 Berit Carow
Berit Carow Martin E. Rottenberg
Martin E. Rottenberg- Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Stockholm, Sweden
In this review, we describe the role of suppressor of cytokine signaling-3 (SOCS3) in modulating the outcome of infections and autoimmune diseases as well as the underlying mechanisms. SOCS3 regulates cytokine or hormone signaling usually preventing, but in some cases aggravating, a variety of diseases. A main role of SOCS3 results from its binding to both the JAK kinase and the cytokine receptor, which results in the inhibition of STAT3 activation. Available data also indicate that SOCS3 can regulate signaling via other STATs than STAT3 and also controls cellular pathways unrelated to STAT activation. SOCS3 might either act directly by hampering JAK activation or by mediating the ubiquitination and subsequent proteasome degradation of the cytokine/growth factor/hormone receptor. Inflammation and infection stimulate SOCS3 expression in different myeloid and lymphoid cell populations as well as in diverse non-hematopoietic cells. The accumulated data suggest a relevant program coordinated by SOCS3 in different cell populations, devoted to the control of immune homeostasis in physiological and pathological conditions such as infection and autoimmunity.
Introduction
Cytokines are secreted proteins central for coordination of the initiation, maintenance, and termination of all types of immune responses, including host responses to infection, inflammation, and trauma. Most cytokines have a short half-life and are released locally at high concentrations. They interact with cell surface receptors triggering responses that include cell survival, activation, coordination of microbicidal effector mechanisms, and proliferation and differentiation depending on the type of cytokine and the nature of the target cell. Cytokines are released in response to environmental clues to ultimately preserve physiological homeostasis. Some of them are pro-inflammatory, and initiate an inflammatory response necessary to fight infection. Other cytokines are anti-inflammatory and serve to reduce inflammation and promote healing once the injury/infection/foreign body has been destroyed.
A tight control of cytokine release and of responses to cytokines is required for the defense against infections, the prevention of infection-associated immunopathology, and the correct development of immune cell populations. A number of different cellular and molecular mechanisms control the magnitude and duration of innate and adaptive immune responses and several of these mechanisms regulate cytokine responses.
Several cytokines, growth factors, and hormones utilize the Janus kinase–signal transducer and activator of transcription (JAK–STAT) pathway to transmit their information into the cell nucleus. In short, the cytokine receptor is activated after binding the cytokine. Binding to the cytokine activates the kinase function of JAK, a tyrosine kinase that binds to the receptor, which auto-phosphorylates itself, cross-phosphorylates a JAK molecule bound to the accompanying heterodimer chain of the cytokine receptor and also different tyrosine sites on the cytokine receptor (1). The STATs will then bind to the phosphorylated receptor though its SH2 domain and be phosphorylated by JAK. The phosphorylated STAT will undergo a conformational change, detach from the receptor, and then bind to another phosphorylated STAT. STAT homo- or hetero-dimers translocate into the cell nucleus, bind to target genes, and promote their transcription (2). It is fascinating that only four JAK and seven STAT molecules mediate a huge diversity of biological effects, in face of their highly specific functions in the control of various immune responses revealed by genetic knockout studies (3). Such specificity is due to their individual patterns of activation by particular cytokine receptors and to some extent by their individual DNA sequence recognition preferences (4).
Janus kinase–signal transducer and activator of transcription pathways are tightly regulated at many steps through distinct mechanisms, including phosphotyrosine phosphatases (PTPs), protein inhibitor of activated STAT (PIAS), and suppressor of cytokine signaling (SOCS) proteins (5).
Phosphotyrosine phosphatases participate in the regulation of the JAK/STAT signaling pathway and have important implications in physiology and diseases (6).
The PIAS regulate the activity of many transcription factors, including STATs (7). Different PIAS bind different STATs and probably act by inhibiting their DNA binding or by recruiting histone deacetylases (8). Neither PTPs nor PIAS exclusively inhibit the JAK/STAT pathways, but are also main regulators of other cellular functions.
Suppressor of cytokine signaling is a protein family of eight members (SOCS1–7 and CIS) that inhibit STAT activation by many, but not all, JAK–STAT activating receptors. Experiments in different genetically manipulated mice have demonstrated a crucial role of SOCS molecules in pathophysiology. For example, SOCS1-deficient mice die within 3 weeks of birth due to severe systemic inflammation resulting from uncontrolled interferon-γ (IFN-γ) signaling (9). SOCS2-deficient mice develop gigantism due to enhanced responses to growth hormone (10). Mice lacking SOCS3, the objective of this review, die perinatally probably due to defective placental formation (11, 12).
Non-canonical ways of STAT activation have been shown (13). For example, unphosphorylated forms of STAT might function as transcription factors, modifiers of transcription factors, regulate the heterochromatin formation, or even possess non-transcriptional and extra-nuclear functions (14–16). The epidermal growth factor receptor (EGFR) catalyzes the tyrosine phosphorylation of STAT3 in response to EGF (17), and the intrinsic kinase activity of the receptor, but not of any JAK, is required for this reaction (18). Of importance for this review, these non-canonical STAT activation pathways might be independent of SOCS control (19).
Suppressor of cytokine signaling-3 regulates STAT3 activation in response to cytokines using the gp130 receptor. gp130 (CD130) forms part of the receptor complex for cytokines belonging to the IL-6 family, including IL-6, IL-11, IL-27, leukemia inhibitory factor (LIF), oncostatin M (OSM), ciliary neurotrophic factor (CNTF), cardiotrophin-1, and cardiotrophin-like cytokine. The functions of these cytokines encompass both unique but also partially redundant activities on multiple cell lineages (20). Gp130 is expressed in almost all organs and targeted deletion of the gp130 gene in mice results in embryonic lethality (21). SOCS3 also regulates the response to cytokines, growth factors, and hormones that are independent of gp130, such as the IL-12R, granulocyte-colony stimulation factor (G-CSF), leptin, insulin, and others, usually inhibiting STAT3 activation (22), but also other receptors that do not activate STAT3 (Table 1).
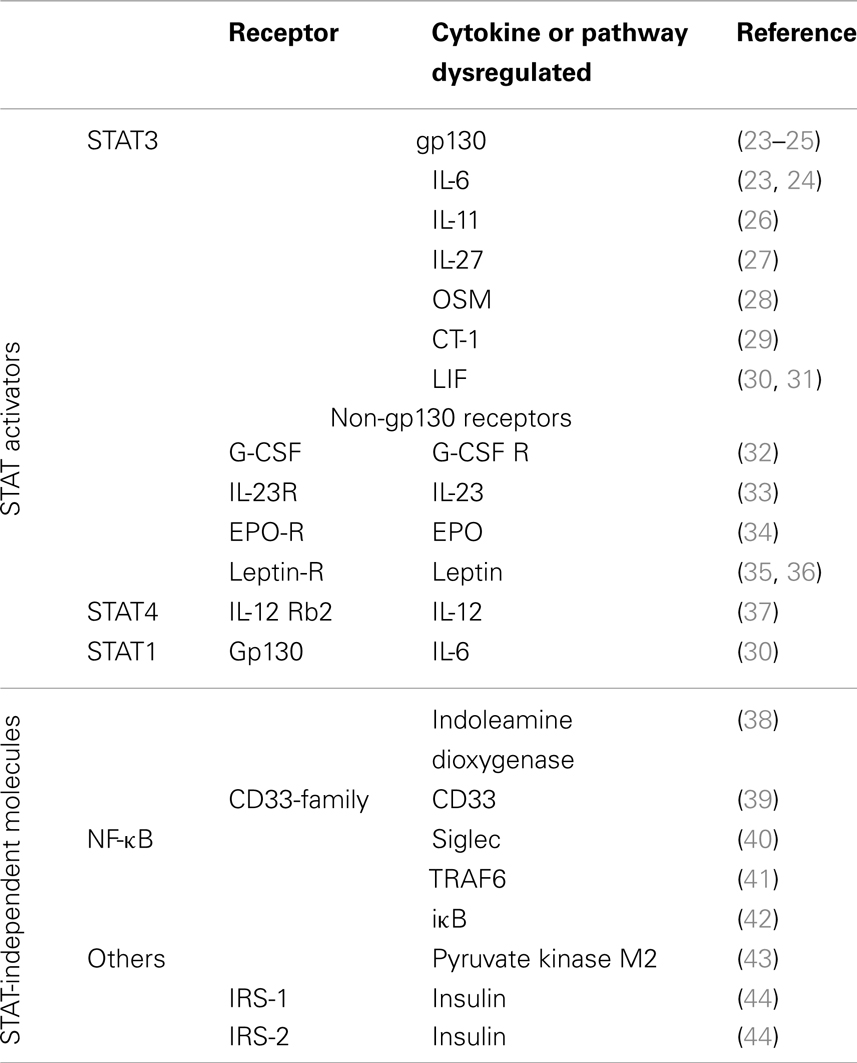
Table 1. Molecules regulated by SOCS3.
Studies in different mouse models have proven the critical importance of SOCS3 in restraining inflammation and allowing optimal levels of protective immune responses against infections. We here review the latest advances in SOCS3 biology, focusing on its role in the control of infection and inflammation.
SOCS Structure and Function
In 1995, Yoshimura et al. identified cytokine-induced STAT inhibitor (CIS), the first member of the SOCS family (45). A couple of years later, SOCS1 was shown to inhibit STAT signaling (46, 47), and the presence of several SOCS proteins with homologous conformations were predicted. Eight of these anticipated molecules in the human genome were subsequently cloned (SOCS1–7, CIS) (48–50).
All SOCS proteins have a central SH2 domain and a short C-terminal domain, the SOCS box as well as an N-terminal domain of varying length. SOCS inhibits the receptor complex by ubiquitination and subsequent proteasome-mediated degradation. SOCS proteins act as substrate adapters: the SOCS box associates with a complex containing elongin B and C and this complex then binds Cullin-5 (51, 52). Since SOCS proteins contain a central SH2 domain, any tyrosine phosphorylated signaling intermediate (phospho-JAK, phospho-STAT, phosphorylated receptors) is a conceivable substrate. Thus, the SH2 domain functions as an adapter bringing ubiquitin ligases close to kinase-activated signaling proteins, mediating their degradation (52).
However, SOCS1 and 3, the most studied molecules of the family, are partially active in absence of their SOCS box domain (53). Moreover, the SOCS box of SOCS1 and SOCS3 binds with lower affinity to the E3 ubiquitin ligase than those of SOCS2, 4–7, and CIS (52).
Instead, SOCS3 and SOCS1, but not the other members of the SOCSs family, bind the JAKs directly inhibiting their kinase activity. Studies using truncated or chimeric forms of SOCS proteins showed that SOCS1 and SOCS3 contained a short N-terminal kinase inhibitory region (KIR) resembling a JAK substrate, which allows them to suppress signaling by direct inhibition of JAK’s catalytic activity (54, 55).
There are four mammalian JAKs (JAK1–3 and TYK2). SOCS3 has been shown to inhibit JAK1, JAK2, and TYK2 but not JAK3 (56). Despite the ability of SOCS3 to bind to and inhibit JAKs, deletion of individual SOCS genes in mice has revealed an exquisite specificity for particular cytokine receptor combinations rather than specific JAKs. This specificity is provided by the binding of the SH2 domain of the SOCS proteins to the gp130 cytokine receptor (57).
The ability of SOCS3 to simultaneously bind to JAK and to the cytokine receptor explains the specificity of the suppression. SOCS3 generates a ternary complex in which each moiety is directly bound to the other two with an overall affinity higher than the individual associations (Figure 1) (58). In other words, SOCS3 inhibits JAK’s enzymatic activity by blocking substrate binding and gains specificity of action by only binding tightly to JAK when the kinase is attached to specific receptors. SOCS3 binds JAK and gp130 receptor simultaneously, using two opposing surfaces: while the phosphotyrosine-binding groove on the SOCS3 SH2 domain is occupied by the gp130 receptor, a subdomain in the SH2 domain of SOCS3 is also required for inhibition of JAK, binding in a phospho-independent manner to a non-canonical surface of JAK2 (58, 59). The KIR of SOCS3 occludes the substrate-binding groove on JAK2.
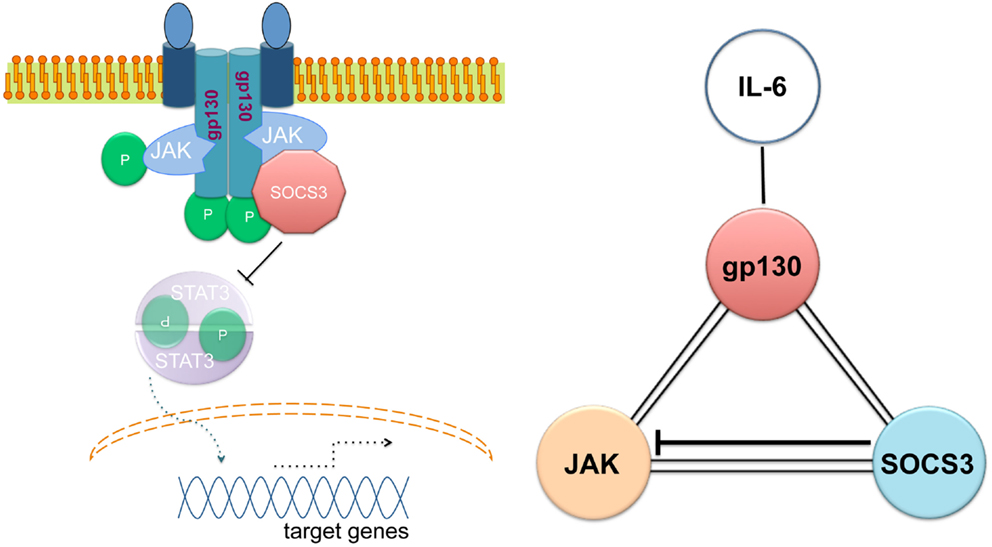
Figure 1. Simultaneous binding of SOCS3 to JAK and the gp130 cytokine receptor. Adapted from Ref. (56).
On the other hand, a relevant role for the SOCS box in the SOCS3-mediated proteasomal degradation resulting in the regulation of cytokine signaling through some receptors (i.e., the G-CSF receptor) has been shown (60, 61). Thereby, SOCS3 regulates a STAT3-mediated chemokine and chemokine receptor function within the bone marrow, and plays a central role in the neutrophil mobilization response (62).
The SOCS3 structure does not exclude an apparent specificity of SOCS3 as an inhibitor of the activation of other STATs than STAT3. As indicated below, SOCS3 also inhibits IL-6-mediated STAT1 and STAT4 activation (30, 63).
SOCS3 and STAT3
Loss of SOCS3 in vivo has profound effects on placental development, inflammation, fat-induced weight gain, and insulin sensitivity.
Genetic deletion of SOCS3 leads to mid-gestational embryonic lethality due to increased STAT3 and MAP kinase activation (11, 12). Lack of suppression of LIF and fetal erythropoiesis signaling had been shown to account for the lethality of Socs3−/− mice (11, 12). LIF belongs to the IL-6 family and is involved in blastocyst implantation. LIFR deficit rescued the Socs3−/− placental defect and embryonic lethality. These double KO mice died by 190 days of age due to neutrophilia accompanied by neutrophil tissular infiltration (64). However, mice with a mutation in the gp130 chain of the IL-6 receptor family that impairs binding of SOCS3 (gp130F/F mice) display no early lethality (65), indicating that altered LIFR signaling is not the only cause of the mid-gestational death of Socs3−/− mice (Table 2).
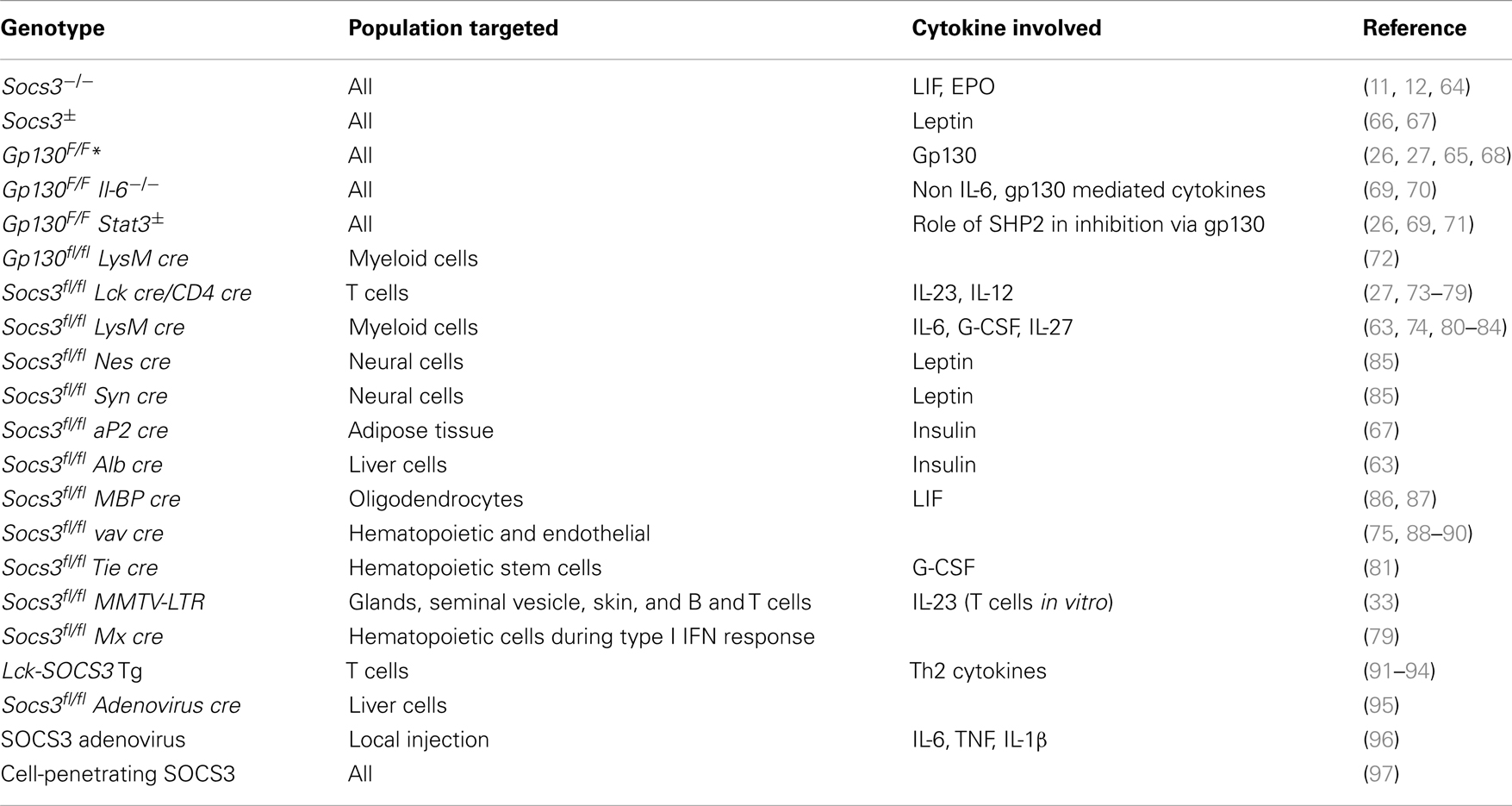
Table 2. Mouse models to study SOCS3 function.
Mice with a deletion of SOCS3 in hematopoietic cells (Socs3fl/fl vav cre) have been shown to develop a severe inflammatory disease during adult life (88). IL-6 was not critical in regulating the severity of this spontaneous inflammatory disease but played a role in the onset (89). Since Socs3−/− but not Socs3fl/fl vav cre mice die during gestation, SOCS3 probably impairs lethal cytokine or growth factor responses in non-hematopoietic cells.
Gp130F/F mice spontaneously develop lymphadenopathy, splenomegaly, and gastric hyperplasia (70). The basis for this phenotype is complex, but it appears that the enhanced ability of IL-11, rather than IL-6, to activate STAT3 and STAT1 in absence of SOCS3 promotes inflammation and cancer (26, 69).
On the other hand, enhanced IL-6 responses accounted for the enhanced susceptibility of Socs3fl/fl vav cre or Socs3fl/fl LysM cre (deficient in SOCS3 in myeloid cells) mice to induced inflammatory diseases like rheumatoid arthritis (RA) or experimental autoimmune encephalomyelitis (EAE) (90, 98). Gp130F/F mice spontaneously develop a RA-like disease that is accelerated by IL-6 administration (99). Accordingly, adenoviral-delivered SOCS3 reduced joint inflammation in mice with arthritis via inhibition of IL-6 signaling (96).
Suppressor of cytokine signaling-3 is not an essential regulator of IL-10 or IFN-γ responses (80). In the absence of SOCS3 in hematopoietic or myeloid cells, IL-6 acts like IL-10 and attenuates macrophage secretion of TNF and IL-12 after LPS stimulation (80). Furthermore, Socs3fl/fl LysM cre mice were protected from the lethal effects of galactosamine and LPS administration, a model that is dependent on TNF-induced liver failure (100). These results were somewhat contrary to the expected: if IL-6 is a pro-inflammatory cytokine, removal of its inhibitor should result in more, rather than in less, inflammation. Paradoxically, the pro-inflammatory IL-6 and the anti-inflammatory IL-10, generating nearly opposing cellular responses, both activate STAT3 after binding to their receptors. The kinetic of STAT3 activation was pointed as the putative cause of SOCS3’s effect: the suppressive effect of IL-6 signaling on TNF and IL-12 secretion in absence of SOCS3 was explained to be due to a sustained STAT3 activation (101, 102). In line with this, a transient activation of the IL-10 receptor elicited an IL-6-like response (102). However, how the duration of STAT3 activation can direct distinct responses is far from being understood.
Interestingly, after the initial phosphorylation of STAT3 in response to IL-6 followed by a subsequent inhibition by SOCS3, a second wave of activation leads to the re-phosphorylation of STAT3 (101). It has been recently shown that re-phosphorylation requires an IL-6-dependent association of IL-6R and EGFR without involvement of gp130. STAT3 phosphorylation thus might continue to be driven for many hours by this two-receptor complex that is immune to inhibition by SOCS3 (19).
The anti-inflammatory responses of SOCS3-deficient macrophages or dendritic cells (DCs) are not restricted to diminished TNF- or IL-12 levels but also have been shown to increase the secretion of IL-10, expand the numbers of regulatory T (Treg) cells, and decrease MHC-II expression levels (81). In line with this, SOCS3 inhibited the TGFβ1/Smad3 signaling pathway, leading to enhanced LPS responses in macrophages (103). In contrast, SOCS3 expression in myeloid cells has been shown to mediate LPS-induced lung injury (82).
Deletion of SOCS3 in hematopoietic cells surprisingly also enhanced the expression of STAT1-stimulated genes in response to IL-6 (30, 63). The activation of STAT1 in SOCS3-deficient cells is probably due to a more dramatic inhibition of STAT1 than of STAT3 by SOCS3. A differential effect of SOCS3 on STAT3 and STAT1 has lately been used to explain the preferential development of either M1 (classically activated) (83, 98) or M2 (alternatively activated) macrophages from Socs3fl/fl Lys M cre mice (104, 105). M1 macrophages are differentiated after IFN-γ-stimulation, while IL-4 and/or IL-10 activate an alternative M2 program. Importantly, in these contradictory studies on the role of SOCS in the regulation of M1 and M2 polarization, Socs3fl/fl LysM cre mice showed either increased resistance or susceptibility to LPS-induced septic shock (83, 104). Altogether, it is still unknown whether SOCS3 determines whether a cellular response to IL-6 is pro- or anti-inflammatory. The fine regulation exerted by SOCS3 needs further understanding.
Mechanisms regulating macrophage polarization via SOCS3 were also studied. The Notch signaling pathway specifies cell differentiation during development (106). Activation of Notch signaling increased M1 macrophage differentiation, no matter whether M1 or M2 inducers were applied. When Notch signaling was blocked, even the M1 inducers induced a M2 response. Interestingly, Notch signaling has been shown to regulate macrophage polarization via SOCS3 (107): in the presence of an inhibitor of Notch signaling, macrophages over-expressing SOCS3 showed restored M1 polarization in response to LPS (107). Notch is also involved in SOCS3 up-regulation following a mycobacterial infection (108).
Perturbed hematopoiesis was observed in mice lacking SOCS3 in myeloid cells (88, 109). IL-6 played a role in the onset of this severe disease (89). These mutant mice were hyper-responsive to injected G-CSF, showing exaggerated neutrophilia, mobilization of progenitor cells into the blood, splenomegaly, and an accelerated disease (88, 110). SOCS3 expression was stimulated by G-CSF and SOCS3 directly bound to a phosphotyrosine on the G-CSF-receptor (32). The effect of SOCS3 in the regulation of neutrophil biology might well underlie its protective activity in development of spontaneous or induced inflammatory diseases.
SOCS3 but Not STAT3
Besides inhibiting JAK–STAT-mediated signals, SOCS3 has been suggested to hamper other signaling pathways (Table 1). SOCS3 has been shown to bind to indoleamine dioxygenase (IDO), targeting the complex for ubiquitination in DCs. Thus, acting at a post-transcriptional level it antagonizes IDO-dependent tolerogenic signals in DCs and converts them into immunogenic (38, 111).
Suppressor of cytokine signaling-3 has also been shown to bind and degrade CD33 or Siglec 3, blocking CD33-mediated inhibition of proliferation (39). Siglec 7, another CD33-family receptor, has also been found to be bound and degraded by SOCS3 (40).
It has also been proposed that SOCS3 can bind to the insulin receptor (IR) or the insulin receptor substrate-1 (IRS-1) targeting them for proteasomal degradation and regulating thereby insulin sensitivity (44, 112–114).
Suppressor of cytokine signaling-3 has also been shown to directly interact with SMAD3 inhibiting the responses to TGF-β (103). On the other hand, TGF-β has been shown to induce the expression of SOCS3, allowing TNF-induced osteoclast formation (115).
Different microorganisms including mycobacteria stimulate the expression of SOCS3. PPE18, associated with mycobacterial virulence has been shown to increase SOCS3 expression and its tyrosine phosphorylation. Surprisingly phospho-SOCS3 was found to bind to iκB hampering its degradation and thereby blocking NF-κB activation (42). Another study indicated that SOCS3 inhibits both the IL-1-induced NF-κB and JNK/p38 pathways by binding the upstream molecule TRAF6 and preventing its function (41). Similarly, IL-25, a member of the IL-17 cytokine family that promotes Th2 responses, has been shown to hamper the LPS-induced, MAP kinase p38-dependent secretion of pro-inflammatory cytokines in a SOCS3-mediated manner (116).
The functional role of a JAK/STAT-independent SOCS3 regulation of these molecules remains to be validated. However, these results suggest that the characterization of SOCS3 as a STAT3 inhibitor is oversimplified.
SOCS3 and T Cells
The role of SOCS3 in T cell development has been somewhat overlooked. SOCS3 is expressed in the double negative (early) stage of thymocyte differentiation (73), a stage at which T cells determine their expression of γδ+ or αβ+ T cell receptors (TCR). Most T cells have a TCR composed of two chains called α and β (so called αβ+ T cells). In contrast, a small subset of T cells has a TCR made up by a γ and a δ chain. These γδ+ T cells are more of an innate T cell subset, with a relatively invariant TCR and lower if any requirement of antigen recognition or processing for activation. Recent experiments from our laboratory showed that the thymus, or secondary immune organs of either neonatal or adult mice with T cells lacking SOCS3 have an increased frequency of γδ+ T cells compared to controls (74). Thus, SOCS3 regulates T cell development in the thymus. However, the precise mechanisms utilized by SOCS3 remain unexplored.
IL-27 has an anti-inflammatory role during immune responses, such as CD28-mediated IL-2 secretion. SOCS3-deficient CD8+ T cells showed higher proliferation in response to TCR ligation than wild-type cells despite a normal activation of signaling pathways downstream the TCR and CD28 receptors. Suppression of IL-27 signaling was found to substantially reduce the increased anti-CD3-induced proliferation of SOCS3-deficient T cells (27). Thus, SOCS3 mediates the anti-proliferative role of IL-27. The expression of SOCS3 is induced by IL-27 in mouse and human cells (117), and mediates the inhibitory effect of IL-27 (118). In line with this, SOCS3 deficiency in donor T cells promoted acute GVHD mortality (75).
Cytokines can direct CD4+ Th0 cells into Th1, Th2, Th17, or Treg cell lineages. Th2 cells contain higher amounts of SOCS3 compared to Th1 cells (119). SOCS3 has been also suggested to inhibit IL-12-induced STAT4 activation by direct binding to IL-12Rβ2 (37), the IL-12R subunit that is not shared with the IL-23R.
In line with this, the increased SOCS3 expression in T cells correlated with the severity of asthma and atopic dermatitis or with the Th1-mediated condition psoriasis (91, 120, 121). Furthermore, defined haplotypes of SOCS3 have been linked with atopic dermatitis in childhood cohorts (121).
Accordingly, over-expression of SOCS3 in T cells inhibits Th1 and promotes Th2 development suggesting that SOCS3 stimulates allergic responses (91). T cell-specific expression of SOCS3 also aggravated allergic conjunctivitis, a Th2-mediated model of disease (92). Inhibition of SOCS3 expression in T cells exhibited markedly suppressed airway hyper-responsiveness and eosinophilia (76, 77). Mice with T cells over-expressing SOCS3 also showed a delayed onset of EAE and restricted Th17 differentiation (122).
In vitro, SOCS3-deficient CD4+ T cells produced more TGF-β and IL-10 but less IL-4 than control T cells (77). TGF-β inhibits IL-6- and IL-21-induced SOCS3 expression, thus enhancing as well as prolonging STAT3 activation in naive T cells (123). Thus, TGF-β production is inhibited by SOCS3 and vice versa.
Th17 cell differentiation is induced by IL-6 and IL-21 in the presence of TGF-β through the activation of STAT3 (124). STAT3 induces the orphan nuclear receptor RORγt, which directs Th17 cell differentiation by producing the IL-23 receptor (124). The survival and expansion of committed murine Th17 cells requires IL-23 (125). The critical role of STAT3 in Th17 differentiation was also confirmed in human patients lacking functional STAT3 (126). SOCS3 was found to be a major negative regulator of IL-23-mediated STAT3 phosphorylation and Th17 generation (33, 78, 123).
Suppressor of cytokine signaling-3 expression in fibroblasts has been shown to participate in Th17 development. IL-17 increased a STAT3-dependent IL-6 expression in fibroblasts. IL-6 secretion was enhanced in mice deficient for SOCS3 in fibroblasts resulting in enhanced Th17 levels (99). Thus, SOCS3-mediated regulation of cytokine responses in T and non-T cell lineages impairs Th17 differentiation.
IL-17 has been also shown to increase collagen fiber formation and fibrous cap development in atherosclerosis models (127). The strength of the cap given by the collagen fibers prevents plaque rupture, a condition that elicits thrombosis and infarction in patients. The increased Th17 frequency among SOCS3-deficient T cells reduced atherosclerotic lesion development, which was dependent on IL-17. Accordingly, the over-expression of SOCS3 in T cells reduced IL-17 levels and accelerated atherosclerosis and severe aortic aneurysm formation (78, 93).
Transplanted neural progenitor cells (NPCs) differentiate into neural cells and provide the means to repair demyelinated nerve fibers in the CNS in neuroinflammatory diseases like multiple sclerosis. Surprisingly, NPC treatment has been shown to suppress self-reactive T cells and control tissue inflammation via the secretion of LIF. LIF inhibits Th17 differentiation by inducing SOCS3 (128). The LIF receptor is expressed on T cells. Thus, two members of the IL-6 family, IL-6 and LIF, show opposing effects on Th17 development: while IL-6 is required for Th17 development, LIF counteracts it. During Th17 development, IL-6 activates while LIF inhibits STAT3 activation. In line, inhibition of SOCS3 with specific siRNA hampered the inhibitory effects of LIF on Th17 development (128).
As stated above, SOCS3 expression in non-T cells can direct T cell differentiation and function. DCs are required for T cell selection, differentiation, and activation. SOCS3-deficient DCs expressed lower levels of MHC II, CD40, CD86, and IL-12 (81, 111). As discussed, SOCS3 expression in DCs antagonized a tolerogenic CTLA-4 activity by direct interaction and degradation of IDO (38).
Contrary to these observations, DCs transduced with SOCS3 significantly inhibited IL-12 and IL-23 activation of STAT4 and STAT3, respectively. Together with an inhibition of MHC-II and CD86 expression, SOCS3 promoted a Th2 differentiation that hampered EAE development (129). SOCS3 might also interact with pyruvate kinase-M2 to decrease ATP production, accounting for such a DC dysfunction (43).
SOCS3 and Infections
Suppressor of cytokine signaling-3 expression is stimulated by cytokine or innate immune receptor agonists present in viruses, bacteria, and parasites (22, 130). Due to the multiple binding partners of SOCS3, it is hard to predict its role in different infections. Of importance, SOCS3 expression is induced by several cytokines to which the receptor has no binding site (i.e., IL-10), probably to inhibit trans-signaling via other cytokines (131). Table 3 provides a summary of the role of SOCS3 in several infections.
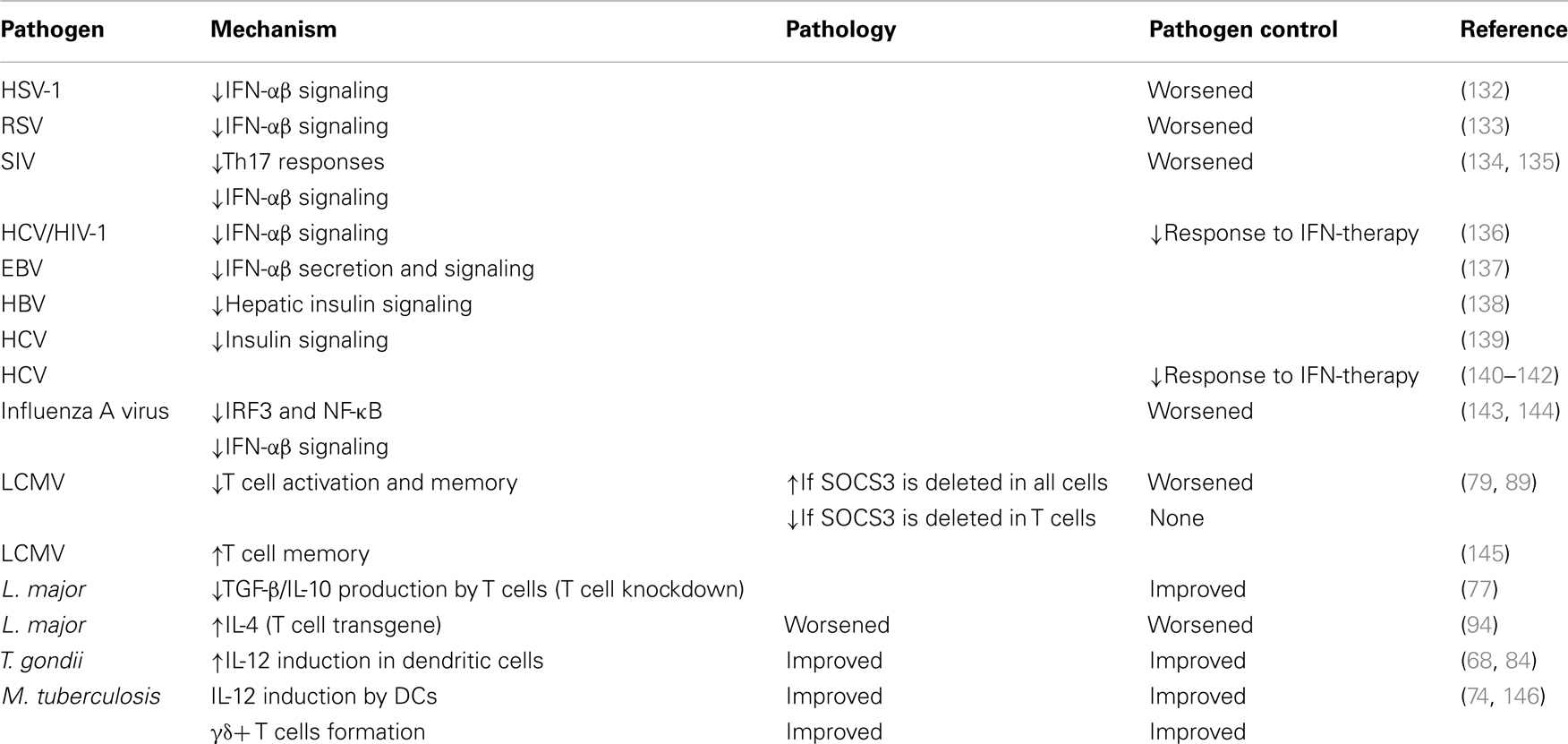
Table 3. Role of SOCS3 in infections.
Viral Infections
The lymphocytic choriomeningitis virus (LCMV) clone 13 triggers insufficient CD8+ T cell responses and thus persists indefinitely in the host. LCMV promotes high expression of SOCS3 in T cells, resulting in impaired antiviral functions and viral persistence. Treatment of LCMV-infected mice with IL-7 repressed SOCS3 expression and enhanced T cell effector functions and viral clearance. Mechanistically, a reduction of SOCS3 allowed the differentiation of Th17 cells. IL-17 stimulates the expression of IL-6 that mediates survival and function of antiviral T cells. IL-7 also promotes IL-22 secretion, which protects against immune tissue destruction (79). On the other hand, the deletion of SOCS3 in all hematopoietic cells induced an IL-6 dependent early lethality in LCMV-infected mice despite the viral clearance (89).
Hepatitis C virus (HCV) infection is a major cause of chronic liver disease, affecting 170 million persons worldwide. The most effective current treatment for chronic HCV is the combination of type I IFN and ribavirin, a nucleoside analog. Hepatic SOCS3 expression is associated with non-response to therapy in human HCV patients (136, 140). Particular SOCS3 (-4874 AA) genotypes express SOCS3 at elevated levels and consequently have a poorer response to therapy (140–142). One microRNA (miR122) modulated the response to type I IFNs: silencing of miR122 enhanced IFN-induced ISRE activity by decreasing the expression of SOCS3. Interestingly, such decrease in SOCS3 levels was not mediated by microRNA target gene suppression, but rather by enhanced methylation at the SOCS3 gene promoter (147).
Alike, the infection of human cells by either Epstein–Barr virus or Herpes simplex virus (HSV) has been shown to stimulate SOCS3 expression that suppresses type I IFN production and responses (132, 137). Silencing of SOCS3 using anti-sense nucleotides significantly hampered replication of HSV-1 (132).
In studies of simian immunodeficiency virus (SIV) infection, a significant increase in markers of microbial translocation that correlate with suppressed Th17 responses was found. Elevated expression of SOCS3 could potentially play a role in suppressing IL-17 expression during an acute SIV infection (134).
In a SIV/macaque model of HIV-associated dementia, SOCS3 expression correlated with recurrence of viral replication and onset of CNS disease. SOCS3 expression attenuated the response of macrophages to IFN-β. Thus, SOCS3 may allow HIV-1 to evade the protective innate immune response in the CNS by overcoming the inhibitory effect of IFN-β on HIV-1 replication within the macrophages (135).
Bacterial and Parasitic Infections
The role of SOCS3 in the outcome of infection with intracellular bacteria and parasite has also been studied.
Toxoplasma gondii is an intracellular eukaryotic pathogen that causes toxoplasmosis, a life-threatening condition, which includes congenital disease and infection in immunocompromised individuals. Toxoplasma possesses a secretory organelle called rhoptry. Infection with T. gondii diminished innate immune responses due to the inoculation of the rhoptry ROP16 kinase (148). ROP16 activated STAT3, which stimulated SOCS3 expression that in turn diminished STAT3 activation (84). Socs3fl/fl LysM cre or gp130F/F mice succumbed to T. gondii infection, and resistance could be restored by neutralization of IL-6 (68, 84). Diminished IL-12 secretion by SOCS3-deficient DCs was suggested to impair IFN-γ secretion by antigen-specific T cells, probably contributing to the increased susceptibility to T. gondii infection of the mutant mice (68).
Mycobacterium tuberculosis causes the highest mortality to a single pathogen worldwide. Only 10% of infected individuals will manifest active tuberculosis while most apparently control the infection by an appropriate immune response. Infection with mycobacteria enhanced the expression of SOCS3 in phagocytes (149).
We demonstrated a critical role for SOCS3 expression by myeloid and lymphoid cells in resistance against M. tuberculosis (74). All Socs3fl/fl LysM cre, Socs3fl/fl lck cre (with SOCS3-deficient T cells), and gp130F/F mice showed increased susceptibility to infection with M. tuberculosis. SOCS3 binding to gp130 in myeloid cells conveyed resistance to M. tuberculosis infection via the regulation of IL-6/STAT3 signaling. SOCS3 was redundant for mycobacterial control by macrophages in vitro. Instead, SOCS3 expression in infected macrophages and DCs prevented the IL-6-mediated inhibition of IL-12 secretion and contributed to a timely CD4+ cell-dependent IFN-γ expression (Figure 2A).
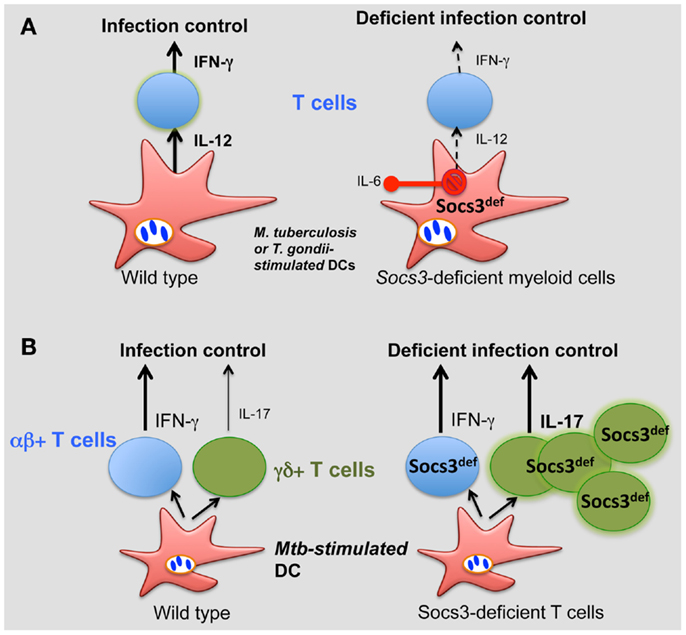
Figure 2. Role of SOCS3 in the control of infection. (A) During M. tuberculosis and T. gondii infection, SOCS3 is induced in macrophages and DCs prevents the IL-6-mediated inhibition of IL-12 secretion, promoting a CD4+ cell-dependent IFN-γ expression. (B) SOCS3 controls the development of γδ+ T cells. Such control is critical for proper protection against M. tuberculosis.
Interestingly, γδ+ rather than αβ+ T cells accounted for the susceptibility to infection of Socs3fl/fl lck cre mice. γδ+ T cells’ numbers in SOCS3-deficient mice were increased (independently of infection) and accounted for the exacerbated susceptibility to disease of these mutant mice (Figure 2B). Surprisingly, opposed to αβ γδ+ T cells have been shown to be redundant in protection of mice against M. tuberculosis. In line with this, severity of tuberculosis in humans was inversely associated with the expression of SOCS3 (146).
Macrophage/neutrophil-specific gp130-deficiency, the over-expression of soluble gp130 (sgp130) or the administration of sgp30 did no affect mycobacterial loads or pathology (72, 150). Similarly, Il-6−/− mice have an unimpaired generation of protective memory responses and control of mycobacterial growth (151).
Socs3fl/fl lck cre mice showed a worsened disease progression after infection with Leishmania major, which associated to the hyper-production of IL-10 and TGF-β (77). On the other hand, transgenic mice over-expressing the SOCS3 gene in T cells (Lck-SOCS3 Tg mice) were also susceptible to infection by L. major due to an increased IL-4 secretion (94), altogether suggesting that a tight regulation of SOCS3 expression in T cells is crucial for disease control during infection by L. major.
Thus, SOCS3 seems to be detrimental for controlling viral infections by impairing proper type I IFN in viral defense. On the other hand, SOCS3 enables suitable IL-12 secretion by DCs for bacterial parasite control. However, a tight regulation of SOCS3 expression in T cells is critical for determining the outcome of infection.
SOCS3 in Non-Hematopoietic Cells
Suppressor of cytokine signaling-3 plays important physiological roles in non-hematopoietic cells such as neurons, muscle cells, hepatocytes, fibroblasts, and adipocytes. For example, SOCS3 controls neurite outgrowth in dorsal root ganglia, insulin resistance in adipocytes, and is associated with age-related blunted muscle stem cell responses (152–154).
During inflammation, SOCS3 is expressed in epithelial and lamina propria cells in the colon of mice with intestinal bowel disease, in human ulcerative colitis and Crohn’s disease patients (155), and in synovial fibroblasts of RA patients (96). In human atherosclerotic lesions, vascular smooth muscle cells and macrophage expressed SOCS3 (156). The over-expression of SOCS3 in T cells reduced IL-17 and accelerated atherosclerosis whereas the in vivo treatment with anti-sense oligodeoxynucleotides targeting SOCS3 exacerbated the atherosclerotic process in ApoE−/− mice (78).
These findings are consistent with the idea that IL-6-related cytokines promote while SOCS3 prevents chronic disease progression. However, no dogmas can be concluded since a pathogenetic involvement of SOCS3 has also been shown: in obesity, chronic JAK–STAT3 activation in the CNS by increased circulating leptin levels lead to the development of leptin resistance, whereas in the peripheral organs chronic IL-6-induced STAT3 activation impairs insulin action (157). Leptin is secreted from adipocytes proportionally to the amount of fat stored in the white adipose tissue and acts on a group of neurons of the hypothalamus to suppress food intake and to increase energy expenditure (158). In obesity, expansion of white adipose tissue increases leptin levels, but the protein does not convey its biological effects. SOCS3 expression in the CNS is largely increased in obesity. SOCS3 binds to the leptin receptor and thereby limits leptin action (35, 36, 159). Mice lacking SOCS3 in this particular population of neurons are protected from the development of diet-induced obesity and maintain central leptin sensitivity (66, 85, 160).
In agreement with these results, circulating levels of cytokines including IL-6 and TNF-α impair insulin signaling in peripheral organs. IL-6 increases SOCS3 levels in adipose tissues, muscle cells, and hepatocytes. Mice with deficient SOCS3 expression in adipose tissues were protected against the development of obesity-associated insulin resistance (67). As indicated above, SOCS3 impairs insulin action by binding to the insulin receptor or the IRS-1 and IRS-2 leading to their ubiquitination and degradation or by inhibition of receptor tyrosine phosphorylation (44, 113).
IL-6-related cytokines are induced by and play a protective role in the injured myocardium. Mice with a SOCS3 deletion in cardiomyocytes showed higher activation of STAT3, AKT, and ERK1/2 pathways, and reduced mitochondrial damage, oxidative stress, and inflammation resulting in the prevention of myocardial injury (161).
Pharmacological Targeting of SOCS3
The multiple effects of SOCS3 in different cell lines and experimental models call for thorough investigations to clarify its main mechanisms and targets. Strategies increasing SOCS3 expression or mimicking its consequences (i.e., hampering STAT3 activation) might be appropriate for immune-prophylaxis or -therapy of several infectious or inflammatory diseases. In line with this, in some inflammation models, SOCS3 over-expression mitigates inflammatory arthritis induced by antigen/IL-1β or collagen, as well as acute inflammation induced by staphylococcal enterotoxin B and LPS (90, 97, 162). On the other hand, when STAT3 plays a protective role for tissue injury, such as in ConA-induced hepatitis, deletion of SOCS3 is anti-inflammatory. As described above, SOCS3 deficiency in macrophages protects mice from LPS-shock because of the enhanced anti-inflammatory effect of STAT3 (80). The down-modulation of SOCS3 expression in CD4+ T cells might be effective in preventing the development of allergic asthma (76).
Therapeutic trials using SOCS3-specific anti-sense oligonucleotides, small hairpin RNAs, or cell-penetrating SOCS3 proteins, have been performed (97, 132, 163). However, to our knowledge the use of small molecules to specifically target SOCS3 have not been reported.
On the other hand, several inhibitors of STAT3, modulating either upstream positive or negative regulators, regulating RNA (DN-STAT3, anti-sense RNA, siRNA) and micro RNA, or small molecules targeting STAT3 at different domains have been approached (164, 165) principally to target constitutive STAT3 activation, which is associated with various human cancers and commonly suggests poor prognosis (166), although attempts to use it in infections or inflammation have not been done. Such inhibitors could also be used in some of the inflammatory or infectious diseases described above to regulate SOCS3 effects.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (1994) 264:1415–21. doi:10.1126/science.8197455
2. Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol (2002) 3:651–62. doi:10.1038/nrm909
3. Ihle JN. STATs: signal transducers and activators of transcription. Cell (1996) 84:331–4. doi:10.1016/S0092-8674(00)81277-5
4. Levy DE, Marie IJ. STATus report on tetramers. Immunity (2012) 36:553–5. doi:10.1016/j.immuni.2012.04.003
5. Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol (2003) 3:900–11. doi:10.1038/nri1226
6. Xu D, Qu CK. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci (2008) 13:4925–32. doi:10.2741/3051
7. Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, et al. Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci U S A (1998) 95:10626–31. doi:10.1073/pnas.95.18.10626
8. Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol (2005) 5:593–605. doi:10.1038/nri1667
9. Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, et al. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci U S A (1998) 95:14395–9. doi:10.1073/pnas.95.24.14395
10. Metcalf D, Greenhalgh CJ, Viney E, Willson TA, Starr R, Nicola NA, et al. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature (2000) 405:1069–73. doi:10.1038/35016611
11. Marine JC, McKay C, Wang D, Topham DJ, Parganas E, Nakajima H, et al. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell (1999) 98:617–27. doi:10.1016/S0092-8674(00)80049-5
12. Roberts AW, Robb L, Rakar S, Hartley L, Cluse L, Nicola NA, et al. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc Natl Acad Sci U S A (2001) 98:9324–9. doi:10.1073/pnas.161271798
13. Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity (2012) 36:503–14. doi:10.1016/j.immuni.2012.03.013
14. Lee JE, Yang YM, Liang FX, Gough DJ, Levy DE, Sehgal PB. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am J Physiol Cell Physiol (2012) 302:C804–20. doi:10.1152/ajpcell.00379.2011
15. Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science (2009) 323:793–7. doi:10.1126/science.1164551
16. Li WX. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol (2008) 18:545–51. doi:10.1016/j.tcb.2008.08.008
17. Han W, Lo HW. Landscape of EGFR signaling network in human cancers: biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett (2012) 318:124–34. doi:10.1016/j.canlet.2012.01.011
18. Leaman DW, Pisharody S, Flickinger TW, Commane MA, Schlessinger J, Kerr IM, et al. Roles of JAKs in activation of STATs and stimulation of c-fos gene expression by epidermal growth factor. Mol Cell Biol (1996) 16:369–75.
19. Wang Y, van Boxel-Dezaire AH, Cheon H, Yang J, Stark GR. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc Natl Acad Sci U S A (2013) 110:16975–80. doi:10.1073/pnas.1315862110
20. Escary JL, Perreau J, Dumenil D, Ezine S, Brulet P. Leukaemia inhibitory factor is necessary for maintenance of haematopoietic stem cells and thymocyte stimulation. Nature (1993) 363:361–4. doi:10.1038/363361a0
21. Yoshida K, Taga T, Saito M, Suematsu S, Kumanogoh A, Tanaka T, et al. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc Natl Acad Sci U S A (1996) 93:407–11. doi:10.1073/pnas.93.1.407
22. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol (2007) 7:454–65. doi:10.1038/nri2093
23. Schmitz J, Weissenbach M, Haan S, Heinrich PC, Schaper F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J Biol Chem (2000) 275:12848–56. doi:10.1074/jbc.275.17.12848
24. Lehmann U, Schmitz J, Weissenbach M, Sobota RM, Hortner M, Friederichs K, et al. SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J Biol Chem (2003) 278:661–71. doi:10.1074/jbc.M210552200
25. Nicholson SE, De Souza D, Fabri LJ, Corbin J, Willson TA, Zhang JG, et al. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc Natl Acad Sci U S A (2000) 97:6493–8. doi:10.1073/pnas.100135197
26. Ernst M, Najdovska M, Grail D, Lundgren-May T, Buchert M, Tye H, et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest (2008) 118:1727–38. doi:10.1172/JCI34944
27. Brender C, Tannahill GM, Jenkins BJ, Fletcher J, Columbus R, Saris CJ, et al. Suppressor of cytokine signaling 3 regulates CD8 T-cell proliferation by inhibition of interleukins 6 and 27. Blood (2007) 110:2528–36. doi:10.1182/blood-2006-08-041541
28. Magrangeas F, Boisteau O, Denis S, Jacques Y, Minvielle S. Negative cross-talk between interleukin-3 and interleukin-11 is mediated by suppressor of cytokine signalling-3 (SOCS-3). Biochem J (2001) 353:223–30. doi:10.1042/0264-6021:3530223
29. Hamanaka I, Saito Y, Yasukawa H, Kishimoto I, Kuwahara K, Miyamoto Y, et al. Induction of JAB/SOCS-1/SSI-1 and CIS3/SOCS-3/SSI-3 is involved in gp130 resistance in cardiovascular system in rat treated with cardiotrophin-1 in vivo. Circ Res (2001) 88:727–32. doi:10.1161/hh0701.088512
30. Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, et al. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol (2003) 4:546–50. doi:10.1038/ni932
31. Auernhammer CJ, Chesnokova V, Bousquet C, Melmed S. Pituitary corticotroph SOCS-3: novel intracellular regulation of leukemia-inhibitory factor-mediated proopiomelanocortin gene expression and adrenocorticotropin secretion. Mol Endocrinol (1998) 12:954–61. doi:10.1210/me.12.7.954
32. Hortner M, Nielsch U, Mayr LM, Johnston JA, Heinrich PC, Haan S. Suppressor of cytokine signaling-3 is recruited to the activated granulocyte-colony stimulating factor receptor and modulates its signal transduction. J Immunol (2002) 169:1219–27.
33. Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A (2006) 103:8137–42. doi:10.1073/pnas.0600666103
34. Hortner M, Nielsch U, Mayr LM, Heinrich PC, Haan S. A new high affinity binding site for suppressor of cytokine signaling-3 on the erythropoietin receptor. Eur J Biochem (2002) 269:2516–26. doi:10.1046/j.1432-1033.2002.02916.x
35. Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, et al. SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem (2000) 275:40649–57. doi:10.1074/jbc.M007577200
36. Eyckerman S, Broekaert D, Verhee A, Vandekerckhove J, Tavernier J. Identification of the Y985 and Y1077 motifs as SOCS3 recruitment sites in the murine leptin receptor. FEBS Lett (2000) 486:33–7. doi:10.1016/S0014-5793(00)02205-5
37. Yamamoto K, Yamaguchi M, Miyasaka N, Miura O. SOCS-3 inhibits IL-12-induced STAT4 activation by binding through its SH2 domain to the STAT4 docking site in the IL-12 receptor beta2 subunit. Biochem Biophys Res Commun (2003) 310:1188–93. doi:10.1016/j.bbrc.2003.09.140
38. Orabona C, Pallotta MT, Volpi C, Fallarino F, Vacca C, Bianchi R, et al. SOCS3 drives proteasomal degradation of indoleamine 2,3-dioxygenase (IDO) and antagonizes IDO-dependent tolerogenesis. Proc Natl Acad Sci U S A (2008) 105:20828–33. doi:10.1073/pnas.0810278105
39. Orr SJ, Morgan NM, Elliott J, Burrows JF, Scott CJ, McVicar DW, et al. CD33 responses are blocked by SOCS3 through accelerated proteasomal-mediated turnover. Blood (2007) 109:1061–8. doi:10.1182/blood-2006-05-023556
40. Orr SJ, Morgan NM, Buick RJ, Boyd CR, Elliott J, Burrows JF, et al. SOCS3 targets Siglec 7 for proteasomal degradation and blocks Siglec 7-mediated responses. J Biol Chem (2007) 282:3418–22. doi:10.1074/jbc.C600216200
41. Frobose H, Ronn SG, Heding PE, Mendoza H, Cohen P, Mandrup-Poulsen T, et al. Suppressor of cytokine signaling-3 inhibits interleukin-1 signaling by targeting the TRAF-6/TAK1 complex. Mol Endocrinol (2006) 20:1587–96. doi:10.1210/me.2005-0301
42. Nair S, Pandey AD, Mukhopadhyay S. The PPE18 protein of Mycobacterium tuberculosis inhibits NF-kappaB/rel-mediated proinflammatory cytokine production by upregulating and phosphorylating suppressor of cytokine signaling 3 protein. J Immunol (2011) 186:5413–24. doi:10.4049/jimmunol.1000773
43. Zhang Z, Liu Q, Che Y, Yuan X, Dai L, Zeng B, et al. Antigen presentation by dendritic cells in tumors is disrupted by altered metabolism that involves pyruvate kinase M2 and its interaction with SOCS3. Cancer Res (2010) 70:89–98. doi:10.1158/0008-5472.CAN-09-2970
44. Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem (2002) 277:42394–8. doi:10.1074/jbc.C200444200
45. Yoshimura A, Ohkubo T, Kiguchi T, Jenkins NA, Gilbert DJ, Copeland NG, et al. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J (1995) 14:2816–26.
46. Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature (1997) 387:921–4. doi:10.1038/43213
47. Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, et al. Structure and function of a new STAT-induced STAT inhibitor. Nature (1997) 387:924–9. doi:10.1038/43219
48. Minamoto S, Ikegame K, Ueno K, Narazaki M, Naka T, Yamamoto H, et al. Cloning and functional analysis of new members of STAT induced STAT inhibitor (SSI) family: SSI-2 and SSI-3. Biochem Biophys Res Commun (1997) 237:79–83. doi:10.1006/bbrc.1997.7080
49. Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, et al. A family of cytokine-inducible inhibitors of signalling. Nature (1997) 387:917–21. doi:10.1038/43206
50. Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, et al. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A (1998) 95:114–9. doi:10.1073/pnas.95.1.114
51. Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci U S A (1999) 96:2071–6. doi:10.1073/pnas.96.5.2071
52. Babon JJ, Sabo JK, Zhang JG, Nicola NA, Norton RS. The SOCS box encodes a hierarchy of affinities for Cullin5: implications for ubiquitin ligase formation and cytokine signalling suppression. J Mol Biol (2009) 387:162–74. doi:10.1016/j.jmb.2009.01.024
53. Fletcher TC, DiGiandomenico A, Hawiger J. Extended anti-inflammatory action of a degradation-resistant mutant of cell-penetrating suppressor of cytokine signaling 3. J Biol Chem (2010) 285:18727–36. doi:10.1074/jbc.M109.095216
54. Sasaki A, Yasukawa H, Suzuki A, Kamizono S, Syoda T, Kinjyo I, et al. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells (1999) 4:339–51. doi:10.1046/j.1365-2443.1999.00263.x
55. Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, et al. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J (1999) 18:1309–20. doi:10.1093/emboj/18.5.1309
56. Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, et al. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity (2012) 36:239–50. doi:10.1016/j.immuni.2011.12.015
57. Bergamin E, Wu J, Hubbard SR. Structural basis for phosphotyrosine recognition by suppressor of cytokine signaling-3. Structure (2006) 14:1285–92. doi:10.1016/j.str.2006.06.011
58. Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol (2013) 20:469–76. doi:10.1038/nsmb.2519
59. Babon JJ, McManus EJ, Yao S, DeSouza DP, Mielke LA, Sprigg NS, et al. The structure of SOCS3 reveals the basis of the extended SH2 domain function and identifies an unstructured insertion that regulates stability. Mol Cell (2006) 22:205–16. doi:10.1016/j.molcel.2006.03.024
60. Irandoust MI, Aarts LH, Roovers O, Gits J, Erkeland SJ, Touw IP. Suppressor of cytokine signaling 3 controls lysosomal routing of G-CSF receptor. EMBO J (2007) 26:1782–93. doi:10.1038/sj.emboj.7601709
61. Boyle K, Egan P, Rakar S, Willson TA, Wicks IP, Metcalf D, et al. The SOCS box of suppressor of cytokine signaling-3 contributes to the control of G-CSF responsiveness in vivo. Blood (2007) 110:1466–74. doi:10.1182/blood-2007-03-079178
62. Nguyen-Jackson H, Panopoulos AD, Zhang H, Li HS, Watowich SS. STAT3 controls the neutrophil migratory response to CXCR2 ligands by direct activation of G-CSF-induced CXCR2 expression and via modulation of CXCR2 signal transduction. Blood (2010) 115:3354–63. doi:10.1182/blood-2009-08-240317
63. Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol (2003) 4:540–5. doi:10.1038/ni931
64. Robb L, Boyle K, Rakar S, Hartley L, Lochland J, Roberts AW, et al. Genetic reduction of embryonic leukemia-inhibitory factor production rescues placentation in SOCS3-null embryos but does not prevent inflammatory disease. Proc Natl Acad Sci U S A (2005) 102:16333–8. doi:10.1073/pnas.0508023102
65. Ohtani T, Ishihara K, Atsumi T, Nishida K, Kaneko Y, Miyata T, et al. Dissection of signaling cascades through gp130 in vivo: reciprocal roles for STAT3- and SHP2-mediated signals in immune responses. Immunity (2000) 12:95–105. doi:10.1016/S1074-7613(00)80162-4
66. Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med (2004) 10:734–8. doi:10.1038/nm1072
67. Palanivel R, Fullerton MD, Galic S, Honeyman J, Hewitt KA, Jorgensen SB, et al. Reduced Socs3 expression in adipose tissue protects female mice against obesity-induced insulin resistance. Diabetologia (2012) 55:3083–93. doi:10.1007/s00125-012-2665-3
68. Silver JS, Stumhofer JS, Passos S, Ernst M, Hunter CA. IL-6 mediates the susceptibility of glycoprotein 130 hypermorphs to Toxoplasma gondii. J Immunol (2011) 187:350–60. doi:10.4049/jimmunol.1004144
69. Jenkins BJ, Grail D, Nheu T, Najdovska M, Wang B, Waring P, et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat Med (2005) 11:845–52. doi:10.1038/nm1282
70. Tebbutt NC, Giraud AS, Inglese M, Jenkins B, Waring P, Clay FJ, et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med (2002) 8:1089–97. doi:10.1038/nm763
71. Nishihara M, Ogura H, Ueda N, Tsuruoka M, Kitabayashi C, Tsuji F, et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int Immunol (2007) 19:695–702. doi:10.1093/intimm/dxm045
72. Sodenkamp J, Behrends J, Forster I, Muller W, Ehlers S, Holscher C. gp130 on macrophages/granulocytes modulates inflammation during experimental tuberculosis. Eur J Cell Biol (2011) 90:505–14. doi:10.1016/j.ejcb.2010.10.010
73. Croom HA, Izon DJ, Chong MM, Curtis DJ, Roberts AW, Kay TW, et al. Perturbed thymopoiesis in vitro in the absence of suppressor of cytokine signalling 1 and 3. Mol Immunol (2008) 45:2888–96. doi:10.1016/j.molimm.2008.01.024
74. Carow B, Reuschl AK, Gavier-Widen D, Jenkins BJ, Ernst M, Yoshimura A, et al. Critical and independent role for SOCS3 in either myeloid or T cells in resistance to Mycobacterium tuberculosis. PLoS Pathog (2013) 9:e1003442. doi:10.1371/journal.ppat.1003442
75. Hill GR, Kuns RD, Raffelt NC, Don AL, Olver SD, Markey KA, et al. SOCS3 regulates graft-versus-host disease. Blood (2010) 116:287–96. doi:10.1182/blood-2009-12-259598
76. Moriwaki A, Inoue H, Nakano T, Matsunaga Y, Matsuno Y, Matsumoto T, et al. T cell treatment with small interfering RNA for suppressor of cytokine signaling 3 modulates allergic airway responses in a murine model of asthma. Am J Respir Cell Mol Biol (2011) 44:448–55. doi:10.1165/rcmb.2009-0051OC
77. Kinjyo I, Inoue H, Hamano S, Fukuyama S, Yoshimura T, Koga K, et al. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J Exp Med (2006) 203:1021–31. doi:10.1084/jem.20052333
78. Taleb S, Romain M, Ramkhelawon B, Uyttenhove C, Pasterkamp G, Herbin O, et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med (2009) 206:2067–77. doi:10.1084/jem.20090545
79. Pellegrini M, Calzascia T, Toe JG, Preston SP, Lin AE, Elford AR, et al. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell (2011) 144:601–13. doi:10.1016/j.cell.2011.01.011
80. Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol (2003) 4:551–6. doi:10.1038/ni938
81. Matsumura Y, Kobayashi T, Ichiyama K, Yoshida R, Hashimoto M, Takimoto T, et al. Selective expansion of foxp3-positive regulatory T cells and immunosuppression by suppressors of cytokine signaling 3-deficient dendritic cells. J Immunol (2007) 179:2170–9.
82. Yan C, Ward PA, Wang X, Gao H. Myeloid depletion of SOCS3 enhances LPS-induced acute lung injury through CCAAT/enhancer binding protein delta pathway. FASEB J (2013) 27:2967–76. doi:10.1096/fj.12-225797
83. Qin H, Holdbrooks AT, Liu Y, Reynolds SL, Yanagisawa LL, Benveniste EN. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol (2012) 189:3439–48. doi:10.4049/jimmunol.1201168
84. Whitmarsh RJ, Gray CM, Gregg B, Christian DA, May MJ, Murray PJ, et al. A critical role for SOCS3 in innate resistance to Toxoplasma gondii. Cell Host Microbe (2011) 10:224–36. doi:10.1016/j.chom.2011.07.009
85. Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med (2004) 10:739–43. doi:10.1038/nm1071
86. Emery B, Butzkueven H, Snell C, Binder M, Kilpatrick TJ. Oligodendrocytes exhibit selective expression of suppressor of cytokine signaling genes and signal transducer and activator of transcription 1 independent inhibition of interferon-gamma-induced toxicity in response to leukemia inhibitory factor. Neuroscience (2006) 137:463–72. doi:10.1016/j.neuroscience.2005.09.022
87. Emery B, Cate HS, Marriott M, Merson T, Binder MD, Snell C, et al. Suppressor of cytokine signaling 3 limits protection of leukemia inhibitory factor receptor signaling against central demyelination. Proc Natl Acad Sci U S A (2006) 103:7859–64. doi:10.1073/pnas.0602574103
88. Croker BA, Metcalf D, Robb L, Wei W, Mifsud S, DiRago L, et al. SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity (2004) 20:153–65. doi:10.1016/S1074-7613(04)00022-6
89. Croker BA, Kiu H, Pellegrini M, Toe J, Preston S, Metcalf D, et al. IL-6 promotes acute and chronic inflammatory disease in the absence of SOCS3. Immunol Cell Biol (2012) 90:124–9. doi:10.1038/icb.2011.29
90. Wong PK, Egan PJ, Croker BA, O’Donnell K, Sims NA, Drake S, et al. SOCS-3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J Clin Invest (2006) 116:1571–81. doi:10.1172/JCI25660
91. Seki Y, Inoue H, Nagata N, Hayashi K, Fukuyama S, Matsumoto K, et al. SOCS-3 regulates onset and maintenance of T(H)2-mediated allergic responses. Nat Med (2003) 9:1047–54. doi:10.1038/nm896
92. Ozaki A, Seki Y, Fukushima A, Kubo M. The control of allergic conjunctivitis by suppressor of cytokine signaling (SOCS)3 and SOCS5 in a murine model. J Immunol (2005) 175:5489–97.
93. Romain M, Taleb S, Dalloz M, Ponnuswamy P, Esposito B, Perez N, et al. Overexpression of SOCS3 in T lymphocytes leads to impaired interleukin-17 production and severe aortic aneurysm formation in mice – brief report. Arterioscler Thromb Vasc Biol (2013) 33:581–4. doi:10.1161/ATVBAHA.112.300516
94. Nakaya M, Hamano S, Kawasumi M, Yoshida H, Yoshimura A, Kobayashi T. Aberrant IL-4 production by SOCS3-over-expressing T cells during infection with Leishmania major exacerbates disease manifestations. Int Immunol (2011) 23:195–202. doi:10.1093/intimm/dxq472
95. Ogata H, Chinen T, Yoshida T, Kinjyo I, Takaesu G, Shiraishi H, et al. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene (2006) 25:2520–30. doi:10.1038/sj.onc.1209281
96. Shouda T, Yoshida T, Hanada T, Wakioka T, Oishi M, Miyoshi K, et al. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J Clin Invest (2001) 108:1781–8. doi:10.1172/JCI200113568
97. Jo D, Liu D, Yao S, Collins RD, Hawiger J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat Med (2005) 11:892–8. doi:10.1038/nm1269
98. Qin H, Yeh WI, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci U S A (2012) 109:5004–9. doi:10.1073/pnas.1117218109
99. Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity (2008) 29:628–36. doi:10.1016/j.immuni.2008.07.018
100. Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol (2003) 4:1169–76. doi:10.1038/ni1012
101. Wormald S, Zhang JG, Krebs DL, Mielke LA, Silver J, Alexander WS, et al. The comparative roles of suppressor of cytokine signaling-1 and -3 in the inhibition and desensitization of cytokine signaling. J Biol Chem (2006) 281:11135–43. doi:10.1074/jbc.M509595200
102. Braun DA, Fribourg M, Sealfon SC. Cytokine response is determined by duration of receptor and signal transducers and activators of transcription 3 (STAT3) activation. J Biol Chem (2013) 288:2986–93. doi:10.1074/jbc.M112.386573
103. Liu X, Zhang Y, Yu Y, Yang X, Cao X. SOCS3 promotes TLR4 response in macrophages by feedback inhibiting TGF-beta1/Smad3 signaling. Mol Immunol (2008) 45:1405–13. doi:10.1016/j.molimm.2007.08.018
104. Spence S, Fitzsimons A, Boyd CR, Kessler J, Fitzgerald D, Elliott J, et al. Suppressors of cytokine signaling 2 and 3 diametrically control macrophage polarization. Immunity (2013) 38:66–78. doi:10.1016/j.immuni.2012.09.013
105. Liu Y, Stewart KN, Bishop E, Marek CJ, Kluth DC, Rees AJ, et al. Unique expression of suppressor of cytokine signaling 3 is essential for classical macrophage activation in rodents in vitro and in vivo. J Immunol (2008) 180:6270–8.
106. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science (1999) 284:770–6. doi:10.1126/science.284.5415.770
107. Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY, et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res (2010) 70:4840–9. doi:10.1158/0008-5472.CAN-10-0269
108. Narayana Y, Balaji KN. NOTCH1 up-regulation and signaling involved in Mycobacterium bovis BCG-induced SOCS3 expression in macrophages. J Biol Chem (2008) 283:12501–11. doi:10.1074/jbc.M709960200
109. Kimura A, Kinjyo I, Matsumura Y, Mori H, Mashima R, Harada M, et al. SOCS3 is a physiological negative regulator for granulopoiesis and granulocyte colony-stimulating factor receptor signaling. J Biol Chem (2004) 279:6905–10. doi:10.1074/jbc.C300496200
110. Croker BA, Mielke LA, Wormald S, Metcalf D, Kiu H, Alexander WS, et al. Socs3 maintains the specificity of biological responses to cytokine signals during granulocyte and macrophage differentiation. Exp Hematol (2008) 36:786–98. doi:10.1016/j.exphem.2008.02.008
111. Orabona C, Belladonna ML, Vacca C, Bianchi R, Fallarino F, Volpi C, et al. Cutting edge: silencing suppressor of cytokine signaling 3 expression in dendritic cells turns CD28-Ig from immune adjuvant to suppressant. J Immunol (2005) 174:6582–6.
112. Senn JJ, Klover PJ, Nowak IA, Zimmers TA, Koniaris LG, Furlanetto RW, et al. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem (2003) 278:13740–6. doi:10.1074/jbc.M210689200
113. Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem (2000) 275:15985–91. doi:10.1074/jbc.275.21.15985
114. Shi H, Tzameli I, Bjorbaek C, Flier JS. Suppressor of cytokine signaling 3 is a physiological regulator of adipocyte insulin signaling. J Biol Chem (2004) 279:34733–40. doi:10.1074/jbc.M403886200
115. Lovibond AC, Haque SJ, Chambers TJ, Fox SW. TGF-beta-induced SOCS3 expression augments TNF-alpha-induced osteoclast formation. Biochem Biophys Res Commun (2003) 309:762–7. doi:10.1016/j.bbrc.2003.08.068
116. Caruso R, Stolfi C, Sarra M, Rizzo A, Fantini MC, Pallone F, et al. Inhibition of monocyte-derived inflammatory cytokines by IL-25 occurs via p38 Map kinase-dependent induction of Socs-3. Blood (2009) 113:3512–9. doi:10.1182/blood-2008-08-172767
117. Schneider R, Yaneva T, Beauseigle D, El-Khoury L, Arbour N. IL-27 increases the proliferation and effector functions of human naive CD8+ T lymphocytes and promotes their development into Tc1 cells. Eur J Immunol (2011) 41:47–59. doi:10.1002/eji.201040804
118. Owaki T, Asakawa M, Kamiya S, Takeda K, Fukai F, Mizuguchi J, et al. IL-27 suppresses CD28-mediated [correction of medicated] IL-2 production through suppressor of cytokine signaling 3. J Immunol (2006) 176:2773–80.
119. Egwuagu CE, Yu CR, Zhang M, Mahdi RM, Kim SJ, Gery I. Suppressors of cytokine signaling proteins are differentially expressed in Th1 and Th2 cells: implications for Th cell lineage commitment and maintenance. J Immunol (2002) 168:3181–7.
120. Horiuchi Y, Bae SJ, Katayama I. Overexpression of the suppressor of cytokine signalling 3 (SOCS3) in severe atopic dermatitis. Clin Exp Dermatol (2006) 31:100–4. doi:10.1111/j.1365-2230.2005.01979.x
121. Ekelund E, Saaf A, Tengvall-Linder M, Melen E, Link J, Barker J, et al. Elevated expression and genetic association links the SOCS3 gene to atopic dermatitis. Am J Hum Genet (2006) 78:1060–5. doi:10.1086/504272
122. Tanaka K, Ichiyama K, Hashimoto M, Yoshida H, Takimoto T, Takaesu G, et al. Loss of suppressor of cytokine signaling 1 in helper T cells leads to defective Th17 differentiation by enhancing antagonistic effects of IFN-gamma on STAT3 and Smads. J Immunol (2008) 180:3746–56.
123. Qin H, Wang L, Feng T, Elson CO, Niyongere SA, Lee SJ, et al. TGF-beta promotes Th17 cell development through inhibition of SOCS3. J Immunol (2009) 183:97–105. doi:10.4049/jimmunol.0801986
124. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammaT directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi:10.1016/j.cell.2006.07.035
125. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24:179–89. doi:10.1016/j.immuni.2006.01.001
126. Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature (2008) 452:773–6. doi:10.1038/nature06764
127. Gistera A, Robertson AK, Andersson J, Ketelhuth DF, Ovchinnikova O, Nilsson SK, et al. Transforming growth factor-beta signaling in T cells promotes stabilization of atherosclerotic plaques through an interleukin-17-dependent pathway. Sci Transl Med (2013) 5:196ra100. doi:10.1126/scitranslmed.3006133
128. Cao W, Yang Y, Wang Z, Liu A, Fang L, Wu F, et al. Leukemia inhibitory factor inhibits T helper 17 cell differentiation and confers treatment effects of neural progenitor cell therapy in autoimmune disease. Immunity (2011) 35:273–84. doi:10.1016/j.immuni.2011.06.011
129. Li Y, Chu N, Rostami A, Zhang GX. Dendritic cells transduced with SOCS-3 exhibit a tolerogenic/DC2 phenotype that directs type 2 Th cell differentiation in vitro and in vivo. J Immunol (2006) 177:1679–88.
130. Carow B, Rottenberg ME. “Suppressor of cytokine signalling” molecules in infection and inflammation. In: Mahin K, editor. Inflammation, Chronic Diseases and Cancer – Cell and Molecular Biology, Immunology and Clinical Bases. InTech (2012). doi:10.5772/25986
131. Babon JJ, Nicola NA. The biology and mechanism of action of suppressor of cytokine signaling 3. Growth Factors (2012) 30:207–19. doi:10.3109/08977194.2012.687375
132. Yokota S, Yokosawa N, Okabayashi T, Suzutani T, Fujii N. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 confers efficient viral replication. Virology (2005) 338:173–81. doi:10.1016/j.virol.2005.04.028
133. Hashimoto K, Ishibashi K, Ishioka K, Zhao D, Sato M, Ohara S, et al. RSV replication is attenuated by counteracting expression of the suppressor of cytokine signaling (SOCS) molecules. Virology (2009) 391:162–70. doi:10.1016/j.virol.2009.06.026
134. Bixler SL, Sandler NG, Douek DC, Mattapallil JJ. Suppressed Th17 levels correlate with elevated PIAS3, SHP2, and SOCS3 expression in CD4 T cells during acute simian immunodeficiency virus infection. J Virol (2013) 87:7093–101. doi:10.1128/JVI.00600-13
135. Akhtar LN, Qin H, Muldowney MT, Yanagisawa LL, Kutsch O, Clements JE, et al. Suppressor of cytokine signaling 3 inhibits antiviral IFN-beta signaling to enhance HIV-1 replication in macrophages. J Immunol (2010) 185:2393–404. doi:10.4049/jimmunol.0903563
136. Kim KA, Lin W, Tai AW, Shao RX, Weinberg E, De Sa Borges CB, et al. Hepatic SOCS3 expression is strongly associated with non-response to therapy and race in HCV and HCV/HIV infection. J Hepatol (2009) 50:705–11. doi:10.1016/j.jhep.2008.12.021
137. Michaud F, Coulombe F, Gaudreault E, Paquet-Bouchard C, Rola-Pleszczynski M, Gosselin J. Epstein-Barr virus interferes with the amplification of IFNalpha secretion by activating suppressor of cytokine signaling 3 in primary human monocytes. PLoS One (2010) 5:e11908. doi:10.1371/journal.pone.0011908
138. Kim K, Kim KH, Cheong J. Hepatitis B virus X protein impairs hepatic insulin signaling through degradation of IRS1 and induction of SOCS3. PLoS One (2010) 5:e8649. doi:10.1371/journal.pone.0008649
139. Kawaguchi T, Yoshida T, Harada M, Hisamoto T, Nagao Y, Ide T, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am J Pathol (2004) 165:1499–508. doi:10.1016/S0002-9440(10)63408-6
140. Persico M, Capasso M, Persico E, Svelto M, Russo R, Spano D, et al. Suppressor of cytokine signaling 3 (SOCS3) expression and hepatitis C virus-related chronic hepatitis: insulin resistance and response to antiviral therapy. Hepatology (2007) 46:1009–15. doi:10.1002/hep.21782
141. Persico M, Capasso M, Russo R, Persico E, Croce L, Tiribelli C, et al. Elevated expression and polymorphisms of SOCS3 influence patient response to antiviral therapy in chronic hepatitis C. Gut (2008) 57:507–15. doi:10.1136/gut.2007.129478
142. Funaoka Y, Sakamoto N, Suda G, Itsui Y, Nakagawa M, Kakinuma S, et al. Analysis of interferon signaling by infectious hepatitis C virus clones with substitutions of core amino acids 70 and 91. J Virol (2011) 85:5986–94. doi:10.1128/JVI.02583-10
143. Pothlichet J, Chignard M, Si-Tahar M. Cutting edge: innate immune response triggered by influenza A virus is negatively regulated by SOCS1 and SOCS3 through a RIG-I/IFNAR1-dependent pathway. J Immunol (2008) 180:2034–8.
144. Pauli EK, Schmolke M, Wolff T, Viemann D, Roth J, Bode JG, et al. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog (2008) 4:e1000196. doi:10.1371/journal.ppat.1000196
145. Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity (2011) 35:792–805. doi:10.1016/j.immuni.2011.09.017
146. Masood KI, Rottenberg ME, Salahuddin N, Irfan M, Rao N, Carow B, et al. Expression of M. tuberculosis-induced suppressor of cytokine signaling (SOCS) 1, SOCS3, FoxP3 and secretion of IL-6 associates with differing clinical severity of tuberculosis. BMC Infect Dis (2013) 13:13. doi:10.1186/1471-2334-13-13
147. Yoshikawa T, Takata A, Otsuka M, Kishikawa T, Kojima K, Yoshida H, et al. Silencing of microRNA-122 enhances interferon-alpha signaling in the liver through regulating SOCS3 promoter methylation. Sci Rep (2012) 2:637. doi:10.1038/srep00637
148. Yamamoto M, Standley DM, Takashima S, Saiga H, Okuyama M, Kayama H, et al. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J Exp Med (2009) 206:2747–60. doi:10.1084/jem.20091703
149. Carow B, Ye X, Gavier-Widen D, Bhuju S, Oehlmann W, Singh M, et al. Silencing suppressor of cytokine signaling-1 (SOCS1) in macrophages improves Mycobacterium tuberculosis control in an interferon-{gamma} (IFN-{gamma})-dependent manner. J Biol Chem (2011) 286:26873–87. doi:10.1074/jbc.M111.238287
150. Sodenkamp J, Waetzig GH, Scheller J, Seegert D, Grotzinger J, Rose-John S, et al. Therapeutic targeting of interleukin-6 trans-signaling does not affect the outcome of experimental tuberculosis. Immunobiology (2012) 217:996–1004. doi:10.1016/j.imbio.2012.01.015
151. Saunders BM, Frank AA, Orme IM, Cooper AM. Interleukin-6 induces early gamma interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infect Immun (2000) 68:3322–6. doi:10.1128/IAI.68.6.3322-3326.2000
152. Miao T, Wu D, Zhang Y, Bo X, Subang MC, Wang P, et al. Suppressor of cytokine signaling-3 suppresses the ability of activated signal transducer and activator of transcription-3 to stimulate neurite growth in rat primary sensory neurons. J Neurosci (2006) 26:9512–9. doi:10.1523/JNEUROSCI.2160-06.2006
153. McKay BR, Ogborn DI, Baker JM, Toth KG, Tarnopolsky MA, Parise G. Elevated SOCS3 and altered IL-6 signaling is associated with age-related human muscle stem cell dysfunction. Am J Physiol Cell Physiol (2013) 304:C717–28. doi:10.1152/ajpcell.00305.2012
154. Jiang C, Kim JH, Li F, Qu A, Gavrilova O, Shah YM, et al. Hypoxia-inducible factor 1alpha regulates a SOCS3-STAT3-adiponectin signal transduction pathway in adipocytes. J Biol Chem (2013) 288:3844–57. doi:10.1074/jbc.M112.426338
155. Suzuki A, Hanada T, Mitsuyama K, Yoshida T, Kamizono S, Hoshino T, et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J Exp Med (2001) 193:471–81. doi:10.1084/jem.193.4.471
156. Ortiz-Munoz G, Martin-Ventura JL, Hernandez-Vargas P, Mallavia B, Lopez-Parra V, Lopez-Franco O, et al. Suppressors of cytokine signaling modulate JAK/STAT-mediated cell responses during atherosclerosis. Arterioscler Thromb Vasc Biol (2009) 29:525–31. doi:10.1161/ATVBAHA.108.173781
157. Wunderlich CM, Hovelmeyer N, Wunderlich FT. Mechanisms of chronic JAK-STAT3-SOCS3 signaling in obesity. JAKSTAT (2013) 2:e23878. doi:10.4161/jkst.23878
158. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature (1998) 395:763–70. doi:10.1038/27376
159. Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell (1998) 1:619–25. doi:10.1016/S1097-2765(00)80062-3
160. Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron (2004) 42:983–91. doi:10.1016/j.neuron.2004.06.004
161. Oba T, Yasukawa H, Hoshijima M, Sasaki K, Futamata N, Fukui D, et al. Cardiac-specific deletion of SOCS-3 prevents development of left ventricular remodeling after acute myocardial infarction. J Am Coll Cardiol (2012) 59:838–52. doi:10.1016/j.jacc.2011.10.887
162. Veenbergen S, Bennink MB, de Hooge AS, Arntz OJ, Smeets RL, van den Berg WB, et al. Splenic suppressor of cytokine signaling 3 transgene expression affects T cell responses and prevents development of collagen-induced arthritis. Arthritis Rheum (2008) 58:3742–52. doi:10.1002/art.24072
163. Gu H, Liu L, Ma S, Liu Y, Ren Y, Zhai L, et al. Inhibition of SOCS-3 in adipocytes of rats with diet-induced obesity increases leptin-mediated fatty acid oxidation. Endocrine (2009) 36:546–54. doi:10.1007/s12020-009-9253-4
164. Wang X, Crowe PJ, Goldstein D, Yang JL. STAT3 inhibition, a novel approach to enhancing targeted therapy in human cancers (review). Int J Oncol (2012) 41:1181–91. doi:10.3892/ijo.2012.1568
165. Yu H, Jove R. The STATs of cancer – new molecular targets come of age. Nat Rev Cancer (2004) 4:97–105. doi:10.1038/nrc1275
166. Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res (2006) 66:3162–8. doi:10.1158/0008-5472.CAN-05-3757
Keywords: SOCS, JAK, STAT, STAT3, cytokine, IL-6, infection, autoimmunity
Citation: Carow B and Rottenberg ME (2014) SOCS3, a major regulator of infection and inflammation. Front. Immunol. 5:58. doi: 10.3389/fimmu.2014.00058
Received: 02 December 2013; Accepted: 31 January 2014;
Published online: 19 February 2014.
Edited by:
Fabrizio Mattei, Istituto Superiore di Sanità, ItalyReviewed by:
Carlo Pucillo, University of Udine, ItalyTakahiro Yamazaki, Institute Gustave Roussy, France
Copyright: © 2014 Carow and Rottenberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Berit Carow and Martin E. Rottenberg, Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Nobels Väg 16, Stockholm 171 77, Sweden e-mail:YmVyaXQuY2Fyb3dAa2kuc2U=;bWFydGluLnJvdHRlbmJlcmdAa2kuc2U=