MHox, Prrx-1) as a regulator of osterix expression and mediator of tumor necrosis factor alpha action in osteoblast differentiation. J Bone Miner Res (2011) 26:209–19. doi:10.1002/jbmr.203
36. Lee HL, Yi T, Baek K, Kwon A, Hwang HR, Qadir AS. Tumor necrosis factor-alpha enhances the transcription of smad ubiquitination regulatory factor 1 in an activating protein-1- and Runx2-dependent manner. J Cell Physiol (2013) 228:1076–86. doi:10.1002/jcp.24256
37. Sidney LE, Kirkham GR, Buttery L. Comparison of osteogenic differentiation of embryonic stem cells and primary osteoblasts, revealed by responses to IL-1beta, TNF-alpha and IFN-gamma. Stem Cells Dev (2014). doi:10.1089/scd.2013.0336
38. Huang H, Zhao N, Xu X, Xu Y, Li S, Zhang J. Dose-specific effects of tumor necrosis factor alpha on osteogenic differentiation of mesenchymal stem cells. Cell Prolif (2011) 44:420–7. doi:10.1111/j.1365-2184.2011.00769.x
39. Glass GE, Chan JK, Freidin A, Feldmann M, Horwood NJ, Nanchahal J. TNF-alpha promotes fracture repair by augmenting the recruitment and differentiation of muscle-derived stromal cells. Proc Natl Acad Sci U S A (2011) 108:1585–90. doi:10.1073/pnas.1018501108
40. Hess K, Ushmorov A, Fiedler J, Brenner RE, Wirth T. TNFalpha promotes osteogenic differentiation of human mesenchymal stem cells by triggering the NF-kappaB signaling pathway. Bone (2009) 45:367–76. doi:10.1016/j.bone.2009.04.252
41. Yu RY, Zeng BJ, Liu YS, Zhou YS. Recombinant human tumor necrosis factor-alpha promotes human adipose-derived stromal cells transforming into osteoblast in vitro. Beijing Da Xue Xue Bao (2012) 44:475–80. doi:10.3969/j.issn.1671-167X.2012.03.0.28
42. Lu Z, Wang G, Dunstan CR, Zreiqat H. Short-term exposure to tumor necrosis factor-alpha enables human osteoblasts to direct adipose tissue-derived mesenchymal stem cells into osteogenic differentiation. Stem Cells Dev (2012) 21:2420–9. doi:10.1089/scd.2011.0589
43. Cho HH, Shin KK, Kim YJ, Song JS, Kim JM, Bae YC, et al. NF-kappaB activation stimulates osteogenic differentiation of mesenchymal stem cells derived from human adipose tissue by increasing TAZ expression. J Cell Physiol (2010) 223:168–77. doi:10.1002/jcp.22024
44. Briolay A, Lencel P, Bessueille L, Caverzasio J, Buchet R, Magne D. Autocrine stimulation of osteoblast activity by Wnt5a in response to TNF-alpha in human mesenchymal stem cells. Biochem Biophys Res Commun (2013) 430:1072–7. doi:10.1016/j.bbrc.2012.12.036
45. Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D. Dickkopf-1 is a master regulator of joint remodeling. Nat Med (2007) 13:156–63. doi:10.1038/nm1538
46. Mountziaris PM, Tzouanas SN, Mikos AG. Dose effect of tumor necrosis factor-alpha on in vitro osteogenic differentiation of mesenchymal stem cells on biodegradable polymeric microfiber scaffolds. Biomaterials (2010) 31:1666–75. doi:10.1016/j.biomaterials.2009.11.058
47. Feng X, Feng G, Xing J, Shen B, Li L, Tan W. TNF-alpha triggers osteogenic differentiation of human dental pulp stem cells via the NF-kappaB signalling pathway. Cell Biol Int (2013) 37:1267–75. doi:10.1002/cbin.10141
48. Bonewald LF. Osteoctytes. In: Marcus DFR, Nelson D, Rosen C editors. Osteoporosis, vol. 1, 3rd ed. San Diego: Elsevier (2007) p. 169–90.
49. Tan SD, Kuijpers-Jagtman AM, Semeins CM, Bronckers AL, Maltha JC, Von den Hoff JW. Fluid shear stress inhibits TNFalpha-induced osteocyte apoptosis. J Dent Res (2006) 85:905–9. doi:10.1177/154405910608501006
50. Bakker AD, Silva VC, Krishnan R, Bacabac RG, Blaauboer ME, Lin YC. Tumor necrosis factor alpha and interleukin-1beta modulate calcium and nitric oxide signaling in mechanically stimulated osteocytes. Arthritis Rheum (2009) 60:3336–45. doi:10.1002/art.24920
51. Pacifici R. Estrogen, cytokines, and pathogenesis of postmenopausal osteoporosis. J Bone Miner Res (1996) 11:1043–51. doi:10.1002/jbmr.5650110802
52. Kudo O, Fujikawa Y, Itonaga I, Sabokbar A, Torisu T, Athanasou NA. Proinflammatory cytokine (TNFalpha/IL-1alpha) induction of human osteoclast formation. J Pathol (2002) 198:220–7. doi:10.1002/path.1190
53. Mucci JM, Scian R, De Francesco PN, Garcia FS, Ceci R, Fossati CA. Induction of osteoclastogenesis in an in vitro model of Gaucher disease is medi- ated by T cells via TNF-alpha. Gene (2012) 509:51–9. doi:10.1016/j.gene.2012.07.071
54. Matsubara R, Kukita T, Ichigi Y, Takigawa I, Qu PF, Funakubo N. Characterization and identification of subpopulations of mononuclear preosteoclasts induced by TNF-alpha in combination with TGF-beta in rats. PLoS One (2012) 7:e47930. doi:10.1371/journal.pone.0047930
55. Kagiya T, Nakamura S. Expression profiling of microRNAs in RAW264.7 cells treated with a combination of tumor necrosis factor alpha and RANKL during osteoclast differentiation. J Periodontal Res (2012) 48(3):373–85. doi:10.1111/jre.12017
56. Hofbauer LC, Heufelder AE. Role of receptor activator of nuclear factor-kappaB ligand and osteoprotegerin in bone cell biology. J Mol Med (Berl) (2001) 79:243–53. doi:10.1007/s001090100226
57. Horowitz MC, Xi Y, Wilson K, Kacena MA. Control of osteoclastogenesis and bone resorption by members of the TNF family of receptors and ligands. Cytokine Growth Factor Rev (2001) 12:9–18. doi:10.1016/S1359-6101(00)00030-7
58. Moon SJ, Ahn IE, Jung H, Yi H, Kim J, Kim Y. Temporal differential effects of proinflammatory cytokines on osteoclastogenesis. Int J Mol Med (2013) 31:769–77. doi:10.3892/ijmm.2013.1269
59. Miossec P, Chomarat P, Dechanet J, Moreau JF, Roux JP, Delmas P. Interleukin-4 inhibits bone resorption through an effect on osteoclasts and proinflammatory cytokines in an ex vivo model of bone resorption in rheumatoid arthritis. Arthritis Rheum (1994) 37:1715–22. doi:10.1002/art.1780371202
60. Chabaud M, Miossec P. The combination of tumor necrosis factor alpha blockade with interleukin-1 and interleukin-17 blockade is more effective for controlling synovial inflammation and bone resorption in an ex vivo model. Arthritis Rheum (2001) 44:1293–303. doi:10.1002/1529-0131(200106)44:6<1293::AID-ART221>3.0.CO;2-T
61. Siggelkow H, Eidner T, Lehmann G, Viereck V, Raddatz D, Munzel U. Cytokines, osteoprotegerin, and RANKL in vitro and histomorphometric indices of bone turnover in patients with different bone diseases. J Bone Miner Res (2003) 18:529–38. doi:10.1359/jbmr.2003.18.3.529
62. Lowik CW, Nibbering PH, van de Ruit M, Papapoulos SE. Inducible production of nitric oxide in osteoblast-like cells and in fetal mouse bone explants is associated with suppression of osteoclastic bone resorption. J Clin Invest (1994) 93:1465–72. doi:10.1172/JCI117124
63. Kuratani T, Nagata K, Kukita T, Hotokebuchi T, Nakasima A, Iijima T. Induction of abundant osteoclast-like multinucleated giant cells in adjuvant arthritic rats with accompanying disordered high bone turnover. Histol Histopathol (1998) 13:751–9.
64. Romas E, Bakharevski O, Hards DK, Kartsogiannis V, Quinn JM, Ryan PF. Expression of osteoclast differentiation factor at sites of bone erosion in collagen-induced arthritis. Arthritis Rheum (2000) 43:821–6. doi:10.1002/1529-0131(200004)43:4<821::AID-ANR12>3.0.CO;2-T
65. Marino MW, Dunn A, Grail D, Inglese M, Noguchi Y, Richards E. Characterization of tumor necrosis factor-deficient mice. Proc Natl Acad Sci U S A (1997) 94:8093–8. doi:10.1073/pnas.94.15.8093
66. van den Berg WB. Arguments for interleukin 1 as a target in chronic arthritis. Ann Rheum Dis (2000) 59(Suppl 1):i81–4. doi:10.1136/ard.59.suppl_1.i81
67. Campbell IK, O’Donnell K, Lawlor KE, Wicks IP. Severe inflammatory arthritis and lymphadenopathy in the absence of TNF. J Clin Invest (2001) 107:1519–27. doi:10.1172/JCI12724
68. Braun T, Zwerina J. Positive regulators of osteoclastogenesis and bone resorption in rheumatoid arthritis. Arthritis Res Ther (2011) 13:235. doi:10.1186/ar3380
69. Baraliakos X, Listing J, Rudwaleit M, Sieper J, Braun J. The relationship between inflammation and new bone formation in patients with ankylosing spondylitis. Arthritis Res Ther (2008) 10:R104. doi:10.1186/ar2496
70. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365:2205–19. doi:10.7748/phc2011.11.21.9.29.c8797
71. Gravallese EM, Harada Y, Wang JT, Gorn AH, Thornhill TS, Goldring SR. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am J Pathol (1998) 152:943–51.
72. Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum (2000) 43:250–8. doi:10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P
73. Walsh NC, Reinwald S, Manning CA, Condon KW, Iwata K, Burr DB. Osteoblast function is compromised at sites of focal bone erosion in inflammatory arthritis. J Bone Miner Res (2009) 24:1572–85. doi:10.1359/jbmr.090320
74. Klarenbeek NB, Kerstens PJ, Huizinga TW, Dijkmans BA, Allaart CF. Recent advances in the management of rheumatoid arthritis. BMJ (2010) 341:c6942. doi:10.1136/bmj.c6942
75. Weaver AL. The impact of new biologicals in the treatment of rheumatoid arthritis. Rheumatology (Oxford) (2004) 43(Suppl 3):iii17–23. doi:10.1093/rheumatology/keh203
76. Marotte H, Pallot-Prades B, Grange L, Gaudin P, Alexandre C, Miossec P. A 1-year case-control study in patients with rheumatoid arthritis indicates prevention of loss of bone mineral density in both responders and nonresponders to infliximab. Arthritis Res Ther (2007) 9:R61. doi:10.1186/ar2219
77. Joosten LA, Helsen MM, van de Loo FA, van den Berg WB. Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice. A comparative study using anti-TNF alpha, anti-IL-1 alpha/beta, and IL-1Ra. Arthritis Rheum (1996) 39:797–809. doi:10.1002/art.1780390513
78. Koenders MI, Marijnissen RJ, Devesa I, Lubberts E, Joosten LA, Roth J. Tumor necrosis factor-interleukin-17 interplay induces S100A8, interleukin-1beta, and matrix metalloproteinases, and drives irreversible cartilage destruction in murine arthritis: rationale for combination treatment during arthritis. Arthritis Rheum (2011) 63:2329–39. doi:10.1002/art.30418
79. Hata H, Sakaguchi N, Yoshitomi H, Iwakura Y, Sekikawa K, Azuma Y. Distinct contribution of IL-6, TNF-alpha, IL-1, and IL-10 to T cell-mediated spontaneous autoimmune arthritis in mice. J Clin Invest (2004) 114:582–8. doi:10.1172/JCI21795
80. Matzelle MM, Gallant MA, Condon KW, Walsh NC, Manning CA, Stein GS. Resolution of inflammation induces osteoblast function and regulates the Wnt signaling pathway. Arthritis Rheum (2012) 64:1540–50. doi:10.1002/art.33504
81. Asquith DL, Miller AM, McInnes IB, Liew FY. Animal models of rheumatoid arthritis. Eur J Immunol (2009) 39:2040–4. doi:10.1002/eji.200939578
82. Frank S, Peters MA, Wehmeyer C, Strietholt S, Koers-Wunrau C, Bertrand J. Regulation of matrixmetalloproteinase-3 and matrixmetalloproteinase-13 by SUMO-2/3 through the transcription factor NF-kappaB. Ann Rheum Dis (2013) 72:1874–81. doi:10.1136/annrheumdis-2012-202080
83. Komine M, Kukita A, Kukita T, Ogata Y, Hotokebuchi T, Kohashi O. Tumor necrosis factor-alpha cooperates with receptor activator of nuclear factor kappaB ligand in generation of osteoclasts in stromal cell-depleted rat bone marrow cell culture. Bone (2001) 28:474–83. doi:10.1016/S8756-3282(01)00420-3
84. Kotake S, Udagawa N, Hakoda M, Mogi M, Yano K, Tsuda E. Activated human T cells directly induce osteoclastogenesis from human monocytes: possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis Rheum (2001) 44:1003–12. doi:10.1002/1529-0131(200105)44:5<1003::AID-ANR179>3.3.CO;2-R
85. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev (2010) 233:233–55. doi:10.1111/j.0105-2896.2009.00859.x
86. Banfi G, Iorio EL, Corsi MM. Oxidative stress, free radicals and bone remodeling. Clin Chem Lab Med (2008) 46:1550–5. doi:10.1515/CCLM.2008.302
87. Kageyama Y, Takahashi M, Ichikawa T, Torikai E, Nagano A. Reduction of oxidative stress marker levels by anti-TNF-alpha antibody, infliximab, in patients with rheumatoid arthritis. Clin Exp Rheumatol (2008) 26:73–80.
88. Hreggvidsdottir HS, Noordenbos T, Baeten DL. Inflammatory pathways in spondyloarthritis. Mol Immunol (2014) 57:28–37. doi:10.1016/j.molimm.2013.07.016
89. Braun J, Sieper J. Ankylosing spondylitis. Lancet (2007) 369:1379–90. doi:10.1016/S0140-6736(07)60635-7
90. van der Heijde D, Salonen D, Weissman BN, Landewe R, Maksymowych WP, Kupper H. Assessment of radiographic progression in the spines of patients with ankylosing spondylitis treated with adalimumab for up to 2 years. Arthritis Res Ther (2009) 11:R127. doi:10.1186/ar2794
91. Edwards CK III, Bendele AM, Reznikov LI, Fantuzzi G, Chlipala ES, Li L. Soluble human p55 and p75 tumor necrosis factor receptors reverse spontaneous arthritis in transgenic mice expressing transmembrane tumor necrosis factor alpha. Arthritis Rheum (2006) 54:2872–85. doi:10.1002/art.22077
92. van Duivenvoorde LM, Huriatul Masdar CAA, van Tok MN, Tak PP, Yeremenko NG, Baeten DL. A2.15 relative overexpression of transmembrane versus soluble TNF in human and experimental spondyloarthritis. Ann Rheum Dis (2013) 72:A9–10. doi:10.1136/annrheumdis-2013-203215.15
93. Murariu RV, Macovei LA, Brujbu IC. Osteoporosis in rheumatoid arthritis. Rev Med Chir Soc Med Nat Iasi (2013) 117:394–403.
94. Okano T, Koike T, Tada M, Sugioka Y, Mamoto K, Wakitani S, et al. The limited effects of anti-tumor necrosis factor blockade on bone health in patients with rheumatoid arthritis under the use of glucocorticoid. J Bone Miner Metab (2013). doi:10.1007/s00774-013-0535-9
95. Klingberg E, Lorentzon M, Mellstrom D, Geijer M, Gothlin J, Hilme E. Osteoporosis in ankylosing spondylitis – prevalence, risk factors and methods of assessment. Arthritis Res Ther (2012) 14:R108. doi:10.1186/ar3833
96. Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science (2002) 296:1634–5. doi:10.1126/science.1071924
Keywords: TNF-α, bone homeostasis, osteoblast, osteoclast, rheumatoid arthritis, ankylosing spondylitis, osteoporosis
Citation: Osta B, Benedetti G and Miossec P (2014) Classical and paradoxical effects of TNF-α on bone homeostasis. Front. Immunol. 5:48. doi: 10.3389/fimmu.2014.00048
Received: 03 January 2014; Paper pending published: 11 January 2014;
Accepted: 27 January 2014; Published online: 13 February 2014.
Edited by:
Masaaki Murakami, Osaka University, JapanReviewed by:
Daisuke Kamimura, Osaka University, JapanFrédéric Velard, Université de Reims Champagne-Ardenne, France
Copyright: © 2014 Osta, Benedetti and Miossec. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pierre Miossec, Immunogenomics and Inflammation Research Unit EA 4130, Department of Immunology and Rheumatology, Hôpital Edouard Herriot, University of Lyon 1, 5 place d’Arsonval, 69437 Lyon Cedex 03, France e-mail:bWlvc3NlY0B1bml2LWx5b24xLmZy