 Tatyana Novoyatleva
Tatyana Novoyatleva Amna Sajjad
Amna Sajjad Felix B. Engel
Felix B. Engel- 1Department of Cardiac Development and Remodelling, Max-Planck-Institute for Heart and Lung Research, Bad Nauheim, Germany
- 2Government College University Faisalabad, Faisalabad, Pakistan
- 3Department of Nephropathology, Experimental Renal and Cardiovascular Research, Institute of Pathology, University of Erlangen-Nürnberg, Erlangen, Germany
Tumor necrosis factor (TNF) has been firmly established as a pathogenic factor in heart failure, a significant socio-economic burden. In this review, we will explore the role of other members of the TNF/TNF receptor superfamily (TNFSF/TNFRSF) in cardiovascular diseases (CVDs) focusing on TWEAK and its receptor Fn14, new players in myocardial remodeling and heart failure. The TWEAK/Fn14 pathway controls a variety of cellular activities such as proliferation, differentiation, and apoptosis and has diverse biological functions in pathological mechanisms like inflammation and fibrosis that are associated with CVDs. Furthermore, it has recently been shown that the TWEAK/Fn14 axis is a positive regulator of cardiac hypertrophy and that deletion of Fn14 receptor protects from right heart fibrosis and dysfunction. We discuss the potential use of the TWEAK/Fn14 axis as biomarker for CVDs as well as therapeutic target for future treatment of human heart failure based on supporting data from animal models and in vitro studies. Collectively, existing data strongly suggest the TWEAK/Fn14 axis as a potential new therapeutic target for achieving cardiac protection in patients with CVDs.
Introduction
Most cardiovascular diseases (CVDs) result in heart failure due to the death of heart muscle cells, the cardiomyocytes. This leads to pathological remodeling, which triggers additional cardiomyocyte loss resulting finally in a diminished quality of life and inevitable in heart failure (1, 2). Thus, the most efficient way to prevent most CVDs appears to be preventing cardiomyocyte loss. One aim in cardiovascular medicine is therefore monitoring and targeting risk factors. These efforts resulted, for example, in the addition of antiplatelet therapy (3–6), introduction of reperfusion therapy with thrombolysis (7, 8), and acute percutaneous coronary intervention (9). The current optimal treatment regimen after the discovery that neurohormones contribute to the progression of heart failure is the use of angiotensin-converting-enzyme-inhibitors or angiotensin receptor blockers, beta-blockers, aldosterone antagonists as well as implantable automatic cardiac defibrillators. However, despite these advances, the prevalence of heart failure has increased in the last decades, remaining one of the leading causes of death worldwide (10, 11). This suggests that this treatment regimen does not target all pathological mechanisms in heart failure.
Already two decades ago, the observation of increased tumor necrosis factor (TNF) levels in patients with heart failure linked inflammation to CVDs (12). Meanwhile, a large number of reports have established the essential role of inflammatory cytokines in the progression of heart failure contributing to the processes of cardiac hypertrophy, fibrosis, and apoptosis (13–15). Recently, other TNFSF/TNFRSF members than TNF have been implicated in the pathophysiology of heart failure. Here, we review the role of the members of the TNFSF/TNFRSF in heart failure focusing on TWEAK and its receptor. In addition, we will explore their potential as biomarker for CVDs as well as therapeutic target for the future treatment of human heart failure.
TWEAK and Its Cognate Receptor Fn14
The member TWEAK of the TNFSF was discovered in 1997 (16). Like the other ligands of the TNFSF, TWEAK is primarily synthesized as a type II transmembrane receptor and then further processed by a furin endoprotease into the soluble cytokine sTWEAK (16–18). Cells can co-express both plasma membrane-anchored and soluble TWEAK (19, 20). However, membrane-anchored TWEAK is due to its efficient cleavage, which is rarely detectable (e.g., in monocytes and macrophages) (21). TWEAK expression was reported in a wide variety of different tissues and cells, including tumor cell lines and specimens (16, 19, 22–30).
TWEAK is known to be the sole TNFSF that signals through the cell surface receptor Fn14, an unusual small TNFRSF member (31). Fn14 was discovered in 1999 as Fibroblast Growth Factor 1 (FGF1)-inducible, immediate-early response gene in murine NIH3T3 fibroblasts (32) and is induced by a large variety of other growth factors including FGF2, Platelet-Derived Growth Factor (PDGF), Epidermal Growth Factor (EGF) and Vascular Endothelial Growth Factor (VEGF) as well as cytokines such as tumor necrosis factor alpha (TNFα), Interleukin-1beta (IL-1β), Interferon gamma (IFNγ), and transforming growth factor-beta (TGF-β) (32–35). Fn14 is a type I transmembrane protein expressed on a broad variety of different cell types (18, 32, 33). It contains a single cysteine-rich domain (CRD) in its ectodomain while most other TNF receptors have two to six copies of this characteristic motif (36). The cytoplasmic domain of Fn14 contains only 28 amino acid residues lacking a death domain. Like other TNFRSF members lacking a death domain, Fn14 trimerizes upon ligand binding recruiting subsequently E3 ligase/adapter proteins of the tumor necrosis factor receptor-associated factor (TRAF) family to its cytoplasmic domain (21, 36). Several members of the TRAF family (TRAF1, TRAF2, TRAF3, and TRAF5) have been shown to be able to bind to Fn14 (37, 38).
TWEAK/Fn14 Axis in Cardiomyocyte Proliferation
The heart grows during embryonic development mainly due to cardiomyocyte proliferation. Shortly after birth, however, cardiomyocytes stop to proliferate and the heart continues to grow through the increase in cardiomyocyte cell size (i.e., hypertrophy) (39). Consistent with its role in proliferation in a number of cell types, such as smooth muscle cells (40), myoblasts (28, 41), astrocytes (42), liver progenitor cells (29), epithelial (43), and tubular cells (44), Fn14 expression correlates with the rate of cardiomyocyte proliferation during heart development (45). However, neither Fn14 nor TWEAK knockout mice exhibit a heart phenotype suggesting that the TWEAK/Fn14 axis is not essential for cardiomyocyte proliferation or heart development (28–30). Yet, TWEAK stimulation of neonatal rat cardiomyocytes, expressing Fn14 endogenously, induced cardiomyocyte proliferation (45). TWEAK activated extracellular signal-regulated kinase (ERK) and phosphatidylinositol 3-kinase (PI3K) but not p38 mitogen-activated kinase (p38) signaling. In addition, TWEAK inhibited glycogen synthase kinase-3beta (GSK-3beta) (Figure 1) (45). The effect of TWEAK on several pathways has been described also for other cell types. In tubular cells, TWEAK activated, for example, ERK, p38, PI3K, and NF-κB signaling (44). TWEAK-induced proliferation in tubular cells and cardiomyocytes was prevented by inhibitors of ERK and PI3K (44, 45). In contrast, inhibition of p38 blocked only tubular cell proliferation. A general role of TWEAK-induced NF-κB signaling in cell proliferation remains unclear as it has not yet been determined in cardiomyocytes. In tubular cells, inhibition of NF-κB signaling blocked proliferation.
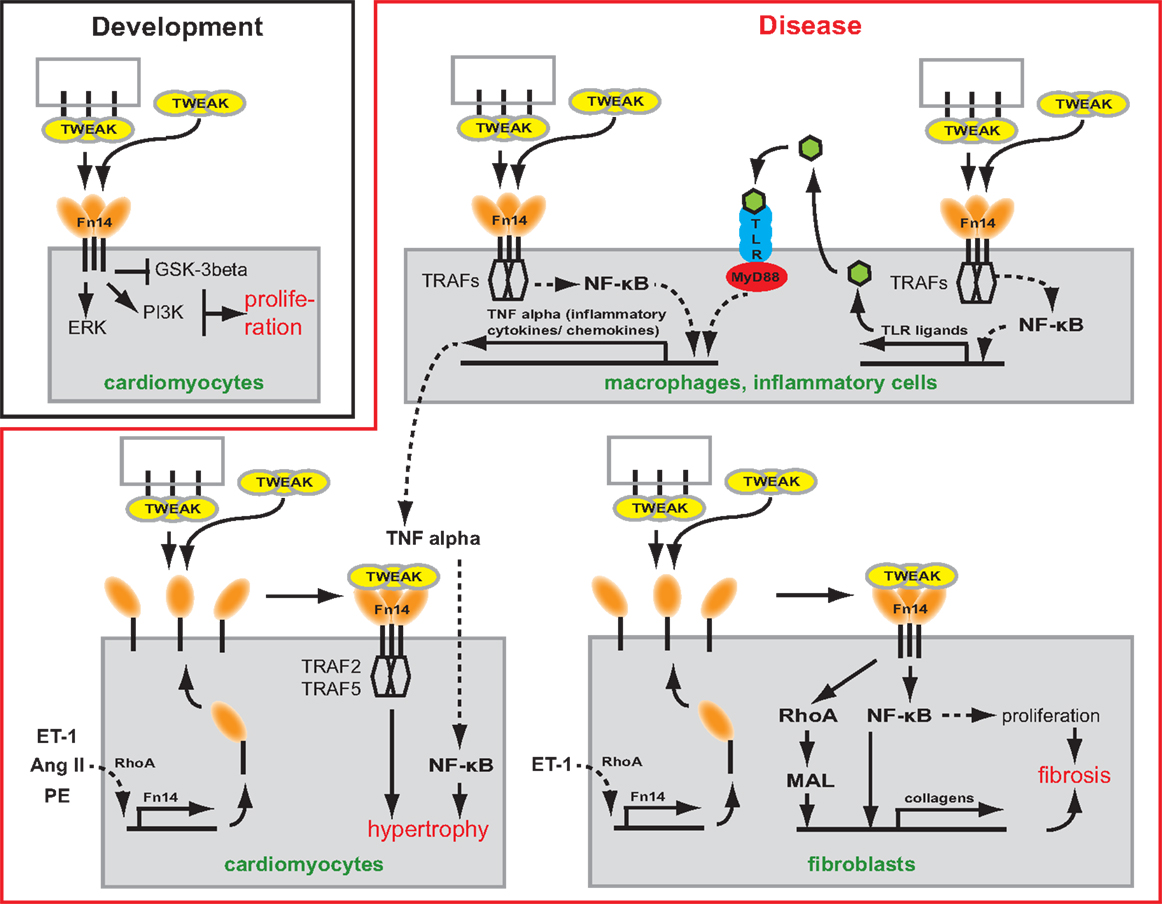
Figure 1. Scheme depicting the potential role of TWEAK/Fn14 signaling in cardiac development and myocardial remodeling and cardiac failure. TWEAK might be presented to Fn14 as membrane-bound or secreted form. TWEAK stimulation induces in vitro proliferation of neonatal cardiomyocytes. Thus, TWEAK/Fn14 signaling might contribute to developmental heart growth. In CVDs, it has been shown that TWEAK has the potential to affect inflammatory cells, cardiomyocytes as well as fibroblasts. In inflammatory cells, TWEAK can enhance secretion of inflammatory cytokines/chemokines by enhancing their expression directly or by increasing the expression of TLR ligands. In cardiomyocytes, TWEAK induces via TRAF hypertrophy. In fibroblast, TWEAK induces the expression of collagens via RhoA and NF-κB and stimulates via NF-κB proliferation leading to cardiac fibrosis.
In contrast to neonatal rat cardiomyocytes, TWEAK had a negligible effect on adult cardiomyocyte proliferation, possibly due to the developmental downregulation of Fn14. However, ectopic expression of Fn14 enabled TWEAK-induced DNA synthesis in adult cardiomyocytes. To date, activation of TWEAK/Fn14 signaling is by far the most potent inducer of adult cardiomyocyte cell cycle re-entry but fails to promote progression into mitosis (45). This is important, as induction of cardiomyocyte proliferation is considered to be a potential future therapy to CVDs. Adult zebrafish and newt as well as newborn mice can all regenerate their heart through cardiomyocyte proliferation (46–48). Moreover, several studies have demonstrated that adult mammalian cardiomyocyte cell division can be induced, even though induction efficiency is relatively low (49, 50). Finally, recent reports utilizing carbon-14 isotope labeling due to atomic bomb tests in the 60s suggest that also human adult mammalian cardiomyocytes, at least a sub-set, might maintain the competence to proliferate (51). Thus, in the future it will be important to elucidate the TWEAK-mediated signaling that induces rat neonatal cardiomyocyte proliferation and to determine if reinstatement of these signaling modalities allows also adult mammalian cardiomyocyte proliferation.
The TWEAK/Fn14 Signaling Promotes Cardiac Hypertrophy and Heart Failure
Pathological cardiac hypertrophy is a key risk factor for heart failure. Cardiac hypertrophy describes the enlargement of the heart due to the increase in cell size of cardiomyocytes. For example, physical exercise and pregnancy can lead to cardiac hypertrophy (52). This form of hypertrophy is considered physiological cardiac hypertrophy as heart function is not affected or improved. In contrast, hypertrophy induced by chronic pressure or volume overload results under certain disease conditions such as hypertension, valvular heart disease, and coronary artery disease, in cardiac dysfunction or heart failure (52). Thus, it is called pathological cardiac hypertrophy.
Tumor necrosis factor alpha was the first member of the TNFSF shown to induce cardiomyocyte hypertrophy (53). Cardiomyocyte-specific overexpression as well as infusion of TNFα causes dilated cardiomyopathy (DCM) suggesting that both circulating and locally produced TNFα induces myocardial dysfunction (54, 55). In recent years, animal experiments have suggested that also other TNFSF ligands can mediate cardiac hypertrophy and heart failure. For example, transgenic overexpression of FasL (TNFSF6) resulted in cardiac hypertrophy with pro-inflammatory consequences (56). That also the TWEAK/Fn14 axis is involved in cardiac hypertrophy was supported by the discovery that transgenic overexpression of full length-TWEAK (fl-TWEAK) in mice resulted in DCM with markedly increased heart to body weight ratio and severe cardiac dysfunction. Moreover, cardiomyocytes from fl-TWEAK-overexpressing mice displayed cellular hypertrophy characterized by pronounced cellular elongation (57). It has also been demonstrated that endogenous Fn14 is required for cardiac hypertrophy. Fn14 deletion attenuated right ventricular (RV) hypertrophy caused by pulmonary artery banding (PAB), a mouse model of pressure-overload-induced RV hypertrophy while TWEAK/Fn14 signaling promoted cardiomyocyte hypertrophy in vitro (58). However, the upstream and downstream signaling pathways regulating TWEAK/Fn14-mediated hypertrophy in vivo remain unclear. It has been shown that hypertrophic agonists including Angiotensin II (Ang II), Phenylephrine (PE), and Endothelin-1 (ET-1) induce Fn14 expression (59). Furthermore, TRAF2 and TRAF5, possible downstream targets of TWEAK/Fn14 signaling, have been implicated in cardiac hypertrophy (Figure 1). Cardiomyocyte-specific TRAF2 transgenic mice developed a time-dependent increase in cardiac hypertrophy, left ventricular (LV) dilation, and adverse LV remodeling, and a significant decrease in heart function (60). Moreover, deficiency of TRAF5 substantially aggravated cardiac hypertrophy and cardiac dysfunction in response to pressure overload after transverse aortic constriction (TAC) (61).
TWEAK is an Extracellular Matrix Modulating Factor in the Heart
Heart failure describes a condition when the heart fails to pump sufficient blood to meet the metabolic demand of the body. It is caused by the loss of cardiomyocytes through necrosis, apoptosis, and autophagy (62, 63), which results in hypertrophy, myocardial fibrosis (fibrillar collagen deposition), and maladaptive extracellular matrix (ECM) remodeling, all characteristics of end-stage heart failure (64). ECM remodeling leads to cardiomyocyte slippage, ventricular dilatation, increased ventricular stiffness as well as impaired diastolic and systolic function (65).
As described above, TNFSF members are involved in the early stages of CVDs such as increased inflammation and hypertrophy (56, 66). However, there is clear evidence that TNFSF members such as TNFα or FasL are also directly involved in myocardial fibrosis (56, 66). First evidence that the TWEAK/Fn14 axis may play a role in ECM remodeling came from the analysis of fl-TWEAK overexpressing mice. These mice develop DCM exhibiting progressive myocardial and perivascular fibrosis (57). Recently, it has been shown that PAB-induced fibrosis was significantly reduced in Fn14 global knockout mice (30). Cell culture experiments demonstrated that TWEAK/Fn14 signaling promotes cardiac mouse fibroblast proliferation and collagen synthesis (30), major sources of fibrillar collagen in the heart under pathophysiological conditions (67, 68). Collagen expression induced by TWEAK/Fn14 signaling was mediated via RhoA-dependent nuclear translocation of the myocardin-related transcription factor-A (MRTF-A)/MAL. Interestingly, upregulation of Fn14 expression in cardiomyocytes due to stretch or stimulation with Ang II or norepinephrine was mediated by RhoA/ROCK signaling, too (69). Furthermore, Chen and colleagues independently demonstrated that TWEAK induces proliferation and collagen synthesis of rat cardiac fibroblasts (70). However, they showed that proliferation and enhancement of collagen synthesis was mediated by the activation of NF-κB signaling (Figure 1). Collectively, these data demonstrate that the TWEAK/Fn14 axis is involved in cardiac ECM remodeling. Importantly, Fn14 knockout mice were protected from PAB-induced RV dysfunction (30, 58) as well as TWEAK-induced cardiac dysfunction and dilation (57).
An essential prerequisite for the formation of fibrotic scar tissue and ECM remodeling is, besides the elevated production of ECM proteins, the expression of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) (71–73). Previously, it has been demonstrated that TNFα-induced cardiac remodeling and dysfunction depends on MMP activation (74). In addition, it has been shown that also FasL and other TNFSF members such as LIGHT, RANKL, and CD40L can potentially regulate MMP activity (75–77). Finally, both TNFα and FasL overexpression is associated with increased levels of TGF-β1, an important inducer of myocardial fibrosis (56, 66). Taken together, these data suggest that besides TNFα several other members of the TNFSF might play important roles in ECM remodeling in cardiac disease.
TWEAK and Other TNFSF Signal via NF-κB
NF-κB has been shown to be cardio-protective (78). However, prolonged activation of NF-κB appears to promote heart failure. For example, cardiomyocyte-specific IκB kinase (IKK)/NF-κB activation induces reversible inflammatory cardiomyopathy and heart failure (79). In addition, it has been demonstrated in animal models, such as TAC-induced pressure-overload (80, 81) and monocrotaline (MCT)-induced RV hypertrophy (82), that inhibition of NF-kB signaling prevents heart failure (cardiac hypertrophy and/or cardiac remodeling).
NF-κB transcription factors are activated through cytokines, pathogens, injuries, and other stressful conditions. Mammalian cells express five NF-κB family members (RelA, RelB, c-Rel, NF-κB2/p100/p52, and NF-κB1/p105/p50) (83–85), that regulate the expression of a large variety of genes which are involved in a number of processes like inflammatory and immune responses of the cell, cell growth, and development. In unstimulated cells, NF-κB is bound to an inhibitory protein, IκB. Binding to IκB masks the nuclear localization signal of NF-κB, sequesters the NF-κB/IκB complex in the cytoplasm, and prevents NF-κB from binding to DNA. Canonical NF-κB signaling culminates in the activation of IKK, which phosphorylates the inhibitory IκB subunit of the NF-κB/IκB complex in the cytoplasm resulting in the proteasomal degradation of IκB. This releases NF-κB resulting in the translocation of NF-κB into the nucleus. In contrast, the non-canonical NF-κB signaling pathway mediates activation of the p52/RelB NF-κB complex. This NF-κB pathway relies on the inducible processing of NF-κB2 precursor protein, as opposed to the degradation of IκBα. A central signaling component of the non-canonical NF-κB pathway is NF-κB-inducing kinase (NIK), which functions together with the inhibitor of NF-κB kinase α (IKKα), to induce phosphorylation-dependent ubiquitination and processing of NF-κB2. Under normal conditions, NIK is continuously degraded. In response to signals mediated by a sub-set of TNFSF members such as Lymphotoxin-β (LT-β), B-cell activating factor (BAFF), and CD40 ligand (CD40L) (86–89), NIK becomes stabilized leading to the activation of non-canonical NF-κB (90, 91).
TWEAK/Fn14 axis has been shown to activate several different signaling cascades, though activation of NF-κB signaling appears to be the major and predominant cellular response through which TWEAK/Fn14 signals. TWEAK/Fn14 has been demonstrated to activate canonical NF-κB signaling in a large variety of cell types (38, 41, 70, 92–96). Interestingly, TWEAK can also signal via Fn14 through the non-canonical NF-κB pathway, which is dependent on the TRAF-binding site of Fn14 as well as TRAF2 and TRAF5 (97). Membrane-bound and oligomerized sTWEAK are superior to soluble TWEAK trimers in regard to the activation of the classical NF-κB pathway. In contrast, both TWEAK variants are equally potent inducers of the non-canonical NF-κB pathway (98). That TWEAK/Fn14 mediates its detrimental effect on heart function at least in part through NF-κB signaling has been supported by several studies. TWEAK-induced proliferation and collagen synthesis of rat cardiac fibroblasts in vitro was mediated by the activation of NF-κB signaling (70). Moreover, DCM induced through elevated circulating TWEAK levels occurred via an FN14-TRAF2-NF-κB-dependent signaling pathway (99). In addition, cardiomyocyte-specific TRAF2 overexpressing mice provoked adverse cardiac remodeling associated with elevated NF-κB signaling (60). These data suggest that the members of the TNFSF mediate their detrimental effects in the heart through TNFRSF members via TRAF2, which is associated directly or indirectly with the majority of TNFSFR members expressed in the heart (TNFR1, TNFR2, RANK, and Fn14), through non-canonical NF-κB signaling (77).
Potential Interactions of Toll-Like Receptors and TWEAK/Fn14 Signaling in CVD
Toll-like receptors (TLRs) are a family of single, membrane-spanning, non-catalytic receptors, which are expressed on various immune cells, such as macrophages, dendritic cells, and neutrophils, as well as on non-immune cells, such as fibroblast cells and epithelial cells. Most commonly, they are known as key activators of the innate immune system as they are responsible for the synthesis and secretion of various inflammatory cytokines by the cells of this system (100). Upon detection of distinct pathogen-associated molecular patterns (PAMPs) of protozoa, virus, and bacteria origin, different members of the TLR family activate signaling pathways that result in the activation of NF-κB-dependent and interferon regulatory factor (IRF)-dependent molecular mechanisms. In addition, TLRs may also be activated by endogenous ligands named damage-associated molecular patterns (DAMPs), which allow the immune system to sense tissue injury in the absence of an infection.
TLR2 and TLR4 activation resulting in NF-κB-dependent release of inflammatory cytokines plays an important role in CVD (101–103). For example, it has been demonstrated that TLR2-deficient mice exhibit higher fractional shortening and survival after myocardial infarction in comparison to wild-type animals (104). In addition, both knockout of TLR2 and inhibition of TLR2 by neutralizing antibodies significantly reduced Ang II-induced cardiac fibrosis, which was associated with a reduction in the infiltration of macrophages, the production of inflammatory cytokines and chemokines, and the activation of NF-κB (103). However, TLR2 deletion in a hypertrophy model (TAC) revealed that TLR2 is required for adaptive cardiac hypertrophy through IL-1β upregulation via NF-κB activation (102). Similar to TLR2, TLR4-deficient mice show after myocardial infarction enhanced LV function and improved remodeling leading to significantly increased survival of TLR4-deficient mice (105, 106). Finally, adenoviral overexpression of dominant-negative MyD88, a common adaptor of TLR2 and TLR4, significantly reduced cardiac hypertrophy and cardiac fibrosis in an aortic constriction model improving cardiac function (107). Taken together there is accumulating evidence for detrimental effects of TLR signaling on cardiac remodeling, cardiac function, and fibrosis upon injury (Figure 1) (108).
Interestingly, it has recently been indicated that TWEAK has the ability to potentiate the pro-inflammatory effects of TLR ligands. For instance, TWEAK has been shown to cooperate with the TLR2 ligand Pam3CysSK4 on the stimulation of IL-8 synthesis by epithelial cells (109). Furthermore, TWEAK is able to stimulate the secretion of HMGB1 (110, 111), another postulated TLR ligand, that contributes to the inflammation in various injury models via signaling through TLR2, TLR4, and RAGE in inflammatory cells (112, 113). Collectively, these data suggest that the TWEAK/Fn14 signaling pathway may also interact with TLR signaling in promoting acute inflammation in CVD.
Prognostic Value of TWEAK/Fn14 Expression for Heart Failure
During recent years, evidence has accumulated that other members of the TNFRSF/TNFSF than TNFα/TNFR might play important roles in the development and progression of heart failure as they are regulated in both experimental and clinical heart failure. Expression analyses of TNFSF ligands and co-stimulatory molecules have revealed that cardiomyocytes of patients with acute myocarditis and DCM express high levels of CD27L, CD30L, and 4-1BBL and exhibit weak to moderate expression of OX40L (114). In heart failure post-myocardial infarction, RANKL was upregulated in both fibroblasts and cardiomyocytes (77). In addition to cardiac cells, elevated expression of TNFSF members were also observed in T lymphocytes in DCM (CD40L) (115) and peripheral blood mononuclear cells in chronic heart failure (4-1BBL, APRIL, CD27L, CD40L, FasL, LIGHT, and TRAIL-receptor 4) (116). Notably, receptors for several of these ligands (e.g., FasL, LIGHT, TNFα, RANKL, and TRAIL) have been reported to be expressed in the heart and enhanced levels of some of these TNF-related molecules also have been found within the failing myocardium (e.g., RANKL, OPG) (77, 117–124). That the members of the TNFSF, can be utilized as prognostic markers, is exemplified by OPG (125), whose plasma level correlated in apparently healthy patients with greater LV mass and lower LV ejection fraction (126). OPG also has been shown to be a reliable predictor of long-term mortality and heart failure development in patients with acute coronary syndrome (127), all-cause mortality in patients with symptomatic severe aortic stenosis (128) or even mixed etiology (129), and with hospitalization of patients with ischemic heart failure due to worsening of heart failure. Another example is BAFF, which is elevated in patients with acute myocardial infarction predicting increased risk of death or recurrent myocardial infarction (130).
In contrast to the poor prognosis found in relation to elevated TNFα levels in heart failure, increased levels of sTWEAK appear to be a good predictor of an adverse short-term outcome after severe type of myocardial infarction (ST-elevation myocardial infarction, STEMI) correlating with hospital duration time of the patients (131). In contrast, TWEAK protein levels were lowered in patients with chronic stable heart failure (132) or advanced non-ischemic heart failure (133). sTWEAK levels were inversely correlated with the severity of the disease and allowed prediction of patient’s mortality, respectively. Importantly, the predictive value was also verified after adjustment for clinical and biochemical variables including the state of the art biomarker, NT-proBNP.
However, sTWEAK alone appears not to be an optimal predictor of heart disease in general as Fn14 gene expression, in contrast to other members of the TNFRSF, is highly regulated in vivo. Under physiological conditions, Fn14 is expressed at relatively low levels but its expression is elevated in several experimental models of injury and inflammation (18, 28, 29, 94, 134, 135). The predictive value of sTWEAK levels is also complicated by the fact that TWEAK and Fn14 can be expressed by a wide variety of cell types. Both are expressed in cardiomyocytes (30, 45, 57, 131) and cardiac fibroblasts (30, 70). In addition, their expression was observed also for macrophages and smooth muscle cells of carotid atherosclerotic plaques (134). Finally, TWEAK is also expressed in endothelial cells of coronary arteries (59) and Fn14 expression was upregulated in proliferating endothelial and smooth muscle cells of injured rat arteries (18). Thus, changed levels of plasma sTWEAK might be difficult to interpret. For example, plasma sTWEAK levels are decreased in patients with pulmonary arterial hypertension (PAH), which results in RV failure (136). This might suggest that sTWEAK has positive adaptive functions. However, animal experiments have demonstrated that Fn14 expression in the heart is highly upregulated after PAB- or MCT-induced PAH (30). In these animal models, TWEAK blood plasma levels were unchanged (PAB) or significantly reduced (MCT) while TWEAK mRNA expression in RVs was elevated. Thus, reduced TWEAK blood levels might be due sequestration of circulating TWEAK by the upregulated Fn14 receptor or might be a compensatory mechanism to protect from the consequences of Fn14 activation. Collectively, sTWEAK appears to be a promising biomarker if combined with clinical parameters.
Blocking of Fn14 Signaling as Potential Therapeutic Approach
In addition to novel candidates for new biomarkers, several TNF-related molecules also could be attractive targets for cardiac therapy. Cell culture as well as in vivo experiments have indicated that TWEAK/Fn14 signaling is involved in cardiac hypertrophy, cardiac remodeling, and heart failure, identifying TWEAK and Fn14 as promising targets to treat CVDs (30, 45, 58, 59, 69, 70, 99, 137). Targeting TWEAK and Fn14 has been also considered in various other pathophysiological conditions. Blocking of TWEAK or Fn14 has successfully been demonstrated to be beneficial in preclinical models of Collagen-Induced Arthritis (138, 139), Experimental Autoimmune Encephalitis (140), Middle Cerebral Artery Occlusion (94, 135, 141), Ischemia Reperfusion Injury (142), and atherosclerosis (143, 144). Furthermore, therapeutical efficacies of TWEAK and Fn14 blocking antibodies were determined in tumor growth inhibition assays, utilizing TWEAK and Fn14-expressing human esophageal and pancreatic cell lines, as well as in a murine gastrointestestinal cancer model (145). Anti-TWEAK and anti-Fn14-specific antibodies are at the moment under clinical investigations in phase I studies in patients suffering on Rheumatoid Arthritis, lupus or solid tumors (http://clinicaltrials.gov/; NCT00771329, NCT001499355 and NCT00738764). Additionally to the usage of antibodies, the employment of the fusion proteins Fn14-Fc and Fc-TWEAK as well as soluble TWEAK provide alternative approaches (137). Taken together, therapies targeting TWEAK and/or Fn14 appear to be a realistic approach and thus warrant future preclinical studies.
Concluding Remarks
The members of the TNFSF and TNFRSF have been shown to be involved in the progression of CVDs to heart failure and thus they appear to be promising prognostic and therapeutic targets. However, the past has shown that correlations of cytokine blood levels to heart disease can be misleading (146). For example, TNFα mediates both adaptive and maladaptive effects on the myocardium. On the one hand it activates via NF-κB expression of anti-apoptotic and cytoprotective genes, but on the other hand it induces also inflammation (147). This explains why clinical trials with anti-TNF therapies were disappointing although overexpression of TNF, which is positively correlated with heart failure in patients, leads to experimental heart failure (146, 148). Thus, it will be important to consider this issue when designing new treatment strategies in heart failure that target members of the TNFSF or TNFRSF.
Based on the disappointing results from anti-TNF trials, the TWEAK/Fn14 axis may represent new targets for heart failure therapies. Fn14 appears to be an excellent therapeutic target as Fn14 knockout mice are viable and show no obvious phenotype under physiological conditions. In addition, Fn14 is upregulated in the myocardium of diseased hearts (30). However, at present the precise role of the TWEAK/Fn14 axis is still poorly understood, and it is unclear whether it has a positive, adaptive role in cardiac disease. One important issue is that the TWEAK/Fn14 axis regulates the behavior of several different cell types. Yet, genetic models inhibiting the TWEAK/Fn14 axis were beneficial in experimental models of heart disease (30, 58). Thus, it is important to test next treatment strategies such as anti-TWEAK antibodies in experimental heart failure models.
Cardiovascular diseases resulting in heart failure are highly complex diseases. Thus, in an ideal case a biomarker should be involved in several pathways of these multiple-pathway diseases reflecting several important pathophysiologies such as hypertrophy, fibrosis, remodeling, and inflammation. As the members of the TNFSF are involved in several of these processes, they appear to be promising biomarkers. However, it might be impudent to assume that a single member of the TNFSF is sufficient. For example, although NT-proBNP is a strong biomarker in heart failure it has recently been shown in patients with symptomatic aortic stenosis that the combination of high levels of both OPG and NT-proBNP was strongly associated with all-cause mortality, thus providing more information together than when either of these markers was used alone (128). Yet, a combination of TNFSF members that provides a “signature of disease” appears likely to be a suitable tool for risk prediction.
Author Contributions
Tatyana Novoyatleva and Felix B. Engel wrote the manuscript. Felix B. Engel generated Figure 1. Amna Sajjad contributed to the literature search. All authors proof read the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Alexander von Humboldt Foundation (Sofja Kovalevskaja Award to Felix B. Engel), the Universities of Giessen and Marburg Lung Center (UGMLC, LOEWE), and the Behrens-Weise-Stiftung. We acknowledge the support by the Deutsche Forschungsgemeinschaft and Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) within the funding programme Open Access Publishing.
References
1. Braunwald E, Bristow MR. Congestive heart failure: fifty years of progress. Circulation (2000) 102(20 Suppl 4):IV14–23. doi: 10.1161/01.CIR.102.suppl_4.IV-14
2. Chien KR. Stress pathways and heart failure. Cell (1999) 98(5):555–8. doi:10.1016/S0092-8674(00)80043-4
3. Brautbar A, Ballantyne CM. Pharmacological strategies for lowering LDL cholesterol: statins and beyond. Nat Rev Cardiol (2011) 8(5):253–65. doi:10.1038/nrcardio.2011.2
4. Brizzio ME, Zapolanski A. Antiplatelet therapy, cardiac surgery, and the risk of bleeding: the surgeon’s perspective. Rev Cardiovasc Med (2011) 12(Suppl 1):S40–6.
5. Zhao D, Qi Y, Zheng Z, Wang Y, Zhang XY, Li HJ, et al. Dietary factors associated with hypertension. Nat Rev Cardiol (2011) 8(8):456–65. doi:10.1038/nrcardio.2011.75
6. Nolan CJ, Damm P, Prentki M. Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet (2011) 378(9786):169–81. doi:10.1016/S0140-6736(11)60614-4
7. Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet (1994) 343(8893):311–22. doi:10.1016/S0140-6736(94)91161-4
8. Antman EM, Morrow DA, McCabe CH, Murphy SA, Ruda M, Sadowski Z, et al. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med (2006) 354(14):1477–88. doi:10.1056/NEJMoa060898
9. Schomig A, Kastrati A, Dirschinger J, Mehilli J, Schricke U, Pache J, et al. Coronary stenting plus platelet glycoprotein IIb/IIIa blockade compared with tissue plasminogen activator in acute myocardial infarction. Stent versus Thrombolysis for Occluded Coronary Arteries in Patients with Acute Myocardial Infarction Study Investigators. N Engl J Med (2000) 343(6):385–91. doi:10.1056/NEJM200008103430602
10. Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol (2011) 8(1):30–41. doi:10.1038/nrcardio.2010.165
11. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics – 2013 update: a report from the American Heart Association. Circulation (2013) 127(1):e6–245. doi:10.1161/CIR.0b013e31828124ad
12. Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med (1990) 323(4):236–41. doi:10.1056/NEJM199007263230405
13. Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci U S A (1995) 92(11):4862–6. doi:10.1073/pnas.92.11.4862
14. Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest (1996) 98(12):2854–65. doi:10.1172/JCI119114
15. Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL. Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J Clin Invest (1993) 92(5):2303–12. doi:10.1172/JCI116834
16. Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, et al. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem (1997) 272(51):32401–10. doi:10.1074/jbc.272.51.32401
17. Bodmer JL, Schneider P, Tschopp J. The molecular architecture of the TNF superfamily. Trends Biochem Sci (2002) 27(1):19–26. doi:10.1016/S0968-0004(01)01995-8
18. Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, et al. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity (2001) 15(5):837–46. doi:10.1016/S1074-7613(01)00232-1
19. Kawakita T, Shiraki K, Yamanaka Y, Yamaguchi Y, Saitou Y, Enokimura N, et al. Functional expression of TWEAK in human colonic adenocarcinoma cells. Int J Oncol (2005) 26(1):87–93.
20. Nakayama M, Ishidoh K, Kayagaki N, Kojima Y, Yamaguchi N, Nakano H, et al. Multiple pathways of TWEAK-induced cell death. J Immunol (2002) 168(2):734–43.
21. Burkly LC, Michaelson JS, Zheng TS. TWEAK/Fn14 pathway: an immunological switch for shaping tissue responses. Immunol Rev (2011) 244(1):99–114. doi:10.1111/j.1600-065X.2011.01054.x
22. Kawakita T, Shiraki K, Yamanaka Y, Yamaguchi Y, Saitou Y, Enokimura N, et al. Functional expression of TWEAK in human hepatocellular carcinoma: possible implication in cell proliferation and tumor angiogenesis. Biochem Biophys Res Commun (2004) 318(3):726–33. doi:10.1016/j.bbrc.2004.04.084
23. Tran NL, McDonough WS, Donohue PJ, Winkles JA, Berens TJ, Ross KR, et al. The human Fn14 receptor gene is up-regulated in migrating glioma cells in vitro and overexpressed in advanced glial tumors. Am J Pathol (2003) 162(4):1313–21. doi:10.1016/S0002-9440(10)63927-2
24. Chacon MR, Richart C, Gomez JM, Megia A, Vilarrasa N, Fernandez-Real JM, et al. Expression of TWEAK and its receptor Fn14 in human subcutaneous adipose tissue. Relationship with other inflammatory cytokines in obesity. Cytokine (2006) 33(3):129–37. doi:10.1016/j.cyto.2005.12.005
25. Chicheportiche Y, Fossati-Jimack L, Moll S, Ibnou-Zekri N, Izui S. Down-regulated expression of TWEAK mRNA in acute and chronic inflammatory pathologies. Biochem Biophys Res Commun (2000) 279(1):162–5. doi:10.1006/bbrc.2000.3913
26. Ho DH, Vu H, Brown SA, Donohue PJ, Hanscom HN, Winkles JA. Soluble tumor necrosis factor-like weak inducer of apoptosis overexpression in HEK293 cells promotes tumor growth and angiogenesis in athymic nude mice. Cancer Res (2004) 64(24):8968–72. doi:10.1158/0008-5472.CAN-04-1879
27. Nakayama M, Harada N, Okumura K, Yagita H. Characterization of murine TWEAK and its receptor (Fn14) by monoclonal antibodies. Biochem Biophys Res Commun (2003) 306(4):819–25. doi:10.1016/S0006-291X(03)01051-9
28. Girgenrath M, Weng S, Kostek CA, Browning B, Wang M, Brown SA, et al. TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J (2006) 25(24):5826–39. doi:10.1038/sj.emboj.7601441
29. Jakubowski A, Ambrose C, Parr M, Lincecum JM, Wang MZ, Zheng TS, et al. TWEAK induces liver progenitor cell proliferation. J Clin Invest (2005) 115(9):2330–40. doi:10.1172/JCI23486
30. Novoyatleva T, Schymura Y, Janssen W, Strobl F, Swiercz JM, Patra C, et al. Deletion of Fn14 receptor protects from right heart fibrosis and dysfunction. Basic Res Cardiol (2013) 108(2):325. doi:10.1007/s00395-012-0325-x
31. Bossen C, Ingold K, Tardivel A, Bodmer JL, Gaide O, Hertig S, et al. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J Biol Chem (2006) 281(20):13964–71. doi:10.1074/jbc.M601553200
32. Meighan-Mantha RL, Hsu DK, Guo Y, Brown SA, Feng SL, Peifley KA, et al. The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J Biol Chem (1999) 274(46):33166–76. doi:10.1074/jbc.274.46.33166
33. Feng SL, Guo Y, Factor VM, Thorgeirsson SS, Bell DW, Testa JR, et al. The Fn14 immediate-early response gene is induced during liver regeneration and highly expressed in both human and murine hepatocellular carcinomas. Am J Pathol (2000) 156(4):1253–61. doi:10.1016/S0002-9440(10)64996-6
34. Kawashima R, Kawamura YI, Oshio T, Son A, Yamazaki M, Hagiwara T, et al. Interleukin-13 damages intestinal mucosa via TWEAK and Fn14 in mice-a pathway associated with ulcerative colitis. Gastroenterology (2011) 141(6):2119.e–29.e. doi:10.1053/j.gastro.2011.08.040
35. Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov (2008) 7(5):411–25. doi:10.1038/nrd2488
36. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell (2001) 104(4):487–501. doi:10.1016/S0092-8674(01)00237-9
37. Brown SA, Hanscom HN, Vu H, Brew SA, Winkles JA. TWEAK binding to the Fn14 cysteine-rich domain depends on charged residues located in both the A1 and D2 modules. Biochem J (2006) 397(2):297–304. doi:10.1042/BJ20051362
38. Han S, Yoon K, Lee K, Kim K, Jang H, Lee NK, et al. TNF-related weak inducer of apoptosis receptor, a TNF receptor superfamily member, activates NF-kappa B through TNF receptor-associated factors. Biochem Biophys Res Commun (2003) 305(4):789–96. doi:10.1016/S0006-291X(03)00852-0
39. Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol (1996) 271(5 Pt 2):H2183–9.
40. Lynch CN, Wang YC, Lund JK, Chen YW, Leal JA, Wiley SR. TWEAK induces angiogenesis and proliferation of endothelial cells. J Biol Chem (1999) 274(13):8455–9. doi:10.1074/jbc.274.13.8455
41. Dogra C, Changotra H, Mohan S, Kumar A. Tumor necrosis factor-like weak inducer of apoptosis inhibits skeletal myogenesis through sustained activation of nuclear factor-kappaB and degradation of MyoD protein. J Biol Chem (2006) 281(15):10327–36. doi:10.1074/jbc.M511131200
42. Saas P, Boucraut J, Walker PR, Quiquerez AL, Billot M, Desplat-Jego S, et al. TWEAK stimulation of astrocytes and the proinflammatory consequences. Glia (2000) 32(1):102–7. doi:10.1002/1098-1136(200010)32:1<102::AID-GLIA100>3.0.CO;2-U
43. Michaelson JS, Cho S, Browning B, Zheng TS, Lincecum JM, Wang MZ, et al. Tweak induces mammary epithelial branching morphogenesis. Oncogene (2005) 24(16):2613–24. doi:10.1038/sj.onc.1208208
44. Sanz AB, Sanchez-Nino MD, Izquierdo MC, Jakubowski A, Justo P, Blanco-Colio LM, et al. Tweak induces proliferation in renal tubular epithelium: a role in uninephrectomy induced renal hyperplasia. J Cell Mol Med (2009) 13(9B):3329–42. doi:10.1111/j.1582-4934.2009.00766.x
45. Novoyatleva T, Diehl F, van Amerongen MJ, Patra C, Ferrazzi F, Bellazzi R, et al. TWEAK is a positive regulator of cardiomyocyte proliferation. Cardiovasc Res (2010) 85(4):681–90. doi:10.1093/cvr/cvp360
46. Jopling C, Sleep E, Raya M, Marti M, Raya A, Izpisua Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature (2010) 464(7288):606–9. doi:10.1038/nature08899
47. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science (2011) 331(6020):1078–80. doi:10.1126/science.1200708
48. Piatkowski T, Muhlfeld C, Borchardt T, Braun T. Reconstitution of the myocardium in regenerating newt hearts is preceded by transient deposition of extracellular matrix components. Stem Cells Dev (2013) 22(13):1921–31. doi:10.1089/scd.2012.0575
49. van Amerongen MJ, Engel FB. Features of cardiomyocyte proliferation and its potential for cardiac regeneration. J Cell Mol Med (2008) 12(6A):2233–44. doi:10.1111/j.1582-4934.2008.00439.x
50. Zebrowski DC, Engel FB. The cardiomyocyte cell cycle in hypertrophy, tissue homeostasis, and regeneration. Rev Physiol Biochem Pharmacol (2013) 165:67–96. doi:10.1007/112_2013_12
51. Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, et al. Evidence for cardiomyocyte renewal in humans. Science (2009) 324(5923):98–102. doi:10.1126/science.1164680
52. Weeks KL, McMullen JR. The athlete’s heart vs. the failing heart: can signaling explain the two distinct outcomes? Physiology (Bethesda) (2011) 26(2):97–105. doi:10.1152/physiol.00043.2010
53. Yokoyama T, Nakano M, Bednarczyk JL, McIntyre BW, Entman M, Mann DL. Tumor necrosis factor-alpha provokes a hypertrophic growth response in adult cardiac myocytes. Circulation (1997) 95(5):1247–52. doi:10.1161/01.CIR.95.5.1247
54. Bozkurt B, Kribbs SB, Clubb FJ Jr, Michael LH, Didenko VV, Hornsby PJ, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation (1998) 97(14):1382–91. doi:10.1161/01.CIR.97.14.1382
55. Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, et al. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res (1997) 81(4):627–35. doi:10.1161/01.RES.81.4.627
56. Nelson DP, Setser E, Hall DG, Schwartz SM, Hewitt T, Klevitsky R, et al. Proinflammatory consequences of transgenic fas ligand expression in the heart. J Clin Invest (2000) 105(9):1199–208. doi:10.1172/JCI8212
57. Jain M, Jakubowski A, Cui L, Shi J, Su L, Bauer M, et al. A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure. Circulation (2009) 119(15):2058–68. doi:10.1161/CIRCULATIONAHA.108.837286
58. Novoyatleva T, Janssen W, Wietelmann A, Schermuly RT, Engel FB. TWEAK/Fn14 axis is a positive regulator of cardiac hypertrophy. Cytokine (2013) 64(1):43–5. doi:10.1016/j.cyto.2013.05.009
59. Mustonen E, Sakkinen H, Tokola H, Isopoussu E, Aro J, Leskinen H, et al. Tumour necrosis factor-like weak inducer of apoptosis (TWEAK) and its receptor Fn14 during cardiac remodelling in rats. Acta Physiol (2010) 199(1):11–22. doi:10.1111/j.1748-1716.2010.02080.x
60. Divakaran VG, Evans S, Topkara VK, Diwan A, Burchfield J, Gao F, et al. Tumor necrosis factor receptor-associated factor 2 signaling provokes adverse cardiac remodeling in the adult mammalian heart. Circ Heart Fail (2013) 6(3):535–43. doi:10.1161/CIRCHEARTFAILURE.112.000080
61. Bian Z, Dai J, Hiroyasu N, Guan H, Yuan Y, Gan L, et al. Disruption of tumor necrosis factor receptor associated factor 5 exacerbates pressure overload cardiac hypertrophy and fibrosis. J Cell Biochem (2013) 115(2):349–58. doi:10.1002/jcb.24669
62. Diwan A, Dorn GW II. Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiology (Bethesda) (2007) 22:56–64. doi:10.1152/physiol.00033.2006
63. Shaw J, Kirshenbaum LA. Molecular regulation of autophagy and apoptosis during ischemic and non-ischemic cardiomyopathy. Autophagy (2008) 4(4):427–34.
65. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling – concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol (2000) 35(3):569–82. doi:10.1016/S0735-1097(99)00630-0
66. Sivasubramanian N, Coker ML, Kurrelmeyer KM, MacLellan WR, DeMayo FJ, Spinale FG, et al. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation (2001) 104(7):826–31. doi:10.1161/hc3401.093154
67. Eghbali M, Blumenfeld OO, Seifter S, Buttrick PM, Leinwand LA, Robinson TF, et al. Localization of types I, III and IV collagen mRNAs in rat heart cells by in situ hybridization. J Mol Cell Cardiol (1989) 21(1):103–13. doi:10.1016/0022-2828(89)91498-3
68. Eghbali M, Czaja MJ, Zeydel M, Weiner FR, Zern MA, Seifter S, et al. Collagen chain mRNAs in isolated heart cells from young and adult rats. J Mol Cell Cardiol (1988) 20(3):267–76. doi:10.1016/S0022-2828(88)80059-2
69. Chorianopoulos E, Heger T, Lutz M, Frank D, Bea F, Katus HA, et al. FGF-inducible 14-kDa protein (Fn14) is regulated via the RhoA/ROCK kinase pathway in cardiomyocytes and mediates nuclear factor-kappaB activation by TWEAK. Basic Res Cardiol (2010) 105(2):301–13. doi:10.1007/s00395-009-0046-y
70. Chen HN, Wang DJ, Ren MY, Wang QL, Sui SJ. TWEAK/Fn14 promotes the proliferation and collagen synthesis of rat cardiac fibroblasts via the NF-small ka, CyrillicB pathway. Mol Biol Rep (2012) 39(8):8231–41. doi:10.1007/s11033-012-1671-3
71. Berry E, Bosonea AM, Wang X, Fernandez-Patron C. Insights into the activity, differential expression, mutual regulation, and functions of matrix metalloproteinases and a disintegrin and metalloproteinases in hypertension and cardiac disease. J Vasc Res (2013) 50(1):52–68. doi:10.1159/000345240
72. Goldsmith EC, Bradshaw AD, Spinale FG. Cellular mechanisms of tissue fibrosis. 2. Contributory pathways leading to myocardial fibrosis: moving beyond collagen expression. Am J Physiol Cell Physiol (2013) 304(5):C393–402. doi:10.1152/ajpcell.00347.2012
73. Iyer RP, Patterson NL, Fields GB, Lindsey ML. The history of matrix metalloproteinases: milestones, myths, and misperceptions. Am J Physiol Heart Circ Physiol (2012) 303(8):H919–30. doi:10.1152/ajpheart.00577.2012
74. Li YY, Kadokami T, Wang P, McTiernan CF, Feldman AM. MMP inhibition modulates TNF-alpha transgenic mouse phenotype early in the development of heart failure. Am J Physiol Heart Circ Physiol (2002) 282(3):H983–9. doi:10.1152/ajpheart.00233.2001
75. Lee WH, Kim SH, Lee Y, Lee BB, Kwon B, Song H, et al. Tumor necrosis factor receptor superfamily 14 is involved in atherogenesis by inducing proinflammatory cytokines and matrix metalloproteinases. Arterioscler Thromb Vasc Biol (2001) 21(12):2004–10. doi:10.1161/hq1201.098945
76. Mach F, Schonbeck U, Fabunmi RP, Murphy C, Atkinson E, Bonnefoy JY, et al. T lymphocytes induce endothelial cell matrix metalloproteinase expression by a CD40L-dependent mechanism: implications for tubule formation. Am J Pathol (1999) 154(1):229–38. doi:10.1016/S0002-9440(10)65269-8
77. Ueland T, Yndestad A, Oie E, Florholmen G, Halvorsen B, Froland SS, et al. Dysregulated osteoprotegerin/RANK ligand/RANK axis in clinical and experimental heart failure. Circulation (2005) 111(19):2461–8. doi:10.1161/01.CIR.0000165119.62099.14
78. Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res (2011) 108(9):1122–32. doi:10.1161/CIRCRESAHA.110.226928
79. Maier HJ, Schips TG, Wietelmann A, Kruger M, Brunner C, Sauter M, et al. Cardiomyocyte-specific IkappaB kinase (IKK)/NF-kappaB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc Natl Acad Sci U S A (2012) 109(29):11794–9. doi:10.1073/pnas.1116584109
80. Liu Q, Chen Y, Auger-Messier M, Molkentin JD. Interaction between NFkappaB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res (2012) 110(8):1077–86. doi:10.1161/CIRCRESAHA.111.260729
81. Tanaka T, Ogawa M, Suzuki J, Sekinishi A, Itai A, Hirata Y, et al. Inhibition of IkappaB phosphorylation prevents load-induced cardiac dysfunction in mice. Am J Physiol Heart Circ Physiol (2012) 303(12):H1435–45. doi:10.1152/ajpheart.00290.2012
82. Kumar S, Wei C, Thomas CM, Kim IK, Seqqat R, Kumar R, et al. Cardiac-specific genetic inhibition of nuclear factor-kappaB prevents right ventricular hypertrophy induced by monocrotaline. Am J Physiol Heart Circ Physiol (2012) 302(8):H1655–66. doi:10.1152/ajpheart.00756.2011
83. Dixit V, Mak TW. NF-kappaB signaling. Many roads lead to madrid. Cell (2002) 111(5):615–9. doi:10.1016/S0092-8674(02)01166-2
84. Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell (2002) 109(Suppl):S81–96. doi:10.1016/S0092-8674(02)00703-1
85. Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol (2002) 3(3):221–7. doi:10.1038/ni0302-221
86. Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol (2002) 3(10):958–65. doi:10.1038/ni842
87. Coope HJ, Atkinson PG, Huhse B, Belich M, Janzen J, Holman MJ, et al. CD40 regulates the processing of NF-kappaB2 p100 to p52. EMBO J (2002) 21(20):5375–85. doi:10.1093/emboj/cdf542
88. Muller JR, Siebenlist U. Lymphotoxin beta receptor induces sequential activation of distinct NF-kappa B factors via separate signaling pathways. J Biol Chem (2003) 278(14):12006–12. doi:10.1074/jbc.M210768200
89. Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is required for Peyer’s patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J (2003) 22(1):121–30. doi:10.1093/emboj/cdg004
90. Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev (2012) 246(1):125–40. doi:10.1111/j.1600-065X.2011.01088.x
91. Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, et al. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell (1998) 93(7):1231–40. doi:10.1016/S0092-8674(00)81466-X
92. De Ketelaere A, Vermeulen L, Vialard J, Van De Weyer I, Van Wauwe J, Haegeman G, et al. Involvement of GSK-3beta in TWEAK-mediated NF-kappaB activation. FEBS Lett (2004) 566(1–3):60–4. doi:10.1016/j.febslet.2004.04.041
93. Donohue PJ, Richards CM, Brown SA, Hanscom HN, Buschman J, Thangada S, et al. TWEAK is an endothelial cell growth and chemotactic factor that also potentiates FGF-2 and VEGF-A mitogenic activity. Arterioscler Thromb Vasc Biol (2003) 23(4):594–600. doi:10.1161/01.ATV.0000062883.93715.37
94. Potrovita I, Zhang W, Burkly L, Hahm K, Lincecum J, Wang MZ, et al. Tumor necrosis factor-like weak inducer of apoptosis-induced neurodegeneration. J Neurosci (2004) 24(38):8237–44. doi:10.1523/JNEUROSCI.1089-04.2004
95. Tanabe K, Bonilla I, Winkles JA, Strittmatter SM. Fibroblast growth factor-inducible-14 is induced in axotomized neurons and promotes neurite outgrowth. J Neurosci (2003) 23(29):9675–86.
96. Tran NL, McDonough WS, Savitch BA, Fortin SP, Winkles JA, Symons M, et al. Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and nuclear factor-kappaB and correlate with poor patient outcome. Cancer Res (2006) 66(19):9535–42. doi:10.1158/0008-5472.CAN-06-0418
97. Saitoh T, Nakayama M, Nakano H, Yagita H, Yamamoto N, Yamaoka S. TWEAK induces NF-kappaB2 p100 processing and long lasting NF-kappaB activation. J Biol Chem (2003) 278(38):36005–12. doi:10.1074/jbc.M304266200
98. Roos C, Wicovsky A, Muller N, Salzmann S, Rosenthal T, Kalthoff H, et al. Soluble and transmembrane TNF-like weak inducer of apoptosis differentially activate the classical and noncanonical NF-kappa B pathway. J Immunol (2010) 185(3):1593–605. doi:10.4049/jimmunol.0903555
99. Shi J, Jiang B, Qiu Y, Guan J, Jain M, Cao X, et al. PGC1alpha plays a critical role in TWEAK-induced cardiac dysfunction. PLoS One (2013) 8(1):e54054. doi:10.1371/journal.pone.0054054
100. Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med (2010) 14(11):2592–603. doi:10.1111/j.1582-4934.2010.01127.x
101. Moghimpour Bijani F, Vallejo JG, Rezaei N. Toll-like receptor signaling pathways in cardiovascular diseases: challenges and opportunities. Int Rev Immunol (2012) 31(5):379–95. doi:10.3109/08830185.2012.706761
102. Higashikuni Y, Tanaka K, Kato M, Nureki O, Hirata Y, Nagai R, et al. Toll-like receptor-2 mediates adaptive cardiac hypertrophy in response to pressure overload through interleukin-1beta upregulation via nuclear factor kappaB activation. J Am Heart Assoc (2013) 2(6):e000267. doi:10.1161/JAHA.113.000267
103. Wang L, Li YL, Zhang CC, Cui W, Wang X, Xia Y, et al. Inhibition of Toll-like receptor 2 reduces cardiac fibrosis by attenuating macrophage-mediated inflammation. Cardiovasc Res (2013). [Epub ahead of print]. doi:10.1093/cvr/cvt258
104. Shishido T, Nozaki N, Yamaguchi S, Shibata Y, Nitobe J, Miyamoto T, et al. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation (2003) 108(23):2905–10. doi:10.1161/01.CIR.0000101921.93016.1C
105. Riad A, Jager S, Sobirey M, Escher F, Yaulema-Riss A, Westermann D, et al. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J Immunol (2008) 180(10):6954–61.
106. Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ, et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res (2008) 102(2):257–64. doi:10.1161/CIRCRESAHA.107.158220
107. Ha T, Hua F, Li Y, Ma J, Gao X, Kelley J, et al. Blockade of MyD88 attenuates cardiac hypertrophy and decreases cardiac myocyte apoptosis in pressure overload-induced cardiac hypertrophy in vivo. Am J Physiol Heart Circ Physiol (2006) 290(3):H985–94. doi:10.1152/ajpheart.00720.2005
108. Navi A, Patel H, Shaw S, Baker D, Tsui J. Therapeutic role of toll-like receptor modification in cardiovascular dysfunction. Vascul Pharmacol (2013) 58(3):231–9. doi:10.1016/j.vph.2012.10.001
109. Han ES, Mekasha S, Ingalls RR. Fibroblast growth factor-inducible 14 (Fn14) is expressed in the lower genital tract and may play a role in amplifying inflammation during infection. J Reprod Immunol (2010) 84(1):16–23. doi:10.1016/j.jri.2009.09.009
110. Moreno JA, Sastre C, Madrigal-Matute J, Munoz-Garcia B, Ortega L, Burkly LC, et al. HMGB1 expression and secretion are increased via TWEAK-Fn14 interaction in atherosclerotic plaques and cultured monocytes. Arterioscler Thromb Vasc Biol (2013) 33(3):612–20. doi:10.1161/ATVBAHA.112.300874
111. Zhou H, Marks JW, Hittelman WN, Yagita H, Cheung LH, Rosenblum MG, et al. Development and characterization of a potent immunoconjugate targeting the Fn14 receptor on solid tumor cells. Mol Cancer Ther (2011) 10(7):1276–88. doi:10.1158/1535-7163.MCT-11-0161
112. Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol (2005) 5(4):331–42. doi:10.1038/nri1594
113. Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol (2010) 28:367–88. doi:10.1146/annurev.immunol.021908.132603
114. Seko Y, Ishiyama S, Nishikawa T, Kasajima T, Hiroe M, Suzuki S, et al. Expression of tumor necrosis factor ligand superfamily costimulatory molecules CD27L, CD30L, OX40L and 4-1BBL in the heart of patients with acute myocarditis and dilated cardiomyopathy. Cardiovasc Pathol (2002) 11(3):166–70. doi:10.1016/S1054-8807(02)00101-1
115. Ueno A, Murasaki K, Hagiwara N, Kasanuki H. Increases in circulating T lymphocytes expressing HLA-DR and CD40 ligand in patients with dilated cardiomyopathy. Heart Vessels (2007) 22(5):316–21. doi:10.1007/s00380-007-0977-x
116. Yndestad A, Damas JK, Geir Eiken H, Holm T, Haug T, Simonsen S, et al. Increased gene expression of tumor necrosis factor superfamily ligands in peripheral blood mononuclear cells during chronic heart failure. Cardiovasc Res (2002) 54(1):175–82. doi:10.1016/S0008-6363(02)00238-9
117. Jeremias I, Kupatt C, Martin-Villalba A, Habazettl H, Schenkel J, Boekstegers P, et al. Involvement of CD95/Apo1/Fas in cell death after myocardial ischemia. Circulation (2000) 102(8):915–20. doi:10.1161/01.CIR.102.8.915
118. Kwon BS, Tan KB, Ni J, Oh KO, Lee ZH, Kim KK, et al. A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J Biol Chem (1997) 272(22):14272–6. doi:10.1074/jbc.272.22.14272
119. MacFarlane M, Ahmad M, Srinivasula SM, Fernandes-Alnemri T, Cohen GM, Alnemri ES. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J Biol Chem (1997) 272(41):25417–20. doi:10.1074/jbc.272.41.25417
120. Madry C, Laabi Y, Callebaut I, Roussel J, Hatzoglou A, Le Coniat M, et al. The characterization of murine BCMA gene defines it as a new member of the tumor necrosis factor receptor superfamily. Int Immunol (1998) 10(11):1693–702. doi:10.1093/intimm/10.11.1693
121. Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science (1997) 277(5327):815–8. doi:10.1126/science.277.5327.815
122. Pan G, Ni J, Yu G, Wei YF, Dixit VM. TRUNDD, a new member of the TRAIL receptor family that antagonizes TRAIL signalling. FEBS Lett (1998) 424(1–2):41–5. doi:10.1016/S0014-5793(98)00135-5
123. Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, et al. The receptor for the cytotoxic ligand TRAIL. Science (1997) 276(5309):111–3. doi:10.1126/science.276.5309.111
124. Torre-Amione G, Kapadia S, Lee J, Durand JB, Bies RD, Young JB, et al. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation (1996) 93(4):704–11. doi:10.1161/01.CIR.93.4.704
125. Nybo M, Rasmussen LM. The capability of plasma osteoprotegerin as a predictor of cardiovascular disease: a systematic literature review. Eur J Endocrinol (2008) 159(5):603–8. doi:10.1530/EJE-08-0554
126. Omland T, Drazner MH, Ueland T, Abedin M, Murphy SA, Aukrust P, et al. Plasma osteoprotegerin levels in the general population: relation to indices of left ventricular structure and function. Hypertension (2007) 49(6):1392–8. doi:10.1161/HYPERTENSIONAHA.107.087742
127. Omland T, Ueland T, Jansson AM, Persson A, Karlsson T, Smith C, et al. Circulating osteoprotegerin levels and long-term prognosis in patients with acute coronary syndromes. J Am Coll Cardiol (2008) 51(6):627–33. doi:10.1016/j.jacc.2007.09.058
128. Ueland T, Dahl CP, Kjekshus J, Hulthe J, Bohm M, Mach F, et al. Osteoprotegerin predicts progression of chronic heart failure: results from CORONA. Circ Heart Fail (2011) 4(2):145–52. doi:10.1161/CIRCHEARTFAILURE.110.957332
129. Roysland R, Masson S, Omland T, Milani V, Bjerre M, Flyvbjerg A, et al. Prognostic value of osteoprotegerin in chronic heart failure: the GISSI-HF trial. Am Heart J (2010) 160(2):286–93. doi:10.1016/j.ahj.2010.05.015
130. Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guerin C, et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med (2013) 19(10):1273–80. doi:10.1038/nm.3284
131. Chorianopoulos E, Jarr K, Steen H, Giannitsis E, Frey N, Katus HA. Soluble TWEAK is markedly upregulated in patients with ST-elevation myocardial infarction and related to an adverse short-term outcome. Atherosclerosis (2010) 211(1):322–6. doi:10.1016/j.atherosclerosis.2010.02.016
132. Chorianopoulos E, Rosenberg M, Zugck C, Wolf J, Katus HA, Frey N. Decreased soluble TWEAK levels predict an adverse prognosis in patients with chronic stable heart failure. Eur J Heart Fail (2009) 11(11):1050–6. doi:10.1093/eurjhf/hfp139
133. Richter B, Rychli K, Hohensinner PJ, Berger R, Mortl D, Neuhold S, et al. Differences in the predictive value of tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in advanced ischemic and non-ischemic heart failure. Atherosclerosis (2010) 213(2):545–8. doi:10.1016/j.atherosclerosis.2010.08.061
134. Munoz-Garcia B, Martin-Ventura JL, Martinez E, Sanchez S, Hernandez G, Ortega L, et al. Fn14 is upregulated in cytokine-stimulated vascular smooth muscle cells and is expressed in human carotid atherosclerotic plaques: modulation by atorvastatin. Stroke (2006) 37(8):2044–53. doi:10.1161/01.STR.0000230648.00027.00
135. Yepes M, Brown SA, Moore EG, Smith EP, Lawrence DA, Winkles JA. A soluble Fn14-Fc decoy receptor reduces infarct volume in a murine model of cerebral ischemia. Am J Pathol (2005) 166(2):511–20. doi:10.1016/S0002-9440(10)62273-0
136. Filusch A, Zelniker T, Baumgartner C, Eschricht S, Frey N, Katus HA, et al. Soluble TWEAK predicts hemodynamic impairment and functional capacity in patients with pulmonary arterial hypertension. Clin Res Cardiol (2011) 100(10):879–85. doi:10.1007/s00392-011-0318-z
137. Wajant H. The TWEAK-Fn14 system as a potential drug target. Br J Pharmacol (2013) 170(4):748–64. doi:10.1111/bph.12337
138. Kamata K, Kamijo S, Nakajima A, Koyanagi A, Kurosawa H, Yagita H, et al. Involvement of TNF-like weak inducer of apoptosis in the pathogenesis of collagen-induced arthritis. J Immunol (2006) 177(9):6433–9.
139. Perper SJ, Browning B, Burkly LC, Weng S, Gao C, Giza K, et al. TWEAK is a novel arthritogenic mediator. J Immunol (2006) 177(4):2610–20.
140. Desplat-Jego S, Creidy R, Varriale S, Allaire N, Luo Y, Bernard D, et al. Anti-TWEAK monoclonal antibodies reduce immune cell infiltration in the central nervous system and severity of experimental autoimmune encephalomyelitis. Clin Immunol (2005) 117(1):15–23. doi:10.1016/j.clim.2005.06.005
141. Zhang X, Winkles JA, Gongora MC, Polavarapu R, Michaelson JS, Hahm K, et al. TWEAK-Fn14 pathway inhibition protects the integrity of the neurovascular unit during cerebral ischemia. J Cereb Blood Flow Metab (2007) 27(3):534–44. doi:10.1038/sj.jcbfm.9600368
142. Hotta K, Sho M, Yamato I, Shimada K, Harada H, Akahori T, et al. Direct targeting of fibroblast growth factor-inducible 14 protein protects against renal ischemia reperfusion injury. Kidney Int (2011) 79(2):179–88. doi:10.1038/ki.2010.379
143. Munoz-Garcia B, Moreno JA, Lopez-Franco O, Sanz AB, Martin-Ventura JL, Blanco J, et al. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) enhances vascular and renal damage induced by hyperlipidemic diet in ApoE-knockout mice. Arterioscler Thromb Vasc Biol (2009) 29(12):2061–8. doi:10.1161/ATVBAHA.109.194852
144. Schapira K, Burkly LC, Zheng TS, Wu P, Groeneweg M, Rousch M, et al. Fn14-Fc fusion protein regulates atherosclerosis in ApoE−/− mice and inhibits macrophage lipid uptake in vitro. Arterioscler Thromb Vasc Biol (2009) 29(12):2021–7. doi:10.1161/ATVBAHA.109.195040
145. Yoriki R, Akashi S, Sho M, Nomi T, Yamato I, Hotta K, et al. Therapeutic potential of the TWEAK/Fn14 pathway in intractable gastrointestinal cancer. Exp Ther Med (2011) 2(1):103–8. doi:10.3892/etm.2010.181
146. Kleinbongard P, Heusch G, Schulz R. TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol Ther (2010) 127(3):295–314. doi:10.1016/j.pharmthera.2010.05.002
147. Wilson EM, Diwan A, Spinale FG, Mann DL. Duality of innate stress responses in cardiac injury, repair, and remodeling. J Mol Cell Cardiol (2004) 37(4):801–11. doi:10.1016/j.yjmcc.2004.05.028
Keywords: cardiovascular disease, fibrosis, proliferation, hypertrophy, extracellular matrix, Toll-like receptors
Citation: Novoyatleva T, Sajjad A and Engel FB (2014) TWEAK-Fn14 cytokine-receptor axis: a new player of myocardial remodeling and cardiac failure. Front. Immunol. 5:50. doi: 10.3389/fimmu.2014.00050
Received: 01 November 2013; Paper pending published: 19 December 2013;
Accepted: 28 January 2014; Published online: 11 February 2014.
Edited by:
Timothy S. Zheng, Biogen Idec, Inc., USAReviewed by:
Hartmut Kleinert, Johannes Gutenberg University Mainz, GermanyKottarappat N. Dileepan, University of Kansas Medical Center, USA
Copyright: © 2014 Novoyatleva, Sajjad and Engel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Felix B. Engel, Department of Nephropathology, Institute of Pathology, Friedrich-Alexander-University Erlangen-Nürnberg, Universitätsstraße 22, 91054 Erlangen, Germany e-mail:ZmVsaXguZW5nZWxAdWstZXJsYW5nZW4uZGU=