 Francesco De Sanctis1
Francesco De Sanctis1 Sara Sandri1
Sara Sandri1 Giovanna Ferrarini1
Giovanna Ferrarini1 Irene Pagliarello1
Irene Pagliarello1 Silvia Sartoris1
Silvia Sartoris1 Stefano Ugel1
Stefano Ugel1 Ilaria Marigo2Barbara Molon3
Ilaria Marigo2Barbara Molon3 Vincenzo Bronte1*
Vincenzo Bronte1*- 1Immunology Section, Department of Pathology and Diagnostics, University of Verona, Verona, Italy
- 2Istituto Oncologico Veneto, Istituto Di Ricovero e Cura a Carattere Scientifico, Padua, Italy
- 3Venetian Institute of Molecular Medicine, Padua, Italy
Under many inflammatory contexts, such as tumor progression, systemic and peripheral immune response is tailored by reactive nitrogen species (RNS)-dependent post-translational modifications, suggesting a biological function for these chemical alterations. RNS modify both soluble factors and receptors essential to induce and maintain a tumor-specific immune response, creating a “chemical barrier” that impairs effector T cell infiltration and functionality in tumor microenvironment and supports the escape phase of cancer. RNS generation during tumor growth mainly depends on nitric oxide production by both tumor cells and tumor-infiltrating myeloid cells that constitutively activate essential metabolic pathways of l-arginine catabolism. This review provides an overview of the potential immunological and biological role of RNS-induced modifications and addresses new approaches targeting RNS either in search of novel biomarkers or to improve anti-cancer treatment.
Introduction
During tumor progression, cancer cells secrete factors, like cytokines, chemokines, and metabolites (1, 2) that promote the development of a flexible microenvironment able to induce a modification in anti-tumor immune response. Tumors, indeed, need to shade and block out the effectors of the immune system in order to keep on growing (3). For this purpose, neoplastic cells release soluble factors that alter the physiological hematopoiesis in bone marrow and secondary lymphoid organs, such as the spleen, by promoting an unusual commitment of highly immunosuppressive immature myeloid cells that sustain tumor growth, neovascularisation, and spreading to local and distant anatomical sites (4, 5). These cells, which have been named myeloid-derived suppressor cells (MDSCs) to highlight their common myeloid origin and immunoregulatory properties on tumor-specific effector T lymphocytes, critically contribute to shape an immune privileged environment (6). MDSCs represent a heterogeneous population, which includes immature monocyte- and granulocyte-like elements, and progressively engrafts the tumor and secondary lymphoid organs in many preclinical contexts, such as in transgenic mouse models of autochthonous pancreatic adenocarcinoma (7). Expansion of MDSCs has been described also in clinical studies demonstrating an inverse correlation between MDSC levels and cancer clinical stage and outcome (8, 9). The other predominant and heterogeneous myeloid cell subset induced by tumor progression is represented by tumor-associated macrophages (TAMs), which are classified in two distinct subgroups: classically activated, type 1 macrophages (M1) and alternatively activated, type 2 macrophages (M2) (10–14). In many solid cancers, tumor-infiltrating macrophages are polarized toward an M2-phenothype and promote tumor progression by sustaining tumor angiogenesis and providing essential growth factors. In agreement with their pro-tumoral activity, the M2-like macrophage presence and location within tumor is often correlated to a poor prognosis (15–17). Tumor-derived soluble factors and tumor-associated myeloid cells build up a “physical barrier” around the tumor cells, which protects them against the attack of cytotoxic T lymphocytes (CTL) and natural killer (NK) cells (18). Moreover, both neoplastic and tumor-associated myeloid cell metabolisms are responsible for the consumption of essential nutrients required to sustain lymphocytic activity, such as l-arginine (l-Arg), l-cysteine (l-Cys), l-phenylalanine (l-Phe), and l-tryptophan (l-Trp) (5, 19): depletion of these amino acids establishes an additional “chemical barrier” around the tumor that affects T cell fitness (20–23). One of the most essential component of this barrier is represented by the diatomic radical nitric oxide (nitrogen monoxide, NO), which is involved in a large spectrum of immune dysfunctions such as T cell inhibition through the blockade of phosphorylation and activation of Janus kinase (JAK) 3, reduction of major histocompatibility complex class II (MHCII) expression, and induction of T cell apoptosis (24). NO is endogenously generated by four isoforms of nitric oxide synthase (NOS): mitochondrial-specific NOS (mtNOS), neuronal NOS (nNOS or NOS1), inducible NOS (iNOS or NOS2), and endothelial NOS (eNOS or NOS3). The mtNOS, nNOS, and eNOS are constitutively expressed in a variety of cell types and are activated following a transient change in intracellular calcium concentration. On the contrary, iNOS is expressed in response to several extracellular stimuli regulating distinct signaling pathways and catalyzes NO production independently from intracellular calcium concentration (25). Interestingly, iNOS enzyme is expressed by M1-like macrophages and can be co-expressed with arginase-1 (ARG1) in MDSCs, resulting in l-Arg reduced availability and production of reactive nitrogen species (RNS) in tumor microenvironment. RNS affect not only the structure of individual proteins but also modify the protein–protein interaction and function (26). Thus, nitrogen-derived post-translational modifications (PTM) result from either direct interaction with NO or after exposure to RNS, a term that includes peroxynitrite (ONOO−), nitrogen dioxide radical (NO2), and other nitrogen oxides and products arising when NO reacts with reactive oxygen species (ROS) such as superoxide anion (O2−) (27). RNS are commonly generated in sub-cellular compartments such as the peroxidase-containing secretory granules of neutrophils and eosinophils, the mitochondria and endoplasmic reticulum of endothelial and smooth muscle cells, and the vasculature (28). Under physiological conditions, cells produce ROS through the mitochondrial electron transport of aerobic respiration. At low levels, ROS are well tolerated by cells thanks to enzymatic detoxifying systems like superoxide dismutases (SOD) and glutathione S-transferase, which neutralize the free radicals (29). Under pathological contexts (such as during sustained inflammation), ROS reach high concentrations and become toxic, thus affecting cellular metabolism by either direct oxidations of macromolecules or after combining with NO to generate RNS. The reaction between NO and O2− is extremely rapid and depends on both redox environment and NO concentrations, producing two distinct PTM: nitration or nitrosylation. An equal concentrations of NO and superoxide leads to peroxynitrite formation, which can nitrate either free or protein-associated tyrosines to form 3-nitrotyrosines; two- to three-fold excess of NO predominantly results in dinitrogen trioxide (N2O3) formation, the major S-nitrosylating agent in the intracellular microenvironment (30).
A representation of the main RNS-generating pathways and derived PTM is depicted in Figure 1.
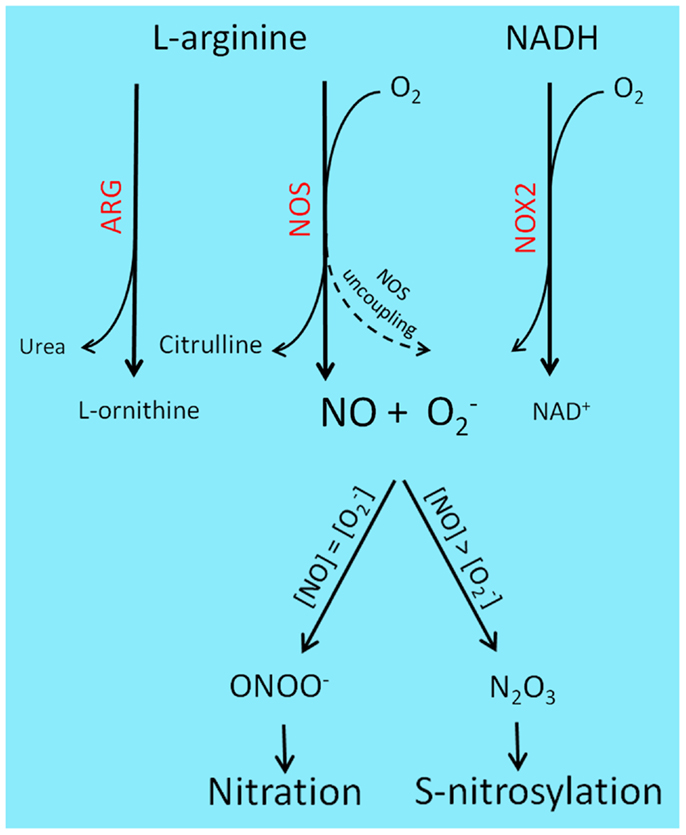
Figure 1. Reactive nitrogen species-generating chemistry and PTM. l-Arginine catabolism involves two main classes of enzymes, ARG and NOS. ARG participates in the urea cycle, producing l-ornithine and urea, while NOS generates nitric oxide and citrulline. l-Arginine deprivation by ARG up-regulation results in NOS uncoupling and consequent production of rather than NO. Superoxide is also produced by NOX enzyme, during the transfer of electrons from NAD(P)H to molecular oxygen. Under pathological contexts, such as cancer and sustained inflammation, NO and levels raise and combine to generate a variety of RNS: equal concentrations of NO and superoxide will lead to ONOO− formation and subsequent protein nitration, while two- to three-fold excess of NO will predominantly result in N2O3 production, a main S-nitrosylating agent.
Protein tyrosine nitration consists in the incorporation of a nitro triatomic molecule (−NO2), generally in ortho position of the phenol hydroxyl group. There are two main mechanisms leading to tyrosine nitration in vivo: the formation of peroxynitrite and the production of NO2 by heme proteins (31). Peroxynitrite chemistry is highly pH dependent. At physiological pH, NO reacts with O2−, producing ONOO−. Nitration is maximal at physiological pH (about pH 7.4) and its yield decreases quickly under more acidic or basic conditions, whereas sulfhydryl oxidation increases from pH 5 to 9 (32). Peroxynitrite-mediated tyrosine nitration is also accelerated in the presence of transition metal ions either in their free form (Cu2+, Fe3+, Fe2+), as complexes involving protoporphyrin IX (hemin), chelators (cyanide (CN−), or ethylenediaminetetraacetic acid (EDTA)) (33).
Nitration is commonly considered an irreversible modification that can be removed only with protein turnover. However, some papers describe the presence of denitrases in different tissues and animal models making controversial this assumption: for example, a denitrase activity was detected in rat and dog tissues (34–36). Moreover, this denitrating activity was described in lipopolysaccharide (LPS)-activated macrophage lines (37, 38). Interestingly a nitration reversible/enzymatic mechanism active in the mitochondria after changes in O2 concentrations was also identified (39). Since RNS-dependent PTM have critical effects on protein function, the reversibility of the process may unveil a crucial biological regulation mechanism for cellular metabolism. The frequency of tyrosine occurrence in proteins is approximately 3–4 mol%, which is similar to other aromatic amino acid such as phenylalanine (40). Tyrosine is a mildly hydrophobic amino acid and so there is a relatively good probability that some tyrosine residues will be exposed to the solvent. Examination of the nitration site in peroxynitrite-modified proteins revealed a preference for tyrosine residues located in loop structures and paucity of nitration in tyrosine residues located on β-strands; however, the primary protein structure does not seem to be a discriminative factor since no consensus sequence for tyrosine nitration has been found so far (40). The addition of a nitro group to tyrosine confers particular physicochemical properties to the modified amino acid, which may have important functional consequences. Three major effects on protein function can be predicted due to tyrosine nitration: no alteration in protein function, loss of function, and gain of function. For example, the plasma proteins α1-antichymotrypsin and transferrin do not undergo any substantial function modification following tyrosine nitration, either in vitro or in vivo (41); on the contrary specific enzymes, as glutathione reductase (GR) and protein kinase C, are inactivated by 3-nytrotyrosine formation (42). On the other hand, cytochrome c (43), fibrinogen (44), protein kinase Cε (45), and microsomal glutathione S-transferase 1 (46) present an increased functionality following PTM.
In addition to nitration, NO could induce other important modifications including protein S-nitrosylation. This is a covalent PTM resulting from the coupling of a NO moiety with the reactive thiol group of a cysteine residue to form an S-nitrosothiol. Contrary to other PTM, such as phosphorylation or ubiquitinylation, S-nitrosylation is not enzyme-dependent. Protein S-nitrosylation is reversibly controlled by S-nitrosylation and denitrosylation reactions, which preserve a dynamic balance in living systems. A number of studies have indicated that abnormal S-nitrosylation is implicated in cancer development and progression as in head and neck squamous cell carcinoma (47), as well as in response to some therapeutic and treatment options (48). Indeed, several S-nitrosylated proteins have been identified to be involved in various cancer-related events. Briefly, the B cell lymphoma (Bcl)-2 S-nitrosylation, which occurs in Cys158 and Cys229 residues, reduces its degradation through the ubiquitin–proteasomal pathway exacerbating the BCL-2 anti-apoptotic effect (49); the S-nitrosylation of apoptosis antigen 1 (Fas) on its Cys304 promotes Fas ligand-mediated apoptosis in cancer cells (50); the S-nitrosylation of p53 prevents normal response to damaged DNA (51); the S-nitrosylation of hypoxia inducible factor (HIF)-1α exerts a pro-angiogenesis function in tumor progression and confers resistance to radiotherapy (52); finally, the S-nitrosylation of O6-alkylguanine-DNA alkyltransferase (AGT) results in a catastrophic and irreversible failure of DNA repair system, suggesting that S-nitrosylation may contribute to cancer development through its indirect role in preventing routine DNA repair in otherwise normal cellular systems (53). RNS-induced PTM may affect important physiological processes such as apoptosis and autophagy that promote tumor onset and neoplastic evolution. Autophagy is a cellular process of turnover for useless proteins and organelles, which is exploited by tumor cells to survive under conditions of metabolic stress, such as oxidizing environments and nutrient starvation (54). Through autophagy cells may survive for a while, by progressively shrinking in size and reducing their metabolism; however, they eventually die if better conditions do not occur; thus autophagy can be considered a temporary and reversible path to cell death. It has been shown that RNS induces autophagy through suppression of mTORC1 pathway decreasing cell viability in MCF-7 breast cancer cell line (55).
All these modifications can also modulate immunological responses and impair any immune-based cancer therapeutic approach; for this reason, in this review, we will focus on the generation, the biological role and the possible strategies to identify proteins subjected to PTM and nitrosative stress in order to design new diagnostic tools and target RNS in tumor microenvironment.
Factors Promoting RNS Generation in Tumor Microenvironment during Cancer Progression
Reactive nitrogen species generation relies on the expression and activity of three main enzymes: NADPH oxidase (NOX), NOS, and ARG. Although contribution of malignant cell to peroxynitrite production is possible, most of the nitrosative stress is considered a reflection of the activation of these specific enzymes in tumor-infiltrating myeloid cells, such as TAMs and MDSCs.
NADPH Oxidase 2
NADPH oxidase 2 (NOX2) is mainly expressed in leukocytes and endothelial cells (56) and it is activated via interaction of vascular endothelial growth factor (VEGF) with VEGFR, tumor necrosis factor (TNF)-α with TNFR1, and angiopoietin with tyrosine kinase with immunoglobulin and epidermal growth factor (EGF) homology domains (Tie) 2 (57). Signal transducer and activator of transcription STAT3 directly regulates the expression of both regulatory (p47phox) and catalytic (gp91phox) NOX2 subunits (58) and controls the synthesis of S100A8 and S100A9, which potentiate NOX2 activity (59). Similarly, IFN-γ up-regulates NOX2 via JAK/STAT1 pathway (60). Interestingly, the protein complex activity is finely tuned by other post-translational mechanisms, such as phosphorylation patterns and subunits availability (61).
iNOS/NOS2
iNOS/NOS2 expression is mainly regulated at the transcription level and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) represents the central switch. In response to specific signals, NF-κB is released from IκB and it can translocate to the nucleus, promoting the transcription of downstream genes (62). As demonstrated by mutational analysis, NF-κB acts through multiple binding sites on iNos promoter, suggesting a modulation of the response proportional to its availability, which in turn is regulated by immune stimulatory signals such as LPS, IL-1β, TNF-α, IFN-γ, and oxidative stress (63). Conversely, TGF-β, antioxidants and glucocorticoids inhibit NF-κB and, consequently, iNOS synthesis (64). iNos promoter comprises binding sites for other transcription factors including CCAAT/enhancer-binding protein (c/EBP), activation protein (AP)-1, interferon regulatory factor (IRF)-1, p53, STAT3, and octamer factors (65–69). Interestingly, cellular iNOS concentration is finely tuned also at a translational and post-translational level by either loss of stabilizing proteins at 3′ untranslated region of iNos mRNA (70, 71) or increase in iNOS turnover in response to caveolin-1, TGF-β, peroxisome proliferator-activated receptor (PPAR)-α agonist, and calpain (72–77).
Arginases
Arginases exist as two isoenzymes in mammals, ARG1 and ARG2 differing for cellular expression/localization and for immunological properties (78). ARG1 takes part in the urea cycle in the liver and it is localized in the cytoplasm, whereas ARG2 is a mitochondrial protein mostly present in kidney, prostate, and small intestine, where the urea cycle is not complete. STAT6 and c/EBPβ are the main transcription factors controlling arginases in myeloid cells (79). ARG1 expression can be regulated by the cytokines IL-4, IL-10, and IL-13 inducing the interaction of PU.1, STAT 6, and c/EBPβ transcription factors on the enhancer element of Arg1 promoter (80). Mouse Arg1 is also regulated by COX2-dependent PGE2 overproduction (81) and by macrophage stimulating protein (MSP) (82). At a post-translational level, ARG1 is stabilized by S-nitrosylation on Cys303 residue, which can depend on an increased iNOS activity (83). All the molecular pathways listed above participate in regulating ROS and RNS-producing enzymes. As further discussed below, cancer exploits these mechanisms pushing the balance toward the expression of NOX, iNOS, and ARG1 with the purpose of increasing ROS and RNS levels tailoring a microenvironment that promotes immune privileged conditions and can sustain its unrestricted growth.
One of the strategies developed by neoplastic cells in tuning the immune system is based on the release of factors that promote the expression of RNS-generating molecules, i.e., HIF1-α, which rises after exposure to hypoxic environments. The synthesis of angiogenic factors (such as VEGF) is promoted by HIF1-α, a transcription factor that is usually quickly degraded in the proteasome after the hydroxylation carried out by the poly hydroxyl domain (PHD) proteins under normal oxygen conditions (84). Current data underline how HIF1-α may trigger broader effects than those related to angiogenesis and support tumor growth also by promoting local immune suppression. The hypoxic tumor microenvironment is indeed able to modify the phenotype of MDSCs by increasing their immunosuppressive properties and promoting their differentiation to TAMs. Exposure to hypoxia was shown to increase ARG1 and iNOS expression in MDSCs with subsequent production of peroxynitrite and their conversion from antigen-specific to non-specific suppressors of T cell function, a phenomenon mediated by HIF1-α (85). Moreover, hypoxia up-regulates NOX2, iNOS, and ARG1 in many cell types (86). The mechanism underneath the modification of iNOS and ARG1 expression in MDSCs is based on the regulation of NF-κB. The iNos promoter contains also a hypoxia response enhancer (HRE) sequence, which potentiates NF-κB-dependent iNOS up-regulation in macrophages (87). Consistent data highlight the role of macrophage-derived-HIF1α in generating a localized immune privileged site within the tumor milieu via iNOS and ARG1 induction (88). Furthermore, the increased production of NO establishes a positive feedback loop on HIF1-α levels: NO-dependent impaired activity of PHD stabilizes HIF1-α and NF-κB, thus amplifying the expression of genes under their control, even under physiologic oxygen environments (89). These observations should be taken into consideration for designing new anti-angiogenic, immunotherapeutic approaches based on HIF1-α-targeting to improve the efficacy of the already available anti-VEGF therapies.
In tumor microenvironment cytokines and chemokines contribute to RNS-mediated myeloid immune suppression by multiple strategies. First of all, they can influence hematopoiesis toward the differentiation of progenitors to MDSCs and TAMs (90) and orchestrate MDSC trafficking to peripheral tissues (i.e., to the tumor) (91). Second, they can tune the expression of iNOS and ARG1. This is the case of granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-6. GM-CSF stimulates the maturation of mouse hematopoietic progenitors to DCs when used alone, and to MDSCs in combination with IL-6 (90). However, GM-CSF binding to its receptor activates different signaling cascades including JAK/STAT, mitogen-activated protein kinase MAPK, and NFκB (92) whereas IL-6 mainly induces MDSC differentiation by STAT3 activation (93). The selective induction of iNOS, ARG1, or both depends on which cytokines are involved. For example, during a pathogen infection, macrophages exclusively up-regulate iNOS to increase their microbicidial activity in response to pathogen-associated molecular patterns and a Th1 inflammatory stimulus like IFN-γ, IL-12, and TNF-α. This phenotype is mediated by NF-κB activation and binding of the IRF-1 element of the iNos promoter (94). Th1 cytokine release is a characteristic of classic macrophage activation and plays a fundamental role in central immunity stimulation. On the contrary, alternative macrophage activation, which promotes humoral immunity, allergic and antiparasitic responses, tissue repair, and fibrosis is linked to release of Th2 cytokines, such as IL-4, IL-10, and IL-13 cytokines, which are responsible for ARG1 up-regulation as well as iNOS down regulation (95). This phenotype is a general feature shared with DCs and granulocytes (96) and is the result of the coordinated action of PU.1 STAT6 and c/EBPβ transcription factors (97) on Arg1 promoter. Cytokines can act also indirectly on myeloid cells modifying their sensitivity to either Th1 or Th2 stimuli. IL-23, for example, can increase Th2-mediated ARG1 induction by up-regulating IL-4-Rα and IL-13-Rα1 chains (98). Moreover, the induction and the activity of l-Arg-metabolizing enzymes are both regulated by other intracellular biochemical pathways and by competition for the same substrate, the amino acid l-Arg (99). Anti-inflammatory cytokines (i.e., TGF-β or IL-10) tune the iNOS/ARG1 balance toward the up-regulation of the latter. TGF-β is a pleiotropic cytokine that possess either inflammatory or regulatory properties depending on the cellular and environmental context (100); however, TGF-β can mediate immune suppression by indirect mechanisms, for example, increasing ARG1 activity and decreasing iNOS expression that limits macrophage-dependent cytostasis of fibrosarcoma cells (101). Interestingly, TGF-β-driven iNOS down regulation is mediated by an increase of ELK3, a transcription repressor which decreases the expression of iNos gene after binding to its promoter sequence (102).
There are some important exceptions in cytokine-mediated tuning of iNOS and ARG1 expression, which make the definition of either classical or alternative macrophage activation in the regulation of the two genes rather simplistic and sometimes inaccurate. In mouse macrophages, LPS, a bacterial product commonly associated with Th1 cytokine release, is able to activate both enzymes. Moreover, in tumor environment myeloid cells can restrain T cell functionality by up-regulating both enzymes. The concomitant activity of ARG1 and iNOS is a peculiar property of MDSCs, or at least of a subset of this heterogeneous population of suppressor cells induced by tumors in mice and humans (26, 103, 104). The molecular bases for the synergism might involve a double cross-talk occurring during the encounter of T cells and MDSCs. IFN-γ released by antigen-activated CD8+ T cells is the primary (but not unique) trigger for autocrine production of IFN-γ and IL-13 by MDSCs. These cytokines are then necessary to provide a sustained expression and activation of both iNOS and ARG1, for a sufficient time to affect T lymphocyte proliferation and survival (103). This scenario reconciles the needs of both limiting the extent of T cell suicide/inactivation only to those cells that are activated by the antigen and fully arm the suppressive program in MDSCs (103); furthermore, IFN-γ produced by activated MDSCs primes other nearby MDSCs, even though this could be limited by the necessity of a cell–cell contact with antigen-activated CD8+ T lymphocytes (13, 103). It is also clear that different T cell responses, biased toward either a Th1 or Th2 type cytokine production, might selectively activate either NOS- or ARG-dependent immune regulation, respectively.
Immune Suppression Promoted by RNS in Tumor Milieu
A number of evidence indicates that peroxynitrite is toxic for lymphocytes and can prime T cells to undergo apoptotic death through different pathways involving inhibition of protein tyrosine phosphorylation via nitration of tyrosine residues (105) or by nitration of the voltage-dependent anion channel protein, a component of mitochondrial permeability transition pore (106). Nitrotyrosine, a marker of nitrative stress in tissues, is found in thymic extracts and thymic sections and co-localized with apoptotic cells, suggesting that RNS are also involved in thymic apoptosis in vivo (107). However, molecular explanations on how peroxynitrite released by myeloid cells can induce tolerance in antigen-specific CD8+ T cells were provided only recently.
Peroxynitrite interferes with peptide binding efficiency of MHC class I molecules as result of MHC class I and ligand–peptide complex nitration/nitrosylation on tumor cells; this aspect is only a part of the immunosuppressive barrier generated by MDSCs to impair tumor-specific immune responses. Neoplastic cell preconditioning with peroxynitrite in vitro reduces immunogenic peptide–MHC (pMHC) complex formation without affecting MHC class I expression on cell surface resulting in impaired recognition of tumor cells by CTLs. This effect was observed either for naturally processed or synthetic peptides. Peroxynitrite modifies the α chain of the MHC class I molecule only if the latter is not already loaded with an epitome peptide. This hypothesis was initially proved using artificially transduced antigen (OVA peptide) and confirmed with naturally processed melanoma antigen gp100 (108). Since nitrated proteins are mainly localized in sub-cellular compartments, like in peroxisome-containing granules, mitochondria, and endoplasmic reticulum (109), it is conceivable that the MHC class I α chain is nitrated/nitrosylated by peroxynitrite in the endoplasmic reticulum, just before assembly with β2 microglobulin and antigenic peptide. Nitration/nitrosylation can also affect immunogenic peptides, reducing their binding to MHC molecules. The conversion of tyrosine residues to nitrotyrosine on peptide epitopes could influence either TCR-contact or MHC class I and II contact positions, with profound impact on T cell responses (110–112). Elevated levels of nitrated proteins have been detected in patients with different type of cancers, i.e., bladder, colorectal, breast, and lung carcinoma, melanoma and Hodgkin lymphoma (111). In addition, in a transplantable model of mouse fibrosarcoma showing extensive myeloid cell infiltration, high levels of nitrotyrosines were found in both cytoplasm and nuclei of tumor cells (113). In the presence of RNS, such as in tumor microenvironment, the likelihood for a peptide epitope to be subjected to nitration/nitrosylation events seems thus quite high. Similarly, nitrogen-induced PTM of proteins can break the tolerance toward self-antigens and initiate autoimmune diseases. A cytochrome C-derived epitope encompassing nitrotyrosine in position 97 was shown to trigger a T cell restricted immune response against the self-protein (110). Accordingly, activated antigen presenting cells (APCs) were capable of stimulating CD4+ T lymphocytes with chemically modified peptides (114). Thus, from a therapeutic point of view, PTM of proteins and peptides may be exploited to break the tolerance against weak tumor-associated antigens.
Alternatively, peroxynitrite can act on the TCR side by nitrating/nitrosylating α and β subunits and the CD8 co-receptor molecule (115): in vitro and in vivo experiments confirmed the molecular modeling predictions of tyrosine residues susceptible to nitration. These modifications impair recognition of pMHC complex and promote dissociation of the CD3 ζ chain from the αβ TCR complex, thus disrupting TCR signaling (116). RNS action on T cells does not appear to be limited to nitration/nitrosylation of the crucial molecules α/β TCR, CD3, and CD8 but it also involves a TCR ligand-independent, transient tyrosine phosphorylation of the CD3 ζ chain, sustained for about 24 h, which delivers an abortive signal preventing further T cell activation by TCR ligands (117). In addition, a rapid down regulation of CD4 and CD8 co-receptors was observed following T cell exposure to RNS (117). In peripheral lymphoid organs, all the inhibitory effects promoted by MDSC-secreted RNS are primarily directed toward naïve T cells bearing the TCRs recognizing the pMHC complexes presented by MDSCs. Some experiments indicate that MDSCs are able to present tumor antigens to specific CD8+ T cells and in this context they should facilitate exposure of TCR to the action of the peroxynitrite they secrete. Failure to respond to stimulation with a specific peptide was observed only for CD8+ T cells nitrated at TCR/CD8 complex. T cells recognizing pMHC complexes different from those expressed on MDSC surface showed less TCR/CD8 nitration and retained the ability to respond to antigens presented by classic APCs. These data indicate that T cells with high affinity TCRs for pMHC complexes presented by MDSCs are more prone to peroxynitrite-dependent inhibition by MDSCs (115, 116).
Another RNS-promoted mechanism of immune suppression relies on the modification of chemokines. As a result of RNS exposure, these molecules might undergo PTM that alter their chemotactic properties, impairing the recruitment of T cells and keeping them at the periphery of the tumor lesion, unable to reach the tumor core and exert their cytotoxic action (118). Chemokines are organized in a complex and dynamic network, which coordinates leukocyte trafficking both under physiological and pathological conditions. To perform their action these proteins bind to specific receptors associated to heterotrimeric G proteins, which are present heterogeneously on all leukocytes (119–121). Receptor activation leads to the triggering of signaling pathways including inositol-phosphate 3-kinase (IP3K) and NF-κB transcription factor. The signaling cascade activates a series of events ending in a rapid remodeling of the cytoskeleton, with increase in integrins and adhesion molecules that promote adhesion, extravasation, and leukocyte migration to the target sites (119). Normal chemokines activity is also regulated by PTM such as proteolytic processing, glycosylation, and deamination (122, 123). RNS inhibit migration of T lymphocytes induced by some chemokines, such as CXCL12, CCL21, CCL2, and CCL5. RNS can down-regulate the expression of CXCR4 on T cells through alteration of the TCR/CD3 signaling complex and reduce CXCL12–induced T cell migration (124). When exposed to the action of RNS, mouse and human CCL2 become nitrated at the level of tyrosine and tryptophan residues respectively; CXCL12, is subjected to tyrosine nitration as well. This PTM reduces the binding affinity of CCL2 to its receptor CCR2, affecting drastically tumor homing of CD8+ T lymphocytes. Nitrotyrosines are found in tissue sections of hyperplastic human lymph nodes obtained from surgical resections of patients with lung, colon, and prostate cancer (105, 124). Surprisingly, the nitrated/nitrosylated CCL2 does not lose the ability to recruit MDSCs to the tumor, likely due to higher expression of CCR2 in myeloid than in T cells (118).
Available Technologies and New Approaches for Identification of Nitrogen-Induced PTM
Nitrogen-induced PTM is a process promoted by oxidative environments in presence of NO and associated with a number of different diseases such as autoimmunity (125), neurodegenerative disorders (126), and cancer (118). Exactly like other PTM (i.e., phosphorylation/dephosphorylation), RNS action on target proteins modifies their biochemical properties. Indeed 3-nitrotyrosine, the most common and studied RNS-derived PTM, shows peculiar chemical characteristics, which makes it different from normal tyrosine (127) and can modify the protein tertiary and quaternary structure, and therefore its activity. Given the clinical relevance of PTM and 3-nitrotyrosine, many efforts have been done in designing and developing tools to identify which proteins are subjected to NO modification, the position of the chemical alteration, and its biological role. Many of them are based on the use of chromatography [i.e., HPLC: Ref. (128)] or mass spectrometry (129), or both (130). These techniques are also reviewed elsewhere (131). However, a major issue in defining the “nitrome” is related to the very low representation of nitrogen-induced PTM within the microenvironment. Indeed, only few tyrosines are modified in vivo under inflammatory conditions: in this context the specificity and resolution of the detection technologies may represent a limiting factor (132). Thus, enrichment of nitrogen-induced PTM (such as with antibodies that specifically bind modified proteins) represents an important technological improvement. Accordingly, the development of 3-nitrotyrosine-specific antibodies opened the way to a body of publications focused on semi-quantitative analysis of 3-nitrotyrosine in complex biological samples. Anti-3-nitrotyrosine antibody improved already available methods for the identification of proteins subjected to nitrogen-induced PTM in complex samples separated through gel-based approaches. Indeed, a semi-quantitative evaluation of 3-nitrotyrosine levels in complex samples can be performed by 2D protein separation followed by transfer on filter and blot with anti-3-nitrotyrosine antibody; furthermore the alignment of the blot with 2D gel identifies the spots that can be excised for further protein identification by MS. However, the strength of this enriching technique strictly relies on the antibody specifications (i.e., affinity). Moreover, some PTM may be ignored depending on the tridimensional availability of the 3-nitrotyrosine residues to the antibody. Recent works provide an example of how these innovative detection tools can be exploited to characterize the biological impact of nitrogen-modified proteins involved in regulating peripheral immune response. Through the implementation of liquid chromatography and MS, in fact, mouse and human CCL2 were found to be nitrated on tyrosine and tryptophan residues upon exposure to RNS, respectively. To confirm the biological impact of nitrogen-induced CCL2 modification, a single-domain recombinant antibody was isolated that selectively binds modified CCL2 and can be used for its detection (118).
Tyrosine is the preferential target of RNS attack; however, a growing body of evidence highlights PTM of other aromatic amino acids, such as tryptophan, which can alter protein function and cellular metabolism as well. Peroxynitrite and oxidant environments modify human Cu, Zn-superoxide dismutase and bovine serum albumin at tryptophan residues (133). Interestingly, these modifications were associated with a 30% decrease in SOD enzymatic activity. Moreover, RNS rich environments promote a broader profile of chemical alterations than those observed on tyrosines. Among them, 1-N-nitrosotryptophan and 6-nitrotryptophan (6-NO2Trp) are the most representative products after peroxynitrite exposure (133). However, the modest presence of tryptophan nitration residues, compared with tyrosine, represents a limiting factor in the identification of proteins that undergo RNS-dependent PTM. This is the result of the lower tryptophan frequency in the primary protein structure, lower exposition to the solvent (hydrophobicity), and higher redox potential than tyrosine. Despite of this technical issue, the identification of nitrotryptophan-modified proteins represents a key task since solvent exposed tryptophan usually participates in molecule interactions. Indeed, the low amount of modified tryptophan does not allow predicting the biological impact that its alteration will cause. A recently developed antibody, which selectively binds 6-NO2Trp, was successfully used in western blot to detect and identify nitrotryptophan-containing proteins in peroxynitrite exposed rat pheochromocytoma cells (134). The access to reagents, which specifically label nitrotryptophan and other nitrogen-dependent modifications, will increase the body of data on nitrogen-dependent PTM and provide a detailed snapshot of the post-translational signature induced by nitrogen under physiologic and pathologic contexts.
Clinical Relevance of RNS
The detection of the nitroxidative stress signature in various human tumors and stages may provide information about its role in tumor formation and progression. The correlation of nitration level with clinical parameters can additionally offer prognostic tools and allow the development of therapeutic strategies.
Expression and distribution of iNOS have been evaluated both at the mRNA and protein level in many tumors and conflicting results of these analyses have been reported (135, 136). Over expression of iNOS is frequent in various human tumors but a correlation with tumor stage, grade, or metastases, as well as with poor prognosis is reported only in certain types of cancer. In gastric cancer (137) and melanoma (138), for example, iNOS level could be a predicting biomarker for poor prognosis. Anyway, other types of cancer show either controversial results such as in colorectal, breast, brain, lung, and cervical cancer (139, 140) or no correlation at all, as in bladder carcinoma, pancreatic and cervical cancer (141). Regarding the other two NOS isoforms, NOS3 expression strongly correlates with iNOS presence in breast carcinoma (142) and NOS1 has been detected in some oligodendrogliomas and neuroblastomas cell lines (143). However, detecting the presence of NOS protein is only an indirect method to evaluate nitroxidative stress (144). In this respect, more accurate information is provided by staining with anti-nitrotyrosine antibodies (145). Table 1 compares nitrotyrosine staining pattern identified in different human tumors. NOS and ARG detection are shown as well, as they are involved both in a synergistic peroxynitrites generation and in independent immunosuppressive activities (26, 146). Immunohistochemistry (IHC) confirms that many tumors generally display higher nitration level than control tissues. Compared to other quantitative approaches that finely measure PTM levels, IHC can provide the localization of the cellular type and compartment subject to nitration: nitrotyrosine staining is usually localized in neoplastic cells, but it is also observed at the level of stromal cells as reported for colon, gastric, breast cancer, and mesothelioma (147–150). Moreover, nitrotyrosine staining is mostly diffused in the cytoplasm; additional focal nuclear pattern of staining is described for breast, colon, gastric, and papillary thyroid cancer (147–149, 151). The presence of the positive staining, however, highlights the localization of tyrosine-nitrated proteins but it does not provide any information about the source of peroxynitrite that generates this modifications. Indeed, RNS can easily diffuse out of the membrane and accomplish their function in a different cellular target.
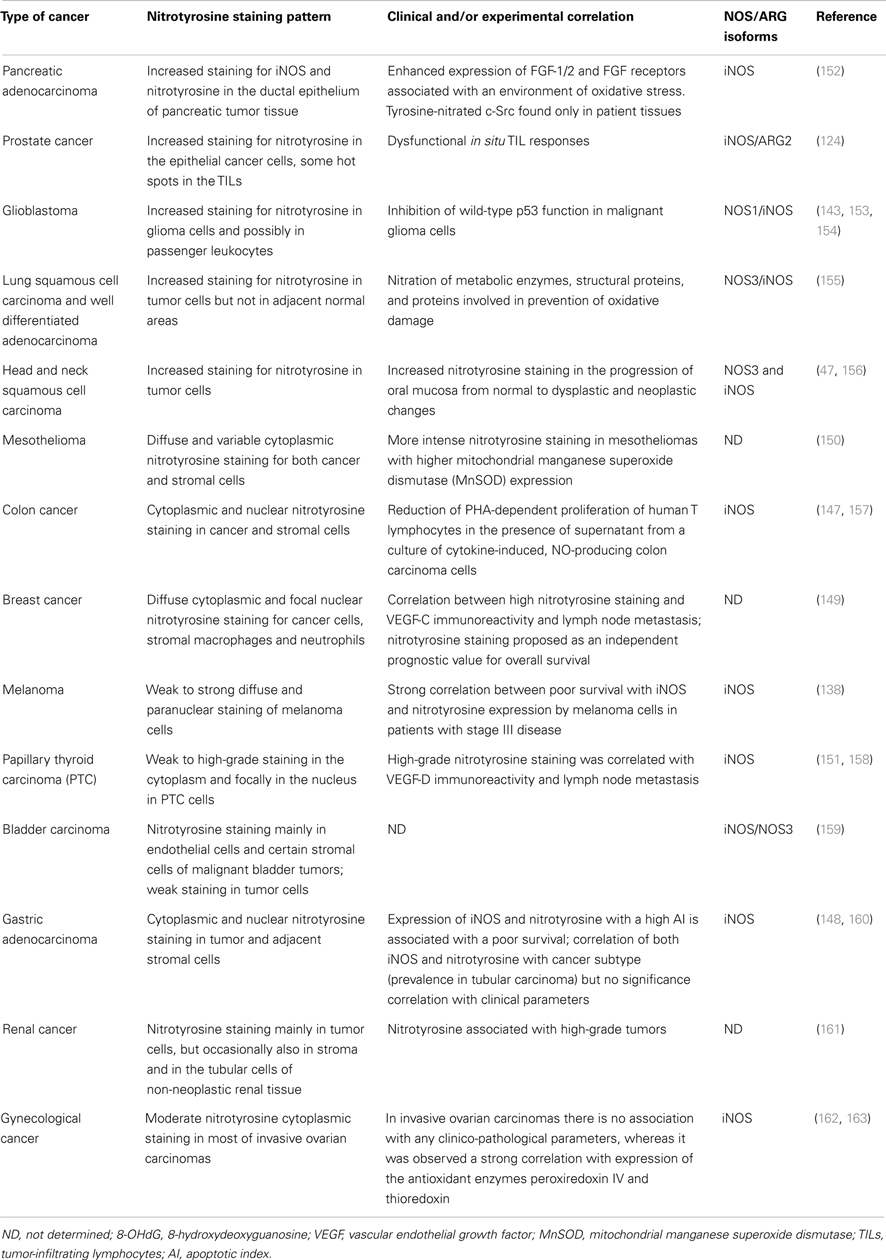
Table 1. Nitrotyrosine presence in human cancers.
Besides qualitative observations, correlations of nitrotyrosine staining with clinical parameters are described only for certain tumor types and positivity is generally associated with worse outcome. In head and neck squamous cell carcinoma, the staining increases with tumor progression, suggesting a possible role of peroxynitrite generation during carcinogenesis (47). In advanced melanomas, a strong correlation exists between poor survival, iNOS expression, and nitrotyrosine presence (138), while in a cohort of patients with invasive breast cancer, nitrotyrosine staining correlates with lymph node metastasis and it may serve as a significant prognostic factor for long-term survival since high nitrotyrosine levels are related with poor overall survival (149). Literature for gastric adenocarcinoma describes heterogeneous and controversial findings: one cohort of patients exhibited a difference in nitrotyrosine staining according to the tumor type, with tubular carcinoma subtype showing the higher positivity (160). In another study, a correlation was described between nitrotyrosine and tumor apoptosis showing that patients with high apoptotic index, elevated iNOS expression, and nitrotyrosine had a poor survival (148). Several factors can potentially explain tumor differences regarding the correlation between RNS and clinical evidences. In first instance, each tumor type can be subjected to a peculiar nitrosative stress level depending on either the abundance or type of RNS-producing cells. Moreover, tumor cells may be more or less resistant to oxidative environments depending on the activity of detoxifying mechanisms such as autophagy and availability of molecules and proteins with scavenging properties. Thus, nitrotyrosine staining is an inflammation indicator that provides useful information on the clinical outcome when used in combination with other parameters such as the immunological context (T cell and myeloid infiltrate, M1/M2 macrophage levels) and local microenvironment (Th1/Th2 cytokines for instance). In addition to clinical parameters, there are few published works describing an association between nitrotyrosine positivity with other biological factors linked to tumor progression. In a cohort of pancreatic adenocarcinoma patients, in fact, the tumor environment was characterized by oxidative stress linked to the expression of mitogen fibroblast growth factor (FGF)-1/2 and their receptors, which all play a role in pancreatic cancer development (164). In a group of advanced breast cancer patients, nitrotyrosine positive staining is associated with the presence of the angiogenic factor VEGF-C (149). In prostatic cancer, NO and peroxynitrites promote tumor growth through the suppression of anti-tumor immune response. Accordingly tumor-infiltrating lymphocytes (TILs) isolated from prostate cancer by using collagen gel matrix-supported organ cultures were unresponsive to mitogen stimuli; ex vivo prostate neoplastic cells generate peroxynitrite responsible for tyrosine nitration in TILs and blockade of peroxynitrite production restored TIL responsiveness (124). Similar dysfunction in T cell proliferation when exposed to NO-producing tumor cell line supernatants was described in colon carcinoma (157).
RNS: Therapeutic Implications
To date, the nitrotyrosine detection in human cancer tissues does not have a uniform relevance with respect to predicting clinical disease progression in cancer patients. The reasons of this lack of homogeneity are not known and might depend, in part, on the limits in the available tools to detect nitrogen-induced PTM and the dichotomic role played by NO (165). Specific locations and targets of tyrosine nitration need to be clearly identified in order to understand in full the biological impact of PTM and find potential candidates for new therapeutic approaches. In glioblastoma multiforme (GBM), for example, the key tumor suppressor protein p53 exhibits peroxynitrite-mediated modifications in vivo and, moreover, malignant glioma cell lines treated with peroxynitrite show wild-type p53 function inhibition (153, 154). Strategies designed to target RNS and restore p53 functionality in tumor cells are thus one example of a promising field of research with translational outcomes.
As discussed above, the l-Arg metabolizing enzymes NOS and ARG are key enzymes for the nitrogen-induced immunosuppressive properties of MDSCs and TAMs. In an in vitro model of human prostate carcinoma, only the contemporary inhibition of ARG and NOS with NG-hydroxyl-l-arginine (NOHA) and l-NG-monomethyl arginine citrate (l-NMMA) respectively, was effective in reducing intratumoral nitrotyrosine staining and this directly correlated with the ability in restoring TIL responsiveness to various stimuli (124). In untreated cultures, in fact, prostate TILs did not react either to antigens or signals bypassing TCR binding, indicating profound state of immune dormancy/anergy (124). Overall, mouse and human studies have clearly recommended that drugs aimed at reverting T lymphocyte unresponsiveness in tumor-bearing hosts should target both ARG and NOS. The pharmacological approach manipulating l-Arg metabolism via ARG1 and iNOS have shown therapeutic potential in preclinical studies, suggesting its use as adjuvant in combination with treatments that directly enhance the immune response. NO-donating drugs such as nitroaspirin (classic aspirin molecule covalently linked to a NO donor group) inhibit iNOS through various mechanisms (166, 167). MDSC-derived immune suppression could be relieved either by nitroaspirin, the combination of ARG and NOS inhibitors, or peroxynitrite scavengers, whereas simple NO-donors or nitroaspirin analogs lacking the NO-donating group were ineffective (168). The salicylic portion (with the attached spacer) of nitroaspirin was necessary for ARG1 blockade whereas NO donation was important for the feedback inhibition of iNOS (166, 167). In vivo administration of nitroaspirin resulted in decreased intratumoral staining with anti-nitrotyrosine and anti-iNOS antibodies, together with a reduced intratumoral ARG activity (168). Despite these positive results, nitroaspirin was not effective as an adjuvant for adoptive cell transfer therapies (ACT; unpublished data). On the contrary, a member of a new class of NO-donating drugs, AT38 ([3-(amino carbonyl) furoxan-4-yl]methyl salicylate), was shown to increase adoptive cell therapy effects. This compound is able to revert RNS-induced PTM of chemokines necessary for T cell tumor invasion (118) and to reduce the in vivo generation of peroxynitrite that can impair T cell migration to the tumor. Interestingly, RNS targeting might represent a new innovative therapeutic strategy for a broad spectrum of RNS-induced diseases, like neurodegenerative and cardiovascular one. AT38 efficacy, in fact, is not limited to the tumor microenvironment but can be useful more in general within an inflammation context. In a mouse model of epicutaneous vaccinia virus infection, AT38 treatment restored CD8+ T cell relocation to epidermal foci, suggesting that peroxynitrite production control CD8+ T cell entry, and consequent activity, into viral lesions (169).
As described above, intratumoral ARG activity can be sustained by other factors, such as COX, acting on tumor-infiltrating myeloid cells. In several preclinical trials the use of COX2 inhibitors, such as celecoxib, down-regulated ARG1 expression, delayed tumor growth, and stimulated anti-tumor response (81, 170–172). In a mouse model of mesothelioma, Celecoxib improved survival when used in combination with DC-based immunotherapy (172). Moreover, inhibition of ARG1 and iNOS activity in MDSCs can be achieved by phosphodiesterase-5 (PDE5) inhibitors (sildenafil, tadalafil, and vardenafil), which can restore the spontaneous anti-tumor T cell response (173). N(6)-(1-iminoethyl)-l-lysine-dihydrochloride (l-nil) is an iNOS-selective small molecule antagonist that showed potent anti-proliferative properties on melanoma cells increasing survival in tumor-bearing mice in combination with chemotherapy (174). To improve cellular antioxidant properties, thus limiting ROS and RNS generation, a group of anti-inflammatory molecules can represent an alternative option: they are synthetic triterpenoids that activate a transcription factor regulating antioxidant genes, the nuclear factor (erythroid-derived 2)-like 2. The synthetic triterpenoid CDDOMe reduced ROS and peroxynitrite levels in MDSCs and abrogated MDSC-dependent immune suppression both in several tumor mouse models and in pancreatic cancer patient; however, ARG activity or NO levels were not affected in MDSCs (175). It is thus possible to exploit in current therapeutic protocols, the association of different drugs affecting either concurrent or alternative steps in the biochemical production of peroxynitrite. Other antioxidant compounds have been validated in vitro and are currently investigated as candidates for cancer therapy. For instance, 3,4′,5-trihydroxy-trans-stilbene (resveratrol), a polyphenolic natural product described as free radical scavenger was able to induce apoptosis in several cell lines by regulating proteins that patrol cell cycle such as p53 and cyclin dependent kinases (176–178). Accordingly, diferuloylmethane (curcumin) showed similar properties in vitro. This phenolic compound can be considered more than just an oxidant scavenger molecule since it inhibits the NF-κB activation reducing iNOS and DNA-dependent protein kinase catalytic subunit levels; additionally curcumin induces the up-regulation of p21, p27, p53, and the checkpoint kinase 2 expression, thus promoting the G2/M cycle arrest and apoptosis in human melanoma cell lines (179). Given their similar biological properties, resveratrol and curcumin have been tested together in different set-ups with promising results; their combination was able to reduce the proliferation of Hepa 1–6 hepatocellular tumor cells in vitro (180) and of HCT-116 colon cancer cells, in vivo (181).
As described above, nitrosative stress plays a role in regulating autophagy in MCF-7 breast tumor cells (55), therefore therapeutic approaches targeting this pathway can be also considered. Autophagy, indeed, is a double-edged sword, which can either promote or control tumor progression; when this process is impaired, cells become more sensitive to RNS and oxidative conditions resulting in higher accumulation of genetic alterations, which increase both apoptosis and neoplastic evolution. From the other side sustained autophagy progressively depletes cells from proteins, membranes, and organelles until death. Therefore this process can be exploited to promote tumor apoptosis by using autophagy activators, such as rapamycin (54).
In conclusion, based on published data, many agents have shown potential benefits in combination with immunotherapy in preclinical studies. l-Arg metabolism manipulation via ARG and NOS can contribute in the regulation of tumor-infiltrating immune cells, reducing MDSC accumulation and promoting T cell infiltration (81, 118, 168, 170), thus representing a potential adjuvant for immune therapeutic approaches. However, before reaching the clinic, further knowledge is necessary, such as the evaluation of systemic side effects and the assessment of cancer types that would receive a real benefit after modulation of l-Arg metabolism.
Figure 2 summarizes MDSC activity in tumor environment (Figure 2A) and the effects of the above mentioned inhibitory compounds (Figure 2B).
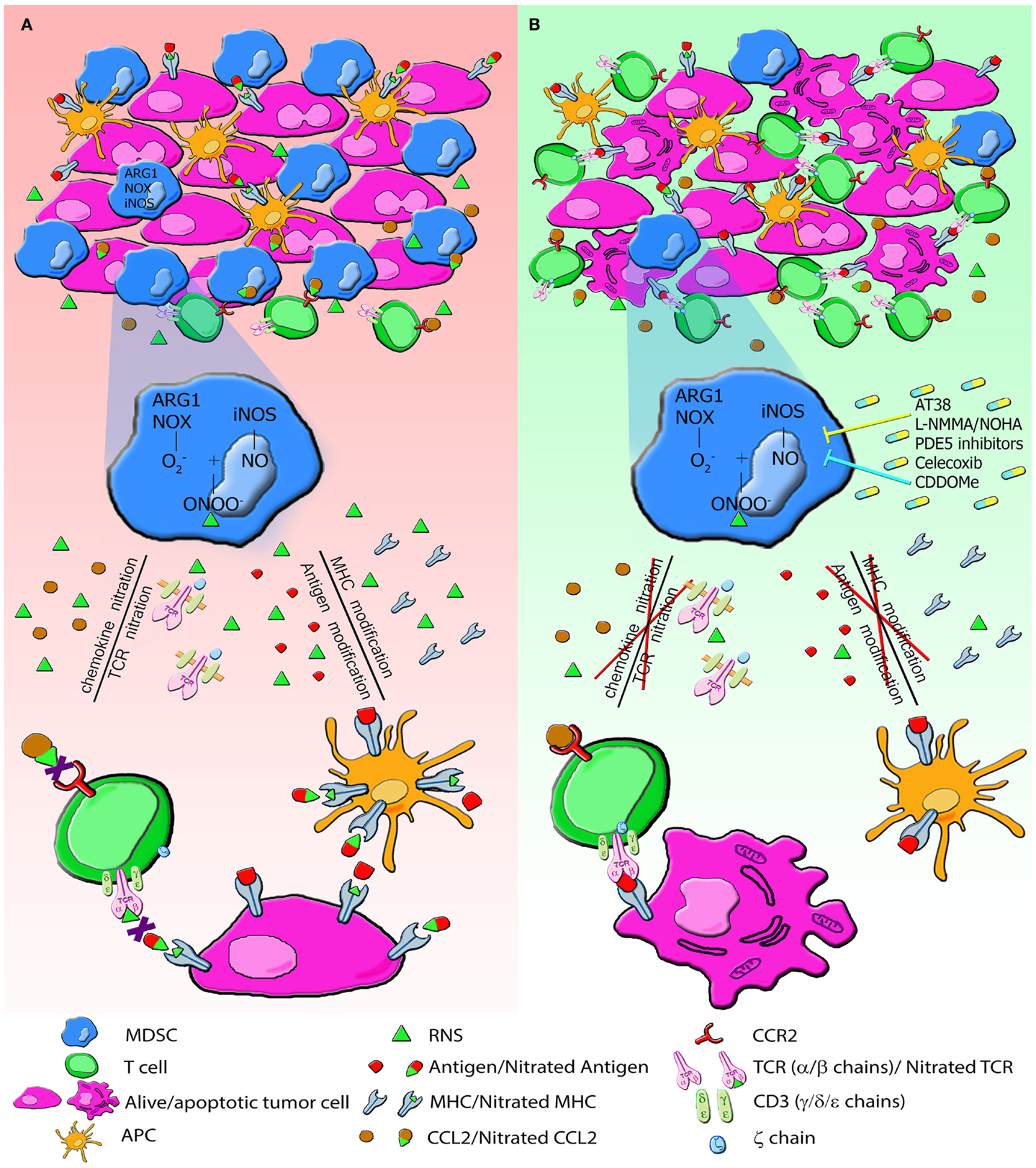
Figure 2. The generation of RNS in tumor environment induces PTM that alter immune responses to tumors. (A) MDSC and tumor cells express high level of NOX, ARG1, iNOS and produce large amount of RNS. RNS induce nitration/nitrosylation of either MHC class I molecules or peptide ligands on tumor cells and TCR chains on T lymphocytes, thus resulting in impaired recognition of tumor cells. RNS also modify chemotactic proprieties of chemokines. CCL2 nitration reduces the binding affinity to its receptor, CCR2, drastically affecting tumor homing of CD8+ T lymphocytes. All these modifications alter the recruitment of T cells and their cytotoxic function. (B) Administration of molecules AT38, l-NMMA, PDE5, Celecoxib, and CDDOMe through the enzymatic inhibition of NOX, ARG and NOS blocks nitration/nitrosylation, restores immune functions, and increases lymphocyte infiltration.
Conclusion
Reactive nitrogen species-induced modifications of proteins play a critical role in distracting the immune system from recognizing and eliminating tumor cells, and most likely act as an important barrier that limits the effectiveness of immunotherapy approaches. Targeting RNS can substantially improve the effect of cancer immune therapy and also their detection could be used as biomarker to define the prognosis and evaluate the clinical outcome of patients. In many types of cancer, tumor-infiltrating myeloid cells, such as MDSCs and TAMs, act as the major source of RNS in tumor tissues. Therefore, their elimination or functional block could also contribute to a fundamental reduction in RNS. Although several mechanisms of RNS-mediated effects on the immune system in cancer have been suggested, the underlying molecular processes need further elucidation. In this context, the development of new tools for the identification of nitrogen-induced PTM will allow clinicians and researchers to provide a signature of nitrated proteins, impaired signaling pathways, and cellular dysfunctions activated by those modifications under specific pathologic conditions, opening the way to a more customized therapeutic intervention. New cancer treatment protocols include combinatorial approaches such as tumor microenvironment preconditioning to eliminate patient immune dormancy. In this scenario, targeting RNS-induced PTM represents a new emerging frontier of combinatorial cancer therapy that can improve the efficacy of either active or passive immunotherapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell (2005) 7(3):211–7. doi: 10.1016/j.ccr.2005.02.013
2. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer (2005) 5(4):263–74. doi:10.1038/nrc1586
3. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (2011) 331(6024):1565–70. doi:10.1126/science.1203486
4. Bronte V, Cingarlini S, Marigo I, De Santo C, Gallina G, Dolcetti L, et al. Leukocyte infiltration in cancer creates an unfavorable environment for antitumor immune responses: a novel target for therapeutic intervention. Immunol Invest (2006) 35(3–4):327–57. doi:10.1080/08820130600754994
5. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi:10.1038/nri3175
6. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res (2007) 67(1):425. doi:10.1158/0008-5472.CAN-06-3037
7. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer cell (2005) 7(5):469–83. doi:10.1016/j.ccr.2005.04.023
8. Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother (2011) 60(10):1419–30. doi:10.1007/s00262-011-1028-0
9. Solito S, Falisi E, Diaz-Montero CM, Doni A, Pinton L, Rosato A, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood (2011) 118(8):2254–65. doi:10.1182/blood-2010-12-325753
10. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity (2010) 32(5):593–604. doi:10.1016/j.immuni.2010.05.007
11. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol (2010) 22(2):231–7. doi:10.1016/j.coi.2010.01.009
12. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23(11):549–55. doi:10.1016/S1471-4906(02)02302-5
13. Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest (2007) 117(5):1155–66. doi:10.1172/JCI31422
14. Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol (2009) 70(5):325–30. doi:10.1016/j.humimm.2009.02.008
15. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141(1):39–51. doi:10.1016/j.cell.2010.03.014
16. Burt BM, Rodig SJ, Tilleman TR, Elbardissi AW, Bueno R, Sugarbaker DJ. Circulating and tumor-infiltrating myeloid cells predict survival in human pleural mesothelioma. Cancer (2011) 117(22):5234–44. doi:10.1002/cncr.26143
17. Chung FT, Lee KY, Wang CW, Heh CC, Chan YF, Chen HW, et al. Tumor-associated macrophages correlate with response to epidermal growth factor receptor-tyrosine kinase inhibitors in advanced non-small cell lung cancer. Int J Cancer (2012) 131(3):E227–35. doi:10.1002/ijc.27403
18. Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol (2011) 11(10):702–11. doi:10.1038/nri3064
19. Grohmann U, Bronte V. Control of immune response by amino acid metabolism. Immunol Rev (2010) 236:243–64. doi:10.1111/j.1600-065X.2010.00915.x
20. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi:10.1158/0008-5472.CAN-04-0465
21. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol (2006) 176(11):6752–61.
22. Boulland ML, Marquet J, Molinier-Frenkel V, Moller P, Guiter C, Lasoudris F, et al. Human IL4I1 is a secreted L-phenylalanine oxidase expressed by mature dendritic cells that inhibits T-lymphocyte proliferation. Blood (2007) 110(1):220–7. doi:10.1182/blood-2006-07-036210
23. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res (2010) 70(1):68–77. doi:10.1158/0008-5472.CAN-09-2587
24. Michel T, Feron O. Nitric oxide synthases: which, where, how, and why? J Clin Invest (1997) 100(9):2146–52. doi:10.1172/JCI119750
25. Spratt DE, Taiakina V, Palmer M, Guillemette JG. Differential binding of calmodulin domains to constitutive and inducible nitric oxide synthase enzymes. Biochemistry (2007) 46(28):8288–300. doi:10.1021/bi062130b
26. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol (2005) 5(8):641–54. doi:10.1038/nri1668
27. Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J (1996) 313(Pt 1):17–29.
28. Heijnen HF, van Donselaar E, Slot JW, Fries DM, Blachard-Fillion B, Hodara R, et al. Subcellular localization of tyrosine-nitrated proteins is dictated by reactive oxygen species generating enzymes and by proximity to nitric oxide synthase. Free Radic Biol Med (2006) 40(11):1903–13. doi:10.1016/j.freeradbiomed.2005.09.006
29. Dalle-Donne I, Milzani A, Gagliano N, Colombo R, Giustarini D, Rossi R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid Redox Signal (2008) 10(3):445–73. doi:10.1089/ars.2007.1716
30. Leon L, Jeannin JF, Bettaieb A. Post-translational modifications induced by nitric oxide (NO): implication in cancer cells apoptosis. Nitric Oxide (2008) 19(2):77–83. doi:10.1016/j.niox.2008.04.014
31. Bian K, Gao Z, Weisbrodt N, Murad F. The nature of heme/iron-induced protein tyrosine nitration. Proc Natl Acad Sci U S A (2003) 100(10):5712–7. doi:10.1073/pnas.0931291100
32. Beckman JS, Chen J, Ischiropoulos H, Crow JP. Oxidative chemistry of peroxynitrite. Methods Enzymol (1994) 233:229–40. doi:10.1016/S0076-6879(94)33026-3
33. Beckman JS, Ischiropoulos H, Zhu L, van der Woerd M, Smith C, Chen J, et al. Kinetics of superoxide dismutase- and iron-catalyzed nitration of phenolics by peroxynitrite. Arch Biochem Biophys (1992) 298(2):438–45. doi:10.1016/0003-9861(92)90432-V
34. Kamisaki Y, Wada K, Bian K, Balabanli B, Davis K, Martin E, et al. An activity in rat tissues that modifies nitrotyrosine-containing proteins. Proc Natl Acad Sci U S A (1998) 95(20):11584–9. doi:10.1073/pnas.95.20.11584
35. Kuo WN, Kanadia RN, Shanbhag VP, Toro R. Denitration of peroxynitrite-treated proteins by ‘protein nitratases’ from rat brain and heart. Mol Cell Biochem (1999) 201(1–2):11–6. doi:10.1023/A:1007024126947
36. Kuo WN, Kanadia RN, Shanbhag VP. Denitration of peroxynitrite-treated proteins by “protein nitratases” from dog prostate. Biochem Mol Biol Int (1999) 47(6):1061–7.
37. Kang M, Akbarali HI. Denitration of L-type calcium channel. FEBS Lett (2008) 582(20):3033–6. doi:10.1016/j.febslet.2008.07.042
38. Smallwood HS, Lourette NM, Boschek CB, Bigelow DJ, Smith RD, Pasa-Tolic L, et al. Identification of a denitrase activity against calmodulin in activated macrophages using high-field liquid chromatography – FTICR mass spectrometry. Biochemistry (2007) 46(37):10498–505. doi:10.1021/bi7009713
39. Koeck T, Fu X, Hazen SL, Crabb JW, Stuehr DJ, Aulak KS. Rapid and selective oxygen-regulated protein tyrosine denitration and nitration in mitochondria. J Biol Chem (2004) 279(26):27257–62. doi:10.1074/jbc.M401586200
40. Ischiropoulos H. Biological selectivity and functional aspects of protein tyrosine nitration. Biochem Biophys Res Commun (2003) 305(3):776–83. doi:10.1016/S0006-291X(03)00814-3
41. Gole MD, Souza JM, Choi I, Hertkorn C, Malcolm S, Foust RF III, et al. Plasma proteins modified by tyrosine nitration in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol (2000) 278(5):L961–7.
42. Knapp LT, Kanterewicz BI, Hayes EL, Klann E. Peroxynitrite-induced tyrosine nitration and inhibition of protein kinase C. Biochem Biophys Res Commun (2001) 286(4):764–70. doi:10.1006/bbrc.2001.5448
43. Cassina AM, Hodara R, Souza JM, Thomson L, Castro L, Ischiropoulos H, et al. Cytochrome c nitration by peroxynitrite. J Biol Chem (2000) 275(28):21409–15. doi:10.1074/jbc.M909978199
44. Vadseth C, Souza JM, Thomson L, Seagraves A, Nagaswami C, Scheiner T, et al. Pro-thrombotic state induced by post-translational modification of fibrinogen by reactive nitrogen species. J Biol Chem (2004) 279(10):8820–6. doi:10.1074/jbc.M306101200
45. Balafanova Z, Bolli R, Zhang J, Zheng Y, Pass JM, Bhatnagar A, et al. Nitric oxide (NO) induces nitration of protein kinase Cepsilon (PKCepsilon), facilitating PKCepsilon translocation via enhanced PKCepsilon -RACK2 interactions: a novel mechanism of no-triggered activation of PKCepsilon. J Biol Chem (2002) 277(17):15021–7. doi:10.1074/jbc.M112451200
46. Ji Y, Neverova I, Van Eyk JE, Bennett BM. Nitration of tyrosine 92 mediates the activation of rat microsomal glutathione S-transferase by peroxynitrite. J Biol Chem (2006) 281(4):1986–91. doi:10.1074/jbc.M509480200
47. Bentz BG, Haines GK III, Radosevich JA. Increased protein nitrosylation in head and neck squamous cell carcinogenesis. Head Neck (2000) 22(1):64–70. doi:10.1002/(SICI)1097-0347(200001)22:1<64::AID-HED10>3.3.CO;2-A
48. Fitzpatrick B, Mehibel M, Cowen RL, Stratford IJ. iNOS as a therapeutic target for treatment of human tumors. Nitric Oxide (2008) 19(2):217–24. doi:10.1016/j.niox.2008.05.001
49. Iyer AK, Azad N, Wang L, Rojanasakul Y. Role of S-nitrosylation in apoptosis resistance and carcinogenesis. Nitric Oxide (2008) 19(2):146–51. doi:10.1016/j.niox.2008.04.019
50. Leon-Bollotte L, Subramaniam S, Cauvard O, Plenchette-Colas S, Paul C, Godard C, et al. S-nitrosylation of the death receptor fas promotes fas ligand-mediated apoptosis in cancer cells. Gastroenterology (2011) 140(7):2009–18. doi:10.1053/j.gastro.2011.02.053
51. Calmels S, Hainaut P, Ohshima H. Nitric oxide induces conformational and functional modifications of wild-type p53 tumor suppressor protein. Cancer Res (1997) 57(16):3365–9.
52. Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, et al. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A (2009) 106(15):6297–302. doi:10.1073/pnas.0901043106
53. Liu L, Xu-Welliver M, Kanugula S, Pegg AE. Inactivation and degradation of O(6)-alkylguanine-DNA alkyltransferase after reaction with nitric oxide. Cancer Res (2002) 62(11):3037–43.
54. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer (2007) 7(12):961–7. doi:10.1038/nrc2254
55. Tripathi DN, Chowdhury R, Trudel LJ, Tee AR, Slack RS, Walker CL, et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci U S A (2013) 110(32):E2950–7. doi:10.1073/pnas.1307736110
56. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev (2007) 87(1):245–313. doi:10.1152/physrev.00044.2005
57. Brandes RP, Schroder K. Differential vascular functions of Nox family NADPH oxidases. Curr Opin Lipidol (2008) 19(5):513–8. doi:10.1097/MOL.0b013e32830c91e3
58. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol (2009) 182(9):5693–701. doi:10.4049/jimmunol.0900092
59. Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med (2008) 205(10):2235–49. doi:10.1084/jem.20080132
60. Manea A, Tanase LI, Raicu M, Simionescu M. Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol (2010) 30(1):105–12. doi:10.1161/ATVBAHA.109.193896
61. Raad H, Paclet MH, Boussetta T, Kroviarski Y, Morel F, Quinn MT, et al. Regulation of the phagocyte NADPH oxidase activity: phosphorylation of gp91phox/NOX2 by protein kinase C enhances its diaphorase activity and binding to Rac2, p67phox, and p47phox. FASEB J (2009) 23(4):1011–22. doi:10.1096/fj.08-114553
62. Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science (1995) 267(5203):1485–8. doi:10.1126/science.7878466
63. Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci (2004) 75(6):639–53. doi:10.1016/j.lfs.2003.10.042
64. Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide (2010) 23(2):75–93. doi:10.1016/j.niox.2010.04.007
65. Lowenstein CJ, Alley EW, Raval P, Snowman AM, Snyder SH, Russell SW, et al. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc Natl Acad Sci U S A (1993) 90(20):9730–4. doi:10.1073/pnas.90.20.9730
66. Kristof AS, Marks-Konczalik J, Moss J. Mitogen-activated protein kinases mediate activator protein-1-dependent human inducible nitric-oxide synthase promoter activation. J Biol Chem (2001) 276(11):8445–52. doi:10.1074/jbc.M009563200
67. Galea E, Feinstein DL. Regulation of the expression of the inflammatory nitric oxide synthase (NOS2) by cyclic AMP. FASEB J (1999) 13(15):2125–37.
68. Mullet D, Fertel RH, Kniss D, Cox GW. An increase in intracellular cyclic AMP modulates nitric oxide production in IFN-gamma-treated macrophages. J Immunol (1997) 158(2):897–904.
69. Park KS, Guo Z, Shao L, Du Q, Geller DA. A far-upstream Oct-1 motif regulates cytokine-induced transcription of the human inducible nitric oxide synthase gene. J Mol Biol (2009) 390(4):595–603. doi:10.1016/j.jmb.2009.05.036
70. Perrella MA, Yoshizumi M, Fen Z, Tsai JC, Hsieh CM, Kourembanas S, et al. Transforming growth factor-beta 1, but not dexamethasone, down-regulates nitric-oxide synthase mRNA after its induction by interleukin-1 beta in rat smooth muscle cells. J Biol Chem (1994) 269(20):14595–600.
71. Rodriguez-Pascual F, Hausding M, Ihrig-Biedert I, Furneaux H, Levy AP, Forstermann U, et al. Complex contribution of the 3’-untranslated region to the expressional regulation of the human inducible nitric-oxide synthase gene. Involvement of the RNA-binding protein HuR. J Biol Chem (2000) 275(34):26040–9. doi:10.1074/jbc.M910460199
72. Walker G, Pfeilschifter J, Kunz D. Mechanisms of suppression of inducible nitric-oxide synthase (iNOS) expression in interferon (IFN)-gamma-stimulated RAW 264.7 cells by dexamethasone. Evidence for glucocorticoid-induced degradation of iNOS protein by calpain as a key step in post-transcriptional regulation. J Biol Chem (1997) 272(26):16679–87.
73. Felley-Bosco E, Bender FC, Courjault-Gautier F, Bron C, Quest AF. Caveolin-1 down-regulates inducible nitric oxide synthase via the proteasome pathway in human colon carcinoma cells. Proc Natl Acad Sci U S A (2000) 97(26):14334–9. doi:10.1073/pnas.250406797
74. Mitani T, Terashima M, Yoshimura H, Nariai Y, Tanigawa Y. TGF-beta1 enhances degradation of IFN-gamma-induced iNOS protein via proteasomes in RAW 264.7 cells. Nitric Oxide (2005) 13(1):78–87. doi:10.1016/j.niox.2005.05.001
75. Paukkeri EL, Leppanen T, Sareila O, Vuolteenaho K, Kankaanranta H, Moilanen E. PPARalpha agonists inhibit nitric oxide production by enhancing iNOS degradation in LPS-treated macrophages. Br J Pharmacol (2007) 152(7):1081–91. doi:10.1038/sj.bjp.0707477
76. Kuang Z, Lewis RS, Curtis JM, Zhan Y, Saunders BM, Babon JJ, et al. The SPRY domain-containing SOCS box protein SPSB2 targets iNOS for proteasomal degradation. J Cell Biol (2010) 190(1):129–41. doi:10.1083/jcb.200912087
77. Nishiya T, Matsumoto K, Maekawa S, Kajita E, Horinouchi T, Fujimuro M, et al. Regulation of inducible nitric-oxide synthase by the SPRY domain- and SOCS box-containing proteins. J Biol Chem (2011) 286(11):9009–19. doi:10.1074/jbc.M110.190678
78. Iyer R, Jenkinson CP, Vockley JG, Kern RM, Grody WW, Cederbaum S. The human arginases and arginase deficiency. J Inherit Metab Dis (1998) 21(Suppl 1):86–100. doi:10.1023/A:1005383919335
79. Gray MJ, Poljakovic M, Kepka-Lenhart D, Morris SM Jr. Induction of arginase I transcription by IL-4 requires a composite DNA response element for STAT6 and C/EBPbeta. Gene (2005) 353(1):98–106. doi:10.1016/j.gene.2005.04.004
80. Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol (2004) 172(12):7565–73.
81. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202(7):931–9. doi:10.1084/jem.20050715
82. Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, et al. Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. J Immunol (2011) 187(5):2181–92. doi:10.4049/jimmunol.1003460
83. Santhanam L, Lim HK, Miriel V, Brown T, Patel M, Balanson S, et al. Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ Res (2007) 101(7):692–702. doi:10.1161/CIRCRESAHA.107.157727
84. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell (2010) 40(2):294–309. doi:10.1016/j.molcel.2010.09.022
85. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207(11):2439–53. doi:10.1084/jem.20100587
86. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer (2003) 3(10):721–32. doi:10.1038/nrc1187
87. Melillo G, Musso T, Sica A, Taylor LS, Cox GW, Varesio L. A hypoxia-responsive element mediates a novel pathway of activation of the inducible nitric oxide synthase promoter. J Exp Med (1995) 182(6):1683–93. doi:10.1084/jem.182.6.1683
88. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res (2010) 70(19):7465–75. doi:10.1158/0008-5472.CAN-10-1439
89. Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell (2003) 14(8):3470–81. doi:10.1091/mbc.E02-12-0791
90. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity (2010) 32(6):790–802. doi:10.1016/j.immuni.2010.05.010
91. Allavena P, Bianchi G, Zhou D, van Damme J, Jilek P, Sozzani S, et al. Induction of natural killer cell migration by monocyte chemotactic protein-1, -2 and -3. Eur J Immunol (1994) 24(12):3233–6. doi:10.1002/eji.1830241249
92. van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood (2012) 119(15):3383–93. doi:10.1182/blood-2011-11-370130
93. Wu L, Du H, Li Y, Qu P, Yan C. Signal transducer and activator of transcription 3 (Stat3C) promotes myeloid-derived suppressor cell expansion and immune suppression during lung tumorigenesis. Am J Pathol (2011) 179(4):2131–41. doi:10.1016/j.ajpath.2011.06.028
94. Martin E, Nathan C, Xie QW. Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J Exp Med (1994) 180(3):977–84. doi:10.1084/jem.180.3.977
95. Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat Immunol (2003) 4(3):269–73. doi:10.1038/ni888
96. Munder M, Mollinedo F, Calafat J, Canchado J, Gil-Lamaignere C, Fuentes JM, et al. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood (2005) 105(6):2549–56. doi:10.1182/blood-2004-07-2521
97. Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol (2009) 158(3):638–51. doi:10.1111/j.1476-5381.2009.00291.x
98. Pesce J, Kaviratne M, Ramalingam TR, Thompson RW, Urban JF Jr, Cheever AW, et al. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J Clin Invest (2006) 116(7):2044–55. doi:10.1172/JCI27727
99. Hecker M, Nematollahi H, Hey C, Busse R, Racke K. Inhibition of arginase by NG-hydroxy-L-arginine in alveolar macrophages: implications for the utilization of L-arginine for nitric oxide synthesis. FEBS Lett (1995) 359(2–3):251–4. doi:10.1016/0014-5793(95)00039-C
100. Sanjabi S, Zenewicz LA, Kamanaka M, Flavell RA. Anti-inflammatory and pro-inflammatory roles of TGF-beta, IL-10, and IL-22 in immunity and autoimmunity. Curr Opin Pharmacol (2009) 9(4):447–53. doi:10.1016/j.coph.2009.04.008
101. Boutard V, Havouis R, Fouqueray B, Philippe C, Moulinoux JP, Baud L. Transforming growth factor-beta stimulates arginase activity in macrophages. Implications for the regulation of macrophage cytotoxicity. J Immunol (1995) 155(4):2077–84.
102. Chen YH, Layne MD, Chung SW, Ejima K, Baron RM, Yet SF, et al. Elk-3 is a transcriptional repressor of nitric-oxide synthase 2. J Biol Chem (2003) 278(41):39572–7. doi:10.1074/jbc.M308179200
103. Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest (2006) 116(10):2777–90. doi:10.1172/JCI28828
104. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi:10.1038/nri2506
105. Brito C, Naviliat M, Tiscornia AC, Vuillier F, Gualco G, Dighiero G, et al. Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J Immunol (1999) 162(6):3356–66.
106. Aulak KS, Miyagi M, Yan L, West KA, Massillon D, Crabb JW, et al. Proteomic method identifies proteins nitrated in vivo during inflammatory challenge. Proc Natl Acad Sci U S A (2001) 98(21):12056–61. doi:10.1073/pnas.221269198
107. Moulian N, Truffault F, Gaudry-Talarmain YM, Serraf A, Berrih-Aknin S. In vivo and in vitro apoptosis of human thymocytes are associated with nitrotyrosine formation. Blood (2001) 97(11):3521–30. doi:10.1182/blood.V97.11.3521
108. Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest (2011) 121(10):4015–29. doi:10.1172/JCI45862
109. Abello N, Kerstjens HA, Postma DS, Bischoff R. Protein tyrosine nitration: selectivity, physicochemical and biological consequences, denitration, and proteomics methods for the identification of tyrosine-nitrated proteins. J Proteome Res (2009) 8(7):3222–38. doi:10.1021/pr900039c
110. Birnboim HC, Lemay AM, Lam DK, Goldstein R, Webb JR. Cutting edge: MHC class II-restricted peptides containing the inflammation-associated marker 3-nitrotyrosine evade central tolerance and elicit a robust cell-mediated immune response. J Immunol (2003) 171(2):528–32.
111. Hardy LL, Wick DA, Webb JR. Conversion of tyrosine to the inflammation-associated analog 3’-nitrotyrosine at either TCR- or MHC-contact positions can profoundly affect recognition of the MHC class I-restricted epitope of lymphocytic choriomeningitis virus glycoprotein 33 by CD8 T cells. J Immunol (2008) 180(9):5956–62.
112. Madhurantakam C, Duru AD, Sandalova T, Webb JR, Achour A. Inflammation-associated nitrotyrosination affects TCR recognition through reduced stability and alteration of the molecular surface of the MHC complex. PLoS One (2012) 7(3):e32805. doi:10.1371/journal.pone.0032805
113. Sandhu JK, Privora HF, Wenckebach G, Birnboim HC. Neutrophils, nitric oxide synthase, and mutations in the mutatect murine tumor model. Am J Pathol (2000) 156(2):509–18. doi:10.1016/S0002-9440(10)64755-4
114. Herzog J, Maekawa Y, Cirrito TP, Illian BS, Unanue ER. Activated antigen-presenting cells select and present chemically modified peptides recognized by unique CD4 T cells. Proc Natl Acad Sci U S A (2005) 102(22):7928–33. doi:10.1073/pnas.0502255102
115. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13(7):828–35. doi:10.1038/nm1609
116. Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol (2010) 184(6):3106–16. doi:10.4049/jimmunol.0902661
117. Kasic T, Colombo P, Soldani C, Wang CM, Miranda E, Roncalli M, et al. Modulation of human T-cell functions by reactive nitrogen species. Eur J Immunol (2011) 41(7):1843–9. doi:10.1002/eji.201040868
118. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208(10):1949–62. doi:10.1084/jem.20101956
119. Curnock AP, Logan MK, Ward SG. Chemokine signalling: pivoting around multiple phosphoinositide 3-kinases. Immunology (2002) 105(2):125–36. doi:10.1046/j.1365-2567.2002.01345.x
120. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med (2006) 354(6):610–21. doi:10.1056/NEJMra052723
121. Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharmacol Toxicol (2008) 48:171–97. doi:10.1146/annurev.pharmtox.48.121806.154841
122. Mortier A, Van Damme J, Proost P. Overview of the mechanisms regulating chemokine activity and availability. Immunol Lett (2012) 145(1–2):2–9. doi:10.1016/j.imlet.2012.04.015
123. Moelants EA, Mortier A, Van Damme J, Proost P. In vivo regulation of chemokine activity by post-translational modification. Immunol Cell Biol (2013) 91(6):402–7. doi:10.1038/icb.2013.16
124. Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med (2005) 201(8):1257–68. doi:10.1084/jem.20042028
125. Nemirovskiy OV, Radabaugh MR, Aggarwal P, Funckes-Shippy CL, Mnich SJ, Meyer DM, et al. Plasma 3-nitrotyrosine is a biomarker in animal models of arthritis: pharmacological dissection of iNOS’ role in disease. Nitric Oxide (2009) 20(3):150–6. doi:10.1016/j.niox.2008.12.005
126. Butterfield DA, Reed T, Sultana R. Roles of 3-nitrotyrosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer’s disease. Free Radic Res (2011) 45(1):59–72. doi:10.3109/10715762.2010.520014
127. Sokolovsky M, Riordan JF, Vallee BL. Conversion of 3-nitrotyrosine to 3-aminotyrosine in peptides and proteins. Biochem Biophys Res Commun (1967) 27(1):20–5. doi:10.1016/S0006-291X(67)80033-0
128. Tsikas D, Caidahl K. Recent methodological advances in the mass spectrometric analysis of free and protein-associated 3-nitrotyrosine in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci (2005) 814(1):1–9. doi:10.1016/j.jchromb.2004.10.003
129. Frost MT, Halliwell B, Moore KP. Analysis of free and protein-bound nitrotyrosine in human plasma by a gas chromatography/mass spectrometry method that avoids nitration artifacts. Biochem J (2000) 345(Pt 3):453–8. doi:10.1042/0264-6021:3450453
130. Bigelow DJ, Qian WJ. Quantitative proteome mapping of nitrotyrosines. Methods Enzymol (2008) 440:191–205. doi:10.1016/S0076-6879(07)00811-7
131. Nuriel T, Deeb RS, Hajjar DP, Gross SS. Protein 3-nitrotyrosine in complex biological samples: quantification by high-pressure liquid chromatography/electrochemical detection and emergence of proteomic approaches for unbiased identification of modification sites. Methods Enzymol (2008) 441:1–17. doi:10.1016/S0076-6879(08)01201-9
132. Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci U S A (2004) 101(12):4003–8. doi:10.1073/pnas.0307446101
133. Yamakura F, Ikeda K. Modification of tryptophan and tryptophan residues in proteins by reactive nitrogen species. Nitric Oxide (2006) 14(2):152–61. doi:10.1016/j.niox.2005.07.009
134. Ikeda K, Yukihiro Hiraoka B, Iwai H, Matsumoto T, Mineki R, Taka H, et al. Detection of 6-nitrotryptophan in proteins by Western blot analysis and its application for peroxynitrite-treated PC12 cells. Nitric Oxide (2007) 16(1):18–28. doi:10.1016/j.niox.2006.04.263
135. Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer (2006) 6(7):521–34. doi:10.1038/nrc1910
136. Rahat MA, Hemmerlein B. Macrophage-tumor cell interactions regulate the function of nitric oxide. Front Physiol (2013) 4:144. doi:10.3389/fphys.2013.00144
137. Zhang W, He XJ, Ma YY, Wang HJ, Xia YJ, Zhao ZS, et al. Inducible nitric oxide synthase expression correlates with angiogenesis, lymphangiogenesis, and poor prognosis in gastric cancer patients. Hum Pathol (2011) 42(9):1275–82. doi:10.1016/j.humpath.2010.09.020
138. Ekmekcioglu S, Ellerhorst JA, Prieto VG, Johnson MM, Broemeling LD, Grimm EA. Tumor iNOS predicts poor survival for stage III melanoma patients. Int J Cancer (2006) 119(4):861–6. doi:10.1002/ijc.21767
139. Eggen T, Sager G, Arnes M, Pettersen I, Orbo A. Expression of iNOS – a favourable prognostic marker for early-stage carcinoma of the uterine cervix. Anticancer Res (2011) 31(6):2319–25.
140. Raspollini MR, Amunni G, Villanucci A, Boddi V, Baroni G, Taddei A, et al. Expression of inducible nitric oxide synthase and cyclooxygenase-2 in ovarian cancer: correlation with clinical outcome. Gynecol Oncol (2004) 92(3):806–12. doi:10.1016/j.ygyno.2003.12.023
141. Oka K, Suzuki Y, Iida H, Nakano T. Pd-ECGF positivity correlates with better survival, while iNOS has no predictive value for cervical carcinomas treated with radiotherapy. Int J Radiat Oncol Biol Phys (2003) 57(1):217–21. doi:10.1016/S0360-3016(03)00436-X
142. Loibl S, von Minckwitz G, Weber S, Sinn HP, Schini-Kerth VB, Lobysheva I, et al. Expression of endothelial and inducible nitric oxide synthase in benign and malignant lesions of the breast and measurement of nitric oxide using electron paramagnetic resonance spectroscopy. Cancer (2002) 95(6):1191–8. doi:10.1002/cncr.10817
143. Cobbs CS, Brenman JE, Aldape KD, Bredt DS, Israel MA. Expression of nitric oxide synthase in human central nervous system tumors. Cancer Res (1995) 55(4):727–30.
144. Daniliuc S, Bitterman H, Rahat MA, Kinarty A, Rosenzweig D, Lahat N. Hypoxia inactivates inducible nitric oxide synthase in mouse macrophages by disrupting its interaction with alpha-actinin 4. J Immunol (2003) 171(6):3225–32.
145. Beckmann JS, Ye YZ, Anderson PG, Chen J, Accavitti MA, Tarpey MM, et al. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol Chem Hoppe Seyler (1994) 375(2):81–8. doi:10.1515/bchm3.1994.375.2.81
146. Raber P, Ochoa AC, Rodriguez PC. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol Invest (2012) 41(6–7):614–34. doi:10.3109/08820139.2012.680634
147. Gochman E, Mahajna J, Shenzer P, Dahan A, Blatt A, Elyakim R, et al. The expression of iNOS and nitrotyrosine in colitis and colon cancer in humans. Acta Histochem (2012) 114(8):827–35. doi:10.1016/j.acthis.2012.02.004
148. Li LG, Xu HM. Inducible nitric oxide synthase, nitrotyrosine and apoptosis in gastric adenocarcinomas and their correlation with a poor survival. World J Gastroenterol (2005) 11(17):2539–44.
149. Nakamura Y, Yasuoka H, Tsujimoto M, Yoshidome K, Nakahara M, Nakao K, et al. Nitric oxide in breast cancer: induction of vascular endothelial growth factor-C and correlation with metastasis and poor prognosis. Clin Cancer Res (2006) 12(4):1201–7. doi:10.1158/1078-0432.CCR-05-1269
150. Kinnula VL, Torkkeli T, Kristo P, Sormunen R, Soini Y, Paakko P, et al. Ultrastructural and chromosomal studies on manganese superoxide dismutase in malignant mesothelioma. Am J Respir Cell Mol Biol (2004) 31(2):147–53. doi:10.1165/rcmb.2003-0409OC
151. Nakamura Y, Yasuoka H, Zuo H, Takamura Y, Miyauchi A, Nakamura M, et al. Nitric oxide in papillary thyroid carcinoma: induction of vascular endothelial growth factor D and correlation with lymph node metastasis. J Clin Endocrinol Metab (2006) 91(4):1582–5. doi:10.1210/jc.2005-1790
152. Vickers SM, MacMillan-Crow LA, Green M, Ellis C, Thompson JA. Association of increased immunostaining for inducible nitric oxide synthase and nitrotyrosine with fibroblast growth factor transformation in pancreatic cancer. Arch Surg (1999) 134(3):245–51. doi:10.1001/archsurg.134.3.245
153. Cobbs CS, Samanta M, Harkins LE, Gillespie GY, Merrick BA, MacMillan-Crow LA. Evidence for peroxynitrite-mediated modifications to p53 in human gliomas: possible functional consequences. Arch Biochem Biophys (2001) 394(2):167–72. doi:10.1006/abbi.2001.2540
154. Cobbs CS, Whisenhunt TR, Wesemann DR, Harkins LE, Van Meir EG, Samanta M. Inactivation of wild-type p53 protein function by reactive oxygen and nitrogen species in malignant glioma cells. Cancer Res (2003) 63(24):8670–3.
155. Masri FA, Comhair SA, Koeck T, Xu W, Janocha A, Ghosh S, et al. Abnormalities in nitric oxide and its derivatives in lung cancer. Am J Respir Crit Care Med (2005) 172(5):597–605. doi:10.1164/rccm.200411-1523OC
156. Bentz BG, Haines GK III, Lingen MW, Pelzer HJ, Hanson DG, Radosevich JA. Nitric oxide synthase type 3 is increased in squamous hyperplasia, dysplasia, and squamous cell carcinoma of the head and neck. Ann Otol Rhinol Laryngol (1999) 108(8):781–7.
157. Kojima M, Morisaki T, Tsukahara Y, Uchiyama A, Matsunari Y, Mibu R, et al. Nitric oxide synthase expression and nitric oxide production in human colon carcinoma tissue. J Surg Oncol (1999) 70(4):222–9. doi:10.1002/(SICI)1096-9098(199904)70:4<222::AID-JSO5>3.0.CO;2-G
158. Kitano H, Kitanishi T, Nakanishi Y, Suzuki M, Takeuchi E, Yazawa Y, et al. Expression of inducible nitric oxide synthase in human thyroid papillary carcinomas. Thyroid (1999) 9(2):113–7. doi:10.1089/thy.1999.9.113
159. Ehsan A, Sommer F, Schmidt A, Klotz T, Koslowski J, Niggemann S, et al. Nitric oxide pathways in human bladder carcinoma. The distribution of nitric oxide synthases, soluble guanylyl cyclase, cyclic guanosine monophosphate, and nitrotyrosine. Cancer (2002) 95(11):2293–301. doi:10.1002/cncr.10942
160. Bancel B, Esteve J, Souquet JC, Toyokuni S, Ohshima H, Pignatelli B. Differences in oxidative stress dependence between gastric adenocarcinoma subtypes. World J Gastroenterol (2006) 12(7):1005–12.
161. Soini Y, Kallio JP, Hirvikoski P, Helin H, Kellokumpu-Lehtinen P, Kang SW, et al. Oxidative/nitrosative stress and peroxiredoxin 2 are associated with grade and prognosis of human renal carcinoma. APMIS (2006) 114(5):329–37. doi:10.1111/j.1600-0463.2006.apm_315.x
162. Karihtala P, Soini Y, Vaskivuo L, Bloigu R, Puistola U. DNA adduct 8-hydroxydeoxyguanosine, a novel putative marker of prognostic significance in ovarian carcinoma. Int J Gynecol Cancer (2009) 19(6):1047–51. doi:10.1111/IGC.0b013e3181ad0f0d
163. Pylvas M, Puistola U, Kauppila S, Soini Y, Karihtala P. Oxidative stress-induced antioxidant enzyme expression is an early phenomenon in ovarian carcinogenesis. Eur J Cancer (2010) 46(9):1661–7. doi:10.1016/j.ejca.2010.02.006
164. Yamanaka Y, Friess H, Buchler M, Beger HG, Uchida E, Onda M, et al. Overexpression of acidic and basic fibroblast growth factors in human pancreatic cancer correlates with advanced tumor stage. Cancer Res (1993) 53(21):5289–96.
165. Aktan B, Taysi S, Gumustekin K, Ucuncu H, Memisogullari R, Save K, et al. Effect of macrolide antibiotics on nitric oxide synthase and xanthine oxidase activities, and malondialdehyde level in erythrocyte of the guinea pigs with experimental otitis media with effusion. Pol J Pharmacol (2003) 55(6):1105–10.
166. Mariotto S, Cuzzolin L, Adami A, Del Soldato P, Suzuki H, Benoni G. Effect of a new non-steroidal anti-inflammatory drug, nitroflurbiprofen, on the expression of inducible nitric oxide synthase in rat neutrophils. Br J Pharmacol (1995) 115(2):225–6. doi:10.1111/j.1476-5381.1995.tb15867.x
167. Griscavage JM, Hobbs AJ, Ignarro LJ. Negative modulation of nitric oxide synthase by nitric oxide and nitroso compounds. Adv Pharmacol (1995) 34:215–34. doi:10.1016/S1054-3589(08)61088-1
168. De Santo C, Serafini P, Marigo I, Dolcetti L, Bolla M, Del Soldato P, et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci U S A (2005) 102(11):4185–90. doi:10.1073/pnas.0409783102
169. Hickman HD, Reynoso GV, Ngudiankama BF, Rubin EJ, Magadan JG, Cush SS, et al. Anatomically restricted synergistic antiviral activities of innate and adaptive immune cells in the skin. Cell Host Microbe (2013) 13(2):155–68. doi:10.1016/j.chom.2013.01.004
170. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res (2007) 67(9):4507–13. doi:10.1158/0008-5472.CAN-06-4174
171. Talmadge JE, Hood KC, Zobel LC, Shafer LR, Coles M, Toth B. Chemoprevention by cyclooxygenase-2 inhibition reduces immature myeloid suppressor cell expansion. Int Immunopharmacol (2007) 7(2):140–51. doi:10.1016/j.intimp.2006.09.021
172. Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer (2010) 10:464. doi:10.1186/1471-2407-10-464
173. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med (2006) 203(12):2691–702. doi:10.1084/jem.20061104
174. Sikora AG, Gelbard A, Davies MA, Sano D, Ekmekcioglu S, Kwon J, et al. Targeted inhibition of inducible nitric oxide synthase inhibits growth of human melanoma in vivo and synergizes with chemotherapy. Clin Cancer Res (2010) 16(6):1834–44. doi:10.1158/1078-0432.CCR-09-3123
175. Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, Cotter MJ, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res (2010) 16(6):1812–23. doi:10.1158/1078-0432.CCR-09-3272
176. Burkitt MJ, Duncan J. Effects of trans-resveratrol on copper-dependent hydroxyl-radical formation and DNA damage: evidence for hydroxyl-radical scavenging and a novel, glutathione-sparing mechanism of action. Arch Biochem Biophys (2000) 381(2):253–63. doi:10.1006/abbi.2000.1973
177. Hsieh TC, Wang Z, Hamby CV, Wu JM. Inhibition of melanoma cell proliferation by resveratrol is correlated with upregulation of quinone reductase 2 and p53. Biochem Biophys Res Commun (2005) 334(1):223–30. doi:10.1016/j.bbrc.2005.06.073
178. Bishayee A. Cancer prevention and treatment with resveratrol: from rodent studies to clinical trials. Cancer Prev Res (Phila) (2009) 2(5):409–18. doi:10.1158/1940-6207.CAPR-08-0160
179. Zheng M, Ekmekcioglu S, Walch ET, Tang CH, Grimm EA. Inhibition of nuclear factor-kappaB and nitric oxide by curcumin induces G2/M cell cycle arrest and apoptosis in human melanoma cells. Melanoma Res (2004) 14(3):165–71. doi:10.1097/01.cmr.0000129374.76399.19
180. Du Q, Hu B, An HM, Shen KP, Xu L, Deng S, et al. Synergistic anticancer effects of curcumin and resveratrol in Hepa1-6 hepatocellular carcinoma cells. Oncol Rep (2013) 29(5):1851–8. doi:10.3892/or.2013.2310
Keywords: RNS, nitrotyrosine, cancer, microenvironment, immune escape
Citation: De Sanctis F, Sandri S, Ferrarini G, Pagliarello I, Sartoris S, Ugel S, Marigo I, Molon B and Bronte V (2014) The emerging immunological role of post-translational modifications by reactive nitrogen species in cancer microenvironment. Front. Immunol. 5:69. doi: 10.3389/fimmu.2014.00069
Received: 12 December 2013; Accepted: 08 February 2014;
Published online: 24 February 2014.
Edited by:
Fabrizio Mattei, Istituto Superiore di Sanità, ItalyReviewed by:
F. Ronchese, Victoria University of Wellington, New ZealandDennis O. Adeegbe, Immunology Frontier Research Center, Japan
Copyright: © 2014 De Sanctis, Sandri, Ferrarini, Pagliarello, Sartoris, Ugel, Marigo, Molon and Bronte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincenzo Bronte, Immunology Section, Department of Pathology and Diagnostics, University of Verona, C/o Policlinico G.B. Rossi, P.le L.A. Scuro 10, Verona 37134, Italy e-mail:dmluY2Vuem8uYnJvbnRlQHVuaXZyLml0