 Veena Roberts
Veena Roberts John Stagg
John Stagg Karen M. Dwyer
Karen M. Dwyer- 1Immunology Research Centre, St. Vincent’s Hospital Melbourne and Department of Medicine, The University of Melbourne, Melbourne, VIC, Australia
- 2Centre de Recherche du Centre Hospitalier de l’Université de Montréal, Faculté de Pharmacie et Institut du Cancer de Montréal, Montréal, QC, Canada
Extracellular adenosine is a potent immunomodulatory molecule that accumulates in states of inflammation. Nucleotides such as adenosine triphosphate and adenosine diphosphate are release from injured and necrotic cells and hydrolyzed to adenosine monophosphate and adenosine by the concerted action of the ectonucleotidases CD39 and CD73. Accumulating evidence suggest that purinergic signaling is involved in the inflammatory response that accompanies acute rejection and chronic allograft dysfunction. Modification of the purinergic pathway has been shown to alter graft survival in a number of solid organ transplant models and the response to ischemia–reperfusion injury (IRI). Furthermore, the purinergic pathway is intrinsically involved in B and T cell biology and function. Although T cells have traditionally been considered the orchestrators of acute allograft rejection, a role for B cells in chronic allograft loss is being increasingly appreciated. This review focuses on the role of the ectonucleotidases CD39 and CD73 and adenosine signaling in solid organ transplantation including the effects on IRI and T and B cell biology.
Introduction
Solid organ transplantation is life sustaining and the preferred treatment for patients with end stage organ disease. Despite improvements in short term graft survival coincident with more potent immunosuppression, long term graft survival has not changed significantly. Although, the cause of chronic allograft dysfunction and failure has not been fully elucidated, evidence implicates recurrent episodes of acute allograft rejection in the pathogenesis. Life-long immunosuppression is a necessity in all but a few patients and a delicate balance exists between sufficient immunosuppressive to preserve graft integrity and unwanted side effects consequent to excess exposure. Immunological tolerance remains the panacea of transplantation medicine enabling long term allograft survival with the avoidance of immunosuppression toxicities.
A number of factors have been identified, which increase graft immunogenicity and the risk of rejection. Vast immunological disparity, evident in xenotransplantation, is a potent activator of the recipient’s immune system inciting an immediate, aggressive, and fulminant response termed hyperacute rejection (HAR). However, lesser antigen mismatches are also sufficient to trigger the alloimmune response, which may be classified histopathologically as cell mediated rejection (CMR) or antibody mediated rejection (AMR). The phenomenon of ischemia–reperfusion injury (IRI), an obligatory event in the transplantation process occurring at the time of organ procurement and engraftment, increases the risk of delayed graft function and immunogenicity of the graft particularly in the setting of extended cold preservation times. Strategies to curb the recipient’s immune response to such events without escalating immunosuppressive requirements remain the focus of intense research.
Current immunosuppressive regimens specifically target T cell activation as a means of preventing allograft rejection. The success of this approach is reflected in the exceptional 1 year graft survival of transplanted organs. However, despite the improvements in short term survival, long term graft survival remains static, and these agents are far less efficacious in combating chronic rejection. Recently, a role for B cells in chronic rejection has been appreciated; however, B cells may also be beneficial to the graft (1). B cells are able to modulate the T cell response by both enhancing the primary T cell response and promoting regulatory T cell (Treg) activity. These data have led researchers to conclude that functionally distinct B cell subsets exist classified as B effectors and B regulatory cells, respectively (2). The exact mechanisms by which B cells exert regulatory function are as yet unknown but putatively involve IL-10 which they produce (3).
Adenosine is an innate immunomodulatory molecule, the pericellular concentration of which rises dramatically in states of inflammation. Nucleotides such as adenosine triphosphate (ATP) and adenosine diphosphate (ADP) are extruded from injured and necrotic cells and hydrolyzed to adenosine monophosphate (AMP) by the ectonucleotidase NTPDase family of which CD39 (NTPDase1) is the prototype. AMP is then hydrolyzed by the 5′ ectonucleotidase CD73 to adenosine (4). Adenosine signals via four G protein coupled receptors namely A1, A2A, A2B, and A3. A1R and A3R are coupled to the G-inhibitory subunit which leads to a reduction in intracellular cAMP upon activation, whereas A2AR and A2BR are coupled to the G-stimulatory subunit resulting in an increase in intracellular cAMP. In addition, A2BR couples to Gq proteins, which stimulate phospholipase C activity and intracellular calcium mobilization (5). Originally described as a marker of B cell activation (6), CD39 expression has been demonstrated on resting B cells (2), T cells including Treg (7, 8), neutrophils (9), NK cells (10) monocytes, and macrophages (11). Similarly, CD73 was originally used as a surface marker to identify individual B cell subsets at specific stages of differentiation (12), but has since been demonstrated on resting B cells (2), T cells including Treg (13), neutrophils (9), NK cells (14) monocytes, and macrophages (11). CD39 and CD73 are co-expressed on resting B cells; however CD73 is down-regulated with activation (2). The ability to generate adenosine by these cells facilitates immunoglobulin diversification via class switch recombination, an essential process in mounting a humoral immune response (15) and which may impact graft survival. Within the T cell population CD39 and CD73 are co-expressed by Treg (8). Further, the adenosine receptor expression pattern has been detailed on both cellular subsets. The A2AR is expressed by T cells under basal conditions and is up regulated following activation (8). Recently, the importance of the A2AR on Treg function has been appreciated (16, 17). On B cells the A1, A2A, and A3R are all expressed although signaling via the A3R inhibits B cell proliferation (2).
There is mounting evidence that purinergic signaling is involved in the inflammatory response that accompanies rejection and in chronic allograft dysfunction. Modification of the purinergic pathway has altered graft survival experimentally in a number of solid organ transplant models and the response to IRI. Furthermore, the purinergic pathway is intrinsically involved in both B and T cell biology, cell subsets critical in maintaining allograft survival.
This review focuses on the role of ectonucleotidases (NTPDase1/CD39 and CD73) and adenosine signaling in solid organ transplantation including the effects on IRI and impact on lymphocyte biology.
Cardiac Transplantation
Chronic cardiac allograft rejection manifests as coronary allograft vasculopathy (CAV), a rapidly progressive form of atherosclerosis that leads to reduced blood flow and ischemia and is a major cause of death in patients surviving more than 1 year after heart transplantation (18). Although numerous immune-mediated and metabolic risk factors have been implicated in the pathogenesis of CAV (19), to date no effective treatment is available to fully eliminate CAV and its related adverse outcomes. The main therapeutic strategy against CAV is the prevention and treatment of the factors known to trigger or accelerate the disease such as repeated episodes of acute allograft rejection and prolonged cold ischemia time associated with severe IRI.
CD39, CD73, and A2BR Mediates Protection in Cardiac Ischemia–Reperfusion Injury
CD39 is a ubiquitously expressed integral immune and vascular ectonucleotidase and manipulation of this ectoenzyme is likely to impact on graft rejection in which inflammation and coagulation predominate. Indeed in the original description of mice deficient in CD39, CD39−/− cardiac xenografts underwent rejection with more rapid vascular occlusion than did the matched wild-type (WT) murine hearts when grafted into rats (20). Thrombotic and inflammatory immunopathological changes were more evident in the mutant vasculature at earlier time-points compared with WT donor hearts. Replacement of CD39 in the form of adenovirus-mediated over-expression (21) or administration of apyrase (22), a soluble form of CD39, prolonged cardiac xenograft survival with reduced vascular thrombosis. Consistent with this the transgenic over-expression of CD39 improved cardiac xenograft survival with less platelet sequestration and preservation of cardiac architecture histologically (23).
NTPDase biochemical activity initially falls dramatically in the donor heart following transplantation but rebounds and CD39 mRNA expression is significantly increased in grafts that survive long term (24). Both human and murine CD39 mRNA expression has been documented to increase in a time dependent manner under hypoxic conditions and is dependent on the transcription factor Sp1 (25). Indeed, the phenomenon of ischemic preconditioning (IP), which involves multiple sublethal episodes of ischemia and reperfusion that augments adenosine concentrations and protects against further ischemia, robustly increases CD39 expression on endothelia and myocytes (26) (Table 1). Preconditioned WT hearts are significantly protected (26), whereas CD39 deficient hearts are more susceptible to myocardial infarction following coronary ischemia due to less ATP hydrolyzing ability (25). Conversely, mice treated with apyrase (26) or over-expressing CD39 (27) are protected against myocardial infarction following coronary artery occlusion through A2BR dependent mechanisms (27). These data implicate purinergic signaling pathways in the innate response to cardiac hypoxia and transplantation.
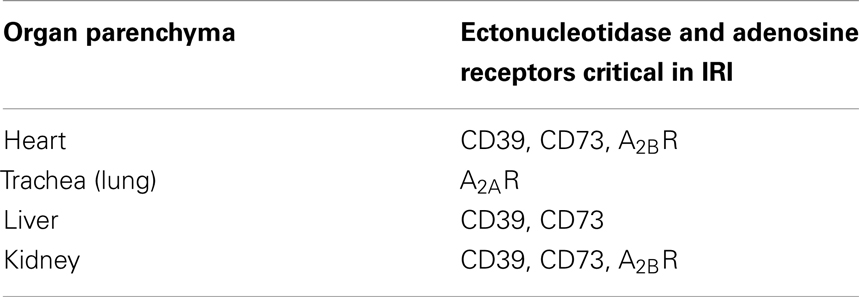
Table 1. Expression of the ectonucleotidases and adenosine receptors on the organ parenchyma during ischemia–reperfusion injury.
The A2BR is critical in mediating cardioprotection against hypoxic injury. Following 60 min of ischemia the area at risk was significantly less in mice treated with the A2BR agonist BAY 60-6583 (28) with the target gene being Per2, a circadian rhythm protein that enhances the glycolytic capacity of the ischemic heart through HIF-1α (29). The A2BR has been shown to increase post cardiac transplantation as early as 4 h following engraftment and parallels the changes in CD73 mRNA expression (14). Deficiency of CD73 in either the donor or the recipient reduced graft survival and accelerated the development of CAV in a murine model of cardiac transplantation and was associated with reduced A2BR expression (14). These data suggest that the expression of CD73 and A2BR are coordinated and dependent under conditions of cardiac hypoxia.
Cardiac transplantation involves a period of extended cold preservation when the donor heart is stored on ice during transportation to the recipient center. In a rat heterotopic heart transplant model administration of an adenosine bolus before cardioplegia and storage reduced myocardial injury and led to faster reanimation following reperfusion. Grafts treated with adenosine were less inflamed with fewer infiltrating cells (30).
CD73 Activity Limits Innate Immunity Following Cardiac IRI
Bonner et al. (31) recently demonstrated that CD73 on circulating immune cells was critical in cardiac healing in a model of cardiac ischemia and reperfusion. Within 3 days of ischemia and reperfusion the myocardium was infiltrated with granulocytes and T cells, which highly express CD73 (32). The generation of adenosine was integral to limiting infarct size, inflammation, and the development of fibrosis (31). Although infarct size and cardiac ejection fraction were similar the day following ischemia, cardiac function continued to deteriorate in CD73 deficient mice whereas some recovery of function was observed WT mice. Lack of CD73 was associated with a sustained leukocytic myocardial infiltrate of a Th1 and M1 phenotype with enhanced expression of TNFα, IL-1β, IL-6, and IL-17 (31). Koeppen et al. (33) demonstrated CD73-generated adenosine mitigated inflammation and fibrosis preserving cardiac function through A2BR signaling specifically on polymorphonuclear cells which limited the release of TNFα.
These data suggest that adenosine is critical in the response of the heart to transplantation and augmenting peri-transplant adenosine levels reduces the impact of IRI and improves graft outcomes.
Lung Transplantation
Chronic pulmonary allograft dysfunction manifests as bronchiolitis obliterans syndrome (BOS), which is characterized by progressive airflow obstruction and deterioration in function. BOS is a major complication following lung transplantation limiting long term survival. Ischemia–reperfusion injury, acute rejection episodes, and CMV infection are independent predictors of progressive BOS (34).
A2AR Activation Attenuates Lung Ischemia–Reperfusion Injury
CD73 dependent generation of adenosine is essential in limiting inflammation occurring early following lung transplantation. Indeed greater inflammation was evident at 1 week in tracheal allografts transplanted into CD73 deficient recipient mice (35). The inflammatory infiltrate demonstrated increased CD3+ T cell infiltration and was accompanied by greater expression of Th1 cytokines IFNγ and IL-2 resulting in luminal narrowing. In the WT allografts, marked upregulation of A2AR mRNA expression was evident such that treatment of CD73 deficient recipients with an A2AR agonist rescued the allograft. In fact A2AR activation potently attenuates lung IRI if given before ischemia (36) or during reperfusion (37). Inflammation and pulmonary edema were maximally decreased and lung function optimized in a blood perfused rabbit-lung model subjected to ischemia followed by treatment with the specific A2AR agonist ATL313. When ischemia was combined with an A2AR inhibitor, this effect was abolished (36). Furthermore, cardiac dysfunction occurring concomitantly with pulmonary IRI is attenuated with A2AR activation (38). In mice A2AR activation reduces CD4+ T cell and neutrophil infiltration with marked reduction in inflammatory cytokines such as TNFα, IL-17, MCP-1, MIP-1, and RANTES (39) significantly improving lung function.
Human lung transplantation includes a period of cold preservation, which slows cellular metabolism limiting ischemic injury however; extended cold storage times increase the incidence of delayed graft function. A2AR activation decreases the inflammatory response and preserves pulmonary function following cold preservation and transplantation. Using a porcine transplant model, Reece et al. (37) demonstrated that treatment of the recipient pig with A2AR agonist beginning 10 min prior to and continuing for the first 3 h of reperfusion improved outcome. In this model, the porcine lung was subjected to 6 h of cold ischemia followed by 4 h of reperfusion. Animals treated with the intravenous infusion of A2AR agonist ATL-146e had improved oxygenation with preserved CO2 levels and acid–base balance. The measured pulmonary artery pressures and pulmonary vascular resistance were also lower in the A2AR agonist treated group. The overall lung injury score was significantly less primarily due to less pulmonary infiltration in the treatment group.
Human donor lungs may be perfused ex vivo with Steen Solution® in order to optimize pulmonary function prior to engraftment. Based on the beneficial effect of A2AR activation in small animal warm ischemia–reperfusion models, Emaminia et al. (40) examined the effect of supplementing Steen Solution® with A2AR agonist on pig explanted lungs which had been stored at 4°C for 5 h. Treated lungs demonstrated less edema, improved oxygenation index and mean airway pressure and lower levels of IFNγ, IL-1β, IL-6, and IL-18 suggesting A2AR agonist supplementation may further optimize lung function prior to transplantation. Indeed using a xenograft model, Westall et al. (41) demonstrated that genetically modified pig lungs lacking the αGal gene and expressing human complement regulatory proteins (CD55, CD59) and CD39 performed better ex vivo following perfusion with human blood. Lungs from genetically modified pigs demonstrated stable pulmonary vascular resistance, better oxygenation, and survived longer than WT lungs. Multiple potential factors may have contributed to the improved outcome including a putative effect of CD39 generated adenosine.
A2AR Activation Attenuates Acute Allograft Rejection in the Lung
Recurrent episodes of acute allograft rejection promote the development of BOS. A2AR signaling is important in modifying the alloimmune response: A2AR activation reduces skin allograft rejection (42) and improves survival and functional engraftment of transplanted islets by inhibiting inflammatory islet damage in the peri-transplant period (43). Complete MHC mismatched tracheal allografts are rejected within 3 weeks manifesting with complete lumen obliteration. The kinetics of rejection was accelerated in A2ARKO mice with more inflammatory infiltrate comprising macrophages, neutrophils, and CD3+ T cells and collagen deposition. Conversely treatment of allograft recipients with A2AR agonist resulted in less leukocytic infiltrate and delay in luminal obliteration supporting an effect of A2AR activation in limiting ischemia–reperfusion injury (44). Intriguingly, a role for A2BR signaling has also been proposed in the development of BOS experimentally. A2BRKO mice recipient of a MHC mismatched tracheal allograft developed less severe BOS at 3 weeks (45). A greater number of FoxP3+ Tregs were present in the tracheal grafts in A2BRKO mice as early as day 3 post-op with concomitant reduction in neutrophils and CD4+ T cells, suggesting that A2BR activation may promote BOS via inhibiting Treg infiltration. The opposing effects of A2AR and A2BR signaling in tracheal transplantation may reflect the cellular expression of each adenosine receptor; the affinity for adenosine being approximately 50 times greater for the A2AR (ref) and that in addition to signaling via the G-stimulatory subunit and increasing intracellular cAMP, A2BR activation promotes calcium mobilization (ref).
Together, these data demonstrate evidence for A2AR activation in limiting acute pulmonary IRI and reducing alloimmune response with significant improvement in lung function following transplantation. On the other hand, A2BR signaling may promote allograft dysfunction indicating early inhibition may be therapeutically advantageous.
Liver Transplantation
CD39 and A2BR Activity is Protective in Liver IRI
Liver transplantation remains the only therapeutic option for patients with end stage liver failure. The prevention of early graft dysfunction principally due to IRI is critical as there are limited supportive options available. As in other solid organs hepatic IP potently induces the transcription of CD39 via Sp1 (46) and CD73 (47), which promotes local adenosine generation protecting against subsequent prolonged ischemia. CD39 deficient (48) and CD73 deficient (47) mice are unable to be preconditioned and remain highly susceptible to the effects of prolonged ischemia with high mortality due to significant hepatic infarction.
Hypoxic preconditioning, like IP, confers protection against subsequent liver ischemia with marked attenuation of serum ALT, TNF-α, and IL-6 (49). This technique involves breathing 10% oxygen for 10 min prior to prolonged ischemia and results in a doubling of plasma adenosine concentrations. Employed in A2BRKO mice or inhibition of the A2BR negated the protective effect of hypoxic preconditioning providing the first evidence for the A2BR pathway in the prevention of warm hepatic IRI (49).
Deceased-donor and living-donor split-liver transplantation is a technique used to increase organ utility amongst potential recipients. The liver has a remarkable ability to regenerate after injury or resection and liver growth occurs at a rapid pace (50). CD39 plays a critical role in the regenerative capacity of the liver and is significantly attenuated in CD39 deficient mice (51). Stem cell mobilization directly correlates with restoration of liver volume and function and this is impaired in mice chimeric with CD39 deficient bone marrow (52).
A2AR Activation and CD39 Activity Reduces Hepatic Inflammation Post IRI
Inflammation is an innate response to hepatic ischemia and reperfusion and T cells rapidly accumulate within the liver parenchyma. Administration of A2AR agonist during reperfusion significantly reduces liver injury following a period of ischemia, an effect lost in A2ARKO mice (53). Through a series of studies using chimeric mice, it was determined that it was the activation of the A2AR on circulating cells that conferred protection with less liver injury, reduced neutrophil infiltration, and induction of proinflammatory cytokine transcripts (54). These data were further refined by Lappas et al. (55), who demonstrated that the NKT cell subset of CD4+ T cells principally mediated early liver injury. Following 72 min of liver ischemia and 2 h of reperfusion CD1d-dependent activation of NKT cells lead to an increase in IFN-γ expression. Injury could be recapitulated in RAG-1 KO mice through the transfer of NKT cells and treatment of these cells prior to transfer with an A2AR agonist mitigated injury (55). However, activation of NKT cells prior to the ischemic insult actually confers protection. Cao et al. (56) activated NKT cells 1 h prior to ischemia and showed a reduction in neutrophilic infiltration and injury, which was both IL-13 and A2AR dependent.
Although these data implicate a pathogenic role for NKT cells in warm hepatic IRI this is not the case with cold IRI. Using a murine liver transplant model where the recipient mouse had undergone a total hepatectomy and was transplanted with either a donor liver from a WT or CD39 over-expressing mouse that had been stored at 4°C for 18 h, Pommey et al. showed significant protection in donor livers from CD39 over-expressing mice (57). In a series of chimeric studies and through detailed immunophenotyping of CD39 over-expressing mice the protective effect was attributed to a decrease in resident hepatic CD4+ T cells and the effect could be replicated through depletion of CD4+ T cells in WT mice. Notably, mice deficient in NKT cells remained susceptible to the effects of cold IRI. These data indicate significant differences in the pathogenetic mechanisms underlying warm and cold IRI associated with liver transplantation.
Renal Transplantation
Renal transplantation remains the optimal form of renal replacement therapy for patients with end stage renal disease. Similar to other solid organ transplants long term survival is limited by the development of chronic allograft dysfunction which in the kidney manifests with interstitial fibrosis and tubular atrophy. Risk factors for late allograft loss include acute rejection that is increased in patients with delayed graft function and which in turn is impacted by IRI particularly with prolonged cold preservation times.
Evidence for the CD39-adenosinergic axis in renal IRI has been appreciated for some time and was recently reviewed (58). CD39 transcript expression is upregulated with IP (59) and CD39 deficient mice are highly susceptible to injury (60). Mice that over-express CD39 (61) or pre-treated with apyrase (59) are robustly protected from injury which is A2BR dependent (unpublished Karen M. Dwyer). As with other solid organs a period of cold ischemia often occurs in renal transplantation. Crikis et al. (61) examined the effect of CD39 over-expression in the donor kidney following a period of 5 h at 4°C. The donor kidney was transplanted into a bilaterally nephrectomized mouse recipient, which was culled 24 h later. Recipients receiving the CD39 over-expressing graft had better renal function as reflected by lower serum creatinine correlating with less tubular injury in the graft. These mice also remained healthy with preserved renal function up to 3 days post transplantation as opposed to mice receiving the WT kidney that did not survive to day 3 post renal transplantation (61).
Like CD39, CD73 is essential for preconditioning prior to renal ischemia (62). Intriguingly, Rajakumar et al. (63) described protection against injury in CD73 deficient mice. In this model, mice underwent 18 min of ischemia followed by 24 h of reperfusion, which induced mild injury in WT mice. Nevertheless, CD73KO mice were protected from injury and a similar result was obtained when WT mice were pre-treated with a CD73 inhibitor. These data implicate direct biological activity of AMP, which has recently been ascribed in vitro at the A1R (64).
With respect to adenosine signaling, A2AR activation mitigates inflammation inherent in this injury and parenchymal A2BR signaling improves post ischemic blood flow limiting injury (65, 66). CD4+ T cells are critical in mediating renal injury and pretreatment of mice with A2AR agonist significantly lessens this burden (67). As demonstrated in liver IRI, the CD4+ subset of NKT cells are activated rapidly after renal IRI, which is followed by the activation of other immune cells (68). Blocking dendritic cell mediated NKT cell activation via A2AR agonist treatment markedly reduced renal injury (69). Indeed, inhibition of adenosine receptor signaling is required for optimal dendritic cell, and subsequent T cell, activation (70). A series of elegant studies by Kinsey et al. (16, 17, 71) have emphasized the role of Tregs in inhibiting CD4+ T cell effector function through upregulation of PD-1 expression, which is augmented by A2AR activation.
Regulatory T cells traditionally have been defined by CD4 and CD25 co-expression and the transcription factor FoxP3. This subset have also been shown to express CD39 (7, 8, 72) and CD73 (8, 13) and the A2AR (8, 17). CD39 is essential for the full suppressive capabilities of these cells and mice deficient in CD39 are immunocompromised being more susceptible to T cell mediated diabetes (73) and experience more rapid skin graft rejection (8). In humans CD4, CD25, and CD39 expression identifies four T cell subsets, which can be followed longitudinally in the peripheral blood of renal transplant recipients (72).
Summary
The process of solid organ transplantation involves ischemia–reperfusion injury that promotes immunogenicity of the donor organ leading to an increased risk of rejection and graft loss. Although T cells have been traditionally considered the orchestrators of these pathological processes, a role for B cells is being increasingly recognized. The recent identification and characterization of B cell subtypes; the recognition of dual but opposing effects on T cells; the putative involvement in allograft rejection make these cells a potentially attractive therapeutic target.
The ectonucleotidases CD39 and CD73 are involved in adenosine generation and expressed by these important immune cell subsets. Adenosine signaling is integral to the function of both B and T cells, is protective in warm and cold ischemic injury and has immunomodulating properties, see Figure 1. There are current adenosine receptor agonists and antagonists being tested [reviewed in Ref. (74)] in non-transplant human trials but the ubiquitous nature of adenosine receptors make targeted therapy difficult. Potentially treatment of the donor organ prior to implantation with adenosine receptor agonists may reduce the impact of ischemia–reperfusion injury, subsequent acute rejection and graft failure.
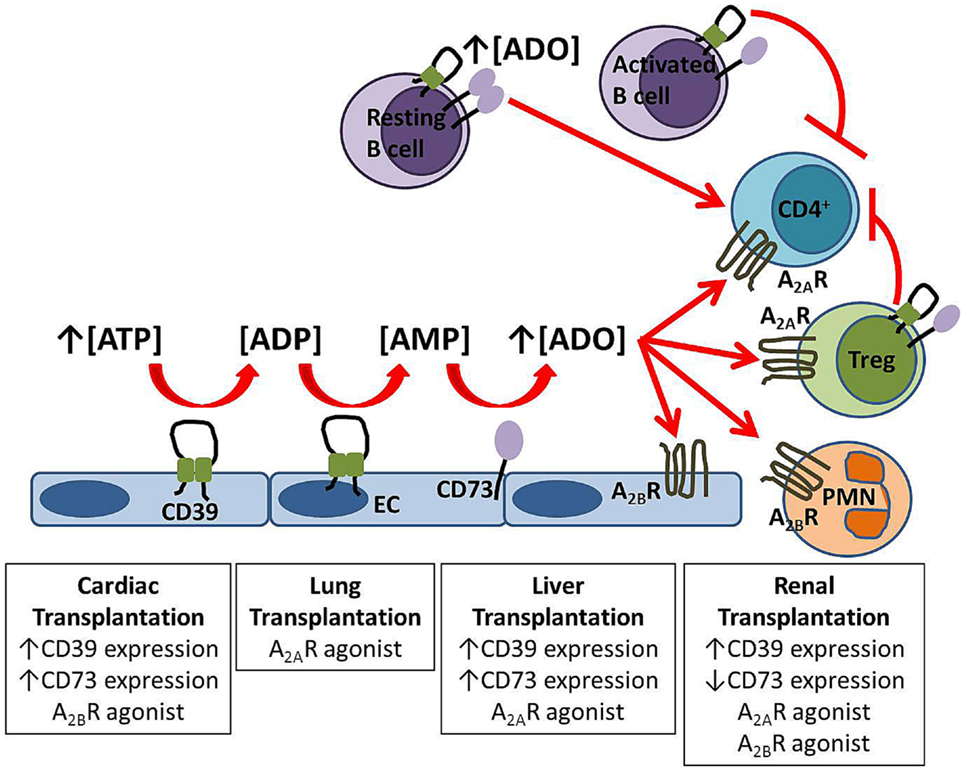
Figure 1. Protective mechanisms in solid organ transplantation. Extracellular adenosine is generated from the enzymatic hydrolysis of nucleotides by the ectoenzymes CD39 and CD73 expressed on endothelial cells (EC) and B cells. Adenosine signals via A2AR on circulating cells including regulatory T cells (Treg) and via A2BR expressed both on the vasculature and inflammatory cells. Experimental strategies which improve graft outcome for each solid organ transplant are listed in boxes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Dalloul AH. B-cell-mediated strategies to fight chronic allograft rejection. Front Immunol (2013) 4:444. doi: 10.3389/fimmu.2013.00444
2. Saze Z, Schuler PJ, Hong C-S, Cheng D, Jackson EK, Whiteside TL. Adenosine production by human B cells and B cell-mediated suppression of activated T cells. Blood (2013) 122(1):9–18. doi:10.1182/blood-2013-02-482406
3. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature (2012) 491(7423):264–8. doi:10.1038/nature11501
4. Deaglio S, Robson SC. Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv Pharmacol (2011) 61:301–32. doi:10.1016/B978-0-12-385526-8.00010-2
5. Eltzschig HK. Adenosine: an old drug newly discovered. Anesthesiology (2009) 111(4):904–15. doi:10.1097/ALN.0b013e3181b060f2
6. Maliszewski CR, Delespesse GJ, Schoenborn MA, Armitage RJ, Fanslow WC, Nakajima T, et al. The CD39 lymphoid cell activation antigen. Molecular cloning and structural characterization. J Immunol (1994) 153(8):3574–83.
7. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood (2007) 110(4):1225–32. doi:10.1182/blood-2006-12-064527
8. Deaglio S, Dwyer K, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204(6):1257–65. doi:10.1084/jem.20062512
9. Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood (2004) 104(13):3986–92. doi:10.1182/blood-2004-06-2066
10. Kaczmarek E, Koziak K, Sevigny J, Siegel J, Anrather J, Beaudoin A, et al. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J Biol Chem (1996) 271(51):33116–33122. doi:10.1074/jbc.271.51.33116
11. Bours MJL, Swennen ELR, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5’-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther (2006) 112(2):358–404. doi:10.1016/j.pharmthera.2005.04.013
12. Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD 7 3. Immunol Rev (1998) 161(1):95–109. doi:10.1111/j.1600-065X.1998.tb01574.x
13. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol (2006) 177(10):6780–6.
14. Hasegawa T, Bouis D, Liao H, Visovatti SH, Pinsky DJ. Ecto-5’ nucleotidase (CD73)-mediated adenosine generation and signaling in murine cardiac allograft vasculopathy. Circ Res (2008) 103(12):1410–21. doi:10.1161/CIRCRESAHA.108.180059
15. Schena F, Volpi S, Caterina E, Faliti F, Penco S, Santi M, et al. Dependence of immunoglobulin class switch recombination in B cells on vesicular release of ATP and CD73 ectonucleotidase activity. Cell Rep (2013) 3(6):1824–31. doi:10.1016/j.celrep.2013.05.022
16. Kinsey GR, Sharma R, Huang L, Li L, Vergis AL, Ye H, et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol (2009) 20(8):1744–53. doi:10.1681/ASN.2008111160
17. Kinsey GR, Huang L, Jaworska K, Khutsishvili K, Becker DA, Ye H, et al. Autocrine adenosine signaling promotes regulatory T cell-mediated renal protection. J Am Soc Nephrol (2012) 23(9):1528–37. doi:10.1681/ASN.2012010070
18. Taylor DO, Edwards LB, Boucek MM, Trulock EP, Waltz DA, Keck BM, et al. Registry of the international society for heart and lung transplantation: twenty-third official adult heart transplantation report-2006. J Heart Lung Transplant (2006) 25(8):869–79. doi:10.1016/j.healun.2006.05.002
19. Valantine HA. Cardiac allograft vasculopathy: central role of endothelial injury leading to transplant atheroma. Transplantation (2003) 76(6):891–9. doi:10.1097/01.TP.0000080981.90718.EB
20. Enjyoji K, Sevigny J, Lin Y, Frenette PS, Christie PD, Esch JS II, et al. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat Med (1999) 5(9):1010–7. doi:10.1038/12447
21. Imai M, Takigami K, Guckelberger O, Kaczmarek E, Csizmadia E, Bach FH, et al. Recombinant adenoviral mediated CD39 gene transfer prolongs cardiac xenograft survival. Transplantation (2000) 70(6):864–70. doi:10.1097/00007890-200009270-00003
22. Koyamada N, Miyatake T, Candinas D, Hechenleitner P, Siegel J, Hancock WW, et al. Apyrase administration prolongs discordant xenograft survival. Transplantation (1996) 62(12):1739–43. doi:10.1097/00007890-199612270-00008
23. Dwyer KM, Robson SC, Nandurkar HH, Campbell DJ, Gock H, Murray-Segal LJ, et al. Thromboregulatory manifestations in human CD39 transgenic mice and the implications for thrombotic disease and transplantation. J Clin Invest (2004) 113(10):1440–6. doi:10.1172/JCI19560
24. Imai M, Takigami K, Guckelberger O, Enjyoji K, Smith RN, Lin Y, et al. Modulation of nucleoside [correction of nucleotide] triphosphate diphosphohydrolase-1 (NTPDase-1)cd39 in xenograft rejection. Mol Med (1999) 5(11):743–52.
25. Eltzschig HK, Kohler D, Eckle T, Kong T, Robson SC, Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood (2009) 113(1):224–32. doi:10.1182/blood-2008-06-165746
26. Kohler D, Eckle T, Faigle M, Grenz A, Mittelbronn M, Laucher S, et al. CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation (2007) 116(16):1784–94. doi:10.1161/CIRCULATIONAHA.107.690180
27. Cai M, Huttinger ZM, He H, Zhang W, Li F, Goodman LA, et al. Transgenic over expression of ectonucleotide triphosphate diphosphohydrolase-1 protects against murine myocardial ischemic injury. J Mol Cell Cardiol (2011) 51(6):927–35. doi:10.1016/j.yjmcc.2011.09.003
28. Eckle T, Krahn T, Grenz A, Kohler D, Mittelbronn M, Ledent C, et al. Cardioprotection by ecto-5’-nucleotidase (CD73) and A2B adenosine receptors. Circulation (2007) 115(12):1581–90. doi:10.1161/CIRCULATIONAHA.106.669697
29. Eckle T, Hartmann K, Bonney S, Reithel S, Mittelbronn M, Walker LA, et al. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat Med (2012) 18(5):774–82. doi:10.1038/nm.2728
30. Lim SH, Lee S, Noda K, Kawamura T, Tanaka Y, Shigemura N, et al. Adenosine injection prior to cardioplegia enhances preservation of senescent hearts in rat heterotopic heart transplantation. Eur J Cardiothorac Surg (2013) 43(6):1202–8. doi:10.1093/ejcts/ezs509
31. Bonner F, Borg N, Jacoby C, Temme S, Ding Z, Flogel U, et al. Ecto-5’-nucleotidase on immune cells protects from adverse cardiac remodeling. Circ Res (2013) 113(3):301–12. doi:10.1161/CIRCRESAHA.113.300180
32. Bonner F, Borg N, Burghoff S, Schrader J. Resident cardiac immune cells and expression of the ectonucleotidase enzymes CD39 and CD73 after ischemic injury. PLoS One (2012) 7(4):e34730. doi:10.1371/journal.pone.0034730
33. Koeppen M, Harter PN, Bonney S, Bonney M, Reithel S, Zachskorn C, et al. Adora2b signaling on bone marrow derived cells dampens myocardial ischemia-reperfusion injury. Anesthesiology (2012) 116(6):1245–57. doi:10.1097/ALN.0b013e318255793c
34. Fiser SM, Tribble CG, Long SM, Kaza AK, Kern JA, Jones DR, et al. Ischemia-reperfusion injury after lung transplantation increases risk of late bronchiolitis obliterans syndrome. Ann Thorac Surg (2002) 73(4):1041–7. doi:10.1016/S0003-4975(01)03606-2
35. Ohtsuka T, Changelian PS, Bouis D, Noon K, Harada H, Lama VN, et al. Ecto-5’-nucleotidase (CD73) attenuates allograft airway rejection through adenosine 2A receptor stimulation. J Immunol (2010) 185(2):1321–9. doi:10.4049/jimmunol.0901847
36. Gazoni LM, Laubach VE, Mulloy DP, Bellizzi A, Unger EB, Linden J, et al. Additive protection against lung ischemia-reperfusion injury by adenosine A2A receptor activation before procurement and during reperfusion. J Thorac Cardiovasc Surg (2008) 135(1):156–65. doi:10.1016/j.jtcvs.2007.08.041
37. Reece TB, Ellman PI, Maxey TS, Crosby IK, Warren PS, Chong TW, et al. Adenosine A2A receptor activation reduces inflammation and preserves pulmonary function in an in vivo model of lung transplantation. J Thorac Cardiovasc Surg (2005) 129(5):1137–43. doi:10.1016/j.jtcvs.2004.11.042
38. Reece TB, Laubach VE, Tribble CG, Maxey TS, Ellman PI, Warren PS, et al. Adenosine A2A receptor agonist improves cardiac dysfunction from pulmonary ischemia-reperfusion injury. Ann Thorac Surg (2005) 79(4):1189–95. doi:10.1016/j.athoracsur.2004.09.038
39. Sharma AK, Laubach VE, Ramos SI, Zhao Y, Stukenborg G, Linden J, et al. Adenosine A2A receptor activation on CD4+ T lymphocytes and neutrophils attenuates lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg (2010) 139(2):474–82. doi:10.1016/j.jtcvs.2009.08.033
40. Emaminia A, Lapar DJ, Zhao Y, Steidle JF, Harris DA, Laubach VE, et al. Adenosine A(2)A agonist improves lung function during ex vivo lung perfusion. Ann Thorac Surg (2011) 92(5):1840–6. doi:10.1016/j.athoracsur.2011.06.062
41. Westall GP, Levvey BJ, Salvaris E, Gooi J, Marasco S, Rosenfeldt F, et al. Sustained function of genetically modified porcine lungs in an ex vivo model of pulmonary xenotransplantation. J Heart Lung Transplant (2013) 32(11):1123–30. doi:10.1016/j.healun.2013.07.001
42. Sevigny CP, Li L, Awad AS, Huang L, McDuffie M, Linden J, et al. Activation of adenosine 2A receptors attenuates allograft rejection and alloantigen recognition. J Immunol (2007) 178(7):4240–9.
43. Chhabra P, Wang K, Zeng Q, Jecmenica M, Langman L, Linden J, et al. Adenosine A(2A) agonist administration improves islet transplant outcome: evidence for the role of innate immunity in islet graft rejection. Cell Transplant (2010) 19(5):597–612. doi:10.3727/096368910X491806
44. Lau CL, Zhao Y, Kron L, Stoler MH, Laubach VE, Ailawadi G, et al. The role of adenosine A2A receptor signaling in bronchiolitis obliterans. Ann Thorac Surg (2009) 88(4):1071–8. doi:10.1016/j.athoracsur.2009.06.032
45. Zhao Y, LaPar DJ, Steidle J, Emaminia A, Kron IL, Ailawadi G, et al. Adenosine signaling via the adenosine 2B receptor is involved in bronchiolitis obliterans development. J Heart Lung Transplant (2010) 29(12):1405–14. doi:10.1016/j.healun.2010.07.005
46. Hart ML, Gorzolla C, Schittenhelm J, Robson SC, Eltzschig HK. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J Immunol (2010) 184(7):4017–24. doi:10.4049/jimmunol.0901851
47. Hart ML, Much C, Gorzolla C, Schittenhelm J, Kloor D, Stahl GL, et al. Extracellular adenosine production by ecto-5’-nucleotidase protects during murine hepatic ischemic preconditioning. Gastroenterology (2008) 135(5):1739–1750e3. doi:10.1053/j.gastro.2008.07.064
48. Sun X, Imai M, Nowak-Machen M, Guckelberger O, Enjyoji K, Wu Y, et al. Liver damage and systemic inflammatory responses are exacerbated by the genetic deletion of CD39 in total hepatic ischemia. Purinergic Signal (2011) 7(4):427–34. doi:10.1007/s11302-011-9239-6
49. Chouker A, Ohta A, Martignoni A, Lukashev D, Zacharia LC, Jackson EK, et al. In vivo hypoxic preconditioning protects from warm liver ischemia-reperfusion injury through the adenosine A2B receptor. Transplantation (2012) 94(9):894–902. doi:10.1097/TP.0b013e31826a9a46
50. Humar A, Kosari K, Sielaff TD, Glessing B, Gomes M, Dietz C, et al. Liver regeneration after adult living donor and deceased donor split-liver transplants. Liver Transpl (2004) 10(3):374–8. doi:10.1002/lt.20096
51. Beldi G, Wu Y, Sun X, Imai M, Enjyoji K, Csizmadia E, et al. Regulated catalysis of extracellular nucleotides by vascular CD39/ENTPD1 is required for liver regeneration. Gastroenterology (2008) 135(5):1751–60. doi:10.1053/j.gastro.2008.07.025
52. Schmelzle M, Duhme C, Junger W, Salhanick SD, Chen Y, Wu Y, et al. CD39 modulates hematopoietic stem cell recruitment and promotes liver regeneration in mice and humans after partial hepatectomy. Ann Surg (2013) 257(4):693–701. doi:10.1097/SLA.0b013e31826c3ec2
53. Day YJ, Marshall MA, Huang L, McDuffie MJ, Okusa MD, Linden J. Protection from ischemic liver injury by activation of A2A adenosine receptors during reperfusion: inhibition of chemokine induction. Am J Physiol Gastrointest Liver Physiol (2004) 286(2):G285–93. doi:10.1152/ajpgi.00348.2003
54. Day YJ, Li Y, Rieger JM, Ramos SI, Okusa MD, Linden J. A2A adenosine receptors on bone marrow-derived cells protect liver from ischemia-reperfusion injury. J Immunol (2005) 174(8):5040–6.
55. Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J Exp Med (2006) 203(12):2639–48. doi:10.1084/jem.20061097
56. Cao Z, Yuan Y, Jeyabalan G, Du Q, Tsung A, Geller DA, et al. Preactivation of NKT cells with alpha-GalCer protects against hepatic ischemia-reperfusion injury in mouse by a mechanism involving IL-13 and adenosine A2A receptor. Am J Physiol Gastrointest Liver Physiol (2009) 297(2):G249–58. doi:10.1152/ajpgi.00041.2009
57. Pommey S, Lu B, McRae J, Stagg J, Hill P, Salvaris E, et al. Liver grafts from CD39-overexpressing mice are protected from ischemia reperfusion injury due to reduced numbers of resident CD4(+) T cells. Hepatology (2013) 57(4):1597–606. doi:10.1002/hep.25985
58. Roberts V, Lu B, Rajakumar S, Cowan PJ, Dwyer KM. The CD39-adenosinergic axis in the pathogenesis of renal ischemia-reperfusion injury. Purinergic Signal (2012) 9(2):135–43. doi:10.1007/s11302-012-9342-3
59. Grenz A, Zhang H, Hermes M, Eckle T, Klingel K, Huang DY, et al. Contribution of E-NTPDase1 (CD39) to renal protection from ischemia-reperfusion injury. FASEB J (2007) 21(11):2863–73. doi:10.1096/fj.06-7947com
60. Lu B, Rajakumar SV, Robson SC, Lee EK, Crikis S, D’Apice AJ, et al. The impact of purinergic signaling on renal ischemia-reperfusion injury. Transplantation (2008) 86(12):1707–12. doi:10.1097/TP.0b013e31819022bc
61. Crikis S, Lu B, Murray-Segal LM, Selan C, Robson SC, D’Apice AJ, et al. Transgenic overexpression of CD39 protects against renal ischemia-reperfusion and transplant vascular injury. Am J Transplant (2010) 10(12):2586–95. doi:10.1111/j.1600-6143.2010.03257.x
62. Grenz A, Zhang H, Eckle T, Mittelbronn M, Wehrmann M, Kohle C, et al. Protective role of ecto-5’-nucleotidase (CD73) in renal ischemia. J Am Soc Nephrol (2007) 18(3):833–45. doi:10.1681/ASN.2006101141
63. Rajakumar SV, Lu B, Crikis S, Robson SC, D’Apice AJ, Cowan PJ, et al. Deficiency or inhibition of CD73 protects in mild kidney ischemia-reperfusion injury. Transplantation (2010) 90(12):1260–4. doi:10.1097/TP.0b013e3182003d9b
64. Rittiner JE, Korboukh I, Hull-Ryde EA, Jin J, Janzen WP, Frye SV, et al. The nucleotide AMP is an adenosine A1 receptor agonist. J Biol Chem (2012) 287(8):5301–9. doi:10.1074/jbc.M111.291666
65. Grenz A, Osswald H, Eckle T, Yang D, Zhang H, Tran ZV, et al. The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med (2008) 5(6):e137. doi:10.1371/journal.pmed.0050137
66. Grenz A, Bauerle JD, Dalton JH, Ridyard D, Badulak A, Tak E, et al. Equilibrative nucleoside transporter 1 (ENT1) regulates postischemic blood flow during acute kidney injury in mice. J Clin Invest (2012) 122(2):693–710. doi:10.1172/JCI60214
67. Day YJ, Huang L, Ye H, Li L, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: the role of CD4+ T cells and IFN-gamma. J Immunol (2006) 176(5):3108–14.
68. Li L, Huang L, Sung S-SJ, Lobo PI, Brown MG, Gregg RK, et al. NKT cell activation mediates neutrophil IFN-γ production and renal ischemia-reperfusion injury. J Immunol (2007) 178(9):5899–911.
69. Li L, Huang L, Ye H, Song SP, Bajwa A, Lee SJ, et al. Dendritic cells tolerized with adenosine A2AR agonist attenuate acute kidney injury. J Clin Invest (2012) 122(11):3931–42. doi:10.1172/JCI63170
70. Linden J, Cekic C. Regulation of lymphocyte function by adenosine. Arterioscler Thromb Vasc Biol (2012) 32(9):2097–103. doi:10.1161/ATVBAHA.111.226837
71. Kinsey GR, Huang L, Vergis AL, Li L, Okusa MD. Regulatory T cells contribute to the protective effect of ischemic preconditioning in the kidney. Kidney Int (2010) 77(9):771–80. doi:10.1038/ki.2010.12
72. Dwyer KM, Hanidziar D, Putheti P, Hill PA, Pommey S, McRae JL, et al. Expression of CD39 by human peripheral blood CD4+ CD25+ T cells denotes a regulatory memory phenotype. Am J Transplant (2010) 10(11):2410–20. doi:10.1111/j.1600-6143.2010.03291.x
73. Chia JS, McRae JL, Thomas H, Fynch S, Elkerbout L, Hill P, et al. The protective effects of CD39 over-expression in multiple low dose streptozotocin-induced diabetes in mice. Diabetes (2013) 62(6):2026–35. doi:10.2337/db12-0625
Keywords: adenosine, CD73, CD39, Treg, B cells
Citation: Roberts V, Stagg J and Dwyer KM (2014) The role of ectonucleotidases CD39 and CD73 and adenosine signaling in solid organ transplantation. Front. Immunol. 5:64. doi: 10.3389/fimmu.2014.00064
Received: 29 October 2013; Paper pending published: 10 December 2013;
Accepted: 03 February 2014; Published online: 18 February 2014.
Edited by:
Sophie Brouard, Centre National de la Recherche Scientifique, FranceReviewed by:
Stanislaw Stepkowski, University of Toledo, USAPhilippe Saas, Etablissement Français du Sang BFC, France
Copyright: © 2014 Roberts, Stagg and Dwyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karen M. Dwyer, Immunology Research Centre, St Vincent’s Hospital, Level 1 Healy Wing, 41 Victoria Parade, Melbourne VIC 3065, Australia e-mail:a2FyZW4uZHd5ZXJAc3ZobS5vcmcuYXU=