 Kevin H. Toomer
Kevin H. Toomer Zhibin Chen
Zhibin Chen- 1Department of Microbiology and Immunology, University of Miami Miller School of Medicine, Miami, FL, USA
- 2Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL, USA
Cancer immunotherapy through manipulation of the immune system holds great potential for the treatment of human cancers. However, recent trials targeting the negative immune regulators cytotoxic T-lymphocyte antigen 4, programed death 1 (PD-1), and PD-1 receptor ligand (PD-L1) demonstrated that clinically significant antitumor responses were often associated with the induction of autoimmune toxicity. This finding suggests that the same immune mechanisms that elicit autoimmunity may also contribute to the destruction of tumors. Given the fact that the immunological identity of tumors might be largely an immunoprivileged self, autoimmunity may not represent a wholly undesirable outcome in the context of cancer immunotherapy. Rather, targeted killing of cancer cells and autoimmune damage to healthy tissues may be intricately linked through molecular mechanisms, in particular inflammatory cytokine signaling. On the other hand, since chronic inflammation is a well-recognized condition that promotes tumor development, it appears that autoimmunity can be a “double agent” in mediating either pro-tumor or antitumor effects. This review surveys the tumor-promoting and tumoricidal activities of several prominent cytokines: IFN-γ, TNF-α, TGF-β, IL-17, IL-23, IL-4, and IL-13, produced by three major subsets of T helper cells that interact with innate immune cells. Many of these cytokines exert divergent and seemingly contradictory effects on cancer development in different human and animal models, suggesting a high degree of context dependence in their functions. We hypothesize that these inflammatory cytokines could mediate a feedback loop of autoimmunity, antitumor immunity, and tumorigenesis. Understanding the diverse and paradoxical roles of cytokines from autoimmune responses in the setting of cancer will advance the long-term goal of improving cancer immunotherapy, while minimizing the hazards of immune-mediated tissue damage and the possibility of de novo tumorigenesis, through proper monitoring and preventive measures.
Introduction: Paradoxical Relationships between Autoimmunity and Tumor
The process of inducing the immune system to selectively destroy tumor tissues in vivo faces numerous conceptual as well as practical hurdles. Foremost among these is the self-derived immunological identity of tumors (1, 2). The prevalence of self-antigen expression on cancer cells implies that many tumors will be protected from cytotoxic immune responses via intrinsic host mechanisms of self-tolerance. It is therefore evident that any biologic therapy capable of provoking a therapeutically useful antitumor immune response will carry some risk of off-target autoimmune toxicity. The resulting destruction of normal host tissues, besides contributing to morbidity and mortality in its own right, can potentially lead to de novo tumorigenesis by initiating chronic inflammation, which is a feature of premalignant states in numerous organs including breast, bladder, prostate, cervix, ovary, stomach, and lungs (3). The molecular sequence that links chronic inflammation to cancer development involves intricate and context-dependent interactions among differentiated tissue cells, immune cells, organ-specific stem cells, and other cell types present at the incipient tumor site (4). In light of these disparate outcomes, autoimmunity may be regarded as a “double agent” implicated in both immune-mediated tumor elimination and the cellular, genetic, and epigenetic changes that underlie carcinogenesis. Given the complexity and interconnections of the associated signaling networks, navigating this new therapeutic realm demands a formidable balancing act: any cancer treatment that seeks to modify immune system function must induce a degree of self-reactivity that leads to immune-mediated tumor killing while containing the destructive and cancer-promoting aspects of that self-reactivity within tolerable limits.
After decades of hard struggle in cancer immunotherapy, exciting opportunities have emerged, especially in monoclonal-antibody-based therapies designed to elicit antitumor immunity either through inhibition of negative immune system regulators or activation of costimulatory receptors (5). Remarkable benefits in patient survival have been demonstrated in clinical trials of novel monoclonal antibodies blocking immune checkpoints, including cytotoxic T-lymphocyte antigen 4 (CTLA4) (6), programed death 1 (PD-1) (7), and PD-1 receptor ligand (PD-L1) (8). In practice, however, many biologic therapies have fallen short of expectations in clinical trials, failing to deliver enhancements in disease-free or overall patient survival (9). A partial explanation for this disappointment is the context-dependent nature of immune signaling pathways themselves. In many cases, a given signal can exert diverse, and often opposing, effects on the progression of cancer depending on a variety of factors including the tissues involved, expression level of the signal molecule(s), tumor stage and antigenic profile, and host genetic background. Thus, there is a remarkable degree of overlap between the signaling mechanisms that mediate the desired outcome of tumor destruction, and those which fuel the detrimental processes of cancer development, tumor progression, and autoimmunity. Many cytokines with therapeutic potential have demonstrated these paradoxical effects, revealing both tumoricidal and tumor-promoting activities under different experimental conditions. The task of eliciting potent cytotoxic immune responses, while managing the concomitant dangers of autoimmunity, therefore requires detailed mechanistic knowledge of immune system signaling. This review will summarize the current understanding of the pro- and antitumor activities of several major cytokines (Table 1): interferon-gamma (IFN-γ); tumor necrosis factor-alpha (TNF-α); transforming growth factor-beta (TGF-β); the Th17 cytokines IL-17 and IL-23; and the Th2 cytokines IL-4 and IL-13. Concluding remarks will address a hypothetical loop of autoimmunity-mediated antitumor immunity, leading to further induction of autoimmune responses, inflammatory cytokine signaling, and tumor promotion, including potential de novo tumorigenesis in solid organs such as the gastrointestinal tract with demonstrated susceptibility to inflammatory carcinogenesis.
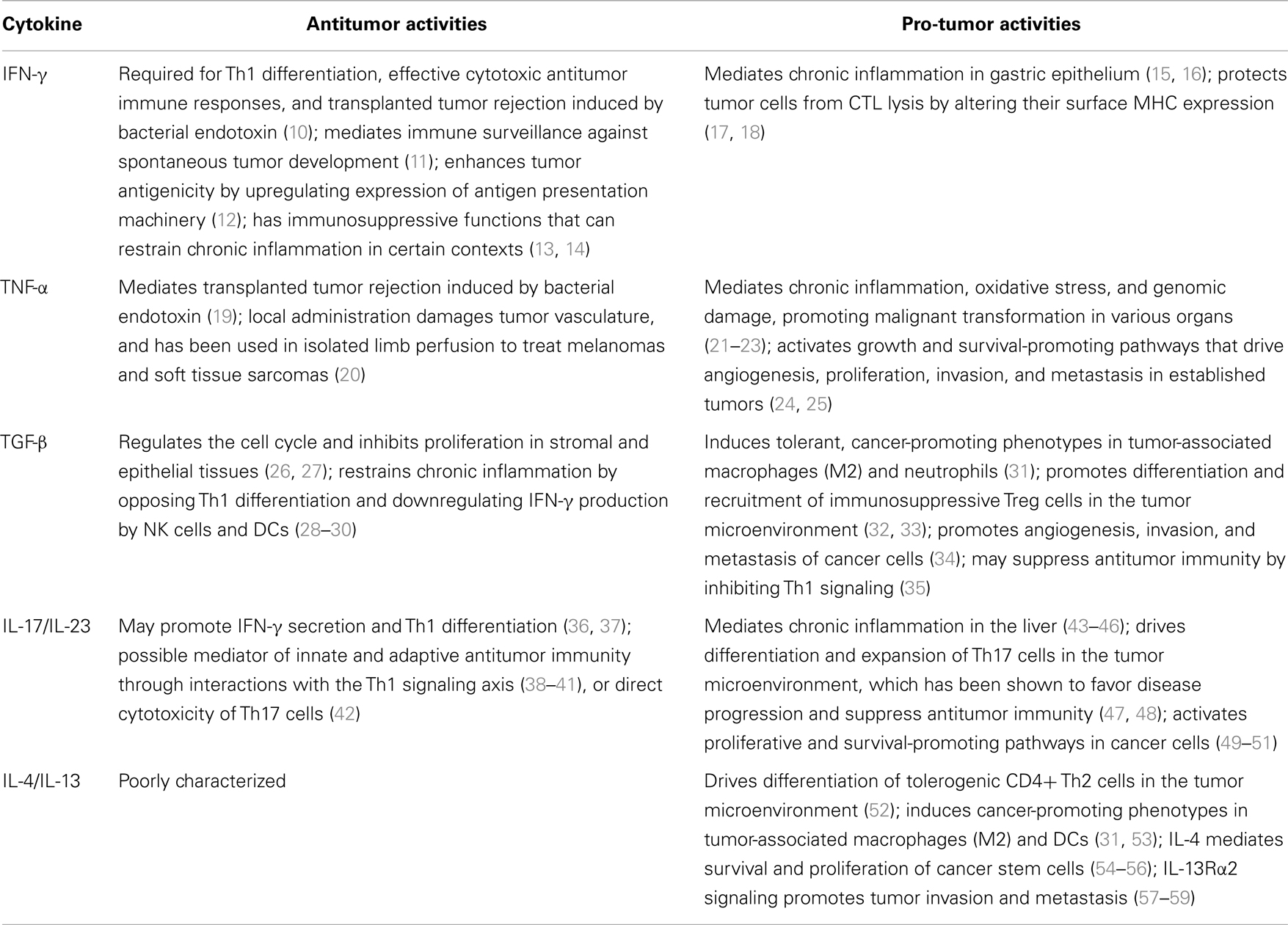
Table 1. Anti- and pro-tumor activities of selected cytokines.
Anti- and Pro-Tumor Roles of Autoimmunity Could be Mediated through Inflammatory Cytokines of Th Cells
The pathological correlation between inflammation and cancer has been known to clinical science for 150 years (60). However, it is only in recent times that our understanding of the immune system has become sophisticated enough to yield practical applications in the realm of cancer biology. For example, ample evidence has been gathered for the roles of inflammatory signals derived from innate immune response in promoting tumor growth and progression [for recent reviews, please see Ref. (4, 61, 62)]. There is emerging evidence for a tumor-promoting role by inflammatory effectors from the adaptive immune system, for example, in the TRAMP model of prostate cancer initiated with transgenic expression of an oncogene (the SV40 large T antigen) (63, 64), or implanted model of melanoma (65). Given the long-lasting nature of memory responses that is characteristic of adaptive immunity and the potency of autoimmune memory T cells [for review see Ref. (66)], one might speculate that inflammatory signals originated from the adaptive immune system, compared to their innate-derived counterparts, might sustain a longer effect, regardless of their pro- or antitumor nature. However, we should also emphasize that in a complex in vivo setting of immune responses, it is likely that the intimate interaction between innate and adaptive immune cells determines an immunological outcome (67–69). Nevertheless, definitive evidence remains to be gathered to show whether an inflammatory signal(s), at a physiologically relevant setting, can initiate de novo tumorigenesis.
Numerous parameters of immune system function have been correlated with clinical outcomes in cancer patients, including the cellular composition of tumor inflammatory infiltrate (70), expression of pro- and anti-apoptotic genes in circulating PBMCs (71), and cytokine profiles measured in peripheral blood (72). It has long been recognized that the same inflammatory cytokine may play a prominent role in either tumor killing or cancer promotion. Although the original stimuli of inflammatory cytokine production are unknown in most cancer settings, given the largely “self” constituents of tumors, we reasoned that autoimmune T cells, especially the three main T helper subsets, Th1, Th2, and Th17, could be a major source of these cytokines, or play a major role in driving other immune cells to produce them.
Early experimental models offered hope that the induction of selective antitumor immune responses, even those mediated by CTL recognition of self-antigens, was possible without clinically significant autoimmunity. One study reported the eradication of established p53-overexpressing tumor cells in mice, achieved through adoptive transfer of a clone of cytotoxic T cells, which recognized wild-type p53. No autoimmune damage was observed in normal tissues, despite the p53+/+ genotype of the treated mice (73). In vitro assays also suggested that epitope-specific CTL clones had the capacity to initiate cytotoxic immune responses against non-mutated, tumor-associated p53, while simultaneously avoiding reactivity with the same antigen when endogenously expressed (74). Other experiments used transgenic models to search for evidence of autoimmune pathology in an organ-specific fashion. In one study, transgenic mice were engineered to express low levels of Friend murine leukemia virus envelope protein (FMuLV) from an immunoglobulin promoter; adoptive transfer of FMuLV-specific T cells mediated complete destruction of leukemia tumor cells, without concurrent autoimmunity. Lymphoid tissues in the treated mice were unharmed, despite the fact that they expressed the targeted cancer-associated antigen in a “self” context (75). Another study employed transgenic mice with tissue-specific expression of influenza virus hemagglutinin (HA) in pancreatic islet β cells. Administration of an anti-HA vaccine to these animals produced CTL-mediated rejection of renal carcinoma cells, which had also been genetically engineered to express HA. Prior to vaccination, the immune systems of the transgenic mice exhibited HA self-tolerance, and supported growth of implanted tumors expressing this antigen. Remarkably, in addition to tumor rejection, immunohistologic analysis of treated mice showed intact structure and function in the pancreatic islets (76).
Despite these favorable preclinical results, most types of cancer immunotherapy tested in human patients have revealed serious and persistent risks of treatment-associated autoimmunity. Reported manifestations include vitiligo, uveitis, psoriasis, and colitis, with potential consequences ranging from cosmetic complaints to permanent disability and death (77). However, it has been suggested that autoimmunity, in addition to mediating these adverse events, makes an essential contribution to antitumor immunity. Indeed, it may be the case that effective cancer immunotherapy requires a significant degree of self-reactivity, since antitumor immune responses must surmount both the preexisting tolerance to self-derived tumor antigens and immunosuppressive signals from the tumor microenvironment (1, 77). For some cancer immunotherapies, such as blockade of the inhibitory T cell signaling molecule CTLA4, autoimmune toxicity shows a positive correlation with therapeutic response (78). Meanwhile, antitumor vaccines, probably the category of cancer immunotherapy least associated with severe autoimmunity in human studies (77), have generally failed to show therapeutic efficacy in large-scale Phase 3 trials (79). Thus, it appears that the future of cancer immunotherapy will not feature the elimination of autoimmunity, which is unlikely to be feasible or wholly desirable. Rather, the cytokines that mediate autoinflammation can be harnessed as effective agents in tumor destruction, if their toxic and tumor-promoting potentials are understood and judiciously managed.
IFN-γ
Interferons (IFNs) are a family of cytokines that bind to cell-surface receptors and mediate numerous functions related to pathogen defenses, immune function, cell survival and differentiation, and angiogenesis (12, 80). Type I interferons are broadly expressed (80), while IFN-γ, the sole member of the Type II interferon family, is produced mainly by T lymphocytes, NKT cells, and natural killer (NK) cells as a crucial component of the inflammatory response (10). IFN-γ is integral to the development of Th1 adaptive immunity, by maintaining IL-12 signaling in CD4+ T lymphocytes while simultaneously suppressing Th2 differentiation (10). Murine models support a range of anti-carcinogenic properties for IFN-γ (11). Antibody-mediated suppression of IFN-γ signaling has revealed that this cytokine is required for LPS-stimulated rejection of transplantable tumors in mice. IFN-γ receptor knockout mice (IFN-γR1−/−) exhibit susceptibility to both spontaneous and chemically induced tumors, in addition to poor resistance to a variety of pathogens (10, 11). IFN-γ is a powerful inducer of Class I and Class II antigen presentation machinery, suggesting its primary importance in the production of specific antitumor immune responses (12). In vitro assays have also revealed pro-apoptotic, antiangiogenic, and antiproliferative effects (10–12, 81). Indeed, reduced IFN-γ production has been observed in a variety of human malignancies, including melanoma, gastric cancer, lung cancer, glioblastoma, nasopharyngeal carcinoma, colorectal cancer, and head and neck cancer (72). Certain human cancers, notably melanoma, are known to develop resistance to IFN-γ through various mutations in downstream molecules in the IFN signal transduction pathway, such as JAK and STAT1 (12, 81).
Despite this promising body of experimental evidence, anticancer therapy with exogenous IFN-γ has generated mixed results. A 2003 clinical trial in malignant melanoma patients showed no positive responses to intratumoral injections of IFN-γ (82). A Phase 3 trial for ovarian cancer and peritoneal carcinoma was prematurely ended in 2006, due to decreased survival and more frequent adverse events in patients treated with subcutaneous IFN-γ, as compared to conventional platinum-based chemotherapy (82). To understand why this cytokine has not demonstrated the broad therapeutic efficacy that prior data would suggest, it is necessary to examine the dual nature of the inflammatory process. While inflammation can promote the cell-mediated destruction of tumor cells, its chronic forms may lead to pathological changes that promote cancer development in a variety of tissues. These changes include accumulation of reactive oxygen species, epithelial hyperplasia, extracellular matrix generation, and angiogenesis (3, 83, 84). Besides mediating anticancer immunity, IFN-γ is a major player in chronic inflammation, as illustrated by its contributions to Th1-driven autoimmune disease. In clinical trials, anti-IFN-γ antibodies have been useful in treating a variety of inflammatory disorders, including rheumatoid arthritis, Crohn’s disease, corneal transplant rejection, and skin diseases such as vitiligo and alopecia areata (82, 85, 86).
The interplay between IFN-γ signaling, aberrant chronic inflammation, and neoplastic disease has been explored in the pathogenesis of gastric cancer, one of the classic models of human malignancy precipitated by chronic inflammation (87–90). With regard to gastric carcinogenesis, the role of Th subsets would also perhaps be best surveyed while keeping in mind the robust evidence of innate immunity in gastric cancer development (91–93). In the natural course of this disease, infection with the bacterium Helicobacter pylori during childhood precipitates a chronic inflammatory response which, in a small portion of patients (<5%), progresses to malignancy decades in the future (84, 94). Studies of mice infected with the related pathogen Helicobacter felis showed that a Th1-biased inflammatory response is involved in gastric cancer development (94). Knockout of the transcription factor T-bet, which controls commitment to the Th1 lineage (95), curtailed gastric tumorigenesis induced in mice infected with H. felis (15). A recent experiment employed a murine model that directly implicates IFN-γ in this course of preneoplastic change. The transgenic mice, which were engineered to overexpress IFN-γ from a stomach-specific, H/K ATPase β promoter, exhibited a prominent inflammatory infiltrate along with accelerated histological changes characteristic of a premalignant phenotype: metaplasia, loss of parietal and chief cells, gastric gland atrophy, and dysplasia beginning as early as 3 months of age (16).
IFN-γ can thus be seen as an essential mediator of both immune-mediated tumor rejection and the destructive chronic inflammation that precedes malignant transformation. There is, however, another vital dimension to this cytokine’s effects, that of a master regulator which restrains inflammation in a variety of contexts. In two key murine model systems of Th1-driven autoimmunity, collagen-induced arthritis (CIA) and experimental autoimmune encephalitis (EAE), it has been established that abrogation of IFN-γ signaling through monoclonal antibody therapy or genetic knockout produces a seemingly paradoxical increase in disease susceptibility and severity (13, 14). The mechanisms for this apparent downregulation of the inflammatory response are not yet clear, but suppression of pro-inflammatory Th17 cytokines, induction of T cell apoptosis, and enhanced Treg cell differentiation appear to play prominent roles (13, 14, 96). Another mechanism of IFN-γ mediated immunosuppression is suggested by its ability to alter tumor cell MHC presentation in a manner that decreases tumor antigenicity and protects tumor cells from CTL killing. One in vitro experiment demonstrated that IFN-γ could protect ovarian carcinoma cell lines from CTL-mediated lysis by upregulating HLA-E on cancer cells, which engaged the inhibitory CD94/NKG2A receptor on CD8+ effector T cells (18). A similar cancer-promoting role for IFN-γ was revealed in a mouse model of melanoma, where tumor protection from CTL killing was associated with IFN-γ-stimulated upregulation of non-cognate MHC-I molecules (17).
Given the evidence above, IFN-γ-mediated downregulation of the inflammatory response can be regarded as an obstacle to effective cancer immunotherapy. However, it remains an important physiological mechanism for preserving tolerance to self-antigens and protecting tissues from the damaging effects of autoimmunity (80, 97). In keeping with this fact, experimental data also suggest that IFN-γ mediated suppression of inflammation may be protective in certain contexts. One recent experiment employed a transgenic mouse line engineered to overexpress IFN-γ in a stomach-specific manner, using the same H/K ATPase β promoter as Syu et al. (16). In this case, however, no spontaneous gastritis or metaplasia occurred (98). Moreover, IFN-γ was reported to be protective against gastric dysplasia produced by either H. felis infection or stomach-specific overexpression of the cytokine IL-1β (a well-established mediator of gastric carcinogenesis). IFN-γ was also shown to inhibit proliferation of gastric epithelial cells, enhance autophagy in a manner recognized as protective against tumor development (99), and diminish expression of pro-inflammatory Th1 and Th17 cytokines (98). As noted by Syu et al. (16), the seemingly contradictory results of these two studies can most likely be explained by differing degrees of overexpression. These findings underscore the challenges of discerning which aspect of IFN-γ will prevail in a particular patient, tumor type, and stage of malignancy. Attempts to broaden the therapeutic use of this cytokine must take into account these intricate and context-dependent multidirectional effects.
TNF-α
Activated macrophages and T lymphocytes produce TNF-α in response to pro-inflammatory stimuli. This cytokine stimulates inflammation through multiple mechanisms, including recruitment of neutrophils and monocytes and induction of cell adhesion molecule expression on the endothelial surface (72, 100). TNF-α is involved in the classical pathway of macrophage activation (M1), which plays a central role in immune defenses against tumors and intracellular parasites (31). Although usually undetectable in the tissues and circulation of healthy people, TNF-α exerts important effects on immune homeostasis during states of disease. It is known, for instance, that systemic TNF-α mediates the wasting observed in humans and animals afflicted with chronic illness (24, 100, 101). As with IFN-γ, aberrant TNF-α signaling is associated with a variety of autoimmune disorders. Five TNF inhibitors have been approved for clinical use in the United States for the treatment of inflammatory illnesses such as Crohn’s disease, ankylosing spondylitis, and rheumatoid arthritis (100, 102).
The observation that the human body’s response to infection is capable of producing regression of tumors has been recorded by clinicians since at least the eighteenth century (103). It was not until 1975, however, that TNF-α was identified as one of the principal mediators of this anticancer immune response. The name “tumor necrosis factor” reflects early experiments, which demonstrated this cytokine’s capacity to induce hemorrhagic necrosis of subcutaneously transplanted sarcomas, leukemias, and mastocytomas in mice whose immune systems were primed by exposure to bacterial endotoxin (19). However, 38 years of data subsequent to this discovery have not fulfilled the early promise of TNF-α as a safe, potent, or selective tumoricidal agent. Unlike mice with abrogated IFN-γ signaling, TNF-α-knockout animals do not develop spontaneous tumors, and peritoneal tumors transplanted into them do not show accelerated growth (104). These knockout animals have also shown an unexpected resistance to chemically induced skin tumors (105). Indeed, there seem to be numerous contexts in which TNF-α signaling helps to initiate carcinogenesis and sustain tumor growth. In obese patients, TNF-α is suggested to be a key mediator of cancer-promoting inflammation in various organs, including the pancreas (22), colon (21), and liver (23). Picogram amounts of TNF-α are constitutively secreted by many tumor types, and appear to stimulate cancer growth, although the underlying signaling mechanisms are not completely understood (24, 106). The pro-carcinogenic functions of TNF-α appear to be mediated predominantly through downstream activation of the proliferative and survival-promoting pathways NF-κB and AP-1 (20, 21, 100, 104). Elevated serum concentrations of TNF-α have been described as a clinical feature of eight independent cancer types (72). A direct link between the pro-inflammatory effects of TNF-α and carcinogenesis can be seen by returning to the example of gastric cancer. The presence of a specific, inflammation-associated single nucleotide polymorphism in the TNF-A gene has been found to increase the odds ratio of non-cardia gastric cancer (83). The importance of this association has been further established by examining the genome of the initiating pathogen: H. pylori harbors the Tipα gene family, whose members are known to induce high levels of TNF-α expression in the gastric epithelium (24, 106). Experimental transfection of a transformed BALB/3T3 cell line with H. pylori genes revealed that tumor progression was dependent on Tipα-induced production of TNF-α (106).
TNF-α has also been implicated as a disease promoter in hepatocellular carcinoma (HCC), from chronic inflammation to tumor progression including invasion, metastasis, and angiogenesis in established HCC tumors (107). In vitro studies of numerous cell types, including murine hepatocytes, have demonstrated that TNF-α-mediated inflammation leads to an increase in markers of oxidative stress, which in turn can lead to genomic damage (108–111). Moreover, the development of obesity-induced hepatosteatosis and steatohepatitis in mice is dependent on signaling by TNF-α, along with IL-6 (21). In the Mdr2-knockout mouse, an experimental model of inflammation-induced HCC, the spontaneous development of hepatic malignancy is dependent on TNF signaling, and can be attenuated through downstream inhibition of NF-κB (23). Overall, experiments on HCC and many other tumor types have established that TNF-α is intimately involved in all aspects of cancer development, including transformation, proliferation, angiogenesis, invasion, and metastasis (25).
Owing to a high frequency of inflammation-related adverse events, the therapeutic applications of exogenous TNF-α in cancer have been quite limited (24). Localized TNF-α infusion has demonstrated antitumor efficacy against melanomas and soft tissue sarcomas in human patients. In this context, high concentrations of TNF-α were shown to induce hyperpermeability and structural disruption in tumor vasculature, thus promoting tumor necrosis and enhancing the efficacy of traditional cytostatic drugs. However, this intervention was aimed at limb sparing and had no impact on patient survival (20). Given the broad range of cancer-promoting activities of TNF-α, blockade of its signaling remains a tempting therapeutic approach. Unfortunately, the fundamental role of TNF-α in pathogen defense represents a formidable obstacle to implementation (24). The feasibility of modifying TNF-α signaling without triggering destructive autoimmunity on one hand, or vulnerability to opportunistic infections on the other, remains to be seen.
TGF-β
Transforming growth factor-beta mediates a vast array of functions related to wound healing, immune responses, cell proliferation and differentiation, and carcinogenesis, via receptors expressed on nearly all human cells. It plays a crucial role in T cell tolerance (112). Its classical signal transducers are transcription factors known as Smads, which combine with each other as well as additional cofactors to form a variety of DNA-binding complexes. These intricate assemblies of transcriptional regulators allow TGF-β to implement a versatile, yet precisely controlled, range of downstream effects. Additional “Smad-independent” signaling pathways are known to further augment this functional repertoire (27, 29, 34, 35). This cytokine has been extensively characterized as a negative regulator of immune responses, with anti-inflammatory, antiproliferative, and immunosuppressive activities (27, 29, 34, 35). Thus, TGF-β helps restrain the destructive effects of uncontrolled inflammation and proliferation that might otherwise occur in the context of infection or tissue damage. Mouse models reveal that knockout of TGF-β or its receptor produces a phenotype characterized by lethal autoimmune disease (113, 114). One recent study featured a murine model with the TGF-β receptor gene deleted in stromal fibroblasts. The transgenic animals experienced an excessive, aberrant inflammatory response in adjacent epithelial tissue, characterized by molecular markers of DNA damage, oxidative stress, cell cycle dysregulation, and death from invasive squamous cell carcinoma by the age of 7 weeks (26). Other models have examined the specific effects of TGF-β silencing in the innate immune and T cell compartments. One experiment blockaded TGF-β signaling in mouse NK cells through transgenic expression of a dominant negative receptor. NK cells were more numerous in lymphatic tissues of the transgenic mice, and showed enhanced secretion of IFN-γ. Consistent with the Th1-promoting effects of IFN-γ signaling, transgenic mice also demonstrated an enhanced Th1 inflammatory response, which protected against infection with cutaneous Leishmaniasis (28). These effects suggest the importance of TGF-β in maintaining NK cell homeostasis, as well as downregulating Th1 differentiation. Another study reported a dendritic cell-specific deletion of the TGF-β receptor gene, which resulted in multiorgan autoimmunity, a pro-inflammatory DC phenotype characterized by IFN-γ overproduction, and reduced Foxp3 expression in Treg cells (30). Accumulating evidence suggests that TGF-β is directly involved in many aspects of T cell homeostasis, including differentiation of Treg cells and CD8+ effectors, maintenance of peripheral tolerance, and preservation of naïve T cell populations (112). The precise molecular mechanisms behind these effects are largely unresolved, and represent an area of active investigation.
In the early stages of carcinogenesis, TGF-β is known to suppress tumor growth through induction of cell cycle inhibitors and promotion of apoptosis. However, in many advanced cancers, paracrine and autocrine TGF-β signaling drives tumor progression and metastasis. TGF-β is produced in large quantities by a variety of human cancers, to the extent that it is arguably the most ubiquitous immunosuppressive mediator in cancer progression (34). Elevated systemic levels of TGF-β have been reported in breast cancer, lung cancer, pancreatic cancer, glioblastoma multiforme, colorectal carcinoma, HCC, renal cell carcinoma, and gastric carcinoma (72). TGF-β is a crucial inducer of pro-tumor phenotypes in both tumor-associated macrophages (TAMs) and tumor-associated neutrophils (TANs), leading to cancer cell proliferation and the curtailing of antitumor immune responses (31). The capacity of TGF-β to promote the differentiation of Treg cells appears to be highly deleterious in this context, since Treg cells are key inducers of immune tolerance in the tumor microenvironment. The association between TGF-β signaling and Treg cell recruitment has been experimentally demonstrated in a lung cancer cell line (115), as well as in animal models of pancreatic cancer (32) and HCC (33). One key observation from these studies is that a tolerogenic immune microenvironment is not exclusively the result of tumor-secreted cytokines. Rather, it requires elaboration of TGF-β and other anti-inflammatory signals from immune cell populations such as dendritic cells and TAMs.
Several other mechanisms are believed to underlie the pro-carcinogenic role of TGF-β, including enhanced extracellular matrix formation, cytoskeletal rearrangements to facilitate epithelial-to-mesenchymal transition (EMT), angiogenesis, and cell cycle dysregulation (27, 29, 34, 35). In a clinical context, TGF-β serves as a marker of metastasis and poor prognosis for many malignancies (35, 72). The transition of TGF-β from a protective role early in tumor development to a tumor-promoting one in more advanced disease appears to be a watershed moment in many cancers, reflecting global derangement of signal transduction through genetic and epigenetic mechanisms. Indeed, in vitro experiments have linked specific forms of oncogenic transformation to alterations in TGF-β responsiveness. One study found that engineered overexpression of HER2 in mesenchymal human breast cancer cells caused a loss of sensitivity to the antiproliferative effects of TGF-β (116). Another reported that loss of TGF-β growth inhibition correlated with the loss of c-myc downregulation in ovarian carcinoma cells (117).
The prevalence of TGF-β overexpression across a broad range of human malignancies has made this cytokine a tantalizing therapeutic target. Four classes of TGF-β inhibiting molecules have already been tested in clinical trials, with responses that generally fell short of hopes (118). At least one class of TGF-β inhibitor has also shown the capacity to elicit biochemical resistance in mouse models (119). This observation, coupled with the integral roles of TGF-β in wound healing and tissue homeostasis, suggests that long-term inhibition of TGF-β signaling may be a dangerous prospect. Rather, it has been suggested that TGF-β inhibition will find its first clinical applications as part of a combined drug regimen, administered to cancer patients over relatively brief spans of time to minimize resistance and adverse events (118).
IL-17 and IL-23
The Th17 subset of CD4+ T cells has added a new dimension of complexity to the Th1/Th2 paradigm since its initial discovery in 2005. Extensive studies in murine models have implicated this T cell population in a number of pro-inflammatory functions, including the pathogenesis of autoimmune diseases of the brain (EAE) and joints (CIA), mediated by the characteristic cytokine IL-17 (36, 120–122). Knockout mice have also revealed an important role for IL-17 signaling in the defense of mucosal surfaces against a variety of bacterial and fungal pathogens, including Staphylococcus aureus, Klebsiella pneumoniae, Citrobacter rodentium, and Candida albicans (36, 120). Th17 cells also engage in physiologically important interactions with key cytokines of the Th1 immune system. One attribute, noted in the initial characterization of this helper T cell subset, is the inhibitory effect of IFN-γ on Th17 differentiation and production of IL-17 (123, 124). Meanwhile, IL-23, a cytokine that is essential to sustain the survival and proliferation of Th17 populations, appears to participate in divergent regulatory pathways with the Th1-associated cytokine IL-12, whose release is promoted by IFN-γ (125–127). The dichotomous signaling of IL-12/IL-23 is especially intriguing, as these two cytokines share a common subunit: IL-12 is formed through the covalent linkage of p40 and p35 subunits, whereas IL-23 is a combination of p40 and p19 (128). TGF-β signaling also appears to make a major contribution to Th17 differentiation, although the precise nature and mechanism of this association remains a subject of intense controversy (122, 129).
Th17 signaling through the canonical IL-23/IL-17 pathway has been shown to contribute to cancer development in a variety of experimental contexts. During the preneoplastic stage, excessive Th17-mediated signaling is a probable contributor to the chronic inflammation that can precipitate cancer in a variety of tissues. In the case of Hepatitis B-induced inflammation, evidence from human and animal experiments suggests that Th17, not Th1, signaling is the primary mechanism underlying liver immunopathology and eventual malignant transformation to HCC (43–46). Th17 cells have been discovered in human tumors in many different organs, and a growing body of evidence suggests that their presence, like that of immunosuppressive Treg cells, may be a general feature of the tumor microenvironment (37, 121). One comprehensive examination of gene expression profiles in human cancers revealed overexpression of IL-23 in the vast majority of human carcinomas, when compared to expression profiles of adjacent, non-cancerous tissue (130). A recent quantitative PCR analysis revealed statistically significant IL-23 upregulation across a panel of non-small cell lung carcinoma (NSCLC) patient samples, in comparison to matched normal tissue controls (49). In mouse models, knockout of IL-23 conferred resistance to both chemically induced and transplanted epithelial tumors, whereas knockout of IL-12 produced the opposite effects (130). Moreover, there is mounting evidence that tumor cells produce chemokines in order to selectively recruit Th17 lymphocytes (121). In the 4T1 mouse model of breast cancer, Qian et al. reported that signaling via mammary tumor-derived prostaglandin E2 (PGE2) caused overexpression of IL-23 (but not IL-12) in the tumor microenvironment (48). This overexpression, in turn, was associated with expansion of Th17 cell populations in the tumor tissue, spleens, and peripheral blood of experimental animals (48). This discovery is likely to have significant implications in a variety of human cancers, since PGE2 is the most abundant prostanoid in epithelial cell tumors (127). In the same study, exposure of murine dendritic cells to tumor-conditioned medium enhanced their expression of the IL-23 subunit p19, and also reduced expression of p40 and the IL-12 subunit p35, in a dose-dependent manner (48). These changes collectively indicate a shift from Th1 to Th17 immune phenotype, which seems to be favorable for cancer persistence. As in the case of experimental models of TGF-β signaling, cells of the innate immune system were key participants in the generation of an immune microenvironment promoting carcinogenesis.
The pathways through which Th17 signaling exerts its tumor-promoting effects are not yet clear, but a variety of relevant observations have emerged. One probable mechanism is the stimulation of angiogenesis and cell proliferation: the p19 subunit of IL-23 is transcriptionally upregulated by growth-promoting signals from NF-κB and AP-1 (48). Administration of recombinant IL-23 has been shown to enhance proliferation in an IL-23 receptor-positive lung adenocarcinoma cell line (49, 51). One study reported IL-23-mediated enhancement of cancer growth and proliferation in cultures of human oral squamous cell carcinoma; in this case, IL-23 exposure was accompanied by enhanced nuclear translocation of NF-κB (50). Mouse models have also furnished valuable mechanistic clues. In one experiment, IL-17 knockout mice showed reduced growth of transplanted B16 melanoma and MB49 bladder carcinoma, whereas acceleration of tumor growth occurred with knockout of IFN-γ. In mice with both knockouts, tumor growth was reduced relative to WT controls (65). In the IFN-γ knockout mice, elevated concentrations of IL-17 were measured in tumor tissue compared to tumors in wild-type controls. Moreover, IL-17 was found to enhance signaling by the pro-survival, pro-angiogenic transcription factor Stat3 in both tumor and stromal cells (65). Another study reported similar results using receptor knockouts: IL-17 receptor-deficient mice showed diminished tumor growth of transplanted melanoma and lymphoma cell lines, while IFN-γ receptor knockout led to growth enhancement. The double-knockout genotype was also protective in comparison to WT controls (47). In addition, IL-17 receptor deficiency was correlated with increased tumor infiltration of CD8+ effector T cells, and decreased numbers of myeloid-derived suppressor cells (MDSCs), both favorable indications for the generation of antitumor immunity (47). Interestingly, these pro- and antitumor effects were successfully reproduced in wild-type mice through administration of either recombinant IL-17 (which accelerated tumor growth) or antibody-mediated IL-17 blockade (which suppressed it), hinting at the viability of anticancer therapies targeting this pathway (47). It is also notable that elimination of IL-17 signaling in both studies produced an antitumor effect that was potent enough to compensate for the concurrent loss of IFN-γ signaling, despite the role of IFN-γ as a potent mediator of Th1-driven antitumor immunity.
The complex and heterogeneous functions of the Th17 signaling axis, and their relationships to cancer progression, are only beginning to be elucidated. Despite the results mentioned above, numerous studies support the existence of antitumor Th17 effects, including those which may be mediated through signals other than IL-17 and IL-23 (121). For instance, subsets of Th17 cells are capable of producing IFN-γ, indicating possible cross-talk with the Th1 pathway of differentiation, as well as the ability to stimulate cytotoxic and tumoricidal immune responses (36, 37). In human pancreatic ductal adenocarcinomas, one recent study found correlation of protective benefit with activity of Th17 cells specific to α-enolase, a pancreatic tumor-associated antigen (42). Another recent study reported that a mouse model deficient in RORγt (a transcription factor required for Th17 differentiation) exhibited accelerated growth of transplanted melanoma tumors, along with a diminished percentage of Th1 CD4+ cells at the tumor site; this phenotype was rescued by adoptive transfer of Th17 cells, a portion of which began to produce measurable quantities of IFN-γ (41). One study of IL-17 knockout mice demonstrated increased susceptibility to lung melanoma. Subsequent treatment by Th17 adoptive transfer served to prevent tumor development by inducing a specific CD8+ antitumor response (40). It has also been found that systemic administration of high-dose IL-23 led to reduced tumor growth and prolonged survival in a mouse fibrosarcoma, due to Th1-mediated activation of cytotoxic T cells, helper T cells, and NK cells (38). Increased growth and lung metastasis of murine colon carcinoma has also been reported in IL-17 deficient animals, with corresponding reductions in IFN-γ+ NK cells and IFN-γ+ tumor-specific T cells (39).
Although characterized less than a decade ago, the Th17 lymphocyte population has already become the focus of a vast and diverse body of scientific literature. However, this expanded knowledge contains apparent contradictions, which will challenge the field of cancer immunology for years to come. As with the pathways and cytokines previously discussed, the tumor-related effects of IL-17 and IL-23 exhibit a high degree of context dependence. Some of these discordant results may therefore be attributable to the source of tumor cells, the tissue involved, the stage of cancer growth, the genetic background of the organism, and other features of the experimental model employed in a particular study. Meanwhile, our understanding of Th17 interactions with other elements of the immune system, including NK cells, antigen-presenting cells and other helper T cell subsets, remains incomplete. Experiments designed to address the cross-talk between Th17 cytokines and other branches of the immune system should help resolve some of the inconsistencies in their reported effects. While a variety of IL-17/IL-23 antagonists are currently being developed for the treatment of autoimmune diseases (131), safe and effective modification of Th17 signaling in cancer therapy will require a more thorough understanding of the forces which underlie Th17 differentiation, recruitment, and interaction with malignant cells.
IL-4 and IL-13
The Th2 subset of CD4+ T cells plays an important physiological role in implementing immune defenses against helminths and other extracellular parasites. Th2-mediated responses include generation of high-affinity IgE antibodies, mucus overproduction, and heightened smooth muscle contractility, all of which function in the clearance of invasive multicellular organisms. However, aberrant and excessive Th2 activation also provides the foundations for allergic disease (132). The major cytokine responsible for differentiation of naïve CD4+ T cells into the Th2 phenotype is IL-4, while Th2 effector functions are mediated through a combination of IL-4, IL-13, and IL-5 (133). All three of these cytokines exert varied effects on cancer development, which remain an area of ongoing investigation. Th2 cytokines appear to be involved in shifting the immune response to forms favorable to tumor growth, particularly in the context of innate immunity. IL-4, IL-13, and IL-5 promote the differentiation of macrophages into an “M2” or alternatively activated form, which displays poor antigen-presenting capacity and local anti-inflammatory effects (31, 83, 134). M2 macrophages play a variety of physiological roles in tissue homeostasis, including wound healing, extracellular matrix remodeling, and scavenging of debris (132, 134). This M2 phenotype contrasts with the classically activated (M1) macrophage, which is specialized for the production of pro-inflammatory cytokines (e.g., IFN-γ), cytotoxic immune responses, and efficient destruction of phagocytosed microbes (132, 134). In the context of cancer, M2 polarization of TAMs is associated with suppression of antitumor immune responses, disease progression, and poor prognosis (31, 83). Moreover, it has been established that tumors are capable of producing Th2 cytokines in order to bias innate and adaptive immune responses toward this more favorable phenotype (134). A study of pancreatic cancer patients demonstrated that tumor-produced cytokines (TNF and IL-1β) triggered activation of a Th2 phenotype in cancer-associated fibroblasts, dendritic cells, and naïve CD4+ T cells. Moreover, the ratio of Th2:Th1 CD4+ T lymphocytes present at the tumor site was negatively correlated with patient survival (52). In a humanized mouse model implanted with human breast carcinoma, Th2 cytokine expression was detected in both cancer cells and tumor-promoting CD4+ T cells within the tumor microenvironment. Dendritic cells isolated from these tumors also potently induced Th2 cytokine secretion from naïve CD4+ T cells in vitro, suggesting that tumor growth is facilitated by a complex network of Th2 paracrine signals (53).
The Th2 cytokine IL-4 is capable of signaling through two distinct cell-surface receptor complexes. The Type I receptor, found on cells of hematopoietic stem cell origin, is composed of IL-4Rα and the common gamma chain γc. The Type II receptor, expressed on cells of non-hematopoietic origin, contains IL-4Rα and IL-13Rα1, and also binds the cytokine IL-13 (135). IL-13Rα2 is a second type of IL-13 binding receptor, whose physiological role remains uncertain. The receptor bears structural similarities to IL-13Rα1, but is expressed in two forms: as soluble IL-13Rα2, and as a transmembrane protein, which interacts with a number of signal transduction pathways (54). IL-4 and IL-13 exert both overlapping and distinct physiological effects by binding these receptors, whose structure and function have been extensively studied as potential therapeutic targets in asthma and other allergic diseases (135, 136). Emerging evidence indicates that these receptors can influence cancer development through pathways other than macrophage polarization, although the molecular details of this process are only starting to become clear.
The Type II IL-4/IL-13 receptor has been found to be overexpressed in a variety of epithelial tumors, and treatment of cancer cell lines with IL-4 is associated with pro-proliferative and anti-apoptotic effects (54, 137). The effects of IL-4 signaling have been studied extensively in the development of colon cancer: one study found pro-proliferative effects of IL-4Rα signaling in mouse colon tumors, as well as human and mouse colon adenocarcinoma cell lines examined in vitro (55). Of particular interest is the relationship between intestinal malignancies and multipotent stem cells, which have been identified in recent experiments as an integral driving force in the growth of both premalignant adenomas and established tumors (138, 139). It now appears that IL-4 signaling is vital to the functioning of at least some of these tumorigenic stem cells. Research on colon cancer has identified a subset of tumor cells with a CD133+ stem-like phenotype, which was found to be necessary and sufficient for the establishment of transplanted human colon tumors in immunodeficient mice (56). In keeping with the cancer stem cell (CSC) hypothesis (4), these cells possess self-renewing capacity, an especially high resistance to death-promoting signals, and the ability to effect regeneration of the overall tumor mass (56, 140). One study revealed that resistance to drug-induced apoptosis in CD133+ colon cancer cells was mediated through increased production of IL-4 (56). Preliminary experiments suggest that this IL-4-mediated, pro-survival pathway may be a promising therapeutic target. In the same study, blockade with either IL-4 neutralizing antibody or a mutant, inhibitory form of IL-4 (IL-4DM) reduced the viability of CD133+ and CD133− tumor cell cultures, while increasing the efficacy of cytotoxic treatment with standard chemotherapeutic agents: oxaliplatin, 5-fluorouracil, and the death receptor ligand TRAIL. IL-4 antagonism also enhanced the effectiveness and duration of chemotherapy response in mice bearing transplanted tumors, suggesting a role for combined therapy in the treatment of human disease (56). Both the in vitro and in vivo effects of IL-4 blockade were mediated by decreases in the anti-apoptotic molecules cFlip, Bcl-xL, and Ped (56, 140).
Another remarkable finding has been the role of IL-13Rα2 in models of cancer development, indicating signaling functions far beyond the previously suggested role of a decoy receptor (141). This receptor is known to be overexpressed in several human cancers. An immunohistochemical analysis of human tissues found IL-13Rα2 overexpression in 71% of pancreatic ductal adenocarcinoma samples in comparison to normal pancreas controls (142). Indeed, experiments in an orthotopic mouse model of pancreatic cancer suggest that IL-13Rα2 is an important mediator of the pro-tumor effects of IL-13, including activation of AP-1 growth signals, production of immunomodulatory cytokines such as TGF-β, and promotion of metastasis (58, 142). Similar IL-13Rα2-dependent effects have been demonstrated in other cancer models, including ovarian carcinoma (59), colorectal cancer (57), head and neck squamous cell carcinoma (143), and malignant glioma (144). As with IL-4, IL-13 signaling has been suggested as a possible target of anticancer therapy. To this end, recombinant cytotoxic proteins have been developed, which consisted of IL-4 or IL-13 joined to a mutant form of Pseudomonas exotoxin. These agents have been found to restrain tumor growth in numerous animal models (142–144). However, in a Phase 3 clinical trial, IL-13 Pseudomonas exotoxin failed to improve median survival time in patients with glioblastoma multiforme when compared to conventional chemotherapy (145). While this agent may still find use as an adjuvant therapy (146), the outcome suggests that the development of Th2-targeted treatments with robust antitumor efficacy will require further exploration.
Despite considerable progress in the field, many Th2-mediated influences on tumor development remain poorly characterized. For instance, populations of eosinophils and mast cells, crucial mediators of Th2-driven allergic responses, also contribute to the inflammatory infiltrate in numerous human tumors. However, experimental data exploring their effects are lacking, and conflicting results have been published regarding their impact on clinical prognosis (83). This ambiguity underscores the fearsomely complex, and incompletely understood, nature of signaling among discrete immune system components in the context of cancer development. Owing to these knowledge gaps, the possibility that an anti-Th2 intervention could impede antitumor immune responses in vivo cannot be prematurely dismissed. Systemic therapies to antagonize Type I or Type II receptors will also confront a high risk of adverse events, owing to the global effects of Th2 cytokines on immune homeostasis and other physiological functions. Nevertheless, a fascinating body of experimental and clinical data suggests that the pro-carcinogenic effects of IL-4 and IL-13 will remain a source of therapeutic interest for years to come.
Potential Feedback between Autoinflammation and Tumor: Implications for Cancer Immunotherapy
Virtually all of the immune signaling pathways relevant to tumor biology play major physiological roles in the maintenance of self-tolerance and tissue homeostasis. Moreover, most well-characterized tumor-associated antigens are self-antigens, meaning that they are also expressed by normal cells in the course of growth and differentiation. Conversely, antigenic proteins expressed exclusively in cancerous tissue, such as viral products or mutated oncogenes, have been characterized in only a small number of tumor types. Overall, it seems that tumors, despite their aberrant characteristics, may remain antigenically “self” entities first and foremost (1, 2). In the clinical context, this suggests that useful antitumor immune responses elicited in patients may be functionally inseparable from those directed against healthy tissues. The seemingly inextricable association between off-target autoimmune damage to healthy tissues and antitumor immunity induced by effective cancer immunotherapy during recent clinical trials can be seen as supporting this hypothesis. Evidence from molecular studies in this regard and a large body of research on inflammation and cancer suggest a feedback loop of autoimmunity, antitumor immunity, inflammation, and de novo tumorigenesis that reinforces the remarkable entanglement between autoimmunity and cancer (Figure 1). In particular, the persistence of antigens in most autoimmune conditions likely leads to the formation of a type of autoimmune memory cells called effector memory T (TEM) cells [for review see Ref. (66)], which could further perpetuate this feedback loop. Advances in not only experimental and clinical research, but also in computational biology tools for large datasets, will be needed to understand the complexity of molecular and cellular interactions in such chronic human disease settings (66, 147).
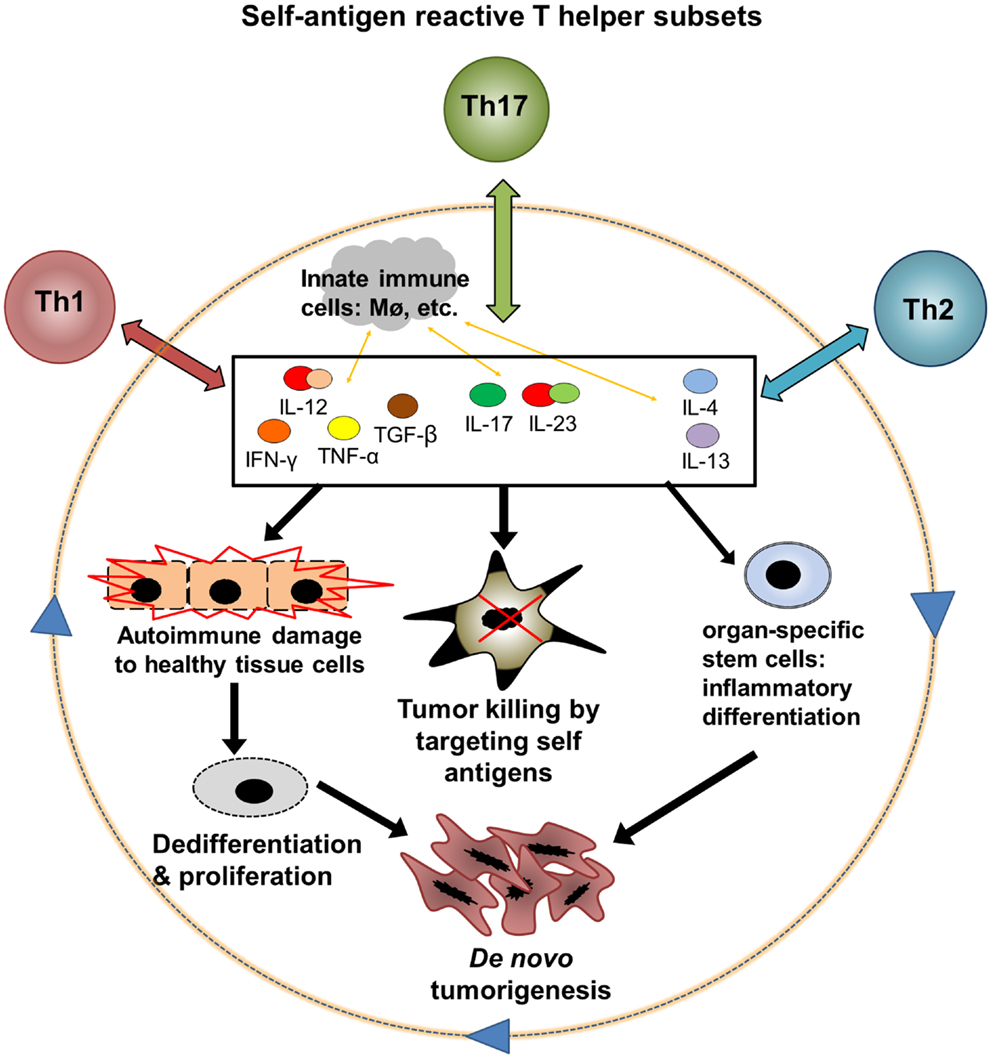
Figure 1. A hypothetic feedback loop between autoimmunity, antitumor immunity, and inflammatory tumorigenesis. Three major subsets of autoantigen reactive T helper cells, Th1, Th2, and Th17, could mediate both antitumor and pro-tumor effects through interaction with innate immune cells and CD8 T cell effectors. (1) Th1, Th2, and Th17 cells release cytokines after encountering autoantigens. These cytokines may exert direct and indirect effects on cell cycle regulation, apoptosis, and differentiation in both tumors and healthy tissues. (2) Cytokines can promote tumor killing by various mechanisms including stimulation of CD8 effectors, innate immune cell activation, and direct toxicity. (3) Inflammation in healthy tissues, particularly when chronic, could contribute to loss of antiproliferative signals and aberrant differentiation in normal cells, leading to a premalignant phenotype. Continued proliferation of these abnormal cells, with accumulation of DNA damage, leads to the emergence of cancer. (4) Chronic cytokine signaling may induce abnormal differentiation of organ-specific stem cells or other precursor cells and drive a cascade of cancer development. The feedback between autoinflammation and cancer may particularly affect solid organs such as the gastrointestinal tract that are demonstrated to be susceptible to inflammatory carcinogenesis.
Along this line, one may argue that a combination of immunomodulation with conventional cancer therapies such as chemotherapy could be used to augment tumor-specific immune responses. Despite the severe immunosuppression produced by many standard chemotherapeutic agents, current data suggest that patients with chemotherapy-induced leukopenia retain a functional T cell compartment that is capable of mediating clinically significant antitumor immunity (148). Within solid tumors, it has been demonstrated that chemotherapy can deplete immune suppressor cells (MDSCs and Treg cells) in the tumor microenvironment, increase tumor antigenicity, and upregulate the expression of costimulatory molecules for CTL activation (79). All of these mechanisms point toward possible synergistic effects that could enhance the clinical efficacy of existing cancer immunotherapies, including agents that have shown meager benefits when administered alone. Indeed, synergy between cancer vaccines and chemotherapy has already been demonstrated in studies of advanced small-cell lung cancer and follicular B-cell lymphoma (149).
Immunological research continues to produce crucial mechanistic insights into the tumor-related effects of major cytokines. For instance, a recent murine experimental model has demonstrated that IFN-γ and TNF-α produced by Th1 cells are capable of inducing prolonged senescence in pancreatic tumors, by inducing expression of the transcription factors JUNB and INK4A (150). The identification of specific signaling pathways for tumor cell growth inhibition and apoptosis heralds a new and improved generation of cytokine-based therapies. More broadly, this knowledge may eventually enable a combinatorial approach to cancer immunotherapy, in which multiple treatments can be jointly administered to yield superior therapeutic outcomes. The current field of cancer immunotherapy is divided between treatments that encourage global activation of cytotoxic immune responses, such as exogenous cytokines and antibodies targeting T cell-inhibitory signals (e.g., anti-PD-1, anti-PD-L1, anti-CTLA4), and treatments based on tumor antigens, which aim to stimulate destruction of cancerous tissues by engaging a specific population of tumor-reactive CTLs (e.g., cancer vaccines and autologous T cell transfers). With further advancement in the clinical and investigative realms, it may become possible for these two approaches to complement each other within the same patient. A cancer vaccine that produces a meager antitumor response in vivo could have enhanced efficacy when administered alongside a treatment that elicits global T cell activation, such as CTLA4 blockade. Moreover, the systemic autoimmune toxicity produced by these broad-acting treatments might be mitigated if a lower dose was combined with a cancer vaccine or other antigen-focused immune stimulus. Adding to the possibilities, multiple immunomodulatory agents from either treatment category could theoretically be combined (e.g., anti-PD-1 in combination with one or more exogenous cytokines) to provide unprecedented control over the targeting, intensity, and duration of the induced immune response. The potential advantages of a combined regimen are already supported by data from numerous preclinical models (5). The true capabilities of cancer immunotherapy may only be realized once multiple treatments can be synthesized into a therapeutic strategy tailored to the pathological and molecular characteristics of every patient’s disease. Although the harmful clinical sequelae of autoimmunity may never be banished entirely, this integrative approach has the potential to harness its tumoricidal functions better than any single agent administered in isolation.
In summary, clinical data suggest that both the anticipated benefits of cancer immunotherapy and its associated adverse events share autoimmunity as a common originating process. Thus autoimmunity, regarded until recently as a “side effect” of cancer immunotherapy, may be more properly considered a correlate of antitumor immunity, or even more appropriately as an antitumor effector in its own right. Despite the lopsided benefit versus risk ratio in cancer immunotherapies that succeed in providing substantial survival benefit, it should be noted that autoimmune damage to healthy tissues is a justifiably dreaded cause of morbidity and mortality in patients receiving these treatments. This hazard may be even more dire if one considers that an immunosuppressive microenvironment effectively designates tumors as an immunoprivileged self, which is more resistant to immune targeting than its healthy counterpart (1, 2). Furthermore, the calculation of benefit versus risk must account for the possibility that inflammatory signals arising from therapeutically induced autoimmunity may ultimately contribute to de novo tumorigenesis in the clinical setting. Therefore, achieving optimal benefit of cancer immunotherapies awaits advances in tumor-specific targeting, either by site or by unique antigens, coupled with proper monitoring and prevention of potentially catastrophic autoimmune damage or long-term risks of de novo tumorigenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by grants from the National Institute of Cancer (1R21CA178675-01 to Zhibin Chen) and the Bankhead-Coley Research Program of Florida Department of Health (09BN-05 to Zhibin Chen). We sincerely apologize to all colleagues whose studies have contributed importantly to the understanding of the topics surveyed in this article, but not cited due to the limit of the length of the article as well as the scope of our knowledge.
References
1. Miska J, Bas E, Devarajan P, Chen Z. Autoimmunity-mediated antitumor immunity: tumor as an immunoprivileged self. Eur J Immunol (2012) 42:2584–2596. doi:10.1002/eji.201242590
2. Miska J, Devarajan P, Chen Z. The immunological identity of tumor: self implications. Oncoimmunology (2013) 2:e23794. doi:10.4161/onci.23794
3. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet (2001) 357:539–45. doi:10.1016/S0140-6736(00)04046-0
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
5. Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer (2007) 7:95–106. doi:10.1038/nrc2051
6. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
7. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi:10.1056/NEJMoa1200690
8. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366:2455–65. doi:10.1056/NEJMoa1200694
9. Whiteside TL. Inhibiting the inhibitors: evaluating agents targeting cancer immunosuppression. Expert Opin Biol Ther (2010) 10:1019–35. doi:10.1517/14712598.2010.482207
10. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol (2004) 75:163–89. doi:10.1189/jlb.0603252
11. Ikeda H, Old LJ, Schreiber RD. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev (2002) 13:95–109. doi:10.1016/S1359-6101(01)00038-7
12. Seliger B, Ruiz-Cabello F, Garrido F. IFN inducibility of major histocompatibility antigens in tumors. Adv Cancer Res (2008) 101:249–76. doi:10.1016/S0065-230X(08)00407-7
13. Kelchtermans H, Billiau A, Matthys P. How interferon-gamma keeps autoimmune diseases in check. Trends Immunol (2008) 29:479–86. doi:10.1016/j.it.2008.07.002
14. Zhang J. Yin and yang interplay of IFN-gamma in inflammation and autoimmune disease. J Clin Invest (2007) 117:871–3. doi:10.1172/JCI31860
15. Stoicov C, Fan X, Liu JH, Bowen G, Whary M, Kurt-Jones E, et al. T-bet knockout prevents Helicobacter felis-induced gastric cancer. J Immunol (2009) 183:642–9. doi:10.4049/jimmunol.0900511
16. Syu LJ, El-Zaatari M, Eaton KA, Liu Z, Tetarbe M, Keeley TM, et al. Transgenic expression of interferon-gamma in mouse stomach leads to inflammation, metaplasia, and dysplasia. Am J Pathol (2012) 181:2114–25. doi:10.1016/j.ajpath.2012.08.017
17. Cho HI, Lee YR, Celis E. Interferon gamma limits the effectiveness of melanoma peptide vaccines. Blood (2011) 117:135–44. doi:10.1182/blood-2010-08-298117
18. Malmberg KJ, Levitsky V, Norell H, de Matos CT, Carlsten M, Schedvins K, et al. IFN-gamma protects short-term ovarian carcinoma cell lines from CTL lysis via a CD94/NKG2A-dependent mechanism. J Clin Invest (2002) 110:1515–23. doi:10.1172/JCI200215564
19. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A (1975) 72:3666–70. doi:10.1073/pnas.72.9.3666
20. van Horssen R, Ten Hagen TL, Eggermont AM. TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist (2006) 11:397–408. doi:10.1634/theoncologist.11-4-397
21. Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis (2011) 70(Suppl 1):i104–8. doi:10.1136/ard.2010.140145
22. Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology (2013) 144:1199.e–209.e. doi:10.1053/j.gastro.2013.02.007
23. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell (2010) 140:197–208. doi:10.1016/j.cell.2009.12.052
24. Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer (2009) 9:361–71. doi:10.1038/nrc2628
25. Wu Y, Zhou BP. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br J Cancer (2010) 102:639–44. doi:10.1038/sj.bjc.6605530
26. Achyut BR, Bader DA, Robles AI, Wangsa D, Harris CC, Ried T, et al. Inflammation-mediated genetic and epigenetic alterations drive cancer development in the neighboring epithelium upon stromal abrogation of TGF-beta signaling. PLoS Genet (2013) 9:e1003251. doi:10.1371/journal.pgen.1003251
27. Chaudhury A, Howe PH. The tale of transforming growth factor-beta (TGFbeta) signaling: a soigne enigma. IUBMB Life (2009) 61:929–39. doi:10.1002/iub.239
28. Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol (2005) 6:600–7. doi:10.1038/ni1197
29. Prud’homme GJ. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab Invest (2007) 87:1077–91. doi:10.1038/labinvest.3700669
30. Ramalingam R, Larmonier CB, Thurston RD, Midura-Kiela MT, Zheng SG, Ghishan FK, et al. Dendritic cell-specific disruption of TGF-beta receptor II leads to altered regulatory T cell phenotype and spontaneous multiorgan autoimmunity. J Immunol (2012) 189:3878–93. doi:10.4049/jimmunol.1201029
31. Galdiero MR, Garlanda C, Jaillon S, Marone G, Mantovani A. Tumor associated macrophages and neutrophils in tumor progression. J Cell Physiol (2013) 228:1404–12. doi:10.1002/jcp.24260
32. Moo-Young TA, Larson JW, Belt BA, Tan MC, Hawkins WG, Eberlein TJ, et al. Tumor-derived TGF-beta mediates conversion of CD4+Foxp3+ regulatory T cells in a murine model of pancreas cancer. J Immunother (2009) 32:12–21. doi:10.1097/CJI.0b013e318189f13c
33. Yang P, Li QJ, Feng Y, Zhang Y, Markowitz GJ, Ning S, et al. TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell (2012) 22:291–303. doi:10.1016/j.ccr.2012.07.023
34. Yang L. TGFbeta and cancer metastasis: an inflammation link. Cancer Metastasis Rev (2010) 29:263–71. doi:10.1007/s10555-010-9226-3
36. Kurebayashi Y, Nagai S, Ikejiri A, Koyasu S. Recent advances in understanding the molecular mechanisms of the development and function of Th17 cells. Genes Cells (2013) 18:247–65. doi:10.1111/gtc.12039
37. Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol (2010) 10:248–56. doi:10.1038/nri2742
38. Kaiga T, Sato M, Kaneda H, Iwakura Y, Takayama T, Tahara H. Systemic administration of IL-23 induces potent antitumor immunity primarily mediated through Th1-type response in association with the endogenously expressed IL-12. J Immunol (2007) 178:7571–80.
39. Kryczek I, Wei S, Szeliga W, Vatan L, Zou W. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood (2009) 114:357–9. doi:10.1182/blood-2008-09-177360
40. Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity (2009) 31:787–98. doi:10.1016/j.immuni.2009.09.014
41. Nuñez S, Saez JJ, Fernandez D, Flores-Santibanez F, Alvarez K, Tejon G, et al. T helper type 17 cells contribute to anti-tumour immunity and promote the recruitment of T helper type 1 cells to the tumour. Immunology (2013) 139:61–71. doi:10.1111/imm.12055
42. Amedei A, Niccolai E, Benagiano M, Della Bella C, Cianchi F, Bechi P, et al. Ex vivo analysis of pancreatic cancer-infiltrating T lymphocytes reveals that ENO-specific Tregs accumulate in tumor tissue and inhibit Th1/Th17 effector cell functions. Cancer Immunol Immunother (2013) 62:1249–60. doi:10.1007/s00262-013-1429-3
43. Huang Z, van Velkinburgh JC, Ni B, Wu Y. Pivotal roles of the interleukin-23/T helper 17 cell axis in hepatitis B. Liver Int (2012) 32:894–901. doi:10.1111/j.1478-3231.2012.02764.x
44. Wu W, Li J, Chen F, Zhu H, Peng G, Chen Z. Circulating Th17 cells frequency is associated with the disease progression in HBV infected patients. J Gastroenterol Hepatol (2010) 25:750–7. doi:10.1111/j.1440-1746.2009.06154.x
45. Yang W, Ding X, Deng J, Lu Y, Matsuda Z, Thiel A, et al. Interferon-gamma negatively regulates Th17-mediated immunopathology during mouse hepatitis virus infection. J Mol Med (Berl) (2011) 89:399–409. doi:10.1007/s00109-010-0711-5
46. Ye Y, Xie X, Yu J, Zhou L, Xie H, Jiang G, et al. Involvement of Th17 and Th1 effector responses in patients with Hepatitis B. J Clin Immunol (2010) 30:546–55. doi:10.1007/s10875-010-9416-3
47. He D, Li H, Yusuf N, Elmets CA, Li J, Mountz JD, et al. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J Immunol (2010) 184:2281–8. doi:10.4049/jimmunol.0902574
48. Qian X, Gu L, Ning H, Zhang Y, Hsueh EC, Fu M, et al. Increased Th17 cells in the tumor microenvironment is mediated by IL-23 via tumor-secreted prostaglandin E2. J Immunol (2013) 190:5894–902. doi:10.4049/jimmunol.1203141
49. Baird AM, Leonard J, Naicker KM, Kilmartin L, O’Byrne KJ, Gray SG. IL-23 is pro-proliferative, epigenetically regulated and modulated by chemotherapy in non-small cell lung cancer. Lung Cancer (2013) 79:83–90. doi:10.1016/j.lungcan.2012.10.003
50. Fukuda M, Ehara M, Suzuki S, Ohmori Y, Sakashita H. IL-23 promotes growth and proliferation in human squamous cell carcinoma of the oral cavity. Int J Oncol (2010) 36:1355–65. doi:10.3892/ijo_00000620
51. Li J, Zhang L, Zhang J, Wei Y, Li K, Huang L, et al. Interleukin 23 regulates proliferation of lung cancer cells in a concentration-dependent way in association with the interleukin-23 receptor. Carcinogenesis (2013) 34:658–66. doi:10.1093/carcin/bgs384
52. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med (2011) 208:469–78. doi:10.1084/jem.20101876
53. Aspord C, Pedroza-Gonzalez A, Gallegos M, Tindle S, Burton EC, Su D, et al. Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. J Exp Med (2007) 204:1037–47. doi:10.1084/jem.20061120
54. Hallett MA, Venmar KT, Fingleton B. Cytokine stimulation of epithelial cancer cells: the similar and divergent functions of IL-4 and IL-13. Cancer Res (2012) 72:6338–43. doi:10.1158/0008-5472.CAN-12-3544
55. Koller FL, Hwang DG, Dozier EA, Fingleton B. Epithelial interleukin-4 receptor expression promotes colon tumor growth. Carcinogenesis (2010) 31:1010–7. doi:10.1093/carcin/bgq044
56. Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell (2007) 1:389–402. doi:10.1016/j.stem.2007.08.001
57. Barderas R, Bartolome RA, Fernandez-Acenero MJ, Torres S, Casal JI. High expression of IL-13 receptor alpha2 in colorectal cancer is associated with invasion, liver metastasis, and poor prognosis. Cancer Res (2012) 72:2780–90. doi:10.1158/0008-5472.CAN-11-4090
58. Fujisawa T, Joshi B, Nakajima A, Puri RK. A novel role of interleukin-13 receptor alpha2 in pancreatic cancer invasion and metastasis. Cancer Res (2009) 69:8678–85. doi:10.1158/0008-5472.CAN-09-2100
59. Fujisawa T, Joshi BH, Puri RK. IL-13 regulates cancer invasion and metastasis through IL-13Ralpha2 via ERK/AP-1 pathway in mouse model of human ovarian cancer. Int J Cancer (2012) 131:344–56. doi:10.1002/ijc.26366
60. Virchow RLK. Cellular Pathology as based upon Physiological and Pathological Histology. Philadelphia: J. B. Lippincott (1863).
61. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
62. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141:39–51. doi:10.1016/j.cell.2010.03.014
63. Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature (2010) 464:302–5. doi:10.1038/nature08782
64. He Y, Zha J, Wang Y, Liu W, Yang X, Yu P. Tissue damage-associated “danger signals” influence T-cell responses that promote the progression of preneoplasia to cancer. Cancer Res (2013) 73:629–39. doi:10.1158/0008-5472.CAN-12-2704
65. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med (2009) 206:1457–64. doi:10.1084/jem.20090207
66. Devarajan P, Chen Z. Autoimmune effector memory T cells: the bad and the good. Immunol Res (2013) 57:12–22. doi:10.1007/s12026-013-8448-1
67. Germain RN, Robey EA, Cahalan MD. A decade of imaging cellular motility and interaction dynamics in the immune system. Science (2012) 336:1676–81. doi:10.1126/science.1221063
68. Miska J, Abdulreda MH, Devarajan P, Lui JB, Suzuki J, Pileggi A, et al. Real-time immune cell interactions in target tissue during autoimmune-induced damage and graft tolerance. J Exp Med (2014) 211:441–56. doi:10.1084/jem.20130785
69. Suzuki J, Ricordi C, Chen Z. Immune tolerance induction by integrating innate and adaptive immune regulators. Cell Transplant (2010) 19:253–68. doi:10.3727/096368909X480314
70. Tomsova M, Melichar B, Sedlakova I, Steiner I. Prognostic significance of CD3+ tumor-infiltrating lymphocytes in ovarian carcinoma. Gynecol Oncol (2008) 108:415–20. doi:10.1016/j.ygyno.2007.10.016
71. Tano T, Okamoto M, Kan S, Nakashiro K, Shimodaira S, Koido S, et al. Prognostic impact of expression of Bcl-2 and Bax genes in circulating immune cells derived from patients with head and neck carcinoma. Neoplasia (2013) 15:305–14. doi:10.1593/neo.121528
72. Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol (2013) 14:e218–28. doi:10.1016/S1470-2045(12)70582-X
73. Vierboom MP, Nijman HW, Offringa R, van der Voort EI, van Hall T, van den Broek L, et al. Tumor eradication by wild-type p53-specific cytotoxic T lymphocytes. J Exp Med (1997) 186:695–704. doi:10.1084/jem.186.5.695
74. Hilburger Ryan M, Abrams SI. Characterization of CD8+ cytotoxic T lymphocyte/tumor cell interactions reflecting recognition of an endogenously expressed murine wild-type p53 determinant. Cancer Immunol Immunother (2001) 49:603–12. doi:10.1007/s002620000156
75. Hu J, Kindsvogel W, Busby S, Bailey MC, Shi YY, Greenberg PD. An evaluation of the potential to use tumor-associated antigens as targets for antitumor T cell therapy using transgenic mice expressing a retroviral tumor antigen in normal lymphoid tissues. J Exp Med (1993) 177:1681–90. doi:10.1084/jem.177.6.1681
76. Morgan DJ, Kreuwel HT, Fleck S, Levitsky HI, Pardoll DM, Sherman LA. Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J Immunol (1998) 160:643–51.
77. Caspi RR. Immunotherapy of autoimmunity and cancer: the penalty for success. Nat Rev Immunol (2008) 8:970–6. doi:10.1038/nri2438
78. Attia P, Phan GQ, Maker AV, Robinson MR, Quezado MM, Yang JC, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol (2005) 23:6043–53. doi:10.1200/JCO.2005.06.205
79. Andersen MH, Junker N, Ellebaek E, Svane IM, Thor Straten P. Therapeutic cancer vaccines in combination with conventional therapy. J Biomed Biotechnol (2010) 2010:237623. doi:10.1155/2010/237623
80. Zaidi MR, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res (2011) 17:6118–24. doi:10.1158/1078-0432.CCR-11-0482
81. Fojtova M, Boudny V, Kovarik A, Lauerova L, Adamkova L, Souckova K, et al. Development of IFN-gamma resistance is associated with attenuation of SOCS genes induction and constitutive expression of SOCS 3 in melanoma cells. Br J Cancer (2007) 97:231–7. doi:10.1038/sj.bjc.6603849
82. Miller CH, Maher SG, Young HA. Clinical use of interferon-gamma. Ann N Y Acad Sci (2009) 1182:69–79. doi:10.1111/j.1749-6632.2009.05069.x
83. Demaria S, Pikarsky E, Karin M, Coussens LM, Chen YC, El-Omar EM, et al. Cancer and inflammation: promise for biologic therapy. J Immunother (2010) 33:335–51. doi:10.1097/CJI.0b013e3181d32e74
84. Moss SF, Blaser MJ. Mechanisms of disease: Inflammation and the origins of cancer. Nat Clin Pract Oncol (2005) 2:90–7. doi:10.1038/ncponc0081 quiz 91 p following 113,
85. Skurkovich B, Skurkovich S. Anti-interferon-gamma antibodies in the treatment of autoimmune diseases. Curr Opin Mol Ther (2003) 5:52–7.
86. Skurkovich S, Skurkovich B, Kelly J. Anticytokine therapy, particularly anti-IFN-gamma, in Th1-mediated autoimmune diseases. Expert Rev Clin Immunol (2005) 1:11–25. doi:10.1586/1744666X.1.1.11
87. Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest (2007) 117:60–9. doi:10.1172/JCI30111
88. Merchant JL. What lurks beneath: IL-11, via Stat3, promotes inflammation-associated gastric tumorigenesis. J Clin Invest (2008) 118:1628–31. doi:10.1172/JCI35344
89. Polk DB, Peek RM Jr. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer (2010) 10:403–14. doi:10.1038/nrc2857
90. Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med (1997) 337:1441–8. doi:10.1056/NEJM199711133372007
91. El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology (2003) 124:1193–201. doi:10.1016/S0016-5085(03)00157-4
92. Judd LM, Alderman BM, Howlett M, Shulkes A, Dow C, Moverley J, et al. Gastric cancer development in mice lacking the SHP2 binding site on the IL-6 family co-receptor gp130. Gastroenterology (2004) 126:196–207. doi:10.1053/j.gastro.2003.10.066
93. Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell (2008) 14:408–19. doi:10.1016/j.ccr.2008.10.011
94. Weis VG, Goldenring JR. Current understanding of SPEM and its standing in the preneoplastic process. Gastric Cancer (2009) 12:189–97. doi:10.1007/s10120-009-0527-6
95. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell (2000) 100:655–69. doi:10.1016/S0092-8674(00)80702-3
96. Li X, McKinstry KK, Swain SL, Dalton DK. IFN-gamma acts directly on activated CD4+ T cells during mycobacterial infection to promote apoptosis by inducing components of the intracellular apoptosis machinery and by inducing extracellular proapoptotic signals. J Immunol (2007) 179:939–49.
97. Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol (2008) 8:74–80. doi:10.1038/nri2233
98. Tu SP, Quante M, Bhagat G, Takaishi S, Cui G, Yang XD, et al. IFN-gamma inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Res (2011) 71:4247–59. doi:10.1158/0008-5472.CAN-10-4009
99. White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res (2009) 15:5308–16. doi:10.1158/1078-0432.CCR-07-5023
100. Bradley JR. TNF-mediated inflammatory disease. J Pathol (2008) 214:149–60. doi:10.1002/path.2287
101. Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL. Anti-TNF-alpha therapies: the next generation. Nat Rev Drug Discov (2003) 2:736–46. doi:10.1038/nrd1175
102. Murdaca G, Colombo BM, Cagnati P, Gulli R, Spano F, Puppo F. Update upon efficacy and safety of TNF-alpha inhibitors. Expert Opin Drug Saf (2012) 11:1–5. doi:10.1517/14740338.2012.630388
104. Kruglov AA, Kuchmiy A, Grivennikov SI, Tumanov AV, Kuprash DV, Nedospasov SA. Physiological functions of tumor necrosis factor and the consequences of its pathologic overexpression or blockade: mouse models. Cytokine Growth Factor Rev (2008) 19:231–44. doi:10.1016/j.cytogfr.2008.04.010
105. Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med (1999) 5:828–31. doi:10.1038/10552
106. Fujiki H, Sueoka E, Suganuma M. Tumor promoters: from chemicals to inflammatory proteins. J Cancer Res Clin Oncol (2013) 139:1603–14. doi:10.1007/s00432-013-1455-8
107. Capece D, Fischietti M, Verzella D, Gaggiano A, Cicciarelli G, Tessitore A, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int (2013) 2013:187204. doi:10.1155/2013/187204
108. Natarajan M, Gibbons CF, Mohan S, Moore S, Kadhim MA. Oxidative stress signalling: a potential mediator of tumour necrosis factor alpha-induced genomic instability in primary vascular endothelial cells. Br J Radiol (2007) 80(1):S13–22. doi:10.1259/bjr/15316848
109. Pierce RH, Campbell JS, Stephenson AB, Franklin CC, Chaisson M, Poot M, et al. Disruption of redox homeostasis in tumor necrosis factor-induced apoptosis in a murine hepatocyte cell line. Am J Pathol (2000) 157:221–36. doi:10.1016/S0002-9440(10)64533-6
110. Suematsu N, Tsutsui H, Wen J, Kang D, Ikeuchi M, Ide T, et al. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation (2003) 107:1418–23. doi:10.1161/01.CIR.0000055318.09997.1F
111. Wheelhouse NM, Chan YS, Gillies SE, Caldwell H, Ross JA, Harrison DJ, et al. TNF-alpha induced DNA damage in primary murine hepatocytes. Int J Mol Med (2003) 12:889–94. doi:10.3892/ijmm.12.6.889
112. Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell (2008) 134:392–404. doi:10.1016/j.cell.2008.07.025
113. Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A (1993) 90:770–4. doi:10.1073/pnas.90.2.770
114. Leveen P, Larsson J, Ehinger M, Cilio CM, Sundler M, Sjostrand LJ, et al. Induced disruption of the transforming growth factor beta type II receptor gene in mice causes a lethal inflammatory disorder that is transplantable. Blood (2002) 100:560–8. doi:10.1182/blood.V100.2.560
115. Ni XY, Sui HX, Liu Y, Ke SZ, Wang YN, Gao FG. TGF-beta of lung cancer microenvironment upregulates B7H1 and GITRL expression in dendritic cells and is associated with regulatory T cell generation. Oncol Rep (2012) 28:615–21. doi:10.3892/or.2012.1822
116. Wilson CA, Cajulis EE, Green JL, Olsen TM, Chung YA, Damore MA, et al. HER-2 overexpression differentially alters transforming growth factor-beta responses in luminal versus mesenchymal human breast cancer cells. Breast Cancer Res (2005) 7:R1058–79. doi:10.1186/bcr1343
117. Baldwin RL, Tran H, Karlan BY. Loss of c-myc repression coincides with ovarian cancer resistance to transforming growth factor beta growth arrest independent of transforming growth factor beta/Smad signaling. Cancer Res (2003) 63:1413–9.
118. Connolly EC, Freimuth J, Akhurst RJ. Complexities of TGF-beta targeted cancer therapy. Int J Biol Sci (2012) 8:964–78. doi:10.7150/ijbs.4564
119. Connolly EC, Saunier EF, Quigley D, Luu MT, De Sapio A, Hann B, et al. Outgrowth of drug-resistant carcinomas expressing markers of tumor aggression after long-term TbetaRI/II kinase inhibition with LY2109761. Cancer Res (2011) 71:2339–49. doi:10.1158/0008-5472.CAN-10-2941
120. Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity (2009) 30:108–19. doi:10.1016/j.immuni.2008.11.009
121. Ye J, Livergood RS, Peng G. The role and regulation of human Th17 cells in tumor immunity. Am J Pathol (2013) 182:10–20. doi:10.1016/j.ajpath.2012.08.041
122. Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature (2013) 496:461–8. doi:10.1038/nature11981
123. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6:1123–32. doi:10.1038/ni1254
124. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6:1133–41. doi:10.1038/ni1261
125. Langowski JL, Kastelein RA, Oft M. Swords into plowshares: IL-23 repurposes tumor immune surveillance. Trends Immunol (2007) 28:207–12. doi:10.1016/j.it.2007.03.006
126. McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol (2006) 27:17–23. doi:10.1016/j.it.2005.10.003
127. Murugaiyan G, Saha B. Protumor vs antitumor functions of IL-17. J Immunol (2009) 183:4169–75. doi:10.4049/jimmunol.0901017
128. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity (2000) 13:715–25. doi:10.1016/S1074-7613(00)00070-4
129. Hatton RD. TGF-beta in Th17 cell development: the truth is out there. Immunity (2011) 34:288–90. doi:10.1016/j.immuni.2011.03.009
130. Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, et al. IL-23 promotes tumour incidence and growth. Nature (2006) 442:461–5. doi:10.1038/nature04808
131. Toussirot E. The IL23/Th17 pathway as a therapeutic target in chronic inflammatory diseases. Inflamm Allergy Drug Targets (2012) 11:159–68. doi:10.2174/187152812800392805
132. Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol (2013) 31:317–43. doi:10.1146/annurev-immunol-032712-095906
133. Chapoval S, Dasgupta P, Dorsey NJ, Keegan AD. Regulation of the T helper cell type 2 (Th2)/T regulatory cell (Treg) balance by IL-4 and STAT6. J Leukoc Biol (2010) 87:1011–8. doi:10.1189/jlb.1209772
134. Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res (2012) 4:376–89.
135. LaPorte SL, Juo ZS, Vaclavikova J, Colf LA, Qi X, Heller NM, et al. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell (2008) 132:259–72. doi:10.1016/j.cell.2007.12.030
136. Oh CK, Geba GP, Molfino N. Investigational therapeutics targeting the IL-4/IL-13/STAT-6 pathway for the treatment of asthma. Eur Respir Rev (2010) 19:46–54. doi:10.1183/09059180.00007609
137. Li Z, Jiang J, Wang Z, Zhang J, Xiao M, Wang C, et al. Endogenous interleukin-4 promotes tumor development by increasing tumor cell resistance to apoptosis. Cancer Res (2008) 68:8687–94. doi:10.1158/0008-5472.CAN-08-0449
138. Humphries A, Cereser B, Gay LJ, Miller DS, Das B, Gutteridge A, et al. Lineage tracing reveals multipotent stem cells maintain human adenomas and the pattern of clonal expansion in tumor evolution. Proc Natl Acad Sci U S A (2013) 110:E2490–9. doi:10.1073/pnas.1220353110
139. Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science (2012) 337:730–5. doi:10.1126/science.1224676
140. Todaro M, Perez Alea M, Scopelliti A, Medema JP, Stassi G. IL-4-mediated drug resistance in colon cancer stem cells. Cell Cycle (2008) 7:309–13. doi:10.4161/cc.7.3.5389
141. Hershey GK. IL-13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol (2003) 111:677–90. doi:10.1067/mai.2003.1333 quiz 691,
142. Shimamura T, Fujisawa T, Husain SR, Joshi B, Puri RK. Interleukin 13 mediates signal transduction through interleukin 13 receptor alpha2 in pancreatic ductal adenocarcinoma: role of IL-13 pseudomonas exotoxin in pancreatic cancer therapy. Clin Cancer Res (2010) 16:577–86. doi:10.1158/1078-0432.CCR-09-2015
143. Hall B, Nakashima H, Sun ZJ, Sato Y, Bian Y, Husain SR, et al. Targeting of interleukin-13 receptor alpha2 for treatment of head and neck squamous cell carcinoma induced by conditional deletion of TGF-beta and PTEN signaling. J Transl Med (2013) 11:45. doi:10.1186/1479-5876-11-45
144. Shimamura T, Husain SR, Puri RK. The IL-4 and IL-13 pseudomonas exotoxins: new hope for brain tumor therapy. Neurosurg Focus (2006) 20:E11. doi:10.3171/foc.2006.20.4.6
145. Mut M, Sherman JH, Shaffrey ME, Schiff D. Cintredekin besudotox in treatment of malignant glioma. Expert Opin Biol Ther (2008) 8:805–12. doi:10.1517/14712598.8.6.805
146. Ohka F, Natsume A, Wakabayashi T. Current trends in targeted therapies for glioblastoma multiforme. Neurol Res Int (2012) 2012:878425. doi:10.1155/2012/878425
147. Han D, Cai X, Wen J, Kenyon NS, Chen Z. From biomarkers to a clue of biology: a computation-aided perspective of immune gene expression profiles in human type 1 diabetes. Front Immunol (2012) 3:320. doi:10.3389/fimmu.2012.00320
148. Liseth K, Ersvaer E, Hervig T, Bruserud O. Combination of intensive chemotherapy and anticancer vaccines in the treatment of human malignancies: the hematological experience. J Biomed Biotechnol (2010) 2010:692097. doi:10.1155/2010/692097
Keywords: autoimmunity, antitumor, tumorigenesis, inflammation, cytokine
Citation: Toomer KH and Chen Z (2014) Autoimmunity as a double agent in tumor killing and cancer promotion. Front. Immunol. 5:116. doi: 10.3389/fimmu.2014.00116
Received: 29 December 2013; Paper pending published: 22 January 2014;
Accepted: 05 March 2014; Published online: 18 March 2014.
Edited by:
Fang-Ping Huang, Imperial College London, UKReviewed by:
Amedeo Amedei, University of Florence, ItalyCarlos Alfaro, Clínica Universidad de Navarra, Spain
Copyright: © 2014 Toomer and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhibin Chen, Department of Microbiology and Immunology, University of Miami Miller School of Medicine, RMSB 3035, 1600 NW 10th Avenue, Miami, FL 33136, USA e-mail:emNoZW5AbWVkLm1pYW1pLmVkdQ==