 Leen Moens1
Leen Moens1 Stuart G. Tangye1,2*
Stuart G. Tangye1,2*- 1Immunology and Immunodeficiency Group, Immunology Research Program, Garvan Institute of Medical Research, Darlinghurst, NSW, Australia
- 2St Vincent’s Clinical School, University of New South Wales, Darlinghurst, NSW, Australia
During our life, we are surrounded by continuous threats from a diverse range of invading pathogens. Our immune system has evolved multiple mechanisms to efficiently deal with these threats so as to prevent them from causing disease. Terminal differentiation of mature B cells into plasma cells (PC) – the antibody (Ab) secreting cells of the immune system – is critical for the generation of protective and long-lived humoral immune responses. Indeed, efficient production of antigen (Ag)-specific Ab by activated B cells underlies the success of most currently available vaccines. The mature B-cell pool is composed of several subsets, distinguished from one according to size, surface marker expression, location, and Ag exposure, and they all have the capacity to differentiate into PCs. For a B-cell to acquire the capacity to produce Abs, it must undergo an extensive differentiation process driven by changes in gene expression. Two broad categories of Ags exist that cause B-cell activation and differentiation: T cell dependent (TD) or T cell independent (TI). In addition to the B-cell subset and nature of the Ag, it is important to consider the cytokine environment that can also influence how B-cell differentiation is achieved. Thus, while many cytokines can induce Ab-secretion by B cells after activation with mimics of TD and TI stimuli in vitro, they can have different efficacies and specificities, and can often preferentially induce production of one particular Ig isotype over another. Here, we will provide an overview of in vitro studies (mouse and human origin) that evaluated the role of different cytokines in inducing the differentiation of distinct B-cell subsets to the PC lineage. We will place particular emphasis on IL-21, which has emerged as the most potent inducer of terminal B-cell differentiation in humans. We will also focus on the role of IL-21 and defects in B-cell function and how these contribute to human immunopathologies such as primary immunodeficiencies and B-cell mediated autoimmune conditions.
Introduction
The humoral arm of the immune system is critical for providing protective antibodies (Abs) against infection pathogens. The Ab pool is maintained by long-lived plasma cells (PCs), which continuously secrete Abs following their formation in response to exposure to specific antigen (Ag). In 1948, Fagraeus was the first to report that PCs are the outcome of terminal B-cell differentiation and demonstrated their importance to Ab production in vitro (1). We now know that B cells are capable of secreting multiple Ig isotypes (IgM, IgG, IgA, IgE) and subclasses of these isotypes (IgG1–4, IgA1–2) following the receipt of appropriate stimulate. However, today – 65 years later – our understanding of the complexities of PC development remains incomplete.
Plasma Cell Formation: The Importance of T Cells, Cytokines, and Transcription Factors
Plasma cells are generated as a result of cognate interactions between Ag-specific B cells, CD4+ T helper cells, and dendritic cells in response to foreign Ags (Figure 1). These interactions can drive B cells to become low-affinity short-lived, predominantly IgM-secreting, plasmablasts that provide an initial wave of protection against invading pathogens. More importantly though, they also lead to the formation of germinal centers (GCs), which are specialized structures in the follicles of secondary lymphoid tissues where somatic hypermutation (SHM) of immunoglobulin (Ig) variable region genes and selection of high-affinity B cells occurs. These selected high-affinity variants can then differentiate into long-lived memory B cells or PCs (2, 3) (Figure 1). This differentiation event is in part mediated by T follicular helper (Tfh) cells, a distinct subset of CD4+ T cells characterized by expression of the transcriptional repressor B-cell lymphoma-6 (Bcl-6), the surface markers CXCR5, PD-1, ICOS, and CD40 ligand (CD40L), and production of various cytokines including interleukin-4 (IL-4), IL-10, and IL-21. Tfh cells localize to follicles and GCs – where they are termed “GC Tfh cells” – where they can interact with B cells and instruct their maturation into memory cells or PCs (4–6).
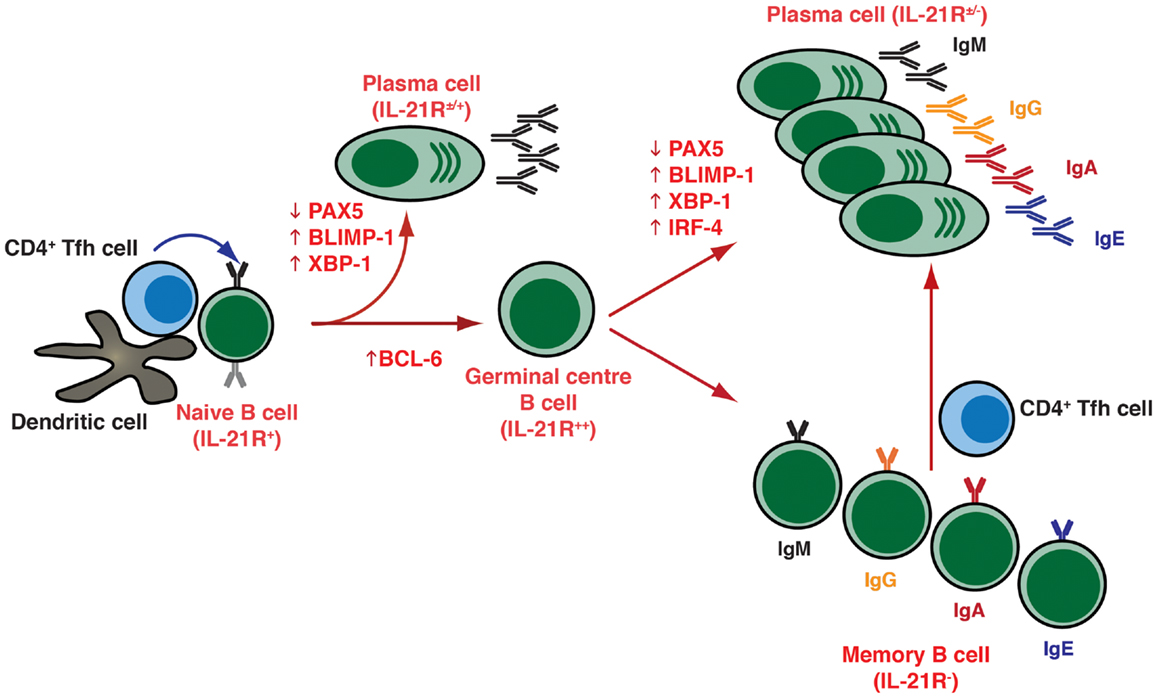
Figure 1. T cell dependent B-cell differentiation. Following the receipt of signals provided by the microenvironment [e.g., Ag, CD4+ T (Tfh) cells, DC], naïve B cells undergo activation and can initially differentiate into either extrafollicular short-lived Ab-secreting plasma cells (secreting predominantly IgM), or can seed a germinal center (GC). Within GCs, B cells undergo somatic hypermutation of their Ig V region genes and only those B cells with the highest affinity are selected to then differentiate into long-lived memory B cells or plasma cells that are capable of secreting a variety of Ig isotypes, including the switched isotypes IgG, IgA, and IgE. The outcome of the GC reaction is heavily influenced by Tfh cells, especially those within the GC itself. These cells are not depicted on the figure but they contribute greatly at this stage of B-cell differentiation. Following re-encounter with the initiating Ag, memory B cells rapidly differentiate into plasma cells. The differentiation of naïve B cells to these distinct effector fates is controlled by the balanced expression and regulated function of various transcription factors, including (but not exclusively) PAX5, BCL-6, BLIMP-1, XBP-1, and IRF4.
The differentiation of activated B cells into PC is regulated by transcriptional programs and networks that are influenced by numerous inputs and microenvironmental factors. These include the nature of the Ag and of the responding B-cell subset, the location in which Ag encounter occurs, and the accessory cells involved (7, 8). The key transcription factors involved in regulating PC formation include the transcriptional repressors Bcl-6 and B-lymphocyte induced maturation protein (BLIMP)-1, encoded by the PRDM1 gene, as well as transcription factors PAX5, X-box-binding protein-1 (XBP-1), and IFN-induced regulatory factor 4 (IRF4) (Figure 1) (7, 8). Thus, while Bcl-6 is expressed in GC B cells and is required for the GC formation (9–11), it blocks PC differentiation and maintains a GC B-cell fate by suppressing expression of BLIMP-1, which is considered the master regulator of PC differentiation, being required for – or at least correlated with – PC commitment in mice and humans (Figure 1) (12–15). BLIMP-1 expression controls PC differentiation by restraining the mature B-cell gene expression program by down-regulating a set of genes including MHC, CIITA, PAX5, and CMYC, which result in a decrease of MHC class II expression, loss of B-cell identity, and cessation of proliferation, respectively (8, 14). BLIMP-1 may also co-ordinate expression of XBP 1, which allows expansion of the secretory apparatus necessary for high-level protein synthesis in PC differentiation (Figure 1) (16).
Cytokines represent a diverse group of small soluble proteins that can function as growth and differentiation factors in autocrine or paracrine ways. Cytokines exhibit considerable redundancy, in that many cytokines share similar functions. Through binding to specific cell surface receptors, they initiate signal transduction pathways that are critical for a diverse spectrum of functions, including induction of immune responses, cell proliferation, differentiation, and apoptosis. The key contribution of cytokines to B-cell differentiation lies in their ability to modulate expression of these transcription factors such that they regulate Ig secretion by B cells activated with mimics of T cell dependent (TD) (e.g., CD40L) or T cell independent (TI) [e.g., engaging the B-cell receptor (BCR), Toll-like receptors (TLRs)] stimuli in vitro and, by extension, in vivo. The effects of cytokines on B-cell differentiation is evidenced not only by the magnitude of the Ab response but also the quality, in terms of the particular Ig isotype(s) induced. Although many cytokines are capable of promoting B-cell differentiation, the relative roles of specific factors, and the hierarchy of the interactions between several cytokines, has only emerged in the last 10 years.
Discovery of T Cell-Derived Factors as Critical Mediators of B-Cell Differentiation and PC Generation
The concept that cross-linking of the BCR initiates B-cell activation and facilitates these cells to respond to T-cell-derived soluble factors and undergo proliferation and differentiation to become Ab-secreting cells was first appreciated in the 1970s (17–20). The different factors were classically grouped as T cell-replacing factors, some of which influence the replication of B cells (B-cell growth factor), while others directly cause B-cell differentiation to Ab-secretion cells (B-cell differentiation factor) (21). While it gradually emerged that these T cell-derived factors are Ag non-specific, genetically non-restricted, and are indeed involved in the differentiation of B cells into Ab-secreting cells, at this time no single factor had been isolated or molecularly cloned, and it remained unknown how many factors were actually involved in, or required for, B-cell terminal differentiation (22, 23).
The molecular revolution of the 1980s saw the cloning and characterization of several cytokines – IL-2, IL-4, IL-5, IL-6, IFNs – which had B-cell growth and differentiation capacity (Table 1). This continued into the 1990s with the discovery of IL-10, IL-12, IL-13, IL-15, TNFα, BAFF, and APRIL, which could promote various aspects of B-cell function (Figure 2; Table 1). Thus, these cytokines enhanced proliferation and induced isotype switching, PC formation, and Ig secretion by activated B cells (22–47) (Table 1; Figure 2). Importantly, this era also saw the identification of CD40L – transiently expressed on the surface of activated CD4+ T cells – which, together with these cytokines, was revealed to be a critical regulator of many facets of B-cell biology (48). Specifically, while CD40L (or anti-CD40 mAb) itself had minimal effect on Ab-secretion by murine and human B cells, Ab-secretion could be induced in an isotype specific manner in the presence of exogenous cytokines (Figure 2; Table 1). Thus, IL-4 and IL-13 directs naïve human B cells to switch to IgG4 and IgE expression and production, while IL-4 exerts a similar effect for inducing IgG1 and IgE by murine B cells (Table 1), with IL-5 acting synergistically with IL-4 in these murine B-cell responses (25, 28, 29, 41, 49–51). The significance of these in vitro findings was underscored by the generation of IL-4 deficient mice, which had significantly reduced production of IgE following nematode infection (52). Interestingly, IL-4-induced IgE production by human B cells could be enhanced by IL-6 or TNFα (33, 45), or inhibited by IL-8 (53), IL-12 (54), or IFN-α or IFN-γ (33, 40). While murine B cells were initially reported to be unresponsive to IL-13 (55), subsequent studies noted that IL-13 could enhance Ab production by murine B cells in vivo and that it acts directly on B cells in vitro to increase survival, thereby increasing Ab production (56). Additional support for a role for IL-13 in modulating murine B cells came from the analysis of IL-13 transgenic mice, which exhibited substantially increased levels of serum IgE, even in the absence of IL-4 (57). Similarly, while deficiency of either IL-4 or IL-13 reduced the levels of Ag-specific IgE, combined deficiency of both IL-4 and IL-13 resulted in undetectable levels of IgE (58). Thus, it is likely that IL-4 and IL-13 co-operate in both mice and humans to regulate Ig class switching, especially to IgE. IL-10 also strongly modulated the behavior of human B cells, significantly increasing the levels of IgM, IgG1, and IgA secreted by human B cells stimulated through CD40 or the BCR (42). IL-10 was also found to induce class switching in human naïve B cells to IgG1 and IgG3 (59), and together with TGF-β promoted switching to IgA (31). IL-10 also mediated the differentiation of GC and memory B cells to PCs (Table 1) (26). The ability of IL-4, IL-10, and IL-13 to induce isotype switching reflected their abilities to upregulate expression of activation induced cytidine deaminase (AICDA), an enzyme critical for class switch recombination, while IL-10 mediated PC generation by inducing BLIMP-1 (7, 8, 60). The effects of IL-10, however, appear to be species specific because serum Ig levels were unaffected in mice that were either deficient for IL-10 or that expressed IL-10 from a transgene (61, 62). Similar to CD40L, the membrane bound form of TNF-α was also found to be transiently expressed on human activated CD4+ T cells, and could co-stimulate polyclonal Ig secretion induced in human B cells co-cultured with mitogen-stimulated CD4+ T cells, or their membranes, together with IL-4 (27, 63) (Table 1).

Table 1. Contribution of different cytokines to the in vitro behavior of human B cells.
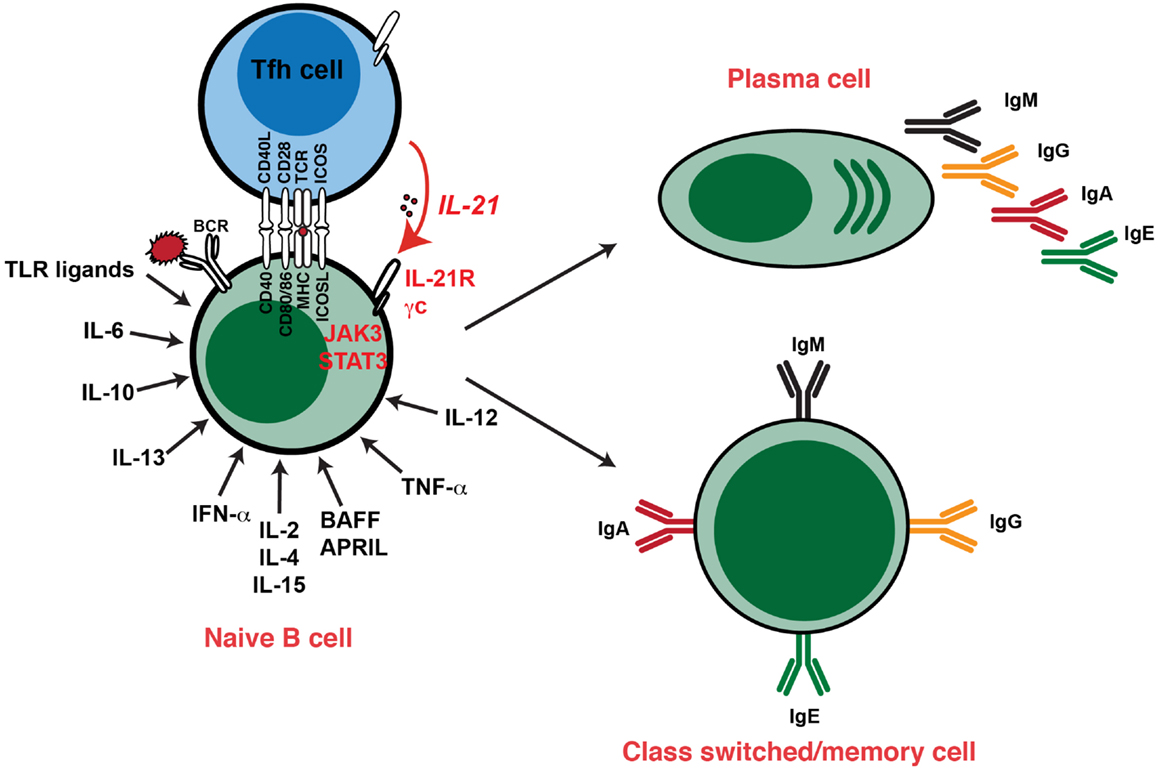
Figure 2. Cytokine-induced differentiation of human B cells in vitro: requirement for IL-21 signaling in vivo. In vitro studies demonstrated that human B cells could undergo events such as Ig class switching and differentiation to become Ig-secreting cells following stimulation with a diverse range of cytokines. However, analysis of individuals with hypomorphic mutations in genes encoding STAT3, γc (IL2RG), JAK3, or IL-21R have revealed that this pathway – activated by IL-21 – is critical for the generation of memory B cells and the establishment of Ag-specific Abs in vivo. Thus, although cytokines such as IL-4, IL-13, IL-10, and BAFF/APRIL are strong B-cell growth and differentiation factors, their function is insufficient to compensate for impaired IL-21/IL-21R signaling in vivo in the setting of generating robust, long-lived Ag-specific Ab, and memory responses. Consequently, IL-21-mediated B-cell activation is a necessary and sufficient step in the generation of protective long-lived humoral immune responses in humans.
IL-2 has had a long history of being documented of enhancing Ig secretion by activated human B cells (Table 1) (38, 39). Consistent with the structural and functional similarities between IL-2 and IL-15, it was not surprising that IL-15 could also stimulate proliferation and induce secretion of IgM, IgG1, and IgA, but not IgG4 or IgE, by CD40L-primed B cells. This activity of IL-15 was comparable to that of IL-2 (24).
More recently, the TNF-related molecule BAFF, and its homolog APRIL, has emerged as a global regulator of B-cell development and function (64–66). While a primary role for BAFF lies in the ability to promote the survival of B cells at the transitional stage of development (65), both BAFF and APRIL can also induce the molecular events associated with isotype switching to IgG and IgA, and to IgE in the presence of IL-4. Furthermore, the secretion of these Ig isotypes occurred when the B cells also received signals through the BCR (64, 66). BAFF and APRIL can also sustain the survival of PCs in vivo and in vitro (66, 67). BAFF functions by binding to the surface receptors BAFF-R, TACI, or BCMA; APRIL can also activate B cells by binding to TACI and BCMA (65, 66) (Table 1). Interestingly, these effects of BAFF and APRIL appear to be mediated through different receptors. Thus, the pro-survival effects of BAFF on transitional and naïve B cells are delivered through BAFF-R, while this effect on PCs occurs predominantly through BCMA. On the other hand, BAFF-R and TACI mediates isotype switching to IgG, IgA, and IgE induced by BAFF and APRIL, respectively (64–66). Lastly, heparan sulfate proteoglycans can also act as a receptor for APRIL, and this appears to be important for mediating the pro-survival effects of APRIL on BM PCs (67–69).
Collectively, it is clear that myriad cytokines and combinations thereof, are capable of eliciting activation and terminal differentiation of human B cells to differing extents. However, with the discover of IL-21 in 2001, and the subsequent characterization of its function on human and murine B cells during the following decade, the physiological significance of many of these factors in initiating humoral immune responses needs to be re-addressed as IL-21 has emerged as the most potent inducer of B cell differentiation.
Pleiotropic Effects of IL-21 on Human and Murine B-Cell Differentiation
IL-21 belongs to the type I family of cytokines that also includes IL-2, IL-4, IL-7, IL-9, and IL-15, all of which bind to and form a complex with the common γ-chain (γc) and their private receptors (70–73). The IL-21 receptor (IL-21R) is expressed by fibroblasts, keratinocytes, and intestinal epithelial cells, but more importantly is also expressed on lymphocytes (T, B, NK cells), macrophages, and dendritic cells, and the levels of expression can be increased following cellular activation (70, 71, 74–77). IL-21 is predominantly produced by activated CD4+ T cells and NKT cells (78–80), with the greatest production being by Tfh and GC Tfh cells (4–6). Akin to most cytokines, IL-21 exerts its effect by activating Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathways, specifically Jak1 and Jak3, and STAT1, STAT3, and to a lesser extent STAT5 (72, 81–84). The initial description of IL-21 hinted at its B-cell tropism, inasmuch that Parrish-Novak et al. showed that IL-21 significantly co-stimulated proliferation of human blood B cells induced by anti-CD40 mAbs (71). Since then, several studies have confirmed that IL-21 is an important regulator of B-cell activation, proliferation, PC differentiation, and Ab-secretion in both mice and humans.
The Role of IL-21 in Murine Activated B-Cell Proliferation, Apoptosis, PC Differentiation, Ab-Secretion, and Memory B-Cell Formation
In a seminal study, Ozaki et al. demonstrated that the IL-21 signaling pathway is involved in regulating Ab production and isotype switching (85). They showed that IL-21R−/− mice, despite having normal lymphoid development, have significantly diminished total serum and Ag-specific IgG1 titers but elevated IgE levels in response to TD Ag immunization compared to wild-type animals. Ag-specific IgG2b and IgG3 serum levels were also decreased whereas IgG2a and IgM titers were largely unaffected in the absence of the IL-21R. The decreased IgG1 response appeared to result from a reduction in the generation of Ag-specific IgG1 producing PCs. These in vivo data established that IL-21 has a critical role in inducing IgG1 production, while concomitantly suppressing IgE responses. Strikingly, IL-4−/−IL-21R−/− double-knockout mice displayed a more severe phenotype, characterized by a more dramatically reduced IgG response. Furthermore, the strong up-regulation of IgE secretion in IL-21R−/− mice was abrogated in IL-4−/−IL-21R−/− mice indicating that the “hyper-IgE” phenotype of IL-21R−/− mice was dependent on IL-4 (85). Importantly, these in vivo findings were complemented by in vitro investigation of the effects of IL-21 on murine B cells. Thus, IL-21 enhanced proliferation of anti-IgM and/or anti-CD40 mAb-stimulated murine B cells and initiated PC differentiation and class switching, as revealed by increased expression of Syndecan-1 (CD138) and surface IgG1 on these cells (86).
These findings provided strong evidence that IL-21 is likely to achieve its potent effect on humoral immune responses in vivo by acting directly on B cells. Indeed, this has been verified in a series of studies where IL-21R-sufficient or deficient B cells were adoptively transferred into recipient mice, and the B-cell response to TD Ags or pathogens then tracked. It was generally found that when B cells were unable to respond to IL-21, humoral immunity was compromised with impaired formation of GC, with respect to magnitude and/or kinetics, and of long-lived Ag-specific PC. The mechanism underlying aberrant GC formation was suboptimal induction of Bcl-6 expression in GC B cells, which attenuated affinity maturation and selection of high-affinity variants. Although memory cells were generated in normal numbers from IL-21R-deficient B cells, the IL-21R-deficient memory cells were unable to respond to secondary challenge with specific Ag, resulting in ineffective recall responses. In contrast to the GC response, the generation of extrafollicular plasmablasts in response to pathogens was unaffected by B-cell specific IL-21R-deficiency (87– 92). IL-21 can activate STAT3 (72, 81– 84). Intriguingly, analysis of STAT3flox/flox CD19cre mice showed some similarities to mice whose B cells lacked IL-21R. Specifically, STAT3flox/flox CD19cre mice have normal levels of serum IgM, IgA, and IgG, but a large reduction in Ag-specific serum IgG1 levels and splenic PCs following immunization with TD Ags (93). This established that expression of STAT3 in B cells is important for TD differentiation of B cells into IgG1-secreting PC (93), with subsequent studies implicating IL-21 as being the key STAT3-activating cytokine potentially involved in this process (87–92). Thus, IL-21/IL-21R signaling, possibly via STAT3, in B cells appears to be required for the generation and maintenance of long-lived PC and humoral memory to TD Ags, but is dispensable for GC-independent Ab responses.
Given the importance of IL-21R expression for normal Ig production in vivo (85, 86), a surprising finding was that murine IL-21 could inhibit B-cell proliferation induced by either anti-IgM and IL-4, or TLR ligands such as LPS or CpG (94, 95). Furthermore, although IL-21 impressively promoted proliferation of CD40-activated B cells, the proportion of B cells that was apoptotic in the presence of IL-21 exceeded that observed in its absence (95). Induction of apoptosis by IL-21 in both resting and activated murine B cells correlated with reduced expression of Bcl-xL and Bcl-2 and elevated expression of Bim (94, 95). Consistent with this, IL-21-induced apoptosis could be prevented by restoring expression of Bcl-xL or Bcl-2 either by overexpressing these proteins or inducing their expression by activation prior to exposure to IL-21 (94). Increased B-cell apoptosis was also observed in vivo in mice either transgenic for IL-21 or that received IL-21 administered via hydrodynamic-based delivery of plasmid DNA. Thus, it appears that IL-21 can differential influence B-cell fate depending on the signaling context (86).
Together, these data show that IL-21 is an important factor for the activation, proliferation, differentiation, Ag production, or death of murine B cells, with the outcome being dependent on the context of co-stimulation. The defect in GC-dependent Ab production in IL-21/IL-21R deficient mice after immunization indicates that differentiation into PCs may be a non-redundant activity of IL-21.
IL-21 and Human B Cells
Initial studies into the stimulatory effect of IL-21 revealed that IL-21 potently enhanced the proliferation of CD40-stimulated human B cells, with memory B cells undergoing a much stronger proliferative response than naïve B cells (Figure 3; Table 1) (96). Despite memory B cells proliferating more than naïve B cells in response to IL-21, the overall effect of IL-21 appeared to be greater on naïve than on memory cells. Thus, naïve B cells stimulated with CD40L/IL-21 exhibited a greater enhancement in their response, as well as a greater reduction in their time to enter cell division, over that induced by CD40L alone than did memory B cells (76). This is probably due to the basal expression of IL-21R on naïve B cells, whereas it is absent from memory cells (Figure 1). Although expression of IL-21R increases following activation on naïve and memory B cells, it remained higher on the naive subset (76).
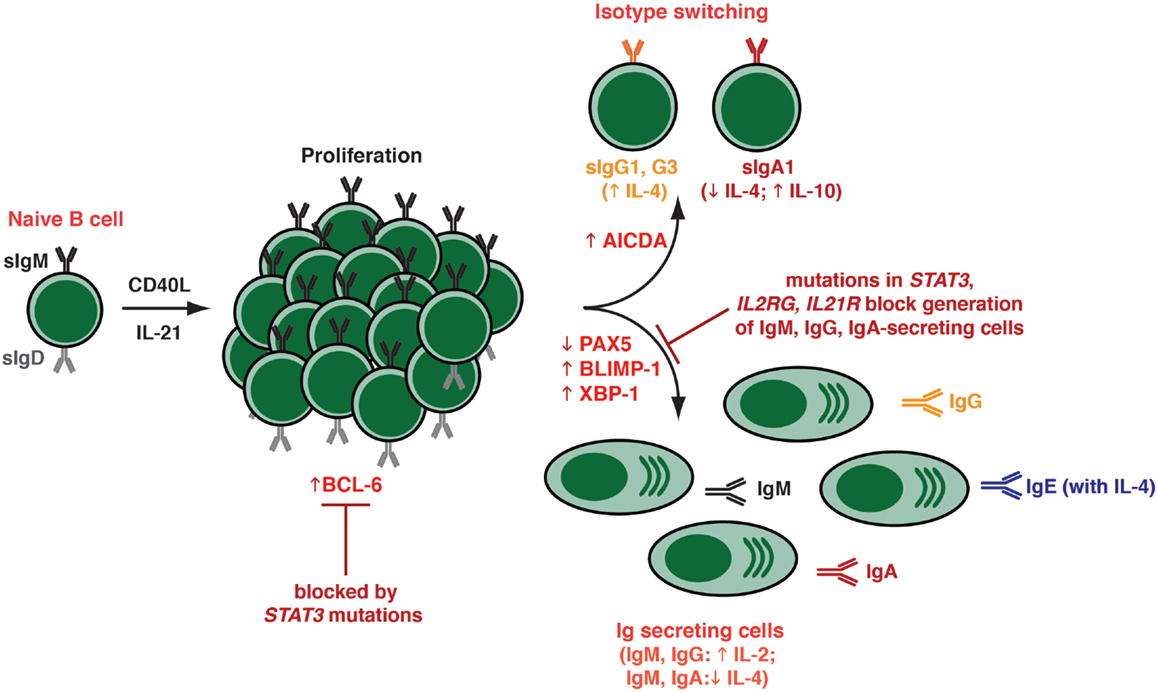
Figure 3. Effects of IL-21 on human naïve B cells in vitro. When human naïve B cells are stimulated with CD40L together with IL-21, they were found to undergo intense proliferation. This was followed by induction of Ig class switching – predominantly to IgG3 and IgG1; a lesser extent to IgA1 – as determined by acquisition of expression of switched isotypes or differentiation to plasma-like cells capable of secreting all major Ig isotypes. The B cells that had undergone switching to become IgG+ or IgA+ were distinct from those that committed to a plasma cell fate – thus, the secreted Ig detected in these cultures was derived from the in vitro-derived plasma cells rather than the surface IgG+/IgA+ class switched cells. Both class switching and plasma cell formation induced by IL-21 could be modulated by additional cytokines, such as IL-4 (promoted switching to IgG; inhibited switching to IgA; increased secretion of IgG and IgE; suppressed secretion of IgM, IgA), IL-10 (increased IgA secretion), and IL-2 (increased secretion of IgM, IgG). These differentiation events coincided with the induction in expression of BCL-6, AICDA (required for class switching), and BLIMP-1/XBP-1 (required for plasma cell formation). The ability of IL-21 to induce naïve B cells to differentiate into plasmablasts/plasma cells in vitro was abolished by hypomorphic mutations in STAT3, as well as null mutations in IL21R or IL2RG.
In terms of differentiation, when total CD19+ splenic B cells were stimulated in vitro with anti-CD40 mAb in the presence of IL-21, they were induced to secrete IgM and IgG in an IL-21 dose-dependent manner (96). Pene et al. also made the important observation that IL-21 specifically induced production of IgG1 and IgG3 by human naïve B cells, demonstrating IL-21 to be a switch factor for these IgG subclasses (96). The findings from this elegant study were confirmed by several groups who also found that IL-21 induced proliferation as well as expression and secretion of IgM, IgG (predominantly IgG3) as well as IgA (mostly IgA1), and IgE by CD40L-stimulated naïve B cells that had been isolated from distinct anatomical sites, including umbilical cord blood, spleen, tonsils, and adult peripheral blood (Figure 3) (60, 76, 97–100) (Table 1). IL-21 also strongly induced Ig secretion from memory and GC B cells isolated from these sites (60, 97, 98, 100). The ability of IL-21 to induce such impressive Ig secretion correlated with the appearance of a substantial proportion of PCs – phenotypically identified as CD19loIgD−CD38hi or CD20loCD38hiCD27hi cells – in cultures of IL-21-stimulated B cells (Figure 3) (60, 98). Interestingly, a recent report also found IL-21 could support the survival of and Ig secretion by PCs in secondary lymphoid organs, but not those in the bone marrow (101). This is consistent with the differential expression of IL-21R on PCs from these diverse sites (76, 98, 102) (Figure 1), and suggests that IL-21 contributes to humoral immunity not only by inducing PC from naïve, memory, and GC B cells, but also promoting the survival and function of these cells in lymphoid tissues before they alter their requirements for survival within niches in bone marrow (67). The ability of IL-21 to sustain survival of normal PCs is reminiscent of the finding that IL-21 can promote growth and survival of malignant PC in multiple myeloma (103).
When compared to other cytokines that have been characterized as B-cell growth and differentiation factors, the effect of IL-21 was found to exceed that of IL-2, IL-4, IL-13, and IL-10 by up to 100-fold (60, 76, 97, 98). However, the actions of IL-21 could be complemented by these cytokines (Table 1; Figure 3). For example, IL-4 increased the frequency of IgG+ cells generated from, and the amount of IgG secreted by, naïve B-cell precursors that had been stimulated with IL-21 (Figure 3) (96, 97). Interestingly, while IL-21 favored the induction of IgG3+ B cells, the combination of IL-4 and IL-21 resulted in the preferential generation of IgG1+ switched B cells, which mirrored the effect of IL-4 alone but the magnitude of the response was greater. IL-4 and IL-21 were also capable of acting synergistically to induce 10- to 100-fold higher levels of IgE by CD40L-stimulated naïve B cells over that observed with either cytokine alone (Figure 3) (96, 99). In contrast, IL-4 abolished not only IL-21-induced IgM secretion but also switching to and secretion of IgA (97, 98); on the other hand, IgA secretion induced by IL-21 was augmented by IL-10 (97). Lastly, IL-2 could enhance PC differentiation induced by IL-21 (Figure 3) (98, 104). This was achieved by IL-21 inducing expression of CD25 – a component of the IL-2R – on activated B cells (104).
The physiological significance of these effects of IL-21 on human B cells has been born from experiments that assessed the relative contribution(s) of CD4+ T cell-derived cytokines to TD B-cell differentiation in vitro. Using an in vitro system whereby human activated CD4+ T cells can induce Ig production by co-cultured B cells (105), several groups have established that neutralization of IL-21 significantly inhibited T cell-induced B-cell activation, proliferation, differentiation, Ig secretion, and PC survival (60, 101, 106). Delayed blockade of IL-21 also inhibited PC differentiation after initial B-cell expansion, indicating that IL-21 is required for B-cell proliferation and PC differentiation (106). The findings that IL-21 is highly expressed by Tfh cells (78), and the IL-21R is upregulated on GC B cells (76) is consistent with a model of Tfh cells interacting with GC B cells to induce their differentiation to memory cells and PC predominantly via the production and delivery of IL-21 (4–6).
Mechanism of Action of IL-21
The ability of IL-21 to guide multiple fates in activated B cells – class switching to express downstream Ig isotypes, commitment to the PC lineage, as well as formation of GCs and memory B cells – reflects the ability of IL-21 to induce the molecular machinery required for these processes. Thus, IL-21 is capable of inducing expression of AICDA, BLIMP1/PRDM1, and XBP-1, as well as reducing expression of PAX5, in both human and murine B cells (Figure 3) (60, 83, 84, 86, 98). Collectively, these factors regulate class switching and PC formation (8). Interestingly, the ability of IL-4 to suppress the stimulatory effects of IL-21 on naïve B cells correlated with a reduction in BLIMP-1 expression (60). IL-21 could also induce BCL-6 (83, 86, 89, 92, 98), which would contribute to GC formation in vivo (Figure 3) (8). Thus, in the setting of TD B-cell activation, Tfh-derived IL-21 can induce B cells to express all of the machinery required to undergo the major fates of differentiation: GC B cells by induction of Bcl-6; PCs following induction of BLIMP-1, and class switched B cells by inducing AICDA. It is likely that IL-21 induces expression of these opposing transcriptional regulators (i.e., BLIMP-1, Bcl-6) in distinct subsets of B cells that will ultimately develop into either PC or memory B cells. However, these outputs will ultimately reflect the balance of signals received and integrated by the B cells, with the effect of IL-21 being influenced by inputs delivered via receptors including the BCR, other complimentary cytokine, and co-stimulatory receptors.
As IL-21 can activate several STATs (73), the relative contribution of individual STAT molecules has been assessed. Diehl et al. demonstrated that constitutive activation of STAT3 in primary human B cells induced BLIMP-1 expression and initiated B cell differentiation, yielding cells with a phenotype (CD38highCD20−CD19lowHLA-DRlowCD138+) consistent with PC as well as enhanced Ab-secretion (84). Importantly, up-regulation of BLIMP-1 alone was not sufficient for differentiation of primary human B cells into PCs; this event also required concomitant down-regulation of BCL-6 (84). This study was the first to propose that STAT3 was the predominant mediator of the differentiation effects that IL-21 has on human B cells. These were largely confirmed by the demonstration that induction of PRDM1, XBP-1, and BCL-6 by IL-21 were abolished in naïve B cells isolated from individuals with hypomorphic mutations in STAT3, while these responses were unaffected by loss-of-function mutations in STAT1 (83, 107). Intriguingly, IL-21-induced expression of AICDA in naïve B cells, as well as of PRDM1 and XBP1 in memory B cells, still occurred despite the presence of hypomorphic STAT3 mutations, suggesting that class switching in naïve B cells and PC differentiation from memory B cells requires less STAT3 function than does the generation of PC from naïve B cells (83, 107).
Interestingly, high-affinity signaling through the BCR on immortalized B-cell lines can activate STAT3 (108). Similarly, CD40L enhanced the expression of BLIMP-1 induced by IL-21/STAT3 signaling in a GC B cell-like human cell line, thereby maximizing PC differentiation (109). Thus, it is possible that signals integrated in B cells through receptors such as CD40 and the BCR can amplify the effects of IL-21 by modulating activating or function of STAT3. It is also worth noting that STAT3 activation is important for the survival of multiple myeloma cells (110). As IL-21 is also anti-apoptotic for myeloma cells, it is tempting to speculate that IL-21 could contribute to STAT3 activation in vivo in the setting of this malignancy. Collectively, these studies have illuminated the pivotal role of IL-21-mediated STAT3 signaling in guiding key events of human B-cell differentiation.
Lessons from Primary Immunodeficiencies
Primary immunodeficiencies (PIDs) result from monogenic mutations that compromise the ability of affected individuals to elicit appropriate immune responses. Consequently, these individuals exhibit susceptibility to infectious diseases and are often unable to respond to vaccination. As the genetic lesion is known in many PIDs, these conditions can reveal the unique functions of specific genes and related signaling pathways in immune cells and the importance of these pathways in productive and protective immune responses. Thus, analysis of PIDs can shed new light on the requirements for lymphocyte development and function. Indeed, several PIDs have confirmed the critical role played by IL-21 in humoral immunity in humans.
Heterozygous mutations in STAT3 are the major cause of autosomal dominant hyper-IgE syndrome (AD-HIES) (111, 112), a multisystem disease affecting the immune and musculoskeletal systems (113, 114). Immunological defects include skin lesions, recurrent mucocutaneous invasive infections with S. aureus and Candida. These patients have normal serum levels of IgM, IgG, and IgA but increased levels of IgE (113, 114). Although the frequencies of total peripheral blood B cells are not significantly different between AD-HIES patients and control individuals, STAT3 deficiency impaired the in vivo generation of human memory B cells as well as the generation of Ag-specific Ab-secreting B cells and high-affinity serum Abs (Figure 2) (83, 107). This reduced number of memory B cells is in line with previously reported defective functional Ab responses in AD-HIES patients (115–118). Cytokines known to be involved in human B cell differentiation are IL-6, IL-10, and IL-21. Consistent with reduced memory B cells and poor induction of Ag-specific Ab responses in AD-HIES, naïve B cells from these patients were unable to respond to the stimulatory effects of IL-10 or IL-21 with respect to differentiation into PC in vitro (Figure 3). STAT3 mutations also compromised the ability of IL-21 to prime B cells to the stimulatory effects of IL-2, inasmuch that induction of CD25 – and subsequent responsiveness to IL-2 – was attenuated on IL-21-stimulated STAT3-deficient human naïve B cells (104). These findings revealed that STAT3 plays a non-redundant role in generating Ag-specific memory B cells and Ab-secreting cells in vivo. However, it remained to be determined which STAT3-activating cytokine was requisite for these effects. This became clearer by examining patients with mutations in IL2RG, encoding γc, or JAK3, which associates with γc and delivers signals downstream of γc-containing cytokine receptors (73), that cause X-linked severe combined immunodeficiency (X-SCID) or one type of autosomal recessive (AR) SCID, respectively (73, 119). These PIDs are fatal unless treated by hematopoietic stem cell transplant (HSCT) (119).
X-linked severe combined immunodeficiency and JAK3 deficiency are characterized by a lack of T and NK cells but normal or increased numbers of B cells. However, due to the lack of CD4+ T cell help, B cell responses are impaired (119). While HSCT corrects the humoral defect in ~50% of patients, the remainder still requires ongoing Ig replacement therapy (120). One of the explanations for this is split chimerism, where donor-derived T cells successfully engraft in the recipient, but autologous host-derived B cells persist (120). Thus, despite the presence of functional CD4+ T cells, the IL2G/JAK3 mutant B cells remain unable to respond to T-cell-derived helper signals, rendering the patient immunodeficient with respect to humoral immune responses (119, 120). We took advantage of this chimeric state to examine the B-cell compartment of X-SCID and JAK3 deficient patients who had undergone HSCT (121). Although IL-2RG/JAK3 mutant naïve B cells responded normally to co-stimulatory signals delivered through the BCR, TLRs, and receptors for IL-10, IL-13, and even IL-4 [which can also signal through the IL13R; (73)], these B cells were completely unresponsive to IL-21. Naïve B cells from these individuals also failed to differentiate into memory cells in vivo (Figures 2 and 3). Thus, despite intact responsiveness to a suite of well-characterized B-cell growth and differentiation factors, the ability to receive signals through a γc-binding/JAK3-activating cytokine is a critical and rate-limiting step for the establishment of humoral immunity in humans (Figure 2) (121). Given the potency that IL-21 exerts on human B-cell differentiation, it was highly likely that this was the key γc-binding/JAK3-activating cytokine involved in human B-cell responses in vivo.
This was confirmed by the recent identification of individuals with homozygous loss-of-function mutations in IL21R that causes a novel PID, features of which include occasionally reduced serum IgG levels, poor Ab responses following vaccination with TD Ags (122), and a paucity of circulating memory B cells, including those expressing class switched Ig isotypes (107, 122). Not surprisingly, IL-21R-deficient naïve B cells exhibited impaired IL-21-induced proliferation, Ig class switching, and PC differentiation in vitro. This is consistent with a failure of IL-21 to mediate the acquisition of expression of AICDA, PRDM1, and XBP1 in these cells, and mirrors the humoral immune defects observed in these patients. The cellular and molecular characterization of these patients has definitively established the criticality of IL-21 in establishing long-lived humoral immune responses. Furthermore, the finding that B cells with mutations in IL2RG, JAK3, or STAT3 phenocopy IL-21R-deficient B cells, with respect to memory cell formation and responsiveness to IL-21, demonstrates that signaling downstream of the IL-21R/γc complex via JAK3 and STAT3 is essential for the effector function of IL-21 on B-cell differentiation in terms of generating efficient Ag-specific humoral immune responses (Figures 2 and 3). However, since serum levels of total IgM, IgG, and IgA are largely normal in most patients with mutations in either STAT3 or IL-21R, it is clear that the production of basal Ig is not dependent on IL-21R/STAT3 signaling. Indeed, as we have previously proposed (83), this is likely achieved by the interplay between ligands that do not signal via STAT3 – these could include many of the cytokines and factors detailed in this review (see Table 1), such as IL-4, IL-13, BAFF/APRIL as well as TLR ligands. Despite the availability of these ligands in STAT3- and IL-21R-deficient patients, and their ability to signal normally in IL-21R/STAT3-deficient B cells, these factors are collectively unable to compensate for impaired IL-21R signaling in order to generate a robust, long lasting Ag-specific Ab response.
Intriguingly, IL-21R-deficient individuals also have elevated levels of serum IgE (122), which is obviously also a feature of AD-HIES (113, 114, 116). Thus, it is likely that IL-21 also plays an important role in regulating IgE production by human B cells. However, whether this is due to a direct effect of IL-21 on B cells, or operates through an intermediate cell type [e.g., by inducing production of IFNγ by T cells and NK cells; (40, 100)] remains to be determined.
Lastly, it is worth commenting that prior to the discovery and subsequent characterization of IL-21, IL-10 was considered to be the most efficient cytokine capable of activating human B cells (31, 42, 48, 59). As IL-10 can also activate STAT3 (73), and STAT3-deficient human naïve B cells are unable to respond to the PC-inducing effects of IL-10 (83), it is possible that the humoral defects in AD-HIES patients reflects an inability to respond to not only IL-21 but also IL-10. However, since individuals with mutations in IL-10 or IL-10R have intact specific Ab responses to vaccines (123), it is possible the IL-10 plays only a minor role in regulating human B-cell function in vivo. There are caveats to this conclusion, however, as most patients examined were young (<10 years old), and they also suffered from early onset inflammatory bowel disease (123). Thus, it remains plausible that IL-10 does contribute to B-cell function in healthy adults.
IL-21/IL-21R and Systemic Autoimmune Diseases
Just as impaired signaling via IL-21 manifests as humoral immunodeficiency, aberrant or excessive IL-21-induced B-cell activation has been associated with the development of Ab-mediated autoimmune states in both murine models and human.
The first indication of a potential involvement of IL-21 in autoimmunity was the finding that IL-21 was overexpressed in several strains of mice (e.g., BXSB-Yaa, B6.Sle1-Yaa, Sanroque, MRL/MpJ-FASlpr/lpr/J) that develop lupus-like disease (86, 124, 125). Furthermore, in vivo blockade of IL-21 ameliorated disease progression and severity in some of these settings (126–128), as well as in animal models of rheumatoid arthritis (129) and Sjogren’s syndrome (130). This was followed by the demonstration of elevated expression and/or production of IL-21 in human autoimmune conditions including SLE (131–134), rheumatoid arthritis (135), and Sjogren’s syndrome (136). Consistent with these findings, as well as with the recognition that IL-21 is predominantly produced by Tfh cells, it was perhaps not surprising that circulating Tfh-like cells have been detected in a broad array of autoimmune conditions including not only SLE, rheumatoid arthritis, and Sjogren’s syndrome, but also multiple sclerosis, autoimmune thyroid disease, myasthenia gravis, and juvenile dermatomyositis [reviewed in Ref. (6, 137)]. Importantly, the increases in Tfh cells generally correlated with numerous indices of disease severity, such as titers of autoAb, numbers of Ab-secreting plasmablasts, clinical scores, and even levels of serum IL-21. Furthermore, the expanded population of Tfh cells, as well as clinical features of each of these diseases, could be reduced following initiation and continuation of immunosuppressive treatments [reviewed in Ref. (6, 137)]. Independent confirmation that IL-21/IL-21R may be involved in the development of autoimmune diseases came from genome-wide association studies. Specifically, polymorphisms in either IL-21 or IL-21R genes have been identified that associated with SLE, RA, and primary Sjogren’s syndrome (138–141). Collectively, there is convincing evidence that IL-21 – most likely produced by Tfh cells – plays a pathological role in the initiation, development, and/or progression of several human autoimmune diseases caused by the production of autoantibodies.
Concluding Comments and Future Perspectives
B cells play myriad fundamental roles in providing protective immunity against infection. However, the most prominent of these is the production of Ag-specific Ab following the terminal differentiation of B cells into long-lived PCs. This event is key to the establishment of long-term humoral immunity and memory, and underlies the success of most currently available vaccines. The criticality of Ab production by B cells to human health is evidenced by the pathological consequences of hypogammaglobulinemia, resulting in immunodeficiency. Conversely, the dysregulated production of excessive quantities of self-reactive Abs can be deleterious in the setting of autoimmunity. The detailed characterization of the effects of cytokines on B cells – from studies in genetically manipulated mice, in vitro cultures of human and murine B cells, and analysis of humans with specific PIDs – have revealed the central role that IL-21 has in generating memory B cells and specific Abs following exposure to TD Ags. Remarkably, alternative signals that could be integrated in B cells through other cytokine or co-stimulatory receptors are insufficient to initiate such B-cell responses when the IL-21/IL-21R signaling pathway is compromised. This paves the way for developing directed therapies to improve immune responses to vaccines or in immunocompromised individuals. Supporting this concept is the finding that administration of IL-21 to macaques increased frequencies of memory B cells as well as titers of virus-specific IgG (142). Conversely, therapies aimed at blocking the action of IL-21, either by directly targeting IL-21 itself or indirectly targeting Tfh cells or appropriate signaling molecules downstream of the IL-21R, so as to restrain the differentiation of rogue, autoreactive B cells into PCs, represents a feasible strategy for the treatment of various autoimmune diseases, as evidenced from numerous murine models (126–129, 143). Hopefully these findings will see successful translation to the clinic, thereby offering new hope for the treatment of these immune dyscrasias.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Tangye lab is supported by research grants and fellowships awarded by the National Health and Medical Research Council of Australia.
References
1. Fagraeus A. The plasma cellular reaction and its relation to the formation of antibodies in vitro. J Immunol (1948) 58:1–13.
2. Tangye SG, Tarlinton DM. Memory B cells: effectors of long-lived immune responses. Eur J Immunol (2009) 39:2065–75. doi:10.1002/eji.200939531
3. Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat Immunol (2010) 11:681–8. doi:10.1038/ni.1900
4. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
5. Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med (2012) 209:1241–53. doi:10.1084/jem.20120994
6. Tangye SG, Ma CS, Brink R, Deenick EK. The good, the bad and the ugly – TFH cells in human health and disease. Nat Rev Immunol (2013) 13:412–26. doi:10.1038/nri3447
7. Oracki SA, Walker JA, Hibbs ML, Corcoran LM, Tarlinton DM. Plasma cell development and survival. Immunol Rev (2010) 237:140–59. doi:10.1111/j.1600-065X.2010.00940.x
8. Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. Semin Immunol (2011) 23:341–9. doi:10.1016/j.smim.2011.08.010
9. Cattoretti G, Chang CC, Cechova K, Zhang J, Ye BH, Falini B, et al. BCL-6 protein is expressed in germinal-center B cells. Blood (1995) 86:45–53.
10. Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science (1997) 276:589–92. doi:10.1126/science.276.5312.589
11. Fukuda T, Yoshida T, Okada S, Hatano M, Miki T, Ishibashi K, et al. Disruption of the Bcl6 gene results in an impaired germinal center formation. J Exp Med (1997) 186:439–48. doi:10.1084/jem.186.3.439
12. Angelin-Duclos C, Cattoretti G, Lin KI, Calame K. Commitment of B lymphocytes to a plasma cell fate is associated with Blimp-1 expression in vivo. J Immunol (2000) 165:5462–71.
13. Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, et al. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J Exp Med (2004) 200:967–77. doi:10.1084/jem.20040973
14. Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity (2002) 17:51–62. doi:10.1016/S1074-7613(02)00335-7
15. Turner CA Jr, Mack DH, Davis MM. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell (1994) 77:297–306. doi:10.1016/0092-8674(94)90321-2
16. Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity (2004) 21:81–93. doi:10.1016/j.immuni.2004.06.010
17. Feldmann M, Basten A. Cell interactions in the immune response in vitro. 3. Specific collaboration across a cell impermeable membrane. J Exp Med (1972) 136:49–67. doi:10.1084/jem.136.4.737
18. Feldmann M, Basten A. Specific collaboration between T and B lymphocytes across a cell impermeable membrane in vitro. Nat New Biol (1972) 237:13–5. doi:10.1038/newbio237013a0
19. Gorczynski RM, Miller RG, Phillips RA. Initiation of antibody production to sheep erythrocytes in vitro: replacement of the requirement for T-cells with a cell-free factor isolated from cultures of lymphoid cells. J Immunol (1972) 108:547–51.
20. Taussig J, Mozes E, Isac R. Antigen-specific thymus cell factors in the genetic control of the immune response to poly-(tyrosyl, glutamyl)-poly-D, L-alanyl – poly-lysyl. J Exp Med (1974) 140:301–12.
21. Kuhara T, Haughton G, Corley RB. Antigen-nonspecific T cell-derived factors in B cell activation: differences in the requirements for interleukin 2 in responses of unprimed and primed B cells. Eur J Immunol (1985) 15:787–93. doi:10.1002/eji.1830150809
22. Jelinek DF, Lipsky PE. The roles of T cell factors in activation, cell cycle progression, and differentiation of human B cells. J Immunol (1985) 134:1690–701.
23. Kishimoto T. Factors affecting B-cell growth and differentiation. Annu Rev Immunol (1985) 3:133–57. doi:10.1146/annurev.iy.03.040185.001025
24. Armitage RJ, Macduff BM, Eisenman J, Paxton R, Grabstein KH. IL-15 has stimulatory activity for the induction of B cell proliferation and differentiation. J Immunol (1995) 154:483–90.
25. Armitage RJ, Macduff BM, Spriggs MK, Fanslow WC. Human B cell proliferation and Ig secretion induced by recombinant CD40 ligand are modulated by soluble cytokines. J Immunol (1993) 150:3671–80.
26. Arpin C, Dechanet J, Van Kooten C, Merville P, Grouard G, Briere F, et al. Generation of memory B cells and plasma cells in vitro. Science (1995) 268:720–2. doi:10.1126/science.7537388
27. Aversa G, Punnonen J, de Vries JE. The 26-kD transmembrane form of tumor necrosis factor alpha on activated CD4+ T cell clones provides a costimulatory signal for human B cell activation. J Exp Med (1993) 177:1575–85. doi:10.1084/jem.177.6.1575
28. Cocks BG, de Waal Malefyt R, Galizzi JP, de Vries JE, Aversa G. IL-13 induces proliferation and differentiation of human B cells activated by the CD40 ligand. Int Immunol (1993) 5:657–63. doi:10.1093/intimm/5.6.657
29. McKenzie AN, Culpepper JA, de Waal Malefyt R, Briere F, Punnonen J, Aversa G, et al. Interleukin 13, a T-cell-derived cytokine that regulates human monocyte and B-cell function. Proc Natl Acad Sci U S A (1993) 90:3735–9. doi:10.1073/pnas.90.8.3735
30. Delfraissy JF, Wallon C, Galanaud P. Interferon-alpha can synergize with interleukin 2 for human in vitro antibody response. Eur J Immunol (1988) 18:1379–84. doi:10.1002/eji.1830180912
31. Defrance T, Vanbervliet B, Briere F, Durand I, Rousset F, Banchereau J. Interleukin 10 and transforming growth factor beta cooperate to induce anti-CD40-activated naive human B cells to secrete immunoglobulin A. J Exp Med (1992) 175:671–82. doi:10.1084/jem.175.3.671
32. Dubois B, Massacrier C, Vanbervliet B, Fayette J, Briere F, Banchereau J, et al. Critical role of IL-12 in dendritic cell-induced differentiation of naive B lymphocytes. J Immunol (1998) 161:2223–31.
33. Gauchat JF, Aversa G, Gascan H, de Vries JE. Modulation of IL-4 induced germline epsilon RNA synthesis in human B cells by tumor necrosis factor-alpha, anti-CD40 monoclonal antibodies or transforming growth factor-beta correlates with levels of IgE production. Int Immunol (1992) 4:397–406.
34. Itoh K, Hirohata S. The role of IL-10 in human B cell activation, proliferation, and differentiation. J Immunol (1995) 154:4341–50.
35. Jelinek DF, Lipsky PE. Inhibitory influence of IL-4 on human B cell responsiveness. J Immunol (1988) 141:164–73.
36. Kehrl JH, Muraguchi A, Fauci AS. The effects of interleukin-1, interleukin-2, alpha-interferon, and gamma-interferon on human B lymphocytes. Trans Assoc Am Physicians (1984) 97:182–9.
37. Lundgren M, Persson U, Larsson P, Magnusson C, Smith CI, Hammarstrom L, et al. Interleukin 4 induces synthesis of IgE and IgG4 in human B cells. Eur J Immunol (1989) 19:1311–5. doi:10.1002/eji.1830190724
38. Mingari MC, Gerosa F, Carra G, Accolla RS, Moretta A, Zubler RH, et al. Human interleukin-2 promotes proliferation of activated B cells via surface receptors similar to those of activated T cells. Nature (1984) 312:641–3. doi:10.1038/312641a0
39. Nakagawa T, Hirano T, Nakagawa N, Yoshizaki K, Kishimoto T. Effect of recombinant IL 2 and gamma-IFN on proliferation and differentiation of human B cells. J Immunol (1985) 134:959–66.
40. Pene J, Rousset F, Briere F, Chretien I, Bonnefoy JY, Spits H, et al. IgE production by normal human lymphocytes is induced by interleukin 4 and suppressed by interferons gamma and alpha and prostaglandin E2. Proc Natl Acad Sci U S A (1988) 85:6880–4. doi:10.1073/pnas.85.18.6880
41. Punnonen J, Aversa G, Cocks BG, McKenzie AN, Menon S, Zurawski G, et al. Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci U S A (1993) 90:3730–4. doi:10.1073/pnas.90.8.3730
42. Rousset F, Garcia E, Defrance T, Peronne C, Vezzio N, Hsu DH, et al. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc Natl Acad Sci U S A (1992) 89:1890–3. doi:10.1073/pnas.89.5.1890
43. Splawski JB, Jelinek DF, Lipsky PE. Immunomodulatory role of IL-4 on the secretion of Ig by human B cells. J Immunol (1989) 142:1569–75.
44. Splawski JB, McAnally LM, Lipsky PE. IL-2 dependence of the promotion of human B cell differentiation by IL-6 (BSF-2). J Immunol (1990) 144: 562–9.
45. Vercelli D, Jabara HH, Arai K, Yokota T, Geha RS. Endogenous interleukin 6 plays an obligatory role in interleukin 4-dependent human IgE synthesis. Eur J Immunol (1989) 19:1419–24. doi:10.1002/eji.1830190811
46. Vernino L, McAnally LM, Ramberg J, Lipsky PE. Generation of nondividing high rate Ig-secreting plasma cells in cultures of human B cells stimulated with anti-CD3-activated T cells. J Immunol (1992) 148:404–10.
47. Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity (2003) 19:225–34. doi:10.1016/S1074-7613(03)00208-5
48. Banchereau J, Bazan F, Blanchard D, Briere F, Galizzi JP, van Kooten C, et al. The CD40 antigen and its ligand. Annu Rev Immunol (1994) 12: 881–922.
49. Aversa G, Punnonen J, Cocks BG, de Waal Malefyt R, Vega F Jr, Zurawski SM, et al. An interleukin 4 (IL-4) mutant protein inhibits both IL-4 or IL-13-induced human immunoglobulin G4 (IgG4) and IgE synthesis and B cell proliferation: support for a common component shared by IL-4 and IL-13 receptors. J Exp Med (1993) 178:2213–8. doi:10.1084/jem.178.6.2213
50. Defrance T, Carayon P, Billian G, Guillemot JC, Minty A, Caput D, et al. Interleukin 13 is a B cell stimulating factor. J Exp Med (1994) 179:135–43. doi:10.1084/jem.179.1.135
51. Maliszewski CR, Grabstein K, Fanslow WC, Armitage R, Spriggs MK, Sato TA. Recombinant CD40 ligand stimulation of murine B cell growth and differentiation: cooperative effects of cytokines. Eur J Immunol (1993) 23:1044–9. doi:10.1002/eji.1830230510
52. Kuhn R, Rajewsky K, Muller W. Generation and analysis of interleukin-4 deficient mice. Science (1991) 254:707–10.
53. Kimata H, Yoshida A, Ishioka C, Lindley I, Mikawa H. Interleukin 8 (IL-8) selectively inhibits immunoglobulin E production induced by IL-4 in human B cells. J Exp Med (1992) 176:1227–31. doi:10.1084/jem.176.4.1227
54. Kiniwa M, Gately M, Gubler U, Chizzonite R, Fargeas C, Delespesse G. Recombinant interleukin-12 suppresses the synthesis of immunoglobulin E by interleukin-4 stimulated human lymphocytes. J Clin Invest (1992) 90:262–6. doi:10.1172/JCI115846
55. Zurawski G, de Vries JE. Interleukin 13, an interleukin 4-like cytokine that acts on monocytes and B cells, but not on T cells. Immunol Today (1994) 15:19–26.
56. Lai YH, Mosmann TR. Mouse IL-13 enhances antibody production in vivo and acts directly on B cells in vitro to increase survival and hence antibody production. J Immunol (1999) 162:78–87.
57. Emson CL, Bell SE, Jones A, Wisden W, McKenzie AN. Interleukin (IL)-4-independent induction of immunoglobulin (Ig)E, and perturbation of T cell development in transgenic mice expressing IL-13. J Exp Med (1998) 188:399–404. doi:10.1084/jem.188.2.399
58. McKenzie GJ, Fallon PG, Emson CL, Grencis RK, McKenzie AN. Simultaneous disruption of interleukin (IL)-4 and IL-13 defines individual roles in T helper cell type 2-mediated responses. J Exp Med (1999) 189:1565–72. doi:10.1084/jem.189.10.1565
59. Briere F, Servet-Delprat C, Bridon JM, Saint-Remy JM, Banchereau J. Human interleukin 10 induces naive surface immunoglobulin D+ (sIgD+) B cells to secrete IgG1 and IgG3. J Exp Med (1994) 179:757–62. doi:10.1084/jem.179.2.757
60. Bryant VL, Ma CS, Avery DT, Li Y, Good KL, Corcoran LM, et al. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J Immunol (2007) 179:8180–90.
61. Groux H, Cottrez F, Rouleau M, Mauze S, Antonenko S, Hurst S, et al. A transgenic model to analyze the immunoregulatory role of IL-10 secreted by antigen-presenting cells. J Immunol (1999) 162:1723–9.
62. Hagenbaugh A, Sharma S, Dubinett SM, Wei SH, Aranda R, Cheroutre H, et al. Altered immune responses in interleukin 10 transgenic mice. J Exp Med (1997) 185:2101–10. doi:10.1084/jem.185.12.2101
63. Macchia D, Almerigogna F, Parronchi P, Ravina A, Maggi E, Romagnani S. Membrane tumour necrosis factor-alpha is involved in the polyclonal B-cell activation induced by HIV-infected human T cells. Nature (1993) 363:464–6. doi:10.1038/363464a0
64. Cerutti A, Puga I, Cols M. Innate control of B cell responses. Trends Immunol (2011) 32:202–11. doi:10.1016/j.it.2011.02.004
65. Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol (2003) 21:231–64. doi:10.1146/annurev.immunol.21.120601.141152
66. Tangye SG, Bryant VL, Cuss AK, Good KL. BAFF, APRIL and human B cell disorders. Semin Immunol (2006) 18:305–17. doi:10.1016/j.smim.2006.04.004
67. Tangye SG. Staying alive: regulation of plasma cell survival. Trends Immunol (2011) 32:595–602. doi:10.1016/j.it.2011.09.001
68. Huard B, McKee T, Bosshard C, Durual S, Matthes T, Myit S, et al. APRIL secreted by neutrophils binds to heparan sulfate proteoglycans to create plasma cell niches in human mucosa. J Clin Invest (2008) 118:2887–95. doi:10.1172/JCI33760
69. Reijmers RM, Groen RW, Kuil A, Weijer K, Kimberley FC, Medema JP, et al. Disruption of heparan sulfate proteoglycan conformation perturbs B-cell maturation and APRIL-mediated plasma cell survival. Blood (2011) 117:6162–71. doi:10.1182/blood-2010-12-325522
70. Ozaki K, Kikly K, Michalovich D, Young PR, Leonard WJ. Cloning of a type I cytokine receptor most related to the IL-2 receptor beta chain. Proc Natl Acad Sci U S A (2000) 97:11439–44. doi:10.1073/pnas.200360997
71. Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature (2000) 408:57–63. doi:10.1038/35040504
72. Asao H, Okuyama C, Kumaki S, Ishii N, Tsuchiya S, Foster D, et al. Cutting edge: the common gamma-chain is an indispensable subunit of the IL-21 receptor complex. J Immunol (2001) 167:1–5.
73. Leonard WJ. Cytokines and immunodeficiency diseases. Nat Rev Immunol (2001) 1:200–8. doi:10.1038/35105066
74. Caruso R, Fina D, Peluso I, Stolfi C, Fantini MC, Gioia V, et al. A functional role for interleukin-21 in promoting the synthesis of the T-cell chemoattractant, MIP-3alpha, by gut epithelial cells. Gastroenterology (2007) 132:166–75. doi:10.1053/j.gastro.2006.09.053
75. Distler JH, Jungel A, Kowal-Bielecka O, Michel BA, Gay RE, Sprott H, et al. Expression of interleukin-21 receptor in epidermis from patients with systemic sclerosis. Arthritis Rheum (2005) 52:856–64. doi:10.1002/art.20883
76. Good KL, Bryant VL, Tangye SG. Kinetics of human B cell behavior and amplification of proliferative responses following stimulation with IL-21. J Immunol (2006) 177:5236–47.
77. Jungel A, Distler JH, Kurowska-Stolarska M, Seemayer CA, Seibl R, Forster A, et al. Expression of interleukin-21 receptor, but not interleukin-21, in synovial fibroblasts and synovial macrophages of patients with rheumatoid arthritis. Arthritis Rheum (2004) 50:1468–76. doi:10.1002/art.20218
78. Chtanova T, Tangye SG, Newton R, Frank N, Hodge MR, Rolph MS, et al. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol (2004) 173:68–78.
79. Coquet JM, Kyparissoudis K, Pellicci DG, Besra G, Berzins SP, Smyth MJ, et al. IL-21 is produced by NKT cells and modulates NKT cell activation and cytokine production. J Immunol (2007) 178:2827–34.
80. Luthje K, Kallies A, Shimohakamada Y, Belz GT, Light A, Tarlinton DM, et al. The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat Immunol (2012) 13:491–8. doi:10.1038/ni.2261
81. Habib T, Nelson A, Kaushansky K. IL-21: a novel IL-2-family lymphokine that modulates B, T, and natural killer cell responses. J Allergy Clin Immunol (2003) 112:1033–45. doi:10.1016/j.jaci.2003.08.039
82. Habib T, Senadheera S, Weinberg K, Kaushansky K. The common gamma chain (gamma c) is a required signaling component of the IL-21 receptor and supports IL-21-induced cell proliferation via JAK3. Biochemistry (2002) 41:8725–31. doi:10.1021/bi0202023
83. Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med (2010) 207:155–71. doi:10.1084/jem.20091706
84. Diehl SA, Schmidlin H, Nagasawa M, van Haren SD, Kwakkenbos MJ, Yasuda E, et al. STAT3-mediated up-regulation of BLIMP1 is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J Immunol (2008) 180:4805–15.
85. Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, et al. A critical role for IL-21 in regulating immunoglobulin production. Science (2002) 298:1630–4. doi:10.1126/science.1077002
86. Ozaki K, Spolski R, Ettinger R, Kim HP, Wang G, Qi CF, et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J Immunol (2004) 173:5361–71.
87. Bessa J, Kopf M, Bachmann MF. Cutting edge: IL-21 and TLR signaling regulate germinal center responses in a B cell-intrinsic manner. J Immunol (2010) 184:4615–9. doi:10.4049/jimmunol.0903949
88. King IL, Mohrs K, Mohrs M. A nonredundant role for IL-21 receptor signaling in plasma cell differentiation and protective type 2 immunity against gastrointestinal helminth infection. J Immunol (2010) 185:6138–45. doi:10.4049/jimmunol.1001703
89. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med (2010) 207:353–63. doi:10.1084/jem.20091738
90. Rankin AL, MacLeod H, Keegan S, Andreyeva T, Lowe L, Bloom L, et al. IL-21 receptor is critical for the development of memory B cell responses. J Immunol (2011) 186:667–74. doi:10.4049/jimmunol.0903207
91. Rasheed MA, Latner DR, Aubert RD, Gourley T, Spolski R, Davis CW, et al. Interleukin-21 is a critical cytokine for the generation of virus-specific long-lived plasma cells. J Virol (2013) 87:7737–46. doi:10.1128/JVI.00063-13
92. Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med (2010) 207:365–78. doi:10.1084/jem.20091777
93. Fornek JL, Tygrett LT, Waldschmidt TJ, Poli V, Rickert RC, Kansas GS. Critical role for Stat3 in T-dependent terminal differentiation of IgG B cells. Blood (2006) 107:1085–91. doi:10.1182/blood-2005-07-2871
94. Mehta DS, Wurster AL, Whitters MJ, Young DA, Collins M, Grusby MJ. IL-21 induces the apoptosis of resting and activated primary B cells. J Immunol (2003) 170:4111–8.
95. Jin H, Carrio R, Yu A, Malek TR. Distinct activation signals determine whether IL-21 induces B cell costimulation, growth arrest, or Bim-dependent apoptosis. J Immunol (2004) 173:657–65.
96. Pene J, Gauchat JF, Lecart S, Drouet E, Guglielmi P, Boulay V, et al. Cutting edge: IL-21 is a switch factor for the production of IgG1 and IgG3 by human B cells. J Immunol (2004) 172:5154–7.
97. Avery DT, Bryant VL, Ma CS, de Waal Malefyt R, Tangye SG. IL-21-induced isotype switching to IgG and IgA by human naive B cells is differentially regulated by IL-4. J Immunol (2008) 181:1767–79.
98. Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Spolski R, et al. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol (2005) 175:7867–79.
99. Avery DT, Ma CS, Bryant VL, Santner-Nanan B, Nanan R, Wong M, et al. STAT3 is required for IL-21-induced secretion of IgE from human naive B cells. Blood (2008) 112:1784–93. doi:10.1182/blood-2008-02-142745
100. Pene J, Guglielmi L, Gauchat JF, Harrer N, Woisetschlager M, Boulay V, et al. IFN-gamma-mediated inhibition of human IgE synthesis by IL-21 is associated with a polymorphism in the IL-21R gene. J Immunol (2006) 177:5006–13.
101. Rodriguez-Bayona B, Ramos-Amaya A, Bernal J, Campos-Caro A, Brieva JA. Cutting edge: IL-21 derived from human follicular helper T cells acts as a survival factor for secondary lymphoid organ, but not for bone marrow, plasma cells. J Immunol (2012) 188:1578–81. doi:10.4049/jimmunol.1102786
102. Rodriguez-Bayona B, Ramos-Amaya A, Lopez-Blanco R, Campos-Caro A, Brieva JA. STAT-3 activation by differential cytokines is critical for human in vivo-generated plasma cell survival and Ig secretion. J Immunol (2013) 191:4996–5004. doi:10.4049/jimmunol.1301559
103. Brenne AT, Ro TB, Waage A, Sundan A, Borset M, Hjorth-Hansen H. Interleukin-21 is a growth and survival factor for human myeloma cells. Blood (2002) 99:3756–62. doi:10.1182/blood.V99.10.3756
104. Berglund LJ, Avery DT, Ma CS, Moens L, Deenick EK, Bustamante J, et al. IL-21 signalling via STAT3 primes human naive B cells to respond to IL-2 to enhance their differentiation into plasmablasts. Blood (2013) 122:3940–50. doi:10.1182/blood-2013-06-506865
105. Hirohata S, Jelinek DF, Lipsky PE. T cell-dependent activation of B cell proliferation and differentiation by immobilized monoclonal antibodies to CD3. J Immunol (1988) 140:3736–44.
106. Kuchen S, Robbins R, Sims GP, Sheng C, Phillips TM, Lipsky PE, et al. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. J Immunol (2007) 179:5886–96.
107. Deenick EK, Avery DT, Chan A, Berglund LJ, Ives ML, Moens L, et al. Naive and memory human B cells have distinct requirements for STAT3 activation to differentiate into antibody-secreting plasma cells. J Exp Med (2013) 210:2739–53. doi:10.1084/jem.20130323
108. Wang L, Kurosaki T, Corey SJ. Engagement of the B-cell antigen receptor activates STAT through Lyn in a Jak-independent pathway. Oncogene (2007) 26:2851–9. doi:10.1038/sj.onc.1210092
109. Ding BB, Bi E, Chen H, Yu JJ, Ye BH. IL-21 and CD40L synergistically promote plasma cell differentiation through upregulation of Blimp-1 in human B cells. J Immunol (2013) 190:1827–36. doi:10.4049/jimmunol.1201678
110. Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity (1999) 10:105–15. doi:10.1016/S1074-7613(00)80011-4
111. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med (2007) 357:1608–19. doi:10.1056/NEJMoa073687
112. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature (2007) 448:1058–62. doi:10.1038/nature06096
113. Freeman AF, Holland SM. The hyper-IgE syndromes. Immunol Allergy Clin North Am (2008) 28:277–291, viii. doi:10.1016/j.iac.2008.01.005
114. Grimbacher B, Holland SM, Puck JM. Hyper-IgE syndromes. Immunol Rev (2005) 203:244–50. doi:10.1111/j.0105-2896.2005.00228.x
115. Dreskin SC, Goldsmith PK, Gallin JI. Immunoglobulins in the hyperimmunoglobulin E and recurrent infection (Job’s) syndrome. Deficiency of anti-Staphylococcus aureus immunoglobulin A. J Clin Invest (1985) 75:26–34. doi:10.1172/JCI111683
116. Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics (1972) 49:59–70.
117. Leung DY, Ambrosino DM, Arbeit RD, Newton JL, Geha RS. Impaired antibody responses in the hyperimmunoglobulin E syndrome. J Allergy Clin Immunol (1988) 81:1082–7. doi:10.1016/0091-6749(88)90873-1
118. Sheerin KA, Buckley RH. Antibody responses to protein, polysaccharide, and phi X174 antigens in the hyperimmunoglobulinemia E (hyper-IgE) syndrome. J Allergy Clin Immunol (1991) 87:803–11. doi:10.1016/0091-6749(91)90126-9
119. Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol (2004) 22:625–55. doi:10.1146/annurev.immunol.22.012703.104614
120. Buckley RH. B-cell function in severe combined immunodeficiency after stem cell or gene therapy: a review. J Allergy Clin Immunol (2010) 125:790–7. doi:10.1016/j.jaci.2010.02.012
121. Recher M, Berglund LJ, Avery DT, Cowan MJ, Gennery AR, Smart J, et al. IL-21 is the primary common gamma chain-binding cytokine required for human B-cell differentiation in vivo. Blood (2011) 118:6824–35. doi:10.1182/blood-2011-06-362533
122. Kotlarz D, Zietara N, Uzel G, Weidemann T, Braun CJ, Diestelhorst J, et al. Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. J Exp Med (2013) 210:433–43. doi:10.1084/jem.20111229
123. Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology (2012) 143:347–55. doi:10.1053/j.gastro.2012.04.045
124. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435:452–8. doi:10.1038/nature03555
125. Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A (2006) 103:9970–5. doi:10.1073/pnas.0603912103
126. Herber D, Brown TP, Liang S, Young DA, Collins M, Dunussi-Joannopoulos K. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J Immunol (2007) 178: 3822–30.
127. Bubier JA, Sproule TJ, Foreman O, Spolski R, Shaffer DJ, Morse HC III, et al. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc Natl Acad Sci U S A (2009) 106:1518–23. doi:10.1073/pnas.0807309106
128. Rankin AL, Guay H, Herber D, Bertino SA, Duzanski TA, Carrier Y, et al. IL-21 receptor is required for the systemic accumulation of activated B and T lymphocytes in MRL/MpJ-Fas(lpr/lpr)/J mice. J Immunol (2012) 188:1656–67. doi:10.4049/jimmunol.1003871
129. Young DA, Hegen M, Ma HL, Whitters MJ, Albert LM, Lowe L, et al. Blockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum (2007) 56:1152–63. doi:10.1002/art.22452
130. Liu H, Liu G, Gong L, Zhang Y, Jiang G. Local suppression of IL-21 in submandibular glands retards the development of Sjogren’s syndrome in non-obese diabetic mice. J Oral Pathol Med (2012) 41:728–35. doi:10.1111/j.1600-0714.2012.01175.x
131. Wong CK, Wong PT, Tam LS, Li EK, Chen DP, Lam CW. Elevated production of B cell chemokine CXCL13 is correlated with systemic lupus erythematosus disease activity. J Clin Immunol (2010) 30:45–52. doi:10.1007/s10875-009-9325-5
132. Dolff S, Abdulahad WH, Westra J, Doornbos-van der Meer B, Limburg PC, Kallenberg CG, et al. Increase in IL-21 producing T-cells in patients with systemic lupus erythematosus. Arthritis Res Ther (2011) 13:R157. doi:10.1186/ar3474
133. Nakou M, Papadimitraki ED, Fanouriakis A, Bertsias GK, Choulaki C, Goulidaki N, et al. Interleukin-21 is increased in active systemic lupus erythematosus patients and contributes to the generation of plasma B cells. Clin Exp Rheumatol (2013) 31:172–9.
134. Terrier B, Costedoat-Chalumeau N, Garrido M, Geri G, Rosenzwajg M, Musset L, et al. Interleukin 21 correlates with T cell and B cell subsetalterations in systemic lupus erythematosus. J Rheumatol (2012) 39:1819–28. doi:10.3899/jrheum.120468
135. Ma J, Zhu C, Ma B, Tian J, Baidoo SE, Mao C, et al. Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin Dev Immunol (2012) 2012:827480. doi:10.1155/2012/827480
136. Kang KY, Kim HO, Kwok SK, Ju JH, Park KS, Sun DI, et al. Impact of interleukin-21 in the pathogenesis of primary Sjogren’s syndrome: increased serum levels of interleukin-21 and its expression in the labial salivary glands. Arthritis Res Ther (2011) 13:R179. doi:10.1186/ar3504
137. Ma CS, Deenick EK. Human T follicular helper (Tfh) cells and disease. Immunol Cell Biol (2014) 92:64–71. doi:10.1038/icb.2013.55
138. Sawalha AH, Kaufman KM, Kelly JA, Adler AJ, Aberle T, Kilpatrick J, et al. Genetic association of interleukin-21 polymorphisms with systemic lupus erythematosus. Ann Rheum Dis (2008) 67:458–61. doi:10.1136/ard.2007.075424
139. Webb R, Merrill JT, Kelly JA, Sestak A, Kaufman KM, Langefeld CD, et al. A polymorphism within IL21R confers risk for systemic lupus erythematosus. Arthritis Rheum (2009) 60:2402–7. doi:10.1002/art.24658
140. Maiti AK, Kim-Howard X, Viswanathan P, Guillen L, Rojas-Villarraga A, Deshmukh H, et al. Confirmation of an association between rs6822844 at the IL2-IL21 region and multiple autoimmune diseases: evidence of a general susceptibility locus. Arthritis Rheum (2010) 62:323–9. doi:10.1002/art.27222
141. Daha NA, Kurreeman FA, Marques RB, Stoeken-Rijsbergen G, Verduijn W, Huizinga TW, et al. Confirmation of STAT4, IL2/IL21, and CTLA4 polymorphisms in rheumatoid arthritis. Arthritis Rheum (2009) 60:1255–60. doi:10.1002/art.24503
142. Pallikkuth S, Rogers K, Villinger F, Dosterii M, Vaccari M, Franchini G, et al. Interleukin-21 administration to rhesus macaques chronically infected with simian immunodeficiency virus increases cytotoxic effector molecules in T cells and NK cells and enhances B cell function without increasing immune activation or viral replication. Vaccine (2011) 29:9229–38. doi:10.1016/j.vaccine.2011.09.118
143. McPhee CG, Bubier JA, Sproule TJ, Park G, Steinbuck MP, Schott WH, et al. IL-21 is a double-edged sword in the systemic lupus erythematosus-like disease of BXSB.Yaa mice. J Immunol (2013) 191:4581–8. doi:10.4049/jimmunol.1300439
144. Itoh K, Inoue T, Ito K, Hirohata S. The interplay of interleukin-10 (IL-10) and interleukin-2 (IL-2) in humoral immune responses: IL-10 synergizes with IL-2 to enhance responses of human B lymphocytes in a mechanism which is different from upregulation of CD25 expression. Cell Immunol (1994) 157:478–88. doi:10.1006/cimm.1994.1243
Keywords: human B cells, differentiation, plasma cells, cytokines, IL-21, immunodeficiency, autoimmune diseases
Citation: Moens L and Tangye SG (2014) Cytokine-mediated regulation of plasma cell generation: IL-21 takes center stage. Front. Immunol. 5:65. doi: 10.3389/fimmu.2014.00065
Received: 12 December 2013; Accepted: 05 February 2014;
Published online: 18 February 2014.
Edited by:
Catherine Pellat-Deceunynck, Centre National de la Recherche Scientifique, FranceReviewed by:
Klaus Warnatz, University of Freiburg, GermanyKarin Tarte, Université Rennes 1, France
Copyright: © 2014 Moens and Tangye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stuart G. Tangye, Immunology and Immunodeficiency Group, Immunology Research Program, Garvan Institute of Medical Research, 384 Victoria Street, Darlinghurst, NSW 2010, Australia e-mail:cy50YW5neWVAZ2FydmFuLm9yZy5hdQ==