 Beatrice Omusiro Ondondo
Beatrice Omusiro Ondondo- The Jenner Institute, Nuffield Department of Medicine, University of Oxford, Oxford, UK
Excessive immune responses directed against foreign pathogens, self-antigens, or commensal microflora can cause cancer establishment and progression if the execution of tight immuno-regulatory mechanisms fails. On the other hand, induction of potent tumor antigen-specific immune responses together with stimulation of the innate immune system is a pre-requisite for effective anti-tumor immunity, and if suppressed by the strong immuno-regulatory mechanisms can lead to cancer progression. Therefore, it is crucial that the inevitable co-existence of these fundamental, yet conflicting roles of immune-regulatory cells is carefully streamlined as imbalances can be detrimental to the host. Infection with chronic persistent viruses is characterized by severe immune dysfunction resulting in T cell exhaustion and sometimes deletion of antigen-specific T cells. More often, this is due to increased immuno-regulatory processes, which are triggered to down-regulate immune responses and limit immunopathology. However, such heightened levels of immune disruption cause a concomitant loss of tumor immune-surveillance and create a permissive microenvironment for cancer establishment and progression, as demonstrated by increased incidences of cancer in immunosuppressed hosts. Paradoxically, while some cancers arise as a consequence of increased immuno-regulatory mechanisms that inhibit protective immune responses and impinge on tumor surveillance, other cancers arise due to impaired immuno-regulatory mechanisms and failure to limit pathogenic inflammatory responses. This intricate complexity, where immuno-regulatory cells can be beneficial in certain immune settings but detrimental in other settings underscores the need for carefully formulated interventions to equilibrate the balance between immuno-stimulatory and immuno-regulatory processes.
Introduction
The observation that a sustained and potent immune response to a foreign pathogen, self-antigen, or normal microflora can be the root cause of uncontrolled cancer outgrowth and progression underscores the need for tight immuno-regulatory interventions that could be harnessed for the development of cancer vaccines and cell-based immunotherapies. On the other hand, inflammatory responses characterized by infiltration of tumor-associated antigen (TAA)-specific T cells and other components of the innate immune system are a pre-requisite for effective anti-tumor immunity. Therefore, it is crucial that the inevitable co-existence of these opposing forces is carefully streamlined as imbalances can be detrimental to the host.
Oncogenic viruses such as Epstein–Barr virus (EBV), human papilloma virus (HPV), and Kaposi sarcoma herpes virus (KSHV) express viral oncogenes, which can directly induce tumorigenic cell transformations and initiate the carcinogenesis process. In the case of non-oncogenic viruses such as hepatitis B (HBV) and hepatitis C (HCV), chronic infection and inflammation can lead to carcinogenic mutations in host cells (1), which are manifested by the increased incidences of liver cancer in chronic HBV and HCV patients. In both of these scenarios, the arising transformed tumor cells are genetically altered in a manner that distinguishes them from ordinary healthy self-cells thus conferring the ability to trigger effector immune responses, which in some cases are capable of controlling tumor growth (2, 3). In other instances, however, such modifications may lead to altered antigenicity and escape from immune-surveillance whereby the newly transformed cells are no longer recognized by their original cognate antigen-specific immune cells, thus leading to uncontrolled cancer progression. On a different platform, continuous antigenic stimulation that occurs during chronic virus infections causes severe immune dysfunction characterized by T cell exhaustion, anergy and in some cases deletion of antigen-specific B and T cells (4–6), and a concomitant induction of immuno-regulatory processes, which all result in the loss of tumor immune-surveillance and lead to cancer establishment. This is indeed supported by epidemiological data showing increased incidences of malignancies such as Kaposi sarcoma (KS) and cervical cancer, as well as EBV-associated malignancies such as non-Hodgkin lymphoma (NHL) and Burkitt lymphoma in immunosuppressed HIV/AIDS (7) and transplant patients.
Cancer can also arise due to dysfunctional immuno-regulatory mechanisms that result in uncontrolled excessive inflammatory immune responses. For example, pathogenic immune responses directed at commensal intestinal microflora during inflammatory bowel disease (IBD) are known to increase the risk of colon cancer (8, 9). Indeed prolonged periods of ulcerative colitis (UC) and Crohn’s disease (CD) are associated with impaired immuno-regulatory mechanisms, which are in turn linked to colitis-associated colon carcinogenesis (10–12). Under normal circumstances both intrinsic and extrinsic regulatory pathways come into force to limit excessive immune activation and inflammation thus preventing tissue pathology and subsequent risk of cancer. However, as in many cases, failures of these control measures, including reduced frequencies or altered phenotype and function of regulatory T cells (Treg) means that this inflammation progresses in leaps and bounds. These paradoxical scenarios highlight a disruption in the natural homeostatic immuno-regulatory mechanisms that can be switched on to prevent excessive immune activation or turned off to allow execution of effector immune responses and tumor immune-surveillance. However, the exact timing of when a “good” immune response aimed at pathogen or tumor clearance can become a very “bad” response that creates an environment conducive for cancer growth and dissemination remains elusive. Understanding the intricate complexities and the timings of these events will be crucial in designing interventions for immune-mediated and viral cancers.
Extrinsic and Intrinsic Immuno-Regulatory Pathways
A complex network of finely tuned immune-regulation pathways exists to actively inhibit excessive immune responses during chronic viral infections and inflammation. This is essential for preventing the hyper-proliferation of antigen-specific T cells that could cause immunopathology due to increased release of inflammatory cytokines and targeted killing of infected or antigen-expressing tumor cells by CD8+ T cells. Immuno-regulatory pathways can broadly be divided into extrinsic or intrinsic pathways as depicted in Figure 1. Intrinsic mechanisms derive from within the effector cell and usually involve down-regulation of activating receptors and up-regulation of inhibitory receptors as well as activation of antagonist mechanisms, as discussed in the next section. Extrinsic pathways on the other hand usually involve other cells, which exert regulatory functions by cell-to-cell contact or via release of suppressive cytokines and biochemical compounds that inhibit cellular functions.
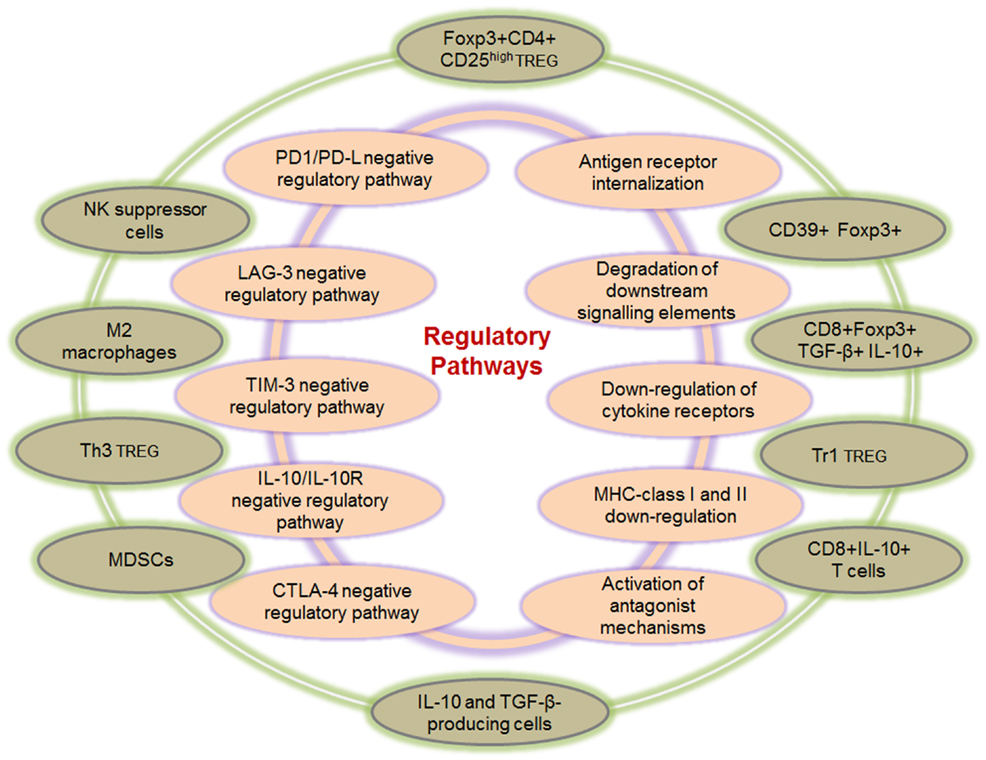
Figure 1. Intrinsic and extrinsic immune-regulatory pathways. Several pathways of immune-regulation exist, and these comprise intrinsic and extrinsic mechanisms. Intrinsic pathways (inner circle) derive from within the effector cell and usually involve up-regulation of inhibitory receptors, down-regulation of cytokine and T cell activation receptors, down-regulation of MHC molecules, as well as the degradation of downstream signaling elements. Although the intrinsic pathways derive mainly from within the effector or antigen presenting cells, interactions with external elements do play a significant role, for instance the down-regulation of MHC class I, which is directly mediated by the HIV-1 Nef protein (13, 14). Extrinsic pathways (outer circle) involve several other cell types that exert immune suppression via cell-to-cell contact or through the release of suppressive cytokines and other biochemical compounds with suppressive activity. These include the various types of regulatory T cells in addition to the Foxp3+ Treg, CD8+ regulatory T cells, MDSCs as well as M2 macrophages and suppressive NK cells.
Of the extrinsic immuno-regulatory pathways, CD4+CD25high Foxp3+ Treg are the most extensively studied and their suppressive mechanisms have been elucidated in greater detail. Existence of other types of immune cells with regulatory functions has been documented, for example CD4+Foxp3− Treg with suppressor functions such as the IL-10 producing Tr1 cells (15) and TGF-β producing Th3 cells (16) have been found in inflammatory environments. Tr1 cells secrete high levels of IL-10 and moderate amounts of TGF-β, and mainly suppress vial IL-10 release, as IL-10 neutralization abrogates their suppressive function (17, 18). On the converse, Th3 cells secrete high levels of TGF-β and low levels of IL-10 and can suppress both Th1 and Th2 responses (16, 17). Other cells with regulatory properties include myeloid-derived suppressor cells (MDSC), which can be induced by cytokines such as IL-6 and growth factors including G-CSF and GM-CSF (19), CD8+Foxp3+ Treg producing both IL-10 and TGF-β, or IL-10-producing CD8+ T cells (20–22), as well as NK cells that possess suppressor functions (23, 24). Activated MDSC can suppress via several mechanisms including IL-10 production as well as via compounds such as arginase 1, reactive oxygen species (ROS), and nitric oxide (NO) among others (19). Moreover, MDSC can indirectly contribute to immuno-regulatory functions by inducing Treg differentiation and expansion.
Immune Dysregulation during Persistent Virus Infections and Chronic Inflammation
T cells are the key players in many infectious diseases and in eradication of malignant cells. This is well-demonstrated in acute infections where T cells become activated and acquire effector functions, with subsequent clearance of infection and formation of stable memory populations. Moreover, tumors heavily infiltrated with fully functional effector T cells progress less rapidly and in some cases regression can be achieved. However, in the case of persistent antigen stimulation in a chronic setting, memory T cell formation and effector functions are altered, resulting in exhausted, functionally impaired defective T cells incapable of conferring protection. The characteristic properties of these defective cells include diminished cytokine production, decreased cytotoxicity, and reduced proliferative and self-renewal potential. In some cases, mutational escape and/or physical deletion of antigen-specific T cells occurs resulting in inadequate immune control, hence chronic persistence of the viruses. Furthermore, some chronic pathogens directly infect the immune cells, e.g., HIV-1 (CD4+ T cells) and EBV (B cells) leading to loss of immune functions. This state of immunological dysfunction is consistently found in chronic virus infections including HIV, HBV, and HCV (25–27) and is also prevalent in cancer patients. Immune dysregulation can be manifested in several forms, some of which are summarized in Figure 2 and described below.
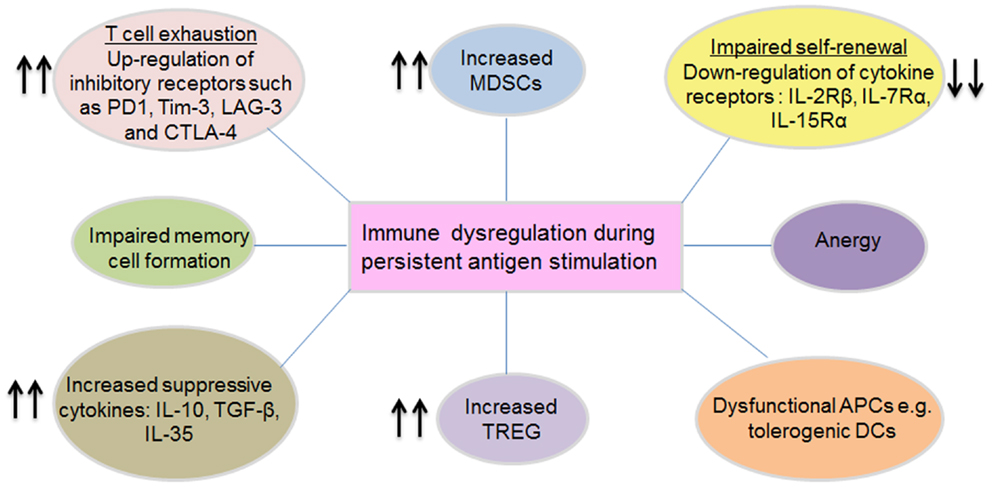
Figure 2. Mechanisms of immune dysregulation during persistent antigen stimulation. Immune dysregulation manifests in several distinct forms, which can occur in isolation or in combination. Persistent antigenic stimulation, especially in settings with high antigen loads can lead to T cell exhaustion (characterized by the up-regulation of several inhibitory receptors and down-regulation of specific T cell activation receptors), anergy (generalized unresponsiveness), impaired memory formation, impaired proliferation, and self-renewal capacity. Besides these, chronic viruses trigger various biochemical pathways that lead to increased frequencies of MDSCs and Treg, which actively suppress effector immune responses via a variety of mechanisms. Immune dysregulation occurring at the T cell priming stage is linked to dysfunctional APCs (for example inadequately activated or tolerogenic DCs), which are likely to skew the immune response toward tolerance. Conceivably, such regulatory mechanisms serve to prevent tissue damage and aberrant immune activation, but they inevitably contribute to the chronic persistence state as a result of inhibiting effector immune responses.
T Cell Exhaustion
T cell exhaustion refers to a state of progressive loss of immune function, which in some cases, can result in physical deletion of responding cells due to imbalances in the expression of pro-apoptotic and anti-apoptotic factors and the inability to respond to IL-7 and IL-15 (26–28). The dominant mechanism of T cell exhaustion is the up-regulation of several inhibitory receptors, although down-regulation of cytokine receptors such as IL-7Rα and IL-15Rα by exhausted memory T cells is frequently observed. Lower levels of IL-7Rα and IL-15Rα can lead to defective cytokine signaling and consequently impaired homeostatic self-renewal and suboptimal numbers of functional memory T cells (27, 28). Up-regulation of inhibitory receptors such as programed-death 1 (PD-1), T cell immunoglobulin mucin 3 (TIM-3), lymphocyte activation gene 3 (LAG-3), and cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) is a characteristic feature of exhausted T cells. PD-1, an inhibitory receptor of the CD28 superfamily is highly expressed on exhausted CD8+ T cells during progressive chronic viral infections and uncontrolled cancer, making it a major factor in T cell exhaustion. Under normal circumstances, PD-1 is induced following T cell activation to inhibit the TCR signaling cascade and prevent excessive T cell activation, but is then down-regulated following pathogen clearance. In peripheral tolerance, PD-1 is important in inhibiting potentially pathogenic self-reactive T cells as well as promoting Treg development (29, 30) and mice lacking PD-1 succumb to autoimmune diseases (31, 32). However, in chronic infection, the PD-1 pathway mediates pathogen-specific CD8+ T cell dysfunction as demonstrated in HIV (33–35), HCV (36, 37), and HBV (38, 39) infections. For example, the frequency of PD-1+CD8+ T cells is highly elevated in HIV-1 patients where it correlates significantly with viral load and declining CD4+ T cell numbers (33, 40). PD-1 is also up-regulated on HIV-specific CD4+ T cells (40, 41) and inhibits CD4+ T cell responses including proliferation. Interestingly, PD-1 levels are significantly reduced in HIV-1 progressors who initiate highly active antiretroviral therapy (HAART) or in long-term non-progressors (LTNPs), suggesting that antigen persistence drives T cells to exhaustion (33, 34). In chronic HCV infection, increased PD-1 expression on HCV-specific CD8+ T cells is associated with impaired proliferation and cytokine production (37). A part from inhibition of T cell function, PD-1 expression can also lead to spontaneous or FAS-mediated apoptosis of virus-specific T cells (42).
Besides PD-1, other inhibitory receptors such as TIM-3, 2B4 (natural killer cell receptor), and LAG-3 are also up-regulated on virus-specific T cells, and the expression of multiple inhibitory receptors correlates with a severely dysfunctional state (43–45). For example, co-expression of PD-1 and TIM-3 is associated with severely exhausted HIV-specific CD8+ T cells (45) and majority of these also co-express PD-1 and 2B4 (46). CTLA-4 is another inhibitory receptor expressed by activated CD4+ and CD8+ T cells. It has a higher affinity for the B7 ligands (CD80 and CD86) allowing it to out-compete CD28, hence it is a powerful negative regulator of CD28-dependent T cell responses. It is significantly up-regulated on CD4+ T cells during HIV-1 and HCV infections where it correlates positively with disease progression and negatively with antigen-specific IL-2 production (41, 47). CTLA-4 is abundantly expressed on Treg as it is required for optimum suppressive function.
Impaired APC Function
The fact that fully functional pathogen-specific T cells are rarely found in chronic infections suggests impaired antigen presentation, which could be attributed to either inadequate priming by non-professional APCs or possibly altered function of professional APCs during the chronic stages of disease. Indeed, functional impairment of DCs has been associated with T cell exhaustion and progression of disease during HIV, HBV, HCV, and LCMV infection (48–51). Decreased expression of co-stimulatory molecules and lower production of immuno-stimulatory cytokines by APCs can result in functionally tolerant or anergic T cells. Furthermore, chronic infections are associated with loss of DCs, possibly due to direct infection by viruses such as HIV and LCMV. Moreover, DCs can induce T cell exhaustion or tolerance by signaling through inhibitory receptors such as PD-1 and CTLA-4, and also acting via indoleamine 2,3-dioxygenase (IDO)-dependent mechanisms to induce Treg, which further suppress immune responses (52). However, other factors such as virus-induced modulation of the expression of MHC or co-stimulatory molecules have been described and may also significantly affect the generation of fully functional T cells (13, 14).
Increased Frequency of Treg and MDSC
Increased frequencies of Treg and MDSC are a common feature of persistent chronic viral infections, which is well-documented in infections with HBV (53–55), HCV (56–58), and HIV (52, 59–61). These chronic persistent viruses trigger the production of IL-10 and TGF-β, which in turn increase the frequency and suppressive function of Treg, such as observed in HCV-infected hepatocytes (62). Alternatively, these cytokines may promote the induction of adaptive Treg further reinforcing the immune barrier at sites of infection. HIV and HCV infections also induce plasmacytoid dendritic cells (pDCs) known to induce IL-10-producing Treg via IDO-dependent mechanisms (52, 58). Additionally, the chronic micro-environments created by virus persistence contribute to enhanced Treg proliferation and suppressive function by secreting cytokines and other factors on which Treg thrive. The high frequencies of Treg and MDSC serve an important role of preventing excessive antigen stimulation, persistent inflammatory responses, and viral mediated immunopathology in the chronic stages of viral disease (56, 63). However, the elevated frequencies and enhanced suppressive capacity of Treg and MDSC also contribute to suppression of effector T cells in an antigen-specific or bystander mechanism (64) thus promoting prolonged viral persistence (65, 66) characterized by secondary T cell impairment and exhaustion (67). Thus, counterintuitively, increased expansion and survival of regulatory cells serve to establish, propagate, and maintain the chronic infection state.
Increased Suppressive Cytokines
Apart from Treg and MDSC, increased IL-10 production is another powerful immuno-regulatory mechanism that negatively impacts on the quantity and quality of antigen-specific immune responses. IL-10 is an immuno-regulatory cytokine produced by many cell types and has multiple functions including inhibition of pro-inflammatory cytokine production, dampening T cell responses, blocking APC functions, and also causing B cell dysregulation. Increased IL-10 production is seen in several chronic viruses including HIV, EBV, HCV, HBV, and LCMV (68–75), and IL-10R blockade can induce rapid virus control indicating that excessive levels of IL-10 have a negative influence on the quality of immune responses and disease course (68, 69). TGF-β is yet another immunosuppressive cytokine whose role in limiting immune responses is documented in a number of disease settings (76). Both IL-10 and TGF-β are known to establish highly suppressive micro-environments that are suitable for cancer progression.
Disruption of Immune-Regulatory T Cells in Inflammatory Environments
Resolution of inflammation requires swift execution of functional regulatory mechanisms such as the expansion of Treg, a lineage of lymphocytes committed to suppressive functions that maintain self-tolerance and immune homeostasis. Dysregulation of Treg function or induction is linked to a number of chronic inflammatory disorders such as IBD and also fatal autoimmune diseases. Thus, interventions which can restore functional regulation without inducing effector immune responses would be beneficial in such settings. Dysfunctional regulation can manifest as reduced Treg numbers (either due to defective Treg induction or loss of Treg), defective suppressive function (due to loss of Foxp3 expression or reduced production of suppressive cytokines), and impaired migration (due to altered expression of adhesion molecules and chemokine receptors). This section gives a brief description of these mechanisms and the various inflammatory conditions that drive phenotypic and functional modification of Treg.
Treg Instability: Phenotypic Alteration and Functional Impairment
Despite the widely held view of thymic imprinting of Treg cell functions, recent studies indicate developmental plasticity and instability, whereby Treg lose Foxp3 expression and convert to Foxp3− helper T cells (exFoxp3) (77, 78) in certain inflammatory or lymphopenic environments (Figure 3). Although exFoxp3 Treg may largely arise from a few promiscuous uncommitted Treg (79), their comparatively higher potential to expand, coupled with the fact that a majority of them are skewed toward self-reactivity suggests potential pathogenicity as a result of altered regulatory functions such as secretion of pro-inflammatory cytokines directed against self-antigens (77). Adoptive transfer studies showed that a large fraction of Treg transferred to lymphopenic recipients lost Foxp3 expression alongside other Treg cell surface markers, and that this was accompanied by acquisition of effector functions including IFN-γ, IL-2, and IL-17-production and a concomitant loss of suppressive function (80–82). Other reports indicated that Foxp3+ Treg effectively lost Foxp3 expression and converted to T helper-type 2 phenotype cells expressing IL-13 and IL-5 (78, 83) or differentiated into follicular helper T cells (Foxp3–TFH-like cells) under the influence of IL-6 and IL-21 (84). Acquisition of T helper features without the simultaneous loss of Foxp3 expression has also been observed. This results in hybrid Treg, which display an activated-memory T cell phenotype and pro-inflammatory properties, such as the IL-17-producing Foxp3+ROR-γt+ IL-17+ (85–90) and IFN-γ-producing Foxp3+T-bet+ IFN-γ+ (91, 92) Treg. Although this hybrid Treg phenotype can exert dual inflammatory and regulatory functions, it has been shown that the phenotypic and transcriptional modifications can reduce their overall suppressive function (81, 93). In other instances however, Treg have been shown to lose their suppressive function without necessarily converting to exFoxp3 or dual function (hybrid) inflammatory Treg (94–100). Such functionally impaired Treg show decreased expression of Foxp3, CTLA-4, and GITR, together with production of very low levels of IL-10 and TGF-β.
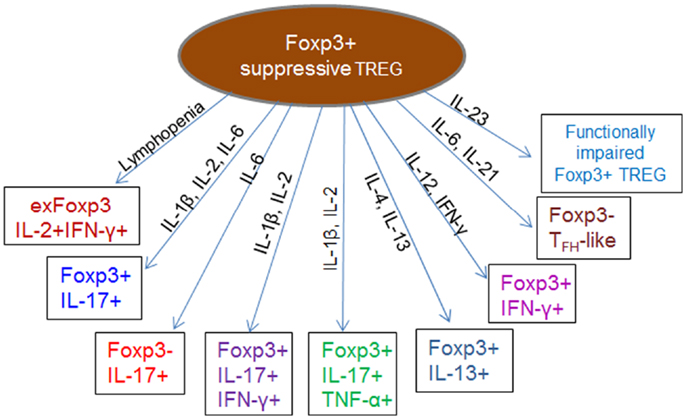
Figure 3. Impaired or altered function of regulatory cells during inflammation. The cytokine and chemokine milieu of inflammatory micro-environments can induce phenotypic and functional modification in Treg, leading to generation of pathogenic exFoxp3 T cells, which express lower levels of Foxp3, CTLA-4, and GITR and produce a combination of Th1, Th2, or Th17 cytokines. Conversion of Foxp3+ Treg into pathogenic IFN-γ-producing Th1 cells or IL-17-producing Th17 exFoxp3 Treg cells is documented in various immunological settings (77, 80, 101). Conversion to a Th2 phenotype expressing IL-13 (Foxp3+IL-13+) and IL-5 (78, 83) as well as differentiation into follicular helper T cells (84) have also been reported. In most cases, the suppressive function of these altered phenotypes is significantly reduced due to decreased Foxp3 expression (81, 82). Certain inflammatory conditions can support the generation of hybrid phenotype Treg, which exhibit dual suppressive and pro-inflammatory functions such as the IL-17-producing Foxp3+IL-17+ (85–90), IFN-γ-producing Foxp3+ IFN-γ+ (91, 92), Foxp3+IL-17+ IFN-γ+, or Foxp3+IL-17+ TNF-α+ (102, 103) Treg. Generally, environments enriched with Th1 cytokines such as IFN-γ, IL-2, and IL-12 favor generation of exFoxp3 Treg producing IFN-γ, those enriched with Th2 cytokines such as IL-4 and IL-13 favor generation of Th2 Treg, while IL-6 favors conversion into the IL-17+Foxp3+ and IL-17+Foxp3− phenotypes.
Several lines of evidence indicate that functional and phenotypic plasticity of Foxp3+ Treg is largely governed by extrinsic signals provided by the inflammatory milieu of their surrounding environments. Increased levels of pro-inflammatory cytokines such as IL-12 or IFN-γ correlate with the frequency of functionally impaired Th1-like Treg (104). In this setting, the Treg suppressive functions were effectively restored by IL-12 withdrawal or IFN-γ blockade suggesting that a pro-inflammatory cytokine milieu not only promotes the Th1-like phenotype, but also inhibits Treg suppressor functions. Overall, inflammatory environments enriched with cytokines such as IL-1β, IL-4, IL-6, IL-21, and IL-23 drive conversion of Foxp3+ Treg into T helper phenotypes (80, 105, 106). As an example, stimulation of peripheral Treg in the presence of IL-6 was shown to result in loss of Foxp3 expression and production of IL-17 (105, 106). Inflammatory environments with IL-1β, IL-2, IL-6, IL-21, IL-23, and TGF-β have been shown to drive conversion of Foxp3+ Treg into IL-17 producing Treg (87, 107), whereas TGF-β, IL-10, and IL-2 help to maintain continued Foxp3 expression, Treg stability, and suppressive function (80, 81, 92, 108–110). Therefore, stable Foxp3 expression and maintenance of optimal Treg suppressive function require the continuous presence of specific signals within the inflammatory environment, without which conversion of Treg into functionally impaired exFoxp3 T cells or hybrid phenotype Treg occurs (102, 103).
Although the various Treg phenomena described above are well-documented in autoimmune settings, it remains possible that the chronic inflammatory environments created by persistent viral infections can also support phenotypic and functional modifications that would render Treg dysfunctional. In favor of this speculation, a recent study has demonstrated that Treg infected with HIV display increased CpG methylation of the Foxp3 locus and a deregulated functional profile, which was characterized by down-regulation of Foxp3 expression, reduced suppressive capacity, and altered cytokine secretion pattern (111). These Treg showed decreased production of TGF-β and increased IL-4 secretion, a characteristic which is thought to orchestrate severe systemic immune hyper-activation that is observed during progressive HIV disease. In chronic infection with HCV, PD-L1 was found to negatively regulate both the function and proliferation of Treg by controlling STAT-5 phosphorylation (112). Although PD-1 was expressed on both Treg and effector T cells, Treg showed significantly higher up-regulation of PD-1, which was correlated with disease progression. These studies highlight the potential of viruses to subvert the induction and function of Treg, but clearly further research is needed to unravel the mechanisms underlying defective regulation during chronic virus infections.
Impaired or Altered Migration of Treg
Another crucial aspect contributing to Treg dysfunction is their ability to migrate to peripheral sites of chronic inflammation such as the skin, urogenital mucosa, gut-associated lymphoid tissues (GALT), transplanted organs, or tumors for appropriate localization, in close proximity with effector immune cells as suppression is mostly contact-dependent. To do this effectively, activated Treg up-regulate distinct site-specific inflammatory chemokine receptors and adhesion ligands, which facilitate their migration into the inflamed tissues, usually in response to a variety of inflammatory chemokines that serve as migrational cues (113–117). Therefore, altered chemokine receptor and adhesion molecule expression can affect the migrational properties of Treg and impact on their ability to access sites of chronic inflammation. Such attenuated Treg migration can in turn lead to sustained inflammation and increased risk of inflammation-driven cancer in the Treg inaccessible areas, owing to reduced frequency and suppressive activities.
The crucial role of chemokine-receptor-dependent migration in functional regulation is demonstrated in several experiments including a mouse model of colitis and IBD, where CCR4-deficient Treg had impaired migration to the mesenteric lymph nodes and therefore failed to prevent colitis (118). In other settings, a number of chemokine receptors including CCR2, CCR4, CCR5, CCR6, CCR7, and CXCR3 have been implicated in the selective and preferential recruitment of Treg to sites of chronic inflammation and/or tumors (115, 119, 120), thus indicating that alteration in chemokine receptor patterns or blockade of chemokine receptor signaling would have a significant impact on their migration and immuno-regulatory activities. Tumors and their associated stroma are known to express elevated levels of specific inflammatory chemokines, which serve to chemoattract various leukocytes including Treg (121, 122). Although the overall recruitment is also significantly influenced by the type of chemokine receptors expressed by the leukocytes, Treg, especially the “inflammation-seeking” phenotype usually up-regulate multiple chemokine receptors (116), which allow them access to a variety of tumors, where they preferentially accumulate (119). Some studies demonstrate that disruption of key chemokine receptor signaling axes such as CCR4 or CCR5, or the depletion of chemokine receptor-specific Treg can significantly inhibit their migration and prevent accumulation in tumors (123, 124), thus influencing the overall prognosis. Conceivably, while impaired migration and reduced Treg access to tumors would be an awesome advantage in the majority of cancer settings where they impinge on anti-tumor immune responses, it may however be a major setback in certain other settings, which require Treg to limit excessive immune responses, such as in IBD and chronic virus infections.
Loss of Treg (Impaired Treg Induction or Treg Deletion)
In certain disease settings, physical deletion of Treg can result in reduced frequencies. For instance, it is postulated that by virtue of their activated nature, Treg express higher levels of CCR5 and CXCR4, the co-receptors for HIV-1 thus making them preferential targets for HIV-1 infection (125, 126). Since Foxp3+ Treg represent a high proportion of CD4+ T cells (up to 50%) found in mucosal lymphoid organs of HIV-infected individuals (60), it is plausible that HIV infection can subsequently lead to significant depletion of these cells and impaired immuno-regulatory functions (127–129). Furthermore, Treg also express both Fas and Fas ligand and can be targeted and killed by effector T cells without necessarily being infected. Several studies indicate that myeloid dendritic cells (mDCs) can contribute to Treg induction by promoting conversion of conventional CD4+ T cells into Treg (130, 131). However, it has been shown that in vitro HIV infection of mDCs not only impairs their capacity to induce Treg but can also trigger preferential targeting and killing of Treg via a caspase-dependent pathway (132), thus contributing to numerical loss of Treg. Changes in the levels of chemokines expressed within certain tissues, together with diminished levels of TGF-β and IL-2 can also result in the loss of Treg in that particular organ. For example, altered expression of ligands for CXCR3, CCR4, and CCR7 was associated with a loss of Treg in lymph nodes during simian immunodeficiency virus (SIV) infection (133). Other mechanisms for reduced Treg frequencies may include increased apoptosis, reduced proliferation and survival, as well as impaired peripheral Treg induction. As discussed earlier, Treg may also be lost by conversion to exFoxp3 T cells under certain inflammatory cytokine milieu.
Imbalances in Immuno-Regulatory and Immuno-Stimulatory Processes Can Cause Cancer
Increased risk of cancer is often associated with poorly regulated immune responses (Figure 4) constituting unresolved inflammation as a result of perturbations in the balance of tumoricidal and tumorigenic activities (134, 135). Treg play a crucial role in maintaining optimum balance between these two arms of the immune response and persistent viruses are known to trigger production of IL-10 and TGF-β (136) to ensure induction and maintenance of adequate numbers of Treg in circulation. In some cases, viruses express homologs of immunosuppressive cytokines or cytokine receptors, such as the well-described human cytomegalovirus (HCMV)-IL-10 and EBV-IL-10 homologs (137, 138), which allow them to directly influence Treg induction or modulate the immune system via other mechanisms including impaired production of pro-inflammatory cytokines and chemokines, as well as MHC class II down-regulation (136). As mentioned earlier, viruses can also promote Treg induction by disrupting the normal activation cascade of dendritic cells and other antigen presenting cells. Furthermore, inflammatory micro-environments are enriched with type 2 macrophages (M2) and MDSC, which also enhance recruitment of Treg, besides directly suppressing antigen-specific effector T cells (19, 139, 140). Additionally, antigen-specific CD8+ Treg are frequently detected in chronic HIV (141, 142), HCV (57, 143), and herpes virus infections (144, 145). The increased numbers of Treg and other immunosuppressive mechanisms serve to actively prevent excessive immune activation and the associated immunopathology, but by so doing, they block antigen-specific effector immune responses that are essential for clearing the pathogen and for tumor immune-surveillance. The resulting immune impairment allows chronic pathogen persistence and an overwhelming state of recurrent inflammation, thus favoring cancer establishment.
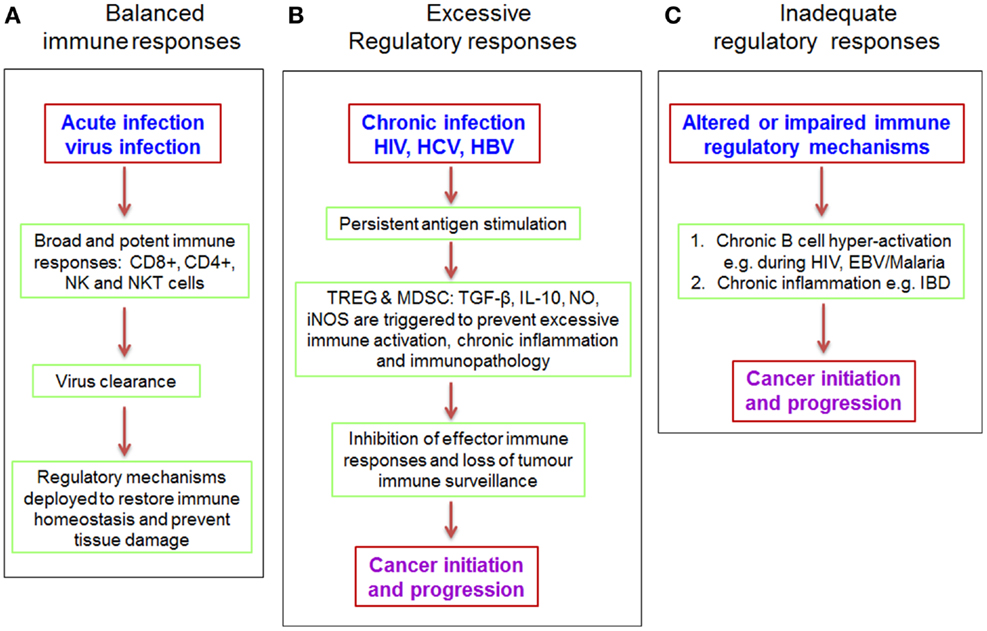
Figure 4. Dysregulated immune responses create a microenvironment suitable for cancer initiation and progression. Perturbations of the balance between effector and regulatory immune responses are often the cause of chronic inflammation and increased risk of cancer (146). Under normal circumstances (A), the immune system mounts a potent and broadly directed immune response following acute virus infection. This immune response, comprising both the innate and adaptive components leads to effective virus clearance and stable memory cell formation, which is effective at rapidly countering subsequent infections. Under this scenario, regulatory mechanisms kick in to prevent tissue damage after virus clearance. However, in the case of persistent antigenic stimulation (B) such as caused by HIV, HCV, and HBV, there is continuous generation of effector immune cells that are incapable of clearing the pathogen. This leads to a state of chronic inflammation that in turn triggers regulatory pathways such as increased production of suppressive cytokines and recruitment of Treg and MDSC to dampen excessive immune responses and prevent tissue damage (136). But as fate would have it, such potent regulatory responses also inhibit anti-tumor effector responses leading to loss of tumor immune-surveillance and subsequently cancer initiation and progression. In certain contrasting scenarios such as IBD, impaired regulatory mechanisms (C) can result in chronic inflammation, which initiates carcinogenesis. Furthermore, uncontrolled B cell activation during HIV and EBV infections is associated with increased risk of non-Hodgkin’s lymphoma (147). NO, nitric oxide; iNOS, nitric oxide synthase.
Besides the direct disruption of tumor immune-surveillance, establishment of chronic inflammation creates a suppressive tumor-promoting microenvironment, which is enriched with IL-10, TGF-β, and other pro-inflammatory cytokines such as IL-17, known to be angiogenic and to contribute to tumor cell survival and growth (148, 149). In the presence of IL-6, TGF-β can further up-regulate ROR-γt expression leading to enhanced Th17 differentiation (150–152) and increased risk of cancer progression (148, 153–155). Moreover, as discussed earlier, Foxp3+ Treg in certain inflammatory environments can express IL-17, which together with hypoxic conditions could play a role in generation of cancer initiating cells (156). As highlighted in earlier sections, inflammatory environments can also induce phenotypic and functional impairments in immuno-regulatory cells thus leading to dysfunctional immune-regulation and increased risk of cancer. However, whether increased incidence of cancer in individuals with chronic virus infection and inflammation is due to increased suppression of tumor immunity as a result of increased frequency and suppressive activity of immuno-regulatory cells, due to failure of regulatory cells to prevent excessive immune activation and inflammation, or due to enhanced oncogenic potential of the carcinogen remains a subject of intense debate. In this review, I will focus on the contradictory roles of immuno-regulatory cells where they can cause cancer by either exerting potent suppression of effector immune responses that inhibit tumor immune-surveillance (e.g., during chronic virus infections) or through their functional impairment and inability to execute effective suppression of pathogenic effector immune responses (e.g., during IBD).
The Delicate Balance between Immunity and Regulation in HIV Infection and Disease
Although HIV can exist in latent reservoirs for many years, it is a chronic persistent virus characterized by the continuous presence of infectious virus and thus chronic immune activation, persistent inflammation, and concurrent CD4+ T cell loss are all observed (157–159). Thus, increased numbers of fully functional regulatory mechanisms become necessary to counteract the ongoing inflammatory processes. However, Treg, which are instrumental in counteracting immune activation and inflammation by actively suppressing effector immune responses can also be detrimental by inhibiting T cell responses that control HIV replication. An appropriate immune response must therefore not disturb this delicate balance, by aiming to maximize the “good” immune responses, which control the virus while minimizing the “bad” immune responses that cause pathology.
Although still a subject of intense debate, a number of studies have demonstrated increased frequencies of Treg during HIV-1 infection and more especially in the chronic stages that mark progression to AIDS (128, 160–163). Studies looking at tissue distribution revealed accumulation of Treg at sites of HIV infection and replication such as the gastrointestinal mucosa and lymph nodes (59, 60, 128, 164). With such increased frequencies and especially given the suppressive role of Treg, it follows that progression of HIV-1 infection to chronic disease could in fact be a consequence of suppressed T cell function. Indeed, robust CD8+ and CD4+ T cell responses (characterized by high proliferation, IFN-γ production, and cytotoxicity), which correlate with HIV control in a minority of infected people usually diminish during chronic infection, coinciding with increased Treg numbers. Depleting Treg was shown to restore the in vitro effector immune functions of these cells (59, 165). Lower levels of Treg and a corresponding higher level of HIV-specific T cell responses have been observed in individuals who naturally control HIV-1 in the absence of HAART, i.e., the LTNPs and Elite controllers (160). Furthermore, depletion of Treg in cord blood samples of HIV-exposed uninfected neonates (166) was shown to augment both CD4+ and CD8+ HIV-specific T cell responses. These findings, together with the observation that Treg frequencies are reduced in HIV-infected patients on HAART (160, 164, 167, 168) provide compelling evidence that Treg impinge on immune control of HIV and strongly support immunotherapeutic interventions that reduce their numbers or impair their functions.
Whereas depleting Treg or interfering with their suppressive function might seem plausible in the context of immune function restoration, in fact several studies indicate that reduced Treg frequencies correlate with increased immune activation, which is in turn significantly associated with higher plasma viral loads (169, 170). Treg can therefore prevent collateral damage during chronic HIV infection by limiting immune activation, while at the same time reducing the pool of activated CD4+ T cell targets that would become susceptible to HIV infection. Accordingly, it is thought that the high frequencies of Treg found in highly exposed persistently sero-negative (HESN) individuals (171) and in the in utero HIV-exposed uninfected neonates (166) contribute to resistance to HIV infection by significantly reducing the numbers of activated target CD4+ T cells. These studies suggest that Treg may be beneficial at least to some extent, not only in HIV-infected individuals where they could limit immune activation, but also in highly exposed individuals with a greater risk of HIV infection. However, given that LTNPs and elite controllers exhibit both lower levels of immune activation and lower Treg frequencies (172), while at the same time mounting robust HIV-specific immune responses that inhibit virus replication, it is plausible to suggest that Treg are dispensable in HIV immunity, although caution must be exercised as LTNPs and elite controllers represent a very small proportion of HIV-infected individuals, in whom protective HLA alleles are over-represented.
Contrary to these findings, many studies document persistence of immune activation in the presence of elevated Treg frequencies, suggesting that perhaps the suppressive activities of the Treg found in chronic HIV infection are not sufficient to completely reverse the state of chronic immune activation. Indeed, it has been demonstrated that higher frequencies of Treg exist in HIV-infected individuals with progressive disease (173), but their ability to suppress HIV-specific T cells is significantly reduced, which in turn leads to inability to control HIV-associated aberrant immune activation (111, 161, 174). This is in fact discredited by studies demonstrating the existence of functionally suppressive Treg in progressive HIV-1 disease (59, 165, 175), hence suggesting that failure to reduce immune activation may be due to overwhelming levels of persistent stimulation rather than functional impairment of Treg. Thus, it is possible that high Treg frequencies found in chronic HIV infection are a result of failed attempts to reduce the state of chronic persistent antigenic stimulation (176, 177).
Faced with this paradox, timings of when to initiate interventions remain critical to achieving desirable outcomes. Whereas, immune-based therapies aimed at increasing the frequencies of Treg such as IL-2 therapy may only serve to suppress anti-HIV immunity and provide more targets for HIV, and thus not offer clinical benefit earlier in HIV infection (178, 179), they might indeed become useful during the chronic stages in order to limit immune activation (169, 170). Conversely, depleting Treg during the early stages of infection will allow for generation of robust immune responses capable of controlling virus replication and preventing establishment of latent reservoirs (126).
HIV-Associated Immune Dysfunction Predisposes to Malignancies
The existence of a few HIV-infected individuals with robust HIV-specific immune responses who maintain very low virus loads for many years without treatment and only progress to AIDS following viral immune escape demonstrates constant immune-surveillance that keeps the virus in check. In these individuals, a normal balance between the effector and regulatory immune responses exists, whereby effective immune responses occur without excessive immune hyper-activation that causes T cell exhaustion and functional impairment. However, in a majority of HIV-infected people, the immune system does not control virus replication, leading to continuous immune stimulation with high antigen loads and generates a large pool of immune-effector cells that are by far inadequate in controlling the virus. This can be either due to anergy, functional exhaustion, or immune escape (157), as described earlier. Furthermore, HIV directly infects CD4+ T cells and this leads to the progressive diminution of T helper functions and immune incapacitation that marks progression to AIDS.
Besides these, a number of immuno-regulatory mechanisms triggered to prevent immune activation and inflammation also suppress immune-effector functions and sustain chronic virus persistence. For example, during chronic HIV infection, the expansion of Treg (180) with potent suppressive activity within mucosal tissues not only contributes to persistence of HIV, but also reduces immune vigilance and predisposes to HPV and cervical cancer. Moreover, HIV-1 gp120 has recently been shown to induce IL-6 and a concomitant expansion of MDSC (181), which contribute to immune suppression by modulating cytokine and cellular responses as well as inducing the differentiation and expansion of Treg (182). Large amounts of B cell activation-associated cytokines such as IL-6 and IL-10 are produced during chronic HIV infection and can also increase the numbers and suppressive capacity of MDSC leading to further suppression of effective immune responses. Indeed, higher levels of MDSC are associated with chronic progressive HIV disease (183). The decline of both IL-6 levels and Treg numbers following HAART-mediated immune restoration strongly supports their role in immune modulation during HIV-1 progression. A highly immunosuppressive environment with increased numbers of Treg, MDSC, and suppressive cytokines such as IL-10 and TGF-β is strongly associated with increased risk of cancer (19, 182, 184, 185). Thus, HIV-1 can be classified as indirect carcinogen that perturbs immune balance through immune suppression and a concomitant loss of tumor immune-surveillance to set the stage for oncogenic tumor viruses (186).
Another consequence of HIV-driven impairment of the immune system is the hyper-activation and uncontrolled proliferation of B cells, which not only favors secondary infection by oncogenic viruses (187) such as KSHV and EBV but also increases significantly the potential of chromosomal translocations and oncogenic mutations. A few studies have linked HIV infection with chronic B cell hyper-activation (147, 188) and lymphomagenesis, for example, increased incidence of Burkitt lymphoma in HIV-infected individuals or those persistently exposed to Plasmodium falciparum in malaria endemic regions where their B cells are in constant stimulation by these antigens (189). HIV-1 can also act directly via gp120 to induce B cell activation and subsequent development of lymphomas (190). Moreover, incorporation of CD40L into HIV virions stimulates B cell activation via interactions with CD40, resulting in production of B cell activating cytokines such as IL-6, IL-8, IL-10, and GM-CSF (191, 192). Indeed, HIV-associated lymphomas are often the aggressive B cell lymphomas, directly supporting a role for HIV in altering the B cell phenotypic and proliferative characteristics.
Therefore, the profound T cell dysfunction, progressive depletion of CD4+ T cells, B cell hyper-activation, together with the increased immuno-regulatory mechanisms all collude to actively impede tumor immune-surveillance and create a permissive environment for cancer initiation and progression. This is a classic example of a “vicious cycle of immune responses” where an effector immune response to a pathogen (in this case HIV) is induced during the initial stages of infection, but somehow fails to eliminate the pathogen, and regulatory mechanisms are triggered in order to restore immune balance and limit excessive inflammation and pathology, yet such regulatory mechanisms actively suppress the anti-tumor immune-surveillance processes and predispose to increased risk of cancer.
HIV-Associated Malignancies
Cancer is a complex multistep process involving many molecular events, which together with the carcinogen or oncogenic virus infection work in concert to generate a transformed cellular phenotype. However, immune response is an important extrinsic factor that determines whether or not cancer occurs following exposure to potential carcinogens. While the immune system of healthy individuals limits proliferation of pre-malignant cells by recognizing and deleting cells that express potentially oncogenic viral proteins, these pre-transformed cells go unchecked and become malignant in immuno-compromised individuals, hence the increased incidence of cancer in transplant patients and those with congenital or secondary immunodeficiency disorders. HIV is not directly oncogenic but it is significantly associated with several lymphoid malignancies known to arise in immuno-compromised individuals who become infected with oncogenic viruses such as HPV, EBV, or KSHV (7). Surveillance data estimates the risk of developing NHL at 60- to 200-fold in people with progressive HIV disease compared to the uninfected population, while that of Hodgkin lymphoma (HL) is 8- to 10-fold, thus supporting the active role of the immune system in controlling cancer. Plausibly, HIV-mediated immune dysregulation contributes to immune escape of these viruses thus allowing for proliferation and emergence of stable populations of virally transformed cells that are not efficiently recognized and eliminated by the host’s immune system (187, 193, 194). A wide body of literature documents several AIDS-defining malignancies in the pre-HAART era, but for the purposes of illustrating how immune dysregulation sets a microenvironment conducive for cancer development, this section will draw examples from HIV-associated predisposition to cervical cancer and KS.
HIV-1, KSHV, and Kaposi Sarcoma
The non-redundant role of host immunity in the control of viral cancers is well-illustrated by KS, which is more prevalent in untreated HIV/AIDS patients, mainly due to immunosuppression (195). KSHV was discovered as the causative agent of KS in 1994 (196), however, infection with this virus alone is not sufficient to cause KS in healthy immuno-competent individuals. Indeed, the incidence of KS in the general population remains very low (around 1/100,000), but increases dramatically to around 1/20 amongst HIV-infected people (197) and almost 1/3 HIV-infected homosexual men in the pre-HAART era (198). Furthermore, countries in which KS was endemic before the AIDS epidemic have seen a sharp increase in the incidence, with almost half of HIV-infected individuals who acquire KSHV infection going on to develop KS (199). However, within the endemic areas or in the high risk groups, most HIV-negative KSHV-infected individuals do not develop KS, indicating that HIV-associated immune impairment predisposes to KS development.
HIV-1, HPV, and Cervical Cancer
Human papilloma viruses are the main etiological factor for cervical cancer (200). Of these, HPV-16 and HPV-18 are linked with cervical and anogenital cancers hence are classified as high risk genotypes. As with other cancers, the immune system is central in the pathogenesis of HPV and cervical cancer. In immuno-competent individuals, robust HPV-specific immune responses comprising B and T cells are generated and these correlate with spontaneous resolution of HPV (201, 202), demonstrating that host immunity can be sufficient to clear HPV infection. In particular, a Th1 cytokine profile is instrumental in HPV clearance and prevention of viral persistence. Thus, detection of both humoral and cellular responses including T helper cells induces regression of cervical lesions (203, 204), whereas T helper cell impairment leads to cancer development (205). Natural killer cells also play a protective role by directly lysing HPV-infected cells and initiating regression of squamous intraepithelial lesions (SIL) (206, 207).
Despite the existence of strong HPV-specific immune responses in HPV-infected individuals, progression to HPV-associated malignancies does occur in some individuals due to escape from immune-surveillance caused by immune dysfunction as discussed earlier. Central to this is the systemic enrichment of Treg, which correlates with HPV persistence and is frequently detected in patients who develop high grade cervical intraepithelial neoplasia (208, 209). Furthermore, mucosal enrichment of Treg, which is often associated with diminished cellular immunity in the cervical mucosa has been observed and is linked with the severe forms of cervical carcinoma (210, 211). Higher frequencies of HPV-specific Treg are found in the stroma, intraepithelial tissues and tumor draining lymph nodes of cervical cancer patients where they suppress alloreactive CD4+ responder T cells (212, 213). Depletion of Treg in the in vitro experiments resulted in increased production of IFN-γ. Besides enhanced Treg-mediated immunosuppression, the profound immune dysfunction resulting from HIV-1 infection and the concomitant loss of CD4+ T cells collude to create an environment permissive for HPV persistence and cervical cancer. This can be directly deduced from the increased incidence of cervical cancer and prolonged persistence of SIL in immunosuppressed women with progressive HIV disease (214–216). In fact, cervical cancer was designated as an AIDS-defining illness in 1993 (217), strongly implicating HIV-driven immune impairment as a major factor favoring the progression from HPV infection to cancer development.
Immune Restoration or HIV Suppression Reduces HIV-Associated Malignancies
There is consensus that HIV-associated malignancies arise mainly due to loss of immune-surveillance caused by a dysfunctional immune system. Indeed, the severity of these malignancies correlates positively with the degree of immune impairment as measured by the extent of CD4+ T cell depletion and HIV viral burden. Moreover, the incidence of AIDS-defining malignancies has significantly reduced since the wide-scale implementation of HAART, strongly suggesting better immune control following reconstitution by HAART or perhaps a direct impact of HAART on the replication of EBV, HPV, and KSHV. Therefore, it seems that interventions which limit virus production and prevent chronic antigenic stimulation can effectively reduce immune activation and inflammation, restore effector immune functions through homeostatic equilibration of immuno-stimulatory and regulatory mechanisms, and lead to reduced incidences of HIV-associated malignancies. Recent studies indicate that the increased frequency and suppressive function of Treg observed during chronic HIV infection decreases significantly following HAART initiation (167). This is accompanied by reduced levels of immune activation and enhanced immune-effector functions, which are in turn associated with decreased prevalence and increased regression of cervical lesions in HAART-treated HPV-infected patients (218–220), thus supporting a role for immune reconstitution in the control of HPV and associated cancers. These observations provide evidence for a strong causative link between HIV-mediated immune dysregulation and the onset of HIV-associated cancers (NHL, KS, and cervical cancer) whose incidence has reduced significantly since the introduction of HAART.
HCV/HBV-Driven Immune Dysregulation Predisposes to Hepatocellular Carcinoma
Unlike HIV, which directly targets the immune cells (CD4+ T cells) causing their deletion and loss of T helper functions, HBV and HCV target the liver and replicate in hepatocytes. These viruses have also evolved multiple mechanisms to escape immune elimination and can establish chronic persistence and replicate in infected hosts for many years. Epidemiological studies indicate a strong link between chronic HBV/HCV persistence with the development of liver disease, initially manifesting as chronic hepatitis, and leading on to nodular fibrosis that can progress to cirrhosis and eventually hepatocellular carcinoma (HCC). These processes are characterized by inflammation and oxidative stress owing to the influx of several cell types including NK, NKT, and PMN leukocytes, which accumulate in inflammatory lesions in the liver and contribute to inflammation and liver damage. In a majority of infected individuals, robust and poly functional T cell responses are generated causing clearance of acute infection, while in a minority of those infected, both low frequencies and narrowly focused virus-specific CD8+ T cell responses in the liver correlate with persistent chronic infection and increased risk of HCC (221). Furthermore, defects in HBV-specific CD8+ T cells characterized by exhaustion and increased expression of pro-apoptotic mediators have been reported (222). Thus, although virus-specific lymphocytes can be readily detected in inflammatory lesions in the liver, they are often defective and not sufficient to clear virus infection (223). Moreover, weaker CD4+ T cell proliferative responses have been reported (224).
Infection with HBV and HCV is known to induce IL-10 and TGF-β (72, 73), which in turn induce the expansion of Treg to maintain a tolerogenic environment in the liver. HCV-specific impairment of dendritic cell function can also lead to increased numbers of Treg, and these have been found in both the blood and liver of patients with chronic HBV and HCV infection and HCC (185), where they correlate with in vitro suppression of antigen-specific effector responses (225). These effector responses were enhanced by depleting Treg (54, 226). Overall, immune function restoration and inhibition of viral replication following treatment with anti-HBV drugs is associated with diminished Treg expression (227). Thus, persistence of weak, defective, and narrowly directed T cell responses coupled with high numbers of immune-regulatory cells and increased levels of suppressive cytokines act to promote chronic liver disease and progression to HCC. Indeed, patients with HCC often have increased Treg numbers in blood and within tumors, and the tumor-infiltrating CD8+ and CD4+ T cells have been found to be dysfunctional (228), suggesting a possible link between immune disruption and the pathogenesis of HCC. Other factors such as chronic unresolved inflammation can further support tumor growth via induction of angiogenic and tumor survival signals (229).
Immune Dysregulation in Inflammatory Bowel Disease and Colorectal Cancer
Inflammatory bowel disease is characterized by an uncontrolled, microbe-induced chronic inflammatory state that increases the risk of colorectal cancer (CRC) by twofold (8, 9). These chronic inflammatory responses also drive carcinogenesis of colitis-associated cancer (230). Various cell types infiltrate the inflamed mucosa including MDSC, M2 macrophages, and Th17 cells, which promote tumor growth, and NK and CD8+ T cells, which either target and destroy or inhibit proliferation of CRC cells. These effects are mediated by cytokines such as IL-17A, IL-21, IL-6, and TNF-α that create a tumor-permissive environment versus IFN-γ, which exerts tumor-suppressive functions (231). IFN-γ protects from carcinogenesis by activating cytotoxic T cells as well as increasing the susceptibility of pre-malignant cells to cell-mediated cytotoxicity, thus IFN-γ-producing Th1 cells correlate with increased immune-surveillance and better prognosis in CRC patients (232).
Although IL-4- and IL-13-producing Th2 cells have been associated with increased tumor growth in humans (233) and in animal models using IFN-γ−/−and IL-4−/− deficient mice (234, 235), Th17 cells seem to be the most aggressive orchestrators of chronic inflammation during IBD and have a significant role in the initiation of CRC. This has been linked to IL-23, a cytokine known to induce high numbers of Th17 cells and a concomitant accumulation of pathogenic IL-17A+ IFN-γ+ effector T cells, which cause intestinal pathology and correlate with poor prognosis in CRC (153, 236–238). Indeed, high frequencies of activated Th17 cells together with their signature cytokines are found in the intestinal and serum samples of patients with IBD, and also within the colon and blood samples from patients with CD. Furthermore, IL-23-mediated accumulation of IL-17+IL-22+ innate lymphoid cells (ILCs) in inflamed colons is associated with development of invasive colon cancer (239–241), while increased frequencies of IL-17+ILCs are often found in the intestines of patients with CD (242). The tumor-promoting feature of Th17 cells largely arises from secretion of large amounts of IL-17, which in turn induces expression of pro-inflammatory factors such as TNF-α, IL-6, IL-1, and iNOS, known to play a role in CRC pathogenesis (243). Mice that are deficient in ROR-γt, the transcription factor of Th17 cells were shown to be resistant to chronic inflammation in models of colitis (244). Thus, immuno-regulatory pathways capable of limiting the induction and function of pathogenic Th17 effectors cells are required.
Treg Play a Critical Role in the Pathogenesis of IBD and CRC
The pro-tumoral role of Treg in cancer establishment and progression is well-documented, and in fact a number of interventions that deplete Treg lead to improved prognosis of cancer patients. Furthermore, Treg depletion increases vaccine-mediated anti-tumor immunity (245) and can lead to eradication of established experimental tumors (210, 246). However, Treg play such a critical role in the maintenance of normal gut mucosal immunity by preventing chronic inflammatory responses to food antigens and commensal microflora (247), that inhibition of their function is associated with development of IBD (11, 12). Most astoundingly, increased infiltration of Treg in CRC correlates with a favorable prognosis (10), with several studies in experimental animal models providing evidence that Treg can prevent establishment of CRC (248, 249). This is thought to be through initiation of potent immuno-regulatory functions that prevent chronic inflammation, which would otherwise predispose to cancer establishment and growth. Under normal homeostatic conditions, high frequencies of Treg are found in the gut as it is a preferential site for peripheral Treg induction due to the abundant commensal micro-biota and CD103-producing DCs, which are specialized in inducing the differentiation of Treg from naïve CD4+ T cells (250, 251). However, inadequate regulatory functions are a major characteristic defect during IBD, suggesting alterations in the induction, maintenance, or even suppressive function of Treg. This section highlights some of the mechanisms of immune dysregulation that exacerbate the inflammatory state of IBD to set a stage for CRC.
Treg Induction and Function are Impaired in IBD and CRC
Impaired frequency and function of Treg is one of the mechanisms of immune dysregulation that plays a central role in the pathogenesis of IBD. This is strongly associated with IL-23, a cytokine whose expression is increased in several human cancers including CRC (252). IL-23R signaling suppresses both the differentiation of Treg and IL-10 production by T cells, hence leading to intestinal pathology (236). Such pathology could be prevented by transfer of Treg or administration of Treg-related cytokines such as IL-10 and TGF-β1 (253). TGF-β signaling in tumor-infiltrating lymphocytes is associated with reduced tumor growth in animal models of CRC (254). Crucially, the frequency of Foxp3+ Treg in the colon increases in the absence of IL-23R signaling, indicating a role for IL-23 in controlling the induction and expansion of Treg (255). Since Treg are a source of both IL-10 and TGF-β, the key cytokines in immuno-regulation, it is plausible that IL-23-driven loss of Treg contributes significantly to immune dysregulation by overriding the immunosuppressive pathways in the intestine and favoring IBD and CRC development via generation of pathogenic Th17 effectors cells. Besides reduced numbers, Treg in IBD show altered phenotype and function, attributed to the local cytokine milieu arising from chronic inflammation of the intestinal mucosa. Perhaps, normal Treg in circulation migrate to the lamina propria during active inflammation in order to maintain homeostasis, but on encountering various cytokines within the inflamed mucosa, they undergo phenotypic and functional modifications turning into dual inflammatory and regulatory Foxp3+IL-17+ Treg, which produce large amounts of IFN-γ and IL-17, and moderate amounts of TNF-α and IL-2 (102, 103).
IL-10 Protects Against IBD and CRC
IL-10 deficiency increases susceptibility to IBD-associated CRC, where it is associated with poor prognosis (256). Mice lacking IL-10 were shown to be highly susceptible to colitis-associated CRC following Helicobacter hepaticus infection, and this could be prevented by exogenous administration of IL-10 (257–259), further demonstrating a critical role for IL-10 in the pathogenesis of CRC. It is thought that IL-10 deficiency leads to elevated levels of TNF-α, IL-6, and IL-17, which in turn allow persistence of chronic inflammation (260) thus promoting tumor growth.
Bi-Functional Immune-Effector Cells Can Promote IBD and CRC
Intriguingly, a single cell type can exhibit bi-functional immune characteristics by co-producing effector and suppressor cytokines, thus may have the potential to exert both tumor-promoting and tumor-suppressive functions, depending on the microenvironment. For example, as mentioned above, CD8+ T cells express cytotoxic molecules, which kill CRC cells in addition to secreting IFN-γ, which augments the anti-tumor response (261, 262). However, in some cases of IBD, infiltration of CD8+ T cells does not correlate with improved prognosis (263) and this is linked to elevated perforin and granzyme levels, which sustain the tumor-promoting chronic inflammation (264). Accordingly, perforin deficient mice develop less severe colitis and much fewer tumors in experimental models of colitis-associated CRC (265). Similar bi-functional characteristics have been observed in NKT cells, which exert protective cytotoxic functions, but also secrete Th1, Th2, and Th17 cytokines that could act as enhancers or suppressors of tumor immunity. Increased infiltration of IFN-γ-secreting NKT cells correlates with tumor immunity, which is reflected in increased disease-free survival of CRC patients (266, 267). Conversely, Th2 NKT cells that secrete the immunosuppressive cytokine IL-13, may contribute to colitis-associated CRC (268, 269). These studies demonstrate that CD8+ T cells and NKT cells can simultaneously exert pro-tumoral and anti-tumoral responses, and that perhaps pro-tumoral responses predominate during progressive IBD and CRC. Arguably, intervention strategies targeted at these bi-functional effector cells may result in undesirable outcomes.
Interventions
As discussed earlier, some settings such as inflammatory autoimmune diseases will require interventions that boost the immuno-regulatory arm of the immune response. Such may include immunotherapeutic agents that expand Treg numbers and enhance suppressive function to effectively curtail chronic inflammation. Therapeutic vaccines to restore immune tolerance could benefit from adjuvants that induce adaptive Treg without generating functional effector cells (270). Other measures such as restoration of TGF-β and IL-10, together with IL-2 administration can help to maintain Treg numbers and Foxp3 expression, thus sustaining functional regulation. In other inflammatory settings such as IBD and colon cancer, measures that enhance Treg differentiation and expansion and restore suppressive function, for example, blockade of IL-23 signaling with the concurrent depletion of IFN-γ and IL-2 to impede generation of pathogenic exFoxp3 Treg might be desirable. Additionally, induction of stable expression of site-specific homing and chemokine receptors in Treg can confer the ability to migrate to preferential sites of chronic inflammation, for example, CCR4 for migration to the lung airways during allergic inflammation, CXCR4 for migration to the bone marrow, and CCR4/CCR9/CD62L/α4β7/αE(CD103)β7 for migration to the intestinal mucosa of IBD patients. However, in cases where immuno-regulatory responses are detrimental then immune deregulation interventions are required. Such can include administration of cytokines and/or antibodies that inhibit Treg induction and expansion, suppressive function, and recruitment via blockade of chemokine receptors (124). Interventions such as concurrent CTLA-4 blockade and vaccination (271–273), combined CTLA-4 and PD-1/PD-L1 blockade (274), and Treg depletion (275–277) have been successfully used to ameliorate Treg-mediated immune pathologies and cancer. Measures to reverse exhaustion and restore immune function in chronic infections include blockade of the PD-1:PD-L1/PD-L2 pathway and MDSC development. PD-1/PD-L1 blockade restores HIV-specific T cell function in vitro (33, 34, 278), and clinical benefit is also documented in cancer patients (279, 280). In some instances, combined blockade of PD-1 and LAG-3 or PD-1 and TIM-3 synergistically improves T cell responses leading to better virus control (43, 45). Very recently, a study utilizing a mouse model of retrovirus infection showed that combining the blockade of inhibitory receptors PD-1 and Tim-3, together with Treg ablation was more efficient in reducing chronic virus load compared with either strategy on its own (281). Functional blockade, developmental inhibition, or physical deletion of MDSC was shown to enhance the efficacy of cancer vaccines in animal models (282–284).
Conclusion
The role of the immune system in inflammation and carcinogenesis is highly influenced by the microenvironment, thus some disease settings can display unique characteristics where immuno-regulatory processes are highly beneficial to the host but in other cases quite detrimental and predispose to pathogen persistence and increased risk of cancer. This calls for tailor-matched interventions, which are quite promising, however caution must be exercised since blocking an inhibitory pathway might re-invigorate the immune system to achieve disease control on one hand, but exacerbate immune activation and inflammation on the other. Overall, the timings of these interventions will be crucial in order to achieve favorable outcomes.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Beatrice Omusiro Ondondo is an immunologist employed on a research grant supported by MRC UK.
References
2. Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol (2003) 21:807–39. doi:10.1146/annurev.immunol.21.120601.141135
3. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature (2001) 410(6832):1107–11. doi:10.1038/35074122
4. Ramsdell F, Fowlkes BJ. Maintenance of in vivo tolerance by persistence of antigen. Science (1992) 257(5073):1130–4. doi:10.1126/science.257.5073.1130
5. Utzschneider DT, Legat A, Fuertes Marraco SA, Carrie L, Luescher I, Speiser DE, et al. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol (2013) 14(6):603–10. doi:10.1038/ni.2606
6. Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell (2009) 138(1):30–50. doi:10.1016/j.cell.2009.06.036
7. Wood C, Harrington W Jr. AIDS and associated malignancies. Cell Res (2005) 15(11–12):947–52. doi:10.1038/sj.cr.7290372
8. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut (2001) 48(4):526–35. doi:10.1136/gut.48.4.526
9. Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer (2001) 91(4):854–62. doi:10.1002/1097-0142(20010215)91:4<854::AID-CNCR1073>3.0.CO;2-Z
10. Ladoire S, Martin F, Ghiringhelli F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunother (2011) 60(7):909–18. doi:10.1007/s00262-011-1046-y
11. Wang Y, Liu XP, Zhao ZB, Chen JH, Yu CG. Expression of CD4+ forkhead box P3 (FOXP3)+ regulatory T cells in inflammatory bowel disease. J Dig Dis (2011) 12(4):286–94. doi:10.1111/j.1751-2980.2011.00505.x
12. Chinen T, Volchkov PY, Chervonsky AV, Rudensky AY. A critical role for regulatory T cell-mediated control of inflammation in the absence of commensal microbiota. J Exp Med (2010) 207(11):2323–30. doi:10.1084/jem.20101235
13. Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature (1998) 391(6665):397–401. doi:10.1038/34929
14. Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med (1996) 2(3):338–42. doi:10.1038/nm0396-338
15. Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature (1997) 389(6652):737–42. doi:10.1038/39614
16. Weiner HL. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol Rev (2001) 182:207–14. doi:10.1034/j.1600-065X.2001.1820117.x
17. Beissert S, Schwarz A, Schwarz T. Regulatory T cells. J Invest Dermatol (2006) 126(1):15–24. doi:10.1038/sj.jid.5700399
18. Vieira PL, Christensen JR, Minaee S, O’Neill EJ, Barrat FJ, Boonstra A, et al. IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J Immunol (2004) 172(10):5986–93.
19. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi:10.1038/nri2506
20. Tang XL, Smith TR, Kumar V. Specific control of immunity by regulatory CD8 T cells. Cell Mol Immunol (2005) 2(1):11–9.
21. Cosmi L, Liotta F, Lazzeri E, Francalanci M, Angeli R, Mazzinghi B, et al. Human CD8+CD25+ thymocytes share phenotypic and functional features with CD4+CD25+ regulatory thymocytes. Blood (2003) 102(12):4107–14. doi:10.1182/blood-2003-04-1320
22. Endharti AT, Rifa IM, Shi Z, Fukuoka Y, Nakahara Y, Kawamoto Y, et al. Cutting edge: CD8+CD122+ regulatory T cells produce IL-10 to suppress IFN-gamma production and proliferation of CD8+ T cells. J Immunol (2005) 175(11):7093–7.
23. Ehlers M, Papewalis C, Stenzel W, Jacobs B, Meyer KL, Deenen R, et al. Immunoregulatory natural killer cells suppress autoimmunity by down-regulating antigen-specific CD8+ T cells in mice. Endocrinology (2012) 153(9):4367–79. doi:10.1210/en.2012-1247
24. Ebata K, Shimizu Y, Nakayama Y, Minemura M, Murakami J, Kato T, et al. Immature NK cells suppress dendritic cell functions during the development of leukemia in a mouse model. J Immunol (2006) 176(7):4113–24.
25. Klenerman P, Hill A. T cells and viral persistence: lessons from diverse infections. Nat Immunol (2005) 6(9):873–9. doi:10.1038/ni1241
26. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol (2003) 77(8):4911–27. doi:10.1128/JVI.77.8.4911-4927.2003
27. Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol (2007) 19(4):408–15. doi:10.1016/j.coi.2007.06.004
28. Shin H, Blackburn SD, Blattman JN, Wherry EJ. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J Exp Med (2007) 204(4):941–9. doi:10.1084/jem.20061937
29. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev (2010) 236:219–42. doi:10.1111/j.1600-065X.2010.00923.x
30. Nishimura H, Honjo T. PD-1: an inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol (2001) 22(5):265–8. doi:10.1016/S1471-4906(01)01888-9
31. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11(2):141–51. doi:10.1016/S1074-7613(00)80089-8
32. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science (2001) 291(5502):319–22. doi:10.1126/science.291.5502.319
33. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443(7109):350–4. doi:10.1038/nature05115
34. Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med (2006) 12(10):1198–202. doi:10.1038/nm1106-1329b
35. Zhang JY, Zhang Z, Wang X, Fu JL, Yao J, Jiao Y, et al. PD-1 up-regulation is correlated with HIV-specific memory CD8+ T-cell exhaustion in typical progressors but not in long-term nonprogressors. Blood (2007) 109(11):4671–8. doi:10.1182/blood-2006-09-044826
36. Maier H, Isogawa M, Freeman GJ, Chisari FV. PD-1:PD-L1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. J Immunol (2007) 178(5):2714–20.
37. Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol (2007) 81(17):9249–58. doi:10.1128/JVI.00409-07
38. Zhang Z, Zhang JY, Wherry EJ, Jin B, Xu B, Zou ZS, et al. Dynamic programmed death 1 expression by virus-specific CD8 T cells correlates with the outcome of acute hepatitis B. Gastroenterology (2008) 134(7):1938–49. doi:10.1053/j.gastro.2008.03.037
39. Fisicaro P, Valdatta C, Massari M, Loggi E, Biasini E, Sacchelli L, et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology (2010) 138(2):e1–4. doi:10.1053/j.gastro.2009.09.052
40. D’Souza M, Fontenot AP, Mack DG, Lozupone C, Dillon S, Meditz A, et al. Programmed death 1 expression on HIV-specific CD4+ T cells is driven by viral replication and associated with T cell dysfunction. J Immunol (2007) 179(3):1979–87.
41. Kaufmann DE, Kavanagh DG, Pereyra F, Zaunders JJ, Mackey EW, Miura T, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol (2007) 8(11):1246–54. doi:10.1038/ni1515
42. Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med (2006) 203(10):2281–92. doi:10.1084/jem.20061496
43. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol (2009) 10(1):29–37. doi:10.1038/ni.1679
44. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med (2008) 205(12):2763–79. doi:10.1084/jem.20081398
45. Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A (2010) 107(33):14733–8. doi:10.1073/pnas.1009731107
46. Yamamoto T, Price DA, Casazza JP, Ferrari G, Nason M, Chattopadhyay PK, et al. Surface expression patterns of negative regulatory molecules identify determinants of virus-specific CD8+ T-cell exhaustion in HIV infection. Blood (2011) 117(18):4805–15. doi:10.1182/blood-2010-11-317297
47. Nakamoto N, Cho H, Shaked A, Olthoff K, Valiga ME, Kaminski M, et al. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog (2009) 5(2):e1000313. doi:10.1371/journal.ppat.1000313
48. Homann D, McGavern DB, Oldstone MB. Visualizing the viral burden: phenotypic and functional alterations of T cells and APCs during persistent infection. J Immunol (2004) 172(10):6239–50.
49. Sevilla N, McGavern DB, Teng C, Kunz S, Oldstone MB. Viral targeting of hematopoietic progenitors and inhibition of DC maturation as a dual strategy for immune subversion. J Clin Invest (2004) 113(5):737–45. doi:10.1172/JCI20243
50. Rodrigue-Gervais IG, Rigsby H, Jouan L, Sauve D, Sekaly RP, Willems B, et al. Dendritic cell inhibition is connected to exhaustion of CD8+ T cell polyfunctionality during chronic hepatitis C virus infection. J Immunol (2010) 184(6):3134–44. doi:10.4049/jimmunol.0902522
51. Chen L, Zhang Z, Chen W, Zhang Z, Li Y, Shi M, et al. B7-H1 up-regulation on myeloid dendritic cells significantly suppresses T cell immune function in patients with chronic hepatitis B. J Immunol (2007) 178(10):6634–41.
52. Manches O, Munn D, Fallahi A, Lifson J, Chaperot L, Plumas J, et al. HIV-activated human plasmacytoid DCs induce Tregs through an indoleamine 2,3-dioxygenase-dependent mechanism. J Clin Invest (2008) 118(10):3431–9. doi:10.1172/JCI34823
53. Franzese O, Kennedy PT, Gehring AJ, Gotto J, Williams R, Maini MK, et al. Modulation of the CD8+-T-cell response by CD4+ CD25+ regulatory T cells in patients with hepatitis B virus infection. J Virol (2005) 79(6):3322–8. doi:10.1128/JVI.79.6.3322-3328.2005
54. Xu D, Fu J, Jin L, Zhang H, Zhou C, Zou Z, et al. Circulating and liver resident CD4+CD25+ regulatory T cells actively influence the antiviral immune response and disease progression in patients with hepatitis B. J Immunol (2006) 177(1):739–47.
55. Barboza L, Salmen S, Goncalves L, Colmenares M, Peterson D, Montes H, et al. Antigen-induced regulatory T cells in HBV chronically infected patients. Virology (2007) 368(1):41–9. doi:10.1016/j.virol.2007.06.030
56. Ebinuma H, Nakamoto N, Li Y, Price DA, Gostick E, Levine BL, et al. Identification and in vitro expansion of functional antigen-specific CD25+ FoxP3+ regulatory T cells in hepatitis C virus infection. J Virol (2008) 82(10):5043–53. doi:10.1128/JVI.01548-07
57. Accapezzato D, Francavilla V, Paroli M, Casciaro M, Chircu LV, Cividini A, et al. Hepatic expansion of a virus-specific regulatory CD8(+) T cell population in chronic hepatitis C virus infection. J Clin Invest (2004) 113(7):963–72. doi:10.1172/JCI20515
58. Dolganiuc A, Paek E, Kodys K, Thomas J, Szabo G. Myeloid dendritic cells of patients with chronic HCV infection induce proliferation of regulatory T lymphocytes. Gastroenterology (2008) 135(6):2119–27. doi:10.1053/j.gastro.2008.07.082
59. Kinter A, McNally J, Riggin L, Jackson R, Roby G, Fauci AS. Suppression of HIV-specific T cell activity by lymph node CD25+ regulatory T cells from HIV-infected individuals. Proc Natl Acad Sci U S A (2007) 104(9):3390–5. doi:10.1073/pnas.0611423104
60. Nilsson J, Boasso A, Velilla PA, Zhang R, Vaccari M, Franchini G, et al. HIV-1-driven regulatory T-cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood (2006) 108(12):3808–17. doi:10.1182/blood-2006-05-021576
61. Bi X, Suzuki Y, Gatanaga H, Oka S. High frequency and proliferation of CD4+ FOXP3+ Treg in HIV-1-infected patients with low CD4 counts. Eur J Immunol (2009) 39(1):301–9. doi:10.1002/eji.200838667
62. Hall CH, Kassel R, Tacke RS, Hahn YS. HCV+ hepatocytes induce human regulatory CD4+ T cells through the production of TGF-beta. PLoS One (2010) 5(8):e12154. doi:10.1371/journal.pone.0012154
63. Ward SM, Fox BC, Brown PJ, Worthington J, Fox SB, Chapman RW, et al. Quantification and localisation of FOXP3+ T lymphocytes and relation to hepatic inflammation during chronic HCV infection. J Hepatol (2007) 47(3):316–24. doi:10.1016/j.jhep.2007.03.023
64. Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol (2000) 164(1):183–90.
65. Cao S, Li Y, Ye J, Yang X, Chen L, Liu X, et al. Japanese encephalitis virus wild strain infection suppresses dendritic cells maturation and function, and causes the expansion of regulatory T cells. Virol J (2011) 8:39. doi:10.1186/1743-422X-8-39
66. Robertson SJ, Hasenkrug KJ. The role of virus-induced regulatory T cells in immunopathology. Springer Semin Immunopathol (2006) 28(1):51–62. doi:10.1007/s00281-006-0019-2
67. Dittmer U, He H, Messer RJ, Schimmer S, Olbrich AR, Ohlen C, et al. Functional impairment of CD8(+) T cells by regulatory T cells during persistent retroviral infection. Immunity (2004) 20(3):293–303. doi:10.1016/S1074-7613(04)00054-8
68. Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med (2006) 12(11):1301–9. doi:10.1038/nm1492
69. Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, et al. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med (2006) 203(11):2461–72. doi:10.1084/jem.20061462
70. Brockman MA, Kwon DS, Tighe DP, Pavlik DF, Rosato PC, Sela J, et al. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood (2009) 114(2):346–56. doi:10.1182/blood-2008-12-191296
71. Clerici M, Wynn TA, Berzofsky JA, Blatt SP, Hendrix CW, Sher A, et al. Role of interleukin-10 in T helper cell dysfunction in asymptomatic individuals infected with the human immunodeficiency virus. J Clin Invest (1994) 93(2):768–75. doi:10.1172/JCI117031
72. Hyodo N, Nakamura I, Imawari M. Hepatitis B core antigen stimulates interleukin-10 secretion by both T cells and monocytes from peripheral blood of patients with chronic hepatitis B virus infection. Clin Exp Immunol (2004) 135(3):462–6.
73. Kaplan DE, Ikeda F, Li Y, Nakamoto N, Ganesan S, Valiga ME, et al. Peripheral virus-specific T-cell interleukin-10 responses develop early in acute hepatitis C infection and become dominant in chronic hepatitis. J Hepatol (2008) 48(6):903–13. doi:10.1016/j.jhep.2008.01.030
74. Maris CH, Chappell CP, Jacob J. Interleukin-10 plays an early role in generating virus-specific T cell anergy. BMC Immunol (2007) 8:8. doi:10.1186/1471-2172-8-8
75. Ohga S, Nomura A, Takada H, Tanaka T, Furuno K, Takahata Y, et al. Dominant expression of interleukin-10 and transforming growth factor-beta genes in activated T-cells of chronic active Epstein-Barr virus infection. J Med Virol (2004) 74(3):449–58. doi:10.1002/jmv.20197
76. Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol (1998) 16:137–61. doi:10.1146/annurev.immunol.16.1.137
77. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol (2009) 10(9):1000–7. doi:10.1038/ni.1774
78. Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature (2007) 445(7129):766–70. doi:10.1038/nature05479
79. Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, et al. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity (2012) 36(2):262–75. doi:10.1016/j.immuni.2011.12.012
80. Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci U S A (2009) 106(6):1903–8. doi:10.1073/pnas.0811556106
81. Duarte JH, Zelenay S, Bergman ML, Martins AC, Demengeot J. Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur J Immunol (2009) 39(4):948–55. doi:10.1002/eji.200839196
82. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature (2007) 445(7129):771–5. doi:10.1038/nature05543
83. Krishnamoorthy N, Khare A, Oriss TB, Raundhal M, Morse C, Yarlagadda M, et al. Early infection with respiratory syncytial virus impairs regulatory T cell function and increases susceptibility to allergic asthma. Nat Med (2012) 18(10):1525–30. doi:10.1038/nm.2896
84. Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science (2009) 323(5920):1488–92. doi:10.1126/science.1169152
85. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature (2008) 453(7192):236–40. doi:10.1038/nature06878
86. Osorio F, LeibundGut-Landmann S, Lochner M, Lahl K, Sparwasser T, Eberl G, et al. DC activated via dectin-1 convert Treg into IL-17 producers. Eur J Immunol (2008) 38(12):3274–81. doi:10.1002/eji.200838950
87. Voo KS, Wang YH, Santori FR, Boggiano C, Wang YH, Arima K, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci U S A (2009) 106(12):4793–8. doi:10.1073/pnas.0900408106
88. Beriou G, Costantino CM, Ashley CW, Yang L, Kuchroo VK, Baecher-Allan C, et al. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood (2009) 113(18):4240–9. doi:10.1182/blood-2008-10-183251
89. Ayyoub M, Deknuydt F, Raimbaud I, Dousset C, Leveque L, Bioley G, et al. Human memory FOXP3+ Tregs secrete IL-17 ex vivo and constitutively express the T(H)17 lineage-specific transcription factor RORgamma t. Proc Natl Acad Sci U S A (2009) 106(21):8635–40. doi:10.1073/pnas.0900621106
90. Lochner M, Peduto L, Cherrier M, Sawa S, Langa F, Varona R, et al. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J Exp Med (2008) 205(6):1381–93. doi:10.1084/jem.20080034
91. Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity (2009) 30(1):155–67. doi:10.1016/j.immuni.2008.12.009
92. Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity (2009) 31(5):772–86. doi:10.1016/j.immuni.2009.10.001
93. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes (2005) 54(1):92–9. doi:10.2337/diabetes.54.1.92
94. Haas J, Hug A, Viehover A, Fritzsching B, Falk CS, Filser A, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol (2005) 35(11):3343–52. doi:10.1002/eji.200526065
95. Huan J, Culbertson N, Spencer L, Bartholomew R, Burrows GG, Chou YK, et al. Decreased FOXP3 levels in multiple sclerosis patients. J Neurosci Res (2005) 81(1):45–52. doi:10.1002/jnr.20522
96. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med (2004) 199(7):971–9. doi:10.1084/jem.20031579
97. Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O’Farrelly C, et al. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol (2009) 183(11):7602–10. doi:10.4049/jimmunol.0901881
98. Astier AL, Meiffren G, Freeman S, Hafler DA. Alterations in CD46-mediated Tr1 regulatory T cells in patients with multiple sclerosis. J Clin Invest (2006) 116(12):3252–7. doi:10.1172/JCI29251
99. Glick AB, Wodzinski A, Fu P, Levine AD, Wald DN. Impairment of regulatory T-cell function in autoimmune thyroid disease. Thyroid (2013) 23(7):871–8. doi:10.1089/thy.2012.0514
100. Arandi N, Mirshafiey A, Jeddi-Tehrani M, Shaghaghi M, Sadeghi B, Abolhassani H, et al. Alteration in frequency and function of CD4+CD25+FOXP3+ regulatory T cells in patients with immune thrombocytopenic purpura. Iran J Allergy Asthma Immunol (2014) 13(2):85–92.
101. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-Hora M, Kodama T, et al. Pathogenic conversion of Foxp3(+) T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20(1):62–8. doi:10.1038/nm.3432
102. Hovhannisyan Z, Treatman J, Littman DR, Mayer L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology (2011) 140(3):957–65. doi:10.1053/j.gastro.2010.12.002
103. Kryczek I, Wu K, Zhao E, Wei S, Vatan L, Szeliga W, et al. IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J Immunol (2011) 186(7):4388–95. doi:10.4049/jimmunol.1003251
104. Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med (2011) 17(6):673–5. doi:10.1038/nm.2389
105. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol (2007) 178(11):6725–9.
106. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity (2008) 29(1):44–56. doi:10.1016/j.immuni.2008.05.007
107. Nyirenda MH, Sanvito L, Darlington PJ, O’Brien K, Zhang GX, Constantinescu CS, et al. TLR2 stimulation drives human naive and effector regulatory T cells into a Th17-like phenotype with reduced suppressive function. J Immunol (2011) 187(5):2278–90. doi:10.4049/jimmunol.1003715
108. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med (2005) 201(7):1061–7. doi:10.1084/jem.20042276
109. Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol (2009) 10(11):1178–84. doi:10.1038/ni.1791
110. Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity (2006) 25(3):455–71. doi:10.1016/j.immuni.2006.07.011
111. Pion M, Jaramillo-Ruiz D, Martinez A, Munoz-Fernandez MA, Correa-Rocha R. HIV infection of human regulatory T cells downregulates Foxp3 expression by increasing DNMT3b levels and DNA methylation in the FOXP3 gene. AIDS (2013) 27(13):2019–29. doi:10.1097/QAD.0b013e32836253fd
112. Franceschini D, Paroli M, Francavilla V, Videtta M, Morrone S, Labbadia G, et al. PD-L1 negatively regulates CD4+CD25+Foxp3+ Tregs by limiting STAT-5 phosphorylation in patients chronically infected with HCV. J Clin Invest (2009) 119(3):551–64. doi:10.1172/JCI36604
113. Iellem A, Colantonio L, D’Ambrosio D. Skin-versus gut-skewed homing receptor expression and intrinsic CCR4 expression on human peripheral blood CD4+CD25+ suppressor T cells. Eur J Immunol (2003) 33(6):1488–96. doi:10.1002/eji.200323658
114. Wysocki CA, Jiang Q, Panoskaltsis-Mortari A, Taylor PA, McKinnon KP, Su L, et al. Critical role for CCR5 in the function of donor CD4+CD25+ regulatory T cells during acute graft-versus-host disease. Blood (2005) 106(9):3300–7. doi:10.1182/blood-2005-04-1632
115. Yurchenko E, Tritt M, Hay V, Shevach EM, Belkaid Y, Piccirillo CA. CCR5-dependent homing of naturally occurring CD4+ regulatory T cells to sites of Leishmania major infection favors pathogen persistence. J Exp Med (2006) 203(11):2451–60. doi:10.1084/jem.20060956
116. Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med (2004) 199(3):303–13. doi:10.1084/jem.20031562
117. Luster AD. The role of chemokines in linking innate and adaptive immunity. Curr Opin Immunol (2002) 14(1):129–35. doi:10.1016/S0952-7915(01)00308-9
118. Yuan Q, Bromley SK, Means TK, Jones KJ, Hayashi F, Bhan AK, et al. CCR4-dependent regulatory T cell function in inflammatory bowel disease. J Exp Med (2007) 204(6):1327–34. doi:10.1084/jem.20062076
119. Ondondo B, Jones E, Godkin A, Gallimore A. Home sweet home: the tumor microenvironment as a haven for regulatory T cells. Front Immunol (2013) 4:197. doi:10.3389/fimmu.2013.00197
120. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10(9):942–9. doi:10.1038/nm1093
121. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoid like stroma and immune escape by tumors that express the chemokine CCL21. Science (2010) 328(5979):749–52. doi:10.1126/science.1185837
122. Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer (2008) 122(10):2286–93. doi:10.1002/ijc.23392
123. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol (2009) 182(3):1746–55.
124. Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci U S A (2013) 110(44):17945–50. doi:10.1073/pnas.1316796110
125. Moreno-Fernandez ME, Zapata W, Blackard JT, Franchini G, Chougnet CA. Human regulatory T cells are targets for human immunodeficiency virus (HIV) infection, and their susceptibility differs depending on the HIV type 1 strain. J Virol (2009) 83(24):12925–33. doi:10.1128/JVI.01352-09
126. Jiang Q, Zhang L, Wang R, Jeffrey J, Washburn ML, Brouwer D, et al. FoxP3+CD4+ regulatory T cells play an important role in acute HIV-1 infection in humanized Rag2-/-gammaC-/- mice in vivo. Blood (2008) 112(7):2858–68. doi:10.1182/blood-2008-03-145946
127. Kinter AL, Hennessey M, Bell A, Kern S, Lin Y, Daucher M, et al. CD25(+)CD4(+) regulatory T cells from the peripheral blood of asymptomatic HIV-infected individuals regulate CD4(+) and CD8(+) HIV-specific T cell immune responses in vitro and are associated with favorable clinical markers of disease status. J Exp Med (2004) 200(3):331–43. doi:10.1084/jem.20032069
128. Andersson J, Boasso A, Nilsson J, Zhang R, Shire NJ, Lindback S, et al. The prevalence of regulatory T cells in lymphoid tissue is correlated with viral load in HIV-infected patients. J Immunol (2005) 174(6):3143–7.
129. Chase AJ, Sedaghat AR, German JR, Gama L, Zink MC, Clements JE, et al. Severe depletion of CD4+ CD25+ regulatory T cells from the intestinal lamina propria but not peripheral blood or lymph nodes during acute simian immunodeficiency virus infection. J Virol (2007) 81(23):12748–57. doi:10.1128/JVI.00841-07
130. Presicce P, Shaw JM, Miller CJ, Shacklett BL, Chougnet CA. Myeloid dendritic cells isolated from tissues of SIV-infected Rhesus macaques promote the induction of regulatory T cells. AIDS (2012) 26(3):263–73. doi:10.1097/QAD.0b013e32834ed8df
131. Banerjee DK, Dhodapkar MV, Matayeva E, Steinman RM, Dhodapkar KM. Expansion of FOXP3high regulatory T cells by human dendritic cells (DCs) in vitro and after injection of cytokine-matured DCs in myeloma patients. Blood (2006) 108(8):2655–61. doi:10.1182/blood-2006-03-011353
132. Presicce P, Moreno-Fernandez ME, Rusie LK, Fichtenbaum C, Chougnet CA. In vitro HIV infection impairs the capacity of myeloid dendritic cells to induce regulatory T cells. PLoS One (2012) 7(8):e42802. doi:10.1371/journal.pone.0042802
133. Qin S, Sui Y, Soloff AC, Junecko BA, Kirschner DE, Murphey-Corb MA, et al. Chemokine and cytokine mediated loss of regulatory T cells in lymph nodes during pathogenic simian immunodeficiency virus infection. J Immunol (2008) 180(8):5530–6.
134. Khatami M. Inflammation, aging, and cancer: tumoricidal versus tumorigenesis of immunity: a common denominator mapping chronic diseases. Cell Biochem Biophys (2009) 55(2):55–79. doi:10.1007/s12013-009-9059-2
135. Khatami M. ‘Yin and Yang’ in inflammation: duality in innate immune cell function and tumorigenesis. Expert Opin Biol Ther (2008) 8(10):1461–72. doi:10.1517/14712598.8.10.1461
136. Rouse BT, Sehrawat S. Immunity and immunopathology to viruses: what decides the outcome? Nat Rev Immunol (2010) 10(7):514–26. doi:10.1038/nri2802
137. Moore KW, Vieira P, Fiorentino DF, Trounstine ML, Khan TA, Mosmann TR. Homology of cytokine synthesis inhibitory factor (IL-10) to the Epstein-Barr virus gene BCRFI. Science (1990) 248(4960):1230–4. doi:10.1126/science.2161559
138. Slobedman B, Barry PA, Spencer JV, Avdic S, Abendroth A. Virus-encoded homologs of cellular interleukin-10 and their control of host immune function. J Virol (2009) 83(19):9618–29. doi:10.1128/JVI.01098-09
139. Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, et al. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS One (2011) 6(4):e19495. doi:10.1371/journal.pone.0019495
140. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res (2010) 70(19):7465–75. doi:10.1158/0008-5472.CAN-10-1439
141. Elrefaei M, Barugahare B, Ssali F, Mugyenyi P, Cao H. HIV-specific IL-10-positive CD8+ T cells are increased in advanced disease and are associated with decreased HIV-specific cytolysis. J Immunol (2006) 176(2):1274–80.
142. Elrefaei M, Ventura FL, Baker CA, Clark R, Bangsberg DR, Cao H. HIV-specific IL-10-positive CD8+ T cells suppress cytolysis and IL-2 production by CD8+ T cells. J Immunol (2007) 178(5):3265–71.
143. Dolganiuc A, Szabo G. T cells with regulatory activity in hepatitis C virus infection: what we know and what we don’t. J Leukoc Biol (2008) 84(3):614–22. doi:10.1189/jlb.1107770
144. Nordstrom I, Nurkkala M, Collins LV, Eriksson K. CD8+ T-cells suppress antigen-specific and allogeneic CD4+ T-cell responses to herpes simplex virus type 2-infected human dendritic cells. Viral Immunol (2005) 18(4):616–26. doi:10.1089/vim.2005.18.616
145. Popescu I, Macedo C, Abu-Elmagd K, Shapiro R, Hua Y, Thomson AW, et al. EBV-specific CD8+ T cell reactivation in transplant patients results in expansion of CD8+ type-1 regulatory T cells. Am J Transplant (2007) 7(5):1215–23. doi:10.1111/j.1600-6143.2007.01740.x
146. Kundu JK, Surh YJ. Inflammation: gearing the journey to cancer. Mutat Res (2008) 659(1–2):15–30. doi:10.1016/j.mrrev.2008.03.002
147. Grulich AE, Wan X, Law MG, Milliken ST, Lewis CR, Garsia RJ, et al. B-cell stimulation and prolonged immune deficiency are risk factors for non-Hodgkin’s lymphoma in people with AIDS. AIDS (2000) 14(2):133–40. doi:10.1097/00002030-200001280-00008
148. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med (2009) 206(7):1457–64. doi:10.1084/jem.20090207
149. Numasaki M, Watanabe M, Suzuki T, Takahashi H, Nakamura A, McAllister F, et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol (2005) 175(9):6177–89.
150. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24(2):179–89. doi:10.1016/j.immuni.2006.01.001
151. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441(7090):235–8. doi:10.1038/nature04753
152. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441(7090):231–4. doi:10.1038/nature04754
153. Liu J, Duan Y, Cheng X, Chen X, Xie W, Long H, et al. IL-17 is associated with poor prognosis and promotes angiogenesis via stimulating VEGF production of cancer cells in colorectal carcinoma. Biochem Biophys Res Commun (2011) 407(2):348–54. doi:10.1016/j.bbrc.2011.03.021
154. Iida T, Iwahashi M, Katsuda M, Ishida K, Nakamori M, Nakamura M, et al. Tumor-infiltrating CD4+ Th17 cells produce IL-17 in tumor microenvironment and promote tumor progression in human gastric cancer. Oncol Rep (2011) 25(5):1271–7. doi:10.3892/or.2011.1201
155. Su Z, Sun Y, Zhu H, Liu Y, Lin X, Shen H, et al. Th17 cell expansion in gastric cancer may contribute to cancer development and metastasis. Immunol Res (2014) 58(1):118–24. doi:10.1007/s12026-013-8483-y
156. Yang S, Wang B, Guan C, Wu B, Cai C, Wang M, et al. Foxp3+IL-17+ T cells promote development of cancer-initiating cells in colorectal cancer. J Leukoc Biol (2011) 89(1):85–91. doi:10.1189/jlb.0910506
157. Bentwich Z, Kalinkovich A, Weisman Z, Grossman Z. Immune activation in the context of HIV infection. Clin Exp Immunol (1998) 111(1):1–2. doi:10.1046/j.1365-2249.1998.00483.x
158. Hazenberg MD, Hamann D, Schuitemaker H, Miedema F. T cell depletion in HIV-1 infection: how CD4+ T cells go out of stock. Nat Immunol (2000) 1(4):285–9. doi:10.1038/79724
159. Hazenberg MD, Otto SA, van Benthem BH, Roos MT, Coutinho RA, Lange JM, et al. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS (2003) 17(13):1881–8. doi:10.1097/00002030-200309050-00006
160. Hunt PW, Landay AL, Sinclair E, Martinson JA, Hatano H, Emu B, et al. A low T regulatory cell response may contribute to both viral control and generalized immune activation in HIV controllers. PLoS One (2011) 6(1):e15924. doi:10.1371/journal.pone.0015924
161. Lim A, Tan D, Price P, Kamarulzaman A, Tan HY, James I, et al. Proportions of circulating T cells with a regulatory cell phenotype increase with HIV-associated immune activation and remain high on antiretroviral therapy. AIDS (2007) 21(12):1525–34. doi:10.1097/QAD.0b013e32825eab8b
162. Suchard MS, Mayne E, Green VA, Shalekoff S, Donninger SL, Stevens WS, et al. FOXP3 expression is upregulated in CD4T cells in progressive HIV-1 infection and is a marker of disease severity. PLoS One (2010) 5(7):e11762. doi:10.1371/journal.pone.0011762
163. Schulze Zur Wiesch J, Thomssen A, Hartjen P, Toth I, Lehmann C, Meyer-Olson D, et al. Comprehensive analysis of frequency and phenotype of T regulatory cells in HIV infection: CD39 expression of FoxP3+ T regulatory cells correlates with progressive disease. J Virol (2011) 85(3):1287–97. doi:10.1128/JVI.01758-10
164. Epple HJ, Loddenkemper C, Kunkel D, Troger H, Maul J, Moos V, et al. Mucosal but not peripheral FOXP3+ regulatory T cells are highly increased in untreated HIV infection and normalize after suppressive HAART. Blood (2006) 108(9):3072–8. doi:10.1182/blood-2006-04-016923
165. Aandahl EM, Michaelsson J, Moretto WJ, Hecht FM, Nixon DF. Human CD4+ CD25+ regulatory T cells control T-cell responses to human immunodeficiency virus and cytomegalovirus antigens. J Virol (2004) 78(5):2454–9. doi:10.1128/JVI.78.5.2454-2459.2004
166. Legrand FA, Nixon DF, Loo CP, Ono E, Chapman JM, Miyamoto M, et al. Strong HIV-1-specific T cell responses in HIV-1-exposed uninfected infants and neonates revealed after regulatory T cell removal. PLoS One (2006) 1:e102. doi:10.1371/journal.pone.0000102
167. Presicce P, Orsborn K, King E, Pratt J, Fichtenbaum CJ, Chougnet CA. Frequency of circulating regulatory T cells increases during chronic HIV infection and is largely controlled by highly active antiretroviral therapy. PLoS One (2011) 6(12):e28118. doi:10.1371/journal.pone.0028118
168. Montes M, Sanchez C, Lewis DE, Graviss EA, Seas C, Gotuzzo E, et al. Normalization of FoxP3(+) regulatory T cells in response to effective antiretroviral therapy. J Infect Dis (2011) 203(4):496–9. doi:10.1093/infdis/jiq073
169. Eggena MP, Barugahare B, Jones N, Okello M, Mutalya S, Kityo C, et al. Depletion of regulatory T cells in HIV infection is associated with immune activation. J Immunol (2005) 174(7):4407–14.
170. Jiao Y, Fu J, Xing S, Fu B, Zhang Z, Shi M, et al. The decrease of regulatory T cells correlates with excessive activation and apoptosis of CD8+ T cells in HIV-1-infected typical progressors, but not in long-term non-progressors. Immunology (2009) 128(1 Suppl):e366–75. doi:10.1111/j.1365-2567.2008.02978.x
171. Card CM, McLaren PJ, Wachihi C, Kimani J, Plummer FA, Fowke KR. Decreased immune activation in resistance to HIV-1 infection is associated with an elevated frequency of CD4(+)CD25(+)FOXP3(+) regulatory T cells. J Infect Dis (2009) 199(9):1318–22. doi:10.1086/597801
172. Hunt PW, Brenchley J, Sinclair E, McCune JM, Roland M, Page-Shafer K, et al. Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis (2008) 197(1):126–33. doi:10.1086/524143
173. Cao W, Jamieson BD, Hultin LE, Hultin PM, Detels R. Regulatory T cell expansion and immune activation during untreated HIV type 1 infection are associated with disease progression. AIDS Res Hum Retroviruses (2009) 25(2):183–91. doi:10.1089/aid.2008.0140
174. Mendez-Lagares G, Pozo-Balado MM, Genebat M, Garcia Perganeda A, Leal M, Pacheco YM. Severe immune dysregulation affects CD4(+)CD25(hi)FoxP3(+) regulatory T cells in HIV-infected patients with low-level CD4 T-cell repopulation despite suppressive highly active antiretroviral therapy. J Infect Dis (2012) 205(10):1501–9. doi:10.1093/infdis/jis230
175. Kwon DS, Angin M, Hongo T, Law KM, Johnson J, Porichis F, et al. CD4+ CD25+ regulatory T cells impair HIV-1-specific CD4 T cell responses by upregulating interleukin-10 production in monocytes. J Virol (2012) 86(12):6586–94. doi:10.1128/JVI.06251-11
176. Shaw JM, Hunt PW, Critchfield JW, McConnell DH, Garcia JC, Pollard RB, et al. Increased frequency of regulatory T cells accompanies increased immune activation in rectal mucosae of HIV-positive noncontrollers. J Virol (2011) 85(21):11422–34. doi:10.1128/JVI.05608-11
177. Fazekas de St Groth B, Landay AL. Regulatory T cells in HIV infection: pathogenic or protective participants in the immune response? AIDS (2008) 22(6):671–83. doi:10.1097/QAD.0b013e3282f466da
178. Weiss L, Letimier FA, Carriere M, Maiella S, Donkova-Petrini V, Targat B, et al. In vivo expansion of naive and activated CD4+CD25+FOXP3+ regulatory T cell populations in interleukin-2-treated HIV patients. Proc Natl Acad Sci U S A (2010) 107(23):10632–7. doi:10.1073/pnas.1000027107
179. Group I-ES, Committee SS, Abrams D, Levy Y, Losso MH, Babiker A, et al. Interleukin-2 therapy in patients with HIV infection. N Engl J Med (2009) 361(16):1548–59. doi:10.1056/NEJMoa0903175
180. Weiss L, Donkova-Petrini V, Caccavelli L, Balbo M, Carbonneil C, Levy Y. Human immunodeficiency virus-driven expansion of CD4+CD25+ regulatory T cells, which suppress HIV-specific CD4 T-cell responses in HIV- infected patients. Blood (2004) 104(10):3249–56. doi:10.1182/blood-2004-01-0365
181. Garg A, Spector SA. HIV type 1 gp120-induced expansion of myeloid derived suppressor cells is dependent on interleukin 6 and suppresses immunity. J Infect Dis (2013) 209(3):441–51. doi:10.1093/infdis/jit469
182. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology (2008) 135(1):234–43. doi:10.1053/j.gastro.2008.03.020
183. Vollbrecht T, Stirner R, Tufman A, Roider J, Huber RM, Bogner JR, et al. Chronic progressive HIV-1 infection is associated with elevated levels of myeloid-derived suppressor cells. AIDS (2012) 26(12):F31–7. doi:10.1097/QAD.0b013e328354b43f
184. Breen EC, Boscardin WJ, Detels R, Jacobson LP, Smith MW, O’Brien SJ, et al. Non-Hodgkin’s B cell lymphoma in persons with acquired immunodeficiency syndrome is associated with increased serum levels of IL10, or the IL10 promoter -592 C/C genotype. Clin Immunol (2003) 109(2):119–29. doi:10.1016/S1521-6616(03)00214-6
185. Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology (2007) 132(7):2328–39. doi:10.1053/j.gastro.2007.03.102
186. Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, et al. A review of human carcinogens – part B: biological agents. Lancet Oncol (2009) 10(4):321–2. doi:10.1016/S1470-2045(09)70096-8
187. van Baarle D, Nanlohy NM, Otto S, Plunkett FJ, Fletcher JM, Akbar AN. Progressive telomere shortening of Epstein-Barr virus-specific memory T cells during HIV infection: contributor to exhaustion? J Infect Dis (2008) 198(9):1353–7. doi:10.1086/592170
188. Martinez-Maza O, Breen EC. B-cell activation and lymphoma in patients with HIV. Curr Opin Oncol (2002) 14(5):528–32. doi:10.1097/00001622-200209000-00009
189. Molyneux EM, Rochford R, Griffin B, Newton R, Jackson G, Menon G, et al. Burkitt’s lymphoma. Lancet (2012) 379(9822):1234–44. doi:10.1016/S0140-6736(11)61177-X
190. He B, Qiao X, Klasse PJ, Chiu A, Chadburn A, Knowles DM, et al. HIV-1 envelope triggers polyclonal Ig class switch recombination through a CD40-independent mechanism involving BAFF and C-type lectin receptors. J Immunol (2006) 176(7):3931–41.
191. Martin G, Roy J, Barat C, Ouellet M, Gilbert C, Tremblay MJ. Human immunodeficiency virus type 1-associated CD40 ligand transactivates B lymphocytes and promotes infection of CD4+ T cells. J Virol (2007) 81(11):5872–81. doi:10.1128/JVI.02542-06
192. Epeldegui M, Thapa DR, De la Cruz J, Kitchen S, Zack JA, Martinez-Maza O. CD40 ligand (CD154) incorporated into HIV virions induces activation-induced cytidine deaminase (AID) expression in human B lymphocytes. PLoS One (2010) 5(7):e11448. doi:10.1371/journal.pone.0011448
193. Killebrew D, Shiramizu B. Pathogenesis of HIV-associated non-Hodgkin lymphoma. Curr HIV Res (2004) 2(3):215–21. doi:10.2174/1570162043351237
194. van Baarle D, Hovenkamp E, Callan MF, Wolthers KC, Kostense S, Tan LC, et al. Dysfunctional Epstein-Barr virus (EBV)-specific CD8(+) T lymphocytes and increased EBV load in HIV-1 infected individuals progressing to AIDS-related non-Hodgkin lymphoma. Blood (2001) 98(1):146–55. doi:10.1182/blood.V98.1.146
195. Engels EA, Pfeiffer RM, Goedert JJ, Virgo P, McNeel TS, Scoppa SM, et al. Trends in cancer risk among people with AIDS in the United States 1980-2002. AIDS (2006) 20(12):1645–54. doi:10.1097/01.aids.0000238411.75324.59
196. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science (1994) 266(5192):1865–9. doi:10.1126/science.7997879
197. Gallo RC. The enigmas of Kaposi’s sarcoma. Science (1998) 282(5395):1837–9. doi:10.1126/science.282.5395.1837
198. Beral V, Peterman TA, Berkelman RL, Jaffe HW. Kaposi’s sarcoma among persons with AIDS: a sexually transmitted infection? Lancet (1990) 335(8682):123–8. doi:10.1016/0140-6736(90)90001-L
199. Martin JN, Ganem DE, Osmond DH, Page-Shafer KA, Macrae D, Kedes DH. Sexual transmission and the natural history of human herpesvirus 8 infection. N Engl J Med (1998) 338(14):948–54. doi:10.1056/NEJM199804023381403
200. Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol (1999) 189(1):12–9. doi:10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F
201. Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med (1998) 338(7):423–8. doi:10.1056/NEJM199802123380703
202. Molano M, van den Brule A, Plummer M, Weiderpass E, Posso H, Arslan A, et al. Determinants of clearance of human papillomavirus infections in Colombian women with normal cytology: a population-based, 5-year follow-up study. Am J Epidemiol (2003) 158(5):486–94. doi:10.1093/aje/kwg171
203. Jenson AB, Kurman RJ, Lancaster WD. Tissue effects of and host response to human papillomavirus infection. Dermatol Clin (1991) 9(2):203–9.
204. Hopfl R, Heim K, Christensen N, Zumbach K, Wieland U, Volgger B, et al. Spontaneous regression of CIN and delayed-type hypersensitivity to HPV-16 oncoprotein E7. Lancet (2000) 356(9246):1985–6. doi:10.1016/S0140-6736(00)03315-8
205. de Jong A, van Poelgeest MI, van der Hulst JM, Drijfhout JW, Fleuren GJ, Melief CJ, et al. Human papillomavirus type 16-positive cervical cancer is associated with impaired CD4+ T-cell immunity against early antigens E2 and E6. Cancer Res (2004) 64(15):5449–55. doi:10.1158/0008-5472.CAN-04-0831
206. Frazer IH, Thomas R, Zhou J, Leggatt GR, Dunn L, McMillan N, et al. Potential strategies utilised by papillomavirus to evade host immunity. Immunol Rev (1999) 168:131–42.
207. Garzetti GG, Ciavattini A, Goteri G, De Nictolis M, Menso S, Muzzioli M, et al. HPV DNA positivity and natural killer cell activity in the clinical outcome of mild cervical dysplasia: integration between virus and immune system. Gynecol Obstet Invest (1995) 39(2):130–5. doi:10.1159/000292394
208. Molling JW, de Gruijl TD, Glim J, Moreno M, Rozendaal L, Meijer CJ, et al. CD4(+)CD25hi regulatory T-cell frequency correlates with persistence of human papillomavirus type 16 and T helper cell responses in patients with cervical intraepithelial neoplasia. Int J Cancer (2007) 121(8):1749–55. doi:10.1002/ijc.22894
209. Visser J, Nijman HW, Hoogenboom BN, Jager P, van Baarle D, Schuuring E, et al. Frequencies and role of regulatory T cells in patients with (pre)malignant cervical neoplasia. Clin Exp Immunol (2007) 150(2):199–209. doi:10.1111/j.1365-2249.2007.03468.x
210. Loddenkemper C, Hoffmann C, Stanke J, Nagorsen D, Baron U, Olek S, et al. Regulatory (FOXP3+) T cells as target for immune therapy of cervical intraepithelial neoplasia and cervical cancer. Cancer Sci (2009) 100(6):1112–7. doi:10.1111/j.1349-7006.2009.01153.x
211. Scott ME, Ma Y, Kuzmich L, Moscicki AB. Diminished IFN-gamma and IL-10 and elevated Foxp3 mRNA expression in the cervix are associated with CIN 2 or 3. Int J Cancer (2009) 124(6):1379–83. doi:10.1002/ijc.24117
212. Piersma SJ, Jordanova ES, van Poelgeest MI, Kwappenberg KM, van der Hulst JM, Drijfhout JW, et al. High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res (2007) 67(1):354–61. doi:10.1158/0008-5472.CAN-06-3388
213. van der Burg SH, Piersma SJ, de Jong A, van der Hulst JM, Kwappenberg KM, van den Hende M, et al. Association of cervical cancer with the presence of CD4+ regulatory T cells specific for human papillomavirus antigens. Proc Natl Acad Sci U S A (2007) 104(29):12087–92. doi:10.1073/pnas.0704672104
214. Chaturvedi AK, Madeleine MM, Biggar RJ, Engels EA. Risk of human papillomavirus-associated cancers among persons with AIDS. J Natl Cancer Inst (2009) 101(16):1120–30. doi:10.1093/jnci/djp205
215. Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet (2007) 370(9581):59–67. doi:10.1016/S0140-6736(07)61050-2
216. Petry KU, Scheffel D, Bode U, Gabrysiak T, Kochel H, Kupsch E, et al. Cellular immunodeficiency enhances the progression of human papillomavirus-associated cervical lesions. Int J Cancer (1994) 57(6):836–40. doi:10.1002/ijc.2910570612
217. From the Centers for Disease Control and Prevention. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. JAMA (1993) 269(6):729–30. doi:10.1001/jama.1993.03500060023008
218. Adler DH, Kakinami L, Modisenyane T, Tshabangu N, Mohapi L, De Bruyn G, et al. Increased regression and decreased incidence of human papillomavirus-related cervical lesions among HIV-infected women on HAART. AIDS (2012) 26(13):1645–52. doi:10.1097/QAD.0b013e32835536a3
219. Ahdieh-Grant L, Li R, Levine AM, Massad LS, Strickler HD, Minkoff H, et al. Highly active antiretroviral therapy and cervical squamous intraepithelial lesions in human immunodeficiency virus-positive women. J Natl Cancer Inst (2004) 96(14):1070–6. doi:10.1093/jnci/djh192
220. Heard I, Schmitz V, Costagliola D, Orth G, Kazatchkine MD. Early regression of cervical lesions in HIV-seropositive women receiving highly active antiretroviral therapy. AIDS (1998) 12(12):1459–64. doi:10.1097/00002030-199812000-00007
221. Freeman AJ, Pan Y, Harvey CE, Post JJ, Law MG, White PA, et al. The presence of an intrahepatic cytotoxic T lymphocyte response is associated with low viral load in patients with chronic hepatitis C virus infection. J Hepatol (2003) 38(3):349–56. doi:10.1016/S0168-8278(02)00424-5
222. Lopes AR, Kellam P, Das A, Dunn C, Kwan A, Turner J, et al. Bim-mediated deletion of antigen-specific CD8 T cells in patients unable to control HBV infection. J Clin Invest (2008) 118(5):1835–45. doi:10.1172/JCI33402
223. Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity (1996) 4(1):25–36. doi:10.1016/S1074-7613(00)80295-2
224. Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med (2001) 194(10):1395–406. doi:10.1084/jem.194.10.1395
225. Boettler T, Spangenberg HC, Neumann-Haefelin C, Panther E, Urbani S, Ferrari C, et al. T cells with a CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J Virol (2005) 79(12):7860–7. doi:10.1128/JVI.79.12.7860-7867.2005
226. Cabrera R, Tu Z, Xu Y, Firpi RJ, Rosen HR, Liu C, et al. An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes in hepatitis C virus infection. Hepatology (2004) 40(5):1062–71. doi:10.1002/hep.20454
227. Nan XP, Zhang Y, Yu HT, Sun RL, Peng MJ, Li Y, et al. Inhibition of viral replication downregulates CD4(+)CD25(high) regulatory T cells and programmed death-ligand 1 in chronic hepatitis B. Viral Immunol (2012) 25(1):21–8. doi:10.1089/vim.2011.0049
228. Unitt E, Rushbrook SM, Marshall A, Davies S, Gibbs P, Morris LS, et al. Compromised lymphocytes infiltrate hepatocellular carcinoma: the role of T-regulatory cells. Hepatology (2005) 41(4):722–30. doi:10.1002/hep.20644
229. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi:10.1016/j.cell.2010.01.025
230. Trinchieri G. Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu Rev Immunol (2012) 30:677–706. doi:10.1146/annurev-immunol-020711-075008
231. Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology (2010) 138(6):2101–14e5. doi:10.1053/j.gastro.2010.01.058
232. Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med (2005) 353(25):2654–66. doi:10.1056/NEJMoa051424
233. Shibata M, Nezu T, Kanou H, Abe H, Takekawa M, Fukuzawa M. Decreased production of interleukin-12 and type 2 immune responses are marked in cachectic patients with colorectal and gastric cancer. J Clin Gastroenterol (2002) 34(4):416–20. doi:10.1097/00004836-200204000-00006
234. Osawa E, Nakajima A, Fujisawa T, Kawamura YI, Toyama-Sorimachi N, Nakagama H, et al. Predominant T helper type 2-inflammatory responses promote murine colon cancers. Int J Cancer (2006) 118(9):2232–6. doi:10.1002/ijc.21639
235. Kettunen HL, Kettunen AS, Rautonen NE. Intestinal immune responses in wild-type and Apcmin/+ mouse, a model for colon cancer. Cancer Res (2003) 63(16):5136–42.
236. Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity (2010) 33(2):279–88. doi:10.1016/j.immuni.2010.08.010
237. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol (2009) 10(3):314–24. doi:10.1038/ni.1698
238. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science (2006) 314(5804):1461–3. doi:10.1126/science.1135245
239. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med (2006) 203(10):2271–9. doi:10.1084/jem.20061308
240. Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med (2013) 210(5):917–31. doi:10.1084/jem.20122308
241. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature (2010) 464(7293):1371–5. doi:10.1038/nature08949
242. Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med (2011) 208(6):1127–33. doi:10.1084/jem.20101712
243. Fouser LA, Wright JF, Dunussi-Joannopoulos K, Collins M. Th17 cytokines and their emerging roles in inflammation and autoimmunity. Immunol Rev (2008) 226:87–102. doi:10.1111/j.1600-065X.2008.00712.x
244. Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology (2009) 136(1):257–67. doi:10.1053/j.gastro.2008.10.018
245. Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest (2005) 115(12):3623–33. doi:10.1172/JCI25947
246. Litzinger MT, Fernando R, Curiel TJ, Grosenbach DW, Schlom J, Palena C. IL-2 immunotoxin denileukin diftitox reduces regulatory T cells and enhances vaccine-mediated T-cell immunity. Blood (2007) 110(9):3192–201. doi:10.1182/blood-2007-06-094615
247. Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell (2010) 140(6):845–58. doi:10.1016/j.cell.2010.02.021
248. Erdman SE, Sohn JJ, Rao VP, Nambiar PR, Ge Z, Fox JG, et al. CD4+CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+ mice. Cancer Res (2005) 65(10):3998–4004. doi:10.1158/0008-5472.CAN-04-3104
249. Sugai M, Aoki K, Osato M, Nambu Y, Ito K, Taketo MM, et al. Runx3 is required for full activation of regulatory T cells to prevent colitis-associated tumor formation. J Immunol (2011) 186(11):6515–20. doi:10.4049/jimmunol.1001671
250. Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med (2007) 204(8):1757–64. doi:10.1084/jem.20070590
251. Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med (2007) 204(8):1775–85. doi:10.1084/jem.20070602
252. Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, et al. IL-23 promotes tumour incidence and growth. Nature (2006) 442(7101):461–5. doi:10.1038/nature04808
253. Li MO, Flavell RA. Contextual regulation of inflammation: a duet by transforming growth factor-beta and interleukin-10. Immunity (2008) 28(4):468–76. doi:10.1016/j.immuni.2008.03.003
254. Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity (2004) 21(4):491–501. doi:10.1016/j.immuni.2004.07.020
255. Izcue A, Hue S, Buonocore S, Arancibia-Carcamo CV, Ahern PP, Iwakura Y, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity (2008) 28(4):559–70. doi:10.1016/j.immuni.2008.02.019
256. Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology (2003) 124(3):762–77. doi:10.1053/gast.2003.50094
257. Erdman SE, Rao VP, Poutahidis T, Ihrig MM, Ge Z, Feng Y, et al. CD4(+)CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res (2003) 63(18):6042–50.
258. Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest (1996) 98(4):1010–20. doi:10.1172/JCI118861
259. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell (1993) 75(2):263–74.
260. Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med (2006) 203(11):2485–94. doi:10.1084/jem.20061082
261. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity (2004) 21(2):137–48. doi:10.1016/j.immuni.2004.07.017
262. Tannenbaum CS, Hamilton TA. Immune-inflammatory mechanisms in IFNgamma-mediated anti-tumor activity. Semin Cancer Biol (2000) 10(2):113–23. doi:10.1006/scbi.2000.0314
263. Michael-Robinson JM, Pandeya N, Walsh MD, Biemer-Huttmann AE, Eri RD, Buttenshaw RL, et al. Characterization of tumour-infiltrating lymphocytes and apoptosis in colitis-associated neoplasia: comparison with sporadic colorectal cancer. J Pathol (2006) 208(3):381–7. doi:10.1002/path.1895
264. Muller S, Lory J, Corazza N, Griffiths GM, Z’Graggen K, Mazzucchelli L, et al. Activated CD4+ and CD8+ cytotoxic cells are present in increased numbers in the intestinal mucosa from patients with active inflammatory bowel disease. Am J Pathol (1998) 152(1):261–8.
265. Waldner MJ, Wirtz S, Becker C, Seidel D, Tubbe I, Cappel K, et al. Perforin deficiency attenuates inflammation and tumor growth in colitis-associated cancer. Inflamm Bowel Dis (2010) 16(4):559–67. doi:10.1002/ibd.21107
266. Berzofsky JA, Terabe M. The contrasting roles of NKT cells in tumor immunity. Curr Mol Med (2009) 9(6):667–72. doi:10.2174/156652409788970706
267. Kitamura H, Iwakabe K, Yahata T, Nishimura S, Ohta A, Ohmi Y, et al. The natural killer T (NKT) cell ligand alpha-galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J Exp Med (1999) 189(7):1121–8. doi:10.1084/jem.189.7.1121
268. Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest (2004) 113(10):1490–7. doi:10.1172/JCI19836
269. Mandal D, Levine AD. Elevated IL-13Ralpha2 in intestinal epithelial cells from ulcerative colitis or colorectal cancer initiates MAPK pathway. Inflamm Bowel Dis (2010) 16(5):753–64. doi:10.1002/ibd.21133
270. Keijzer C, van der Zee R, van Eden W, Broere F. Treg inducing adjuvants for therapeutic vaccination against chronic inflammatory diseases. Front Immunol (2013) 4:245. doi:10.3389/fimmu.2013.00245
271. Hurwitz AA, Foster BA, Kwon ED, Truong T, Choi EM, Greenberg NM, et al. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res (2000) 60(9):2444–8.
272. Hurwitz AA, Yu TF, Leach DR, Allison JP. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Natl Acad Sci U S A (1998) 95(17):10067–71. doi:10.1073/pnas.95.17.10067
273. Bajor DL, Vonderheide RH. Cracking the stone: combination vaccination and CTLA-4 blockade in pancreatic cancer. J Immunother (2013) 36(7):362–4. doi:10.1097/CJI.0b013e31829fb7c8
274. Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res (2013) 19(19):5300–9. doi:10.1158/1078-0432.CCR-13-0143
275. Kadin ME, Vonderheid EC. Targeted therapies: denileukin diftitox – a step towards a ‘magic bullet’ for CTCL. Nat Rev Clin Oncol (2010) 7(8):430–2. doi:10.1038/nrclinonc.2010.105
276. Dao H Jr, Barr PM, Honda K. Regression of CD30+ cutaneous anaplastic large-cell lymphoma with denileukin diftitox. J Am Acad Dermatol (2011) 64(6):e123–5. doi:10.1016/j.jaad.2010.11.001
277. Gritzapis AD, Voutsas IF, Baxevanis CN. Ontak reduces the immunosuppressive tumor environment and enhances successful therapeutic vaccination in HER-2/neu-tolerant mice. Cancer Immunol Immunother (2012) 61(3):397–407. doi:10.1007/s00262-011-1113-4
278. Porichis F, Kwon DS, Zupkosky J, Tighe DP, McMullen A, Brockman MA, et al. Responsiveness of HIV-specific CD4 T cells to PD-1 blockade. Blood (2011) 118(4):965–74. doi:10.1182/blood-2010-12-328070
279. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol (2010) 28(19):3167–75. doi:10.1200/JCO.2009.26.7609
280. Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res (2008) 14(10):3044–51. doi:10.1158/1078-0432.CCR-07-4079
281. Dietze KK, Zelinskyy G, Liu J, Kretzmer F, Schimmer S, Dittmer U. Combining regulatory T cell depletion and inhibitory receptor blockade improves reactivation of exhausted virus-specific CD8+ T cells and efficiently reduces chronic retroviral loads. PLoS Pathog (2013) 9(12):e1003798. doi:10.1371/journal.ppat.1003798
282. Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, et al. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer (2011) 129(9):2158–70. doi:10.1002/ijc.25863
283. Nakashima H, Terabe M, Berzofsky JA, Husain SR, Puri RK. A novel combination immunotherapy for cancer by IL-13Ralpha2-targeted DNA vaccine and immunotoxin in murine tumor models. J Immunol (2011) 187(10):4935–46. doi:10.4049/jimmunol.1102095
284. Ishizaki H, Manuel ER, Song GY, Srivastava T, Sun S, Diamond DJ, et al. Modified vaccinia Ankara expressing survivin combined with gemcitabine generates specific antitumor effects in a murine pancreatic carcinoma model. Cancer Immunol Immunother (2011) 60(1):99–109. doi:10.1007/s00262-010-0923-0
Keywords: regulatory T cells, immune dysfunction, immune-regulation, inflammation, cancer, HIV-1
Citation: Ondondo BO (2014) Fallen angels or risen apes? A tale of the intricate complexities of imbalanced immune responses in the pathogenesis and progression of immune-mediated and viral cancers. Front. Immunol. 5:90. doi: 10.3389/fimmu.2014.00090
Received: 26 December 2013; Accepted: 20 February 2014;
Published online: 06 March 2014.
Edited by:
Fang-Ping Huang, Imperial College London, UKReviewed by:
Hua-Chen Chang, Indiana University-Purdue University Indianapolis, USADennis O. Adeegbe, Immunology Frontier Research Center, Japan
Copyright: © 2014 Ondondo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beatrice Omusiro Ondondo, The Jenner Institute, Nuffield Department of Medicine, University of Oxford, ORCRB, Roosevelt Drive, Oxford OX3 7DQ, UK e-mail:YmVhdHJpY2Uub25kb25kb0BuZG0ub3guYWMudWs=