 Elke Gülden
Elke Gülden Li Wen*
Li Wen*Introduction
Toll-Like Receptors: Key Receptors of the Innate Immune System
The human body constantly encounters a diverse spectrum of pathogens. To defend itself, a complex immune system has evolved consisting of two subdivisions: the innate and the adaptive immune systems. The innate immune system constitutes the so-called “first line of defense” and acts through highly conserved germ-line-encoded pattern recognition receptors (PRRs) (1). These receptors bind to common pathogen-associated molecular patterns (PAMPs), which are vital for the survival of the microorganisms and cannot be altered by mutations.
Pattern recognition receptors were first discovered in the fruit fly Drosophila melanogaster. The PRRs in Drosophila, named Toll, play an important role in the recognition of microorganisms such as fungi as well as coordinating embryonic development of the dorso-ventral axis.
Homologs of Toll in vertebrates are called Toll-like receptors (TLRs). The first TLR in humans was described in 1997 (2). To date, another 9 TLRs have been identified in humans and 13 in mice. Among these, only murine TLR10 is non-functional due to retrovirus insertion (3). Except for TLRs 3, 7, 8, and 9, which are expressed intracellularly, TLRs are located on the surface of innate and adaptive immune cells as well as on non-immune cells such as muscle cells, epithelial cells, adipocytes, and pancreatic beta cells (1). Cell-surface TLRs detect exogenous lipids, lipoproteins, and proteins from microbes; intracellular TLRs recognize bacterial and viral nucleic acids (3). Recognition of their cognate ligand triggers a complex signaling cascade that ultimately results in the induction of various pro-inflammatory chemokines and cytokines and the activation of the adaptive immune system.
However, the activation of TLRs and the subsequent induction of inflammatory immune responses are not always beneficial to the host. TLRs are involved in the pathogenesis of various autoimmune and non-infective inflammatory diseases such as systemic lupus erythematosus, multiple sclerosis, arteriosclerosis, inflammatory bowel disease, diabetes, allergy, and cancer (4, 5). Their activation can either attenuate or boost the course of disease by inducing tolerance or triggering autoreactivity, respectively.
The role of the TLRs in the pathogenesis of autoimmune-mediated diabetes, also referred to as type 1 diabetes (T1D), has been extensively studied (Table 1). T1D is a T-cell-mediated metabolic disorder with progressive destruction of insulin-producing pancreatic β cells (6). During the course of disease development, diabetogenic T-cells, macrophages, and dendritic cells will infiltrate the pancreatic islets and cause islet inflammation and eventually β cell loss.
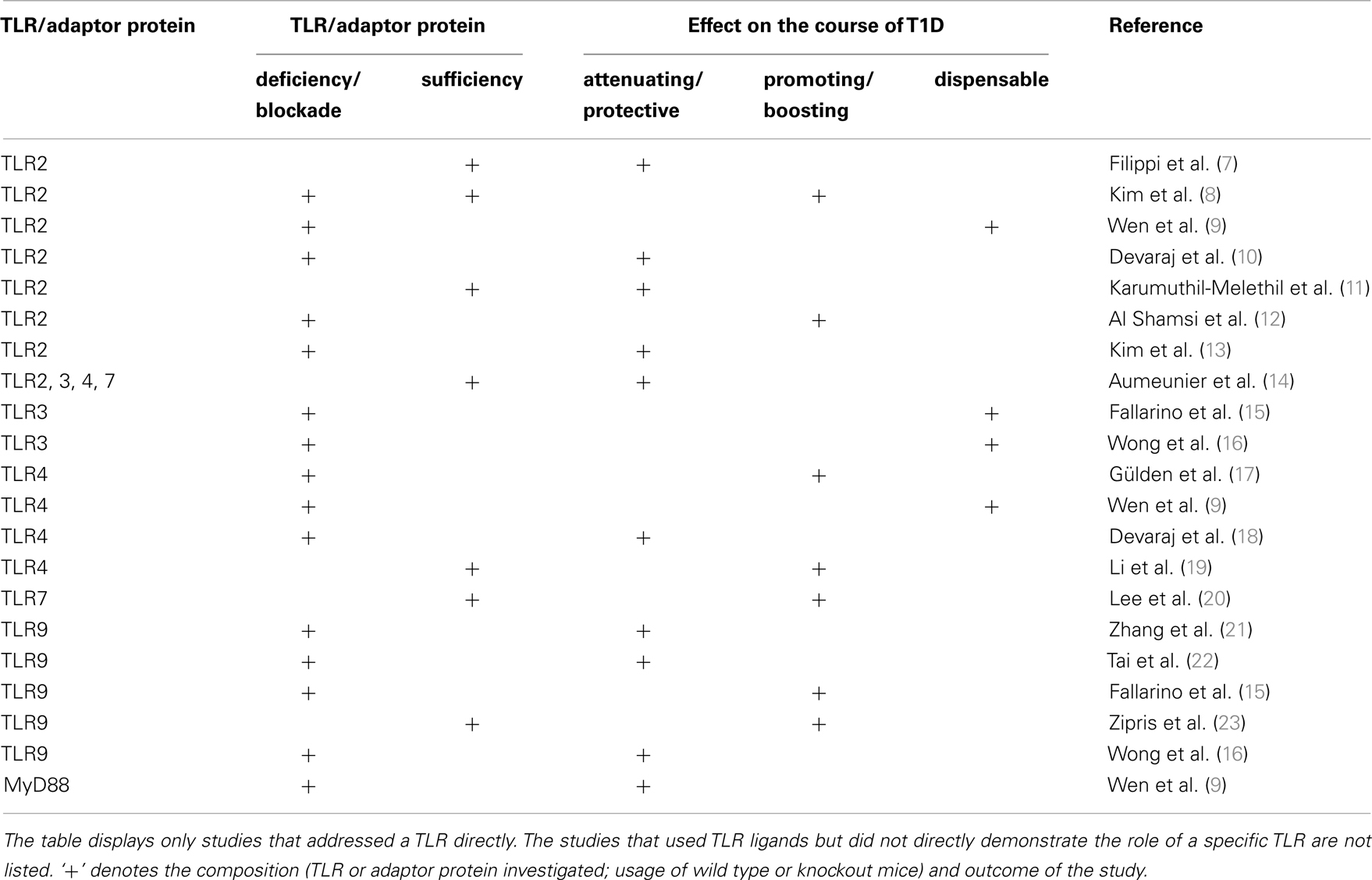
Table 1. TLR-related studies in the field of T1D.
This opinion letter focuses on the beneficial and detrimental aspects of TLR induction in the course of T1D development.
TLR and Their Role in Autoimmune-Mediated Type 1 Diabetes
TLR-Mediated Initial Events in the Induction of Islet-Directed Immune Responses
One of the major functions of the immune system is to distinguish self from non-self in order to fight invaders (pathogens) while sparing endogenous structures. PAMPs, the ligands of TLRs, are conserved molecular patterns that are exclusively expressed by pathogens. Therefore, the innate immune system is able to distinguish self and non-self. This beneficial “discrimination” is further enhanced by the adaptive immune system, in which most of the self-reactive T- and B-cells are deleted by central tolerance through negative selection. However, increasing evidence suggests that TLRs also recognize endogenous molecules including self-DNA released by injured or dying cells. These endogenous molecules act as danger signals and are therefore called “danger-associated molecular patterns (DAMPs).” The exposure of DAMPs by necrotic cells is considered to be a potential trigger of autoimmune diseases such as T1D (24). Studies have shown a defective clearance of dying cells in the NOD mouse (25), currently the best characterized model of human T1D (26). Due to the diabetes-prone genetic background, this defect might essentially contribute to the induction of autoimmunity in these mice. The accumulation of apoptotic β cells, which may undergo secondary necrosis (so-called “late state apoptosis”), could result in the activation of antigen-presenting cells (APC) via TLR engagement by released endogenous molecules and thereby contributing to the induction of diabetogenic T-cells (8, 27). Diabetogenic T-cells are then recruited to the pancreatic islets by chemokines like CCL2 (28, 29), CCL5, CXCL9, and CXCL10 (30) that can be released from β cells upon TLR ligation.
Consistent with this hypothesis is the fact that TLR2 induces apoptosis (31) and promotes diabetes in a streptozotocin-induced diabetic model following activation via the synthetic ligand Pam3CSK4 (12). The observation of delayed or reduced diabetes onset in TLR9-deficient mice further supports the theory (16, 21, 22). However, in a chemically induced diabetes mouse model, Fallarino et al. found that TLR9-deficient C57BL/6 mice were more susceptible to diabetes induction (15). This opposing finding is most likely due to the different choice of animal model. Other investigators have used the NOD mouse model (16, 21, 22) whereas Fallarino and colleagues induced diabetes by injections of β cell-toxic drug streptozotocin to C57BL/6 mice.
Disease Protection vs. Disease Induction
As key receptors of the innate immune system, TLRs trigger inflammatory immune responses upon binding of cognate ligands. On a predisposed genetic background, this event might initiate islet inflammation followed by progressive β cell destruction and finally overt T1D.
However, TLR activation is not necessarily causative for T1D development. T1D-related studies summarized in Table 1, reveal either a protective or detrimental effect on the induction of islet-directed autoimmunity. One reason for this dichotomy might be the point in time during the prediabetic phase when TLR activation is induced. Moreover, the presence or absence of β cell antigens and/or endogenous DAMPs released by late state apoptotic β cells possibly plays a critical role. Studies showed that activation of TLR2 (8), TLR3 (32), or TLR9 (33) by their cognate ligands in the presence of β cell antigens or DAMPs give rise to T1D development, whereas TLR stimulation in the absence of β cell antigens results in tolerogenic immune responses (11, 34–36). Observations by Filippi et al. suggest that the reason for this outcome might be the capacity of immunostimulatory factors to augment immune regulation (7).
The Role of TLRs in Modulating Treg Functions
Regulatory T-cells (Treg) are TLR-expressing adaptive immune cells which control immune responses in order to prevent aberrant immune reactions which could be harmful (37). In the presence of β cell antigens, TLR2 signaling could, while inducing a pro-inflammatory immune response, simultaneously promote suppressive Treg functions.
Dasgupta et al. demonstrated that engagement of TLR2 reverses the suppressor function of conjunctiva Treg in rabbits (38). In mice, in some cases, TLR2 stimulation resulted in a temporary abrogation of the regulatory capacity of Treg (39, 40), while other studies reported that the Treg function was either unchanged (41) or improved (7, 11). On the other hand, studies by other investigators showed a decreased number of Tregs following TLR2 stimulation (12).
One of the explanations for these contrary findings might be the different concentrations of the TLR ligand used in their experimental systems. It has been reported that the concentration of TLR ligands influences the regulatory activity of Tregs (40, 42, 43). For example, Zanin-Zhorov and colleagues showed that low doses of the TLR2 ligand, Hsp60, resulted in enhanced suppression without increased Treg proliferation (42, 43).
Studies by Round and Mazmanian investigating the immunomodulatory effect of polysaccharide A (PSA), a microbial molecule of the commensal bacterium Bacteroides fragilis, reveal that the PSA can signal directly on Treg cells via their TLR2 and promote immune tolerance (44, 45). It is possible that the effect may be accounted for by the anatomical site where TLR2 is engaged. Specifically in the intestine, TLR2 signaling induced by PSA is required for Treg induction and IL-10 expression.
TLR2 ligation does not merely exert influence on Treg; studies also reveal an effect on effector T-cells (46, 47). TLR2 signaling via Pam3Cys achieves resistance of T effector cells toward Tregs (47).
Besides TLR2, other TLRs also modulate Treg functions. TLR4 and TLR5 ligands are capable of boosting the suppressive function of Treg on T effector cells (48, 49). In line with these observations is the finding that TLR4-deficient NOD mice exhibit significantly accelerated diabetes development and impaired suppressive function of Tregs, although the frequency of Tregs remains unchanged (17).
Taken together, TLRs act as important modulators of Treg proliferation and function. Treg function can either be enhanced or attenuated depending on the concentration of TLR ligands and the anatomical site of TLR engagement.
Tolerance Induction by TLR Signaling
As potent activators of inflammatory immune responses, the activation of TLRs must be tightly controlled since over-activation or loss of negative regulation can lead to detrimental or even life-threatening effects as seen in the condition of sepsis (50, 51). Repeated exposure to the same ligand can therefore result in hyporesponsiveness or tolerance through down-regulation of TLRs and simultaneous up-regulation of a negative feedback loop (50, 52).
In contrast to short term TLR stimulation, which results in initiation of immune responses, repeated exposure to TLR ligands might lead to the abrogation of inflammatory immune responses. In such a manner, TLR2 tolerance and inhibition of T1D development in NOD mice could be achieved by repeated administration of the TLR2 agonist Pam3CSK4 or zymosan (11, 13). Decreased T1D incidence was also accomplished following treatment with LPS, poly (I:C) (34), or CpG oligonucleotides (36).
As systemic chronic TLR stimulation suppresses the development of T1D in NOD mice (14, 53), it is possible that the absence of TLR stimulation might facilitate T1D development. This possibility is supported by the so-called hygiene hypothesis (54). The hygiene hypothesis coincides with the increase in allergy and autoimmune diseases over the past decades, possibly due to less exposure to microbial products as the hygiene standard has been significantly improved. Supporting the hygiene hypothesis is a recent study revealing that NOD mice deficient in the innate adaptor protein MyD88 are protected from diabetes development in non-germ-free conditions but the protection is abolished in germ-free conditions (9). Furthermore, introducing gut commensals into germ-free mice re-establishes the protection. This study supports the crucial role of environmental (TLR) stimuli in modulating the pathogenesis of diabetes through commensal bacteria.
Concluding Remarks
The important role of TLRs in the pathogenesis of T1D manifests mainly in their ability to induce APC maturation and to produce inflammatory chemokines and cytokines. These two features will contribute to the priming of autoreactive T-cells, which cause islet cell destruction. However, TLR signaling can also induce immune tolerance that results in diabetes prevention depending on the genetic background and the environment.
Increasing evidence suggests that TLRs also express on tissue cells including pancreatic beta cells. The role of TLRs on islet beta cells is largely unknown. Due to the complexity of the T1D pathogenesis, there is still no cure or ultimate prevention from the disease development. TLRs are critical modulators of islet-directed immune responses and are, therefore, important targets for anti-diabetogenic therapies. However, many functions of TLRs and causal relations are still unknown. Many questions must be answered before we can generate novel and effective therapeutic approaches that target TLRs in treating T1D. Since therapeutic targeting of TLRs can also increase the susceptibility toward infections, safety and efficacy have to be thoroughly balanced when modulating TLRs.
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft fellowship GU 1122/3-1 (to Elke Gülden) and RC1 DK087699 and DK088181 (to Li Wen). We owe a debt of gratitude to Susan Wong for critically reading the manuscript.
References
1. Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol (2001) 2:675–80. doi: 10.1038/90609
2. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature (1997) 388:394–7. doi:10.1038/41131
3. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol (2010) 11:373–84. doi:10.1038/ni.1863
4. Fischer M, Ehlers M. Toll-like receptors in autoimmunity. Ann N Y Acad Sci (2008) 1143:21–34. doi:10.1196/annals.1443.012
5. Montero Vega MT, de Andres Martin A. The significance of Toll-like receptors in human diseases. Allergol Immunopathol (Madr) (2009) 37:252–63. doi:10.1016/j.aller.2009.04.004
6. Jahromi MM, Eisenbarth GS. Cellular and molecular pathogenesis of type 1A diabetes. Cell Mol Life Sci (2007) 64:865–72. doi:10.1007/s00018-007-6469-4
7. Filippi CM, Ehrhardt K, Estes EA, Larsson P, Oldham JE, Von Herrath MG. TLR2 signaling improves immunoregulation to prevent type 1 diabetes. Eur J Immunol (2011) 41:1399–409. doi:10.1002/eji.200939841
8. Kim HS, Han MS, Chung KW, Kim S, Kim E, Kim MJ, et al. Toll-like receptor 2 senses beta-cell death and contributes to the initiation of autoimmune diabetes. Immunity (2007) 27:321–33. doi:10.1016/j.immuni.2007.06.010
9. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature (2008) 455:1109–13. doi:10.1038/nature07336
10. Devaraj S, Tobias P, Kasinath BS, Ramsamooj R, Afify A, Jialal I. Knockout of Toll-like receptor-2 attenuates both the proinflammatory state of diabetes and incipient diabetic nephropathy. Arterioscler Thromb Vasc Biol (2011) 31:1796–804. doi:10.1161/ATVBAHA.111.228924
11. Karumuthil-Melethil S, Perez N, Li R, Vasu C. Induction of innate immune response through TLR2 and dectin 1 prevents type 1 diabetes. J Immunol (2008) 181:8323–34.
12. Al Shamsi M, Shahin A, Iwakura Y, Lukic ML, Mensah-Brown EP. Pam3CSK4 enhanced beta cell loss and diabetogenesis: the roles of IFN-gamma and IL-17. Clin Immunol (2013) 149:86–96. doi:10.1016/j.clim.2013.06.001
13. Kim DH, Lee JC, Kim S, Oh SH, Lee MK, Kim KW, et al. Inhibition of autoimmune diabetes by TLR2 tolerance. J Immunol (2011) 187:5211–20. doi:10.4049/jimmunol.1001388
14. Aumeunier A, Grela F, Ramadan A, Pham Van L, Bardel E, Gomez Alcala A, et al. Systemic Toll-like receptor stimulation suppresses experimental allergic asthma and autoimmune diabetes in NOD mice. PLoS One (2010) 5:e11484. doi:10.1371/journal.pone.0011484
15. Fallarino F, Volpi C, Zelante T, Vacca C, Calvitti M, Fioretti MC, et al. IDO mediates TLR9-driven protection from experimental autoimmune diabetes. J Immunol (2009) 183:6303–12. doi:10.4049/jimmunol.0901577
16. Wong FS, Hu C, Zhang L, Du W, Alexopoulou L, Flavell RA, et al. The role of Toll-like receptors 3 and 9 in the development of autoimmune diabetes in NOD mice. Ann N Y Acad Sci (2008) 1150:146–8. doi:10.1196/annals.1447.039
17. Gülden E, Ihira M, Ohashi A, Reinbeck AL, Freudenberg MA, Kolb H, et al. Toll-like receptor 4 deficiency accelerates the development of insulin-deficient diabetes in non-obese diabetic mice. PLoS One (2013) 8:e75385. doi:10.1371/journal.pone.0075385
18. Devaraj S, Tobias P, Jialal I. Knockout of Toll-like receptor-4 attenuates the pro-inflammatory state of diabetes. Cytokine (2011) 55:441–5. doi:10.1016/j.cyto.2011.03.023
19. Li M, Song L, Gao X, Chang W, Qin X. Toll-like receptor 4 on islet beta cells senses expression changes in high-mobility group box 1 and contributes to the initiation of type 1 diabetes. Exp Mol Med (2012) 44:260–7. doi:10.3858/emm.2012.44.4.021
20. Lee AS, Ghoreishi M, Cheng WK, Chang TY, Zhang YQ, Dutz JP. Toll-like receptor 7 stimulation promotes autoimmune diabetes in the NOD mouse. Diabetologia (2011) 54:1407–16. doi:10.1007/s00125-011-2083-y
21. Zhang Y, Lee AS, Shameli A, Geng X, Finegood D, Santamaria P, et al. TLR9 blockade inhibits activation of diabetogenic CD8+ T cells and delays autoimmune diabetes. J Immunol (2010) 184:5645–53. doi:10.4049/jimmunol.0901814
22. Tai N, Wong FS, Wen L. TLR9 deficiency promotes CD73 expression in T cells and diabetes protection in nonobese diabetic mice. J Immunol (2013) 191:2926–37. doi:10.4049/jimmunol.1300547
23. Zipris D, Lien E, Nair A, Xie JX, Greiner DL, Mordes JP, et al. TLR9-signaling pathways are involved in Kilham rat virus-induced autoimmune diabetes in the biobreeding diabetes-resistant rat. J Immunol (2007) 178:693–701.
24. Trudeau JD, Dutz JP, Arany E, Hill DJ, Fieldus WE, Finegood DT. Neonatal beta-cell apoptosis: a trigger for autoimmune diabetes? Diabetes (2000) 49:1–7. doi:10.2337/diabetes.49.1.1
25. O’Brien BA, Huang Y, Geng X, Dutz JP, Finegood DT. Phagocytosis of apoptotic cells by macrophages from NOD mice is reduced. Diabetes (2002) 51:2481–8. doi:10.2337/diabetes.51.8.2481
26. Ding Y, Subramanian S, Montes VN, Goodspeed L, Wang S, Han C, et al. Toll-like receptor 4 deficiency decreases atherosclerosis but does not protect against inflammation in obese low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol (2012) 32:1596–604. doi:10.1161/ATVBAHA.112.249847
27. Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol (2006) 6:823–35. doi:10.1038/nri1957
28. Chen MC, Schuit F, Eizirik DL. Identification of IL-1 beta-induced messenger RNAs in rat pancreatic beta cells by differential display of messenger RNA. Diabetologia (1999) 42:1199–203. doi:10.1007/s001250051292
29. Frigerio S, Junt T, Lu B, Gerard C, Zumsteg U, Hollander GA, et al. Beta cells are responsible for CXCR3-mediated T-cell infiltration in insulitis. Nat Med (2002) 8:1414–20. doi:10.1038/nm1202-792
30. Schulthess FT, Paroni F, Sauter NS, Shu L, Ribaux P, Haataja L, et al. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metab (2009) 9:125–39. doi:10.1016/j.cmet.2009.01.003
31. Aliprantis AO, Yang RB, Weiss DS, Godowski P, Zychlinsky A. The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J (2000) 19:3325–36. doi:10.1093/emboj/19.13.3325
32. Moriyama H, Wen L, Abiru N, Liu E, Yu L, Miao D, et al. Induction and acceleration of insulitis/diabetes in mice with a viral mimic (polyinosinic-polycytidylic acid) and an insulin self-peptide. Proc Natl Acad Sci U S A (2002) 99:5539–44. doi:10.1073/pnas.082120099
33. Lang KS, Recher M, Junt T, Navarini AA, Harris NL, Freigang S, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med (2005) 11:138–45. doi:10.1038/nm1105-1256
34. Serreze DV, Hamaguchi K, Leiter EH. Immunostimulation circumvents diabetes in NOD/Lt mice. J Autoimmun (1989) 2:759–76. doi:10.1016/0896-8411(89)90003-6
35. Iguchi M, Inagawa H, Nishizawa T, Okutomi T, Morikawa A, Soma GI, et al. Homeostasis as regulated by activated macrophage. V. Suppression of diabetes mellitus in non-obese diabetic mice by LPSw (a lipopolysaccharide from wheat flour). Chem Pharm Bull (Tokyo) (1992) 40:1004–6. doi:10.1248/cpb.40.1004
36. Quintana FJ, Rotem A, Carmi P, Cohen IR. Vaccination with empty plasmid DNA or CpG oligonucleotide inhibits diabetes in nonobese diabetic mice: modulation of spontaneous 60-kDa heat shock protein autoimmunity. J Immunol (2000) 165:6148–55.
37. Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol (2005) 6:345–52. doi:10.1038/ni1178
38. Dasgupta G, Chentoufi AA, You S, Falatoonzadeh P, Urbano LA, Akhtarmalik A, et al. Engagement of TLR2 reverses the suppressor function of conjunctiva CD4+CD25+ regulatory T cells and promotes herpes simplex virus epitope-specific CD4+CD25- effector T cell responses. Invest Ophthalmol Vis Sci (2011) 52:3321–33. doi:10.1167/iovs.10-6522
39. Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci U S A (2006) 103:7048–53. doi:10.1073/pnas.0601554103
40. Sutmuller R, Garritsen A, Adema GJ. Regulatory T cells and Toll-like receptors: regulating the regulators. Ann Rheum Dis (2007) 66(Suppl 3):91–5. doi:10.1136/ard.2007.078535
41. Chen Q, Davidson TS, Huter EN, Shevach EM. Engagement of TLR2 does not reverse the suppressor function of mouse regulatory T cells, but promotes their survival. J Immunol (2009) 183:4458–66. doi:10.4049/jimmunol.0901465
42. Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. J Clin Invest (2006) 116:2022–32. doi:10.1172/JCI28423
43. Zanin-Zhorov A, Cohen IR. Signaling via TLR2 and TLR4 directly down-regulates T cell effector functions: the regulatory face of danger signals. Front Immunol (2013) 4:211. doi:10.3389/fimmu.2013.00211
44. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A (2010) 107:12204–9. doi:10.1073/pnas.0909122107
45. Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, et al. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science (2011) 332:974–7. doi:10.1126/science.1206095
46. van Maren WW, Nierkens S, Toonen LW, Bolscher JM, Sutmuller RP, Adema GJ. Multifaceted effects of synthetic TLR2 ligand and Legionella pneumophila on Treg-mediated suppression of T cell activation. BMC Immunol (2011) 12:23. doi:10.1186/1471-2172-12-23
47. Amiset L, Fend L, Gatard-Scheikl T, Rittner K, Duong V, Rooke R, et al. TLR2 ligation protects effector T cells from regulatory T-cell mediated suppression and repolarizes T helper responses following MVA-based cancer immunotherapy. Oncoimmunology (2012) 1:1271–80. doi:10.4161/onci.21479
48. Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express Toll-like receptors and are activated by lipopolysaccharide. J Exp Med (2003) 197:403–11. doi:10.1084/jem.20021633
49. Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol (2005) 175:8051–9.
50. Cavaillon JM, Adrie C, Fitting C, Adib-Conquy M. Endotoxin tolerance: is there a clinical relevance? J Endotoxin Res (2003) 9:101–7. doi:10.1177/09680519030090020501
51. Knapp S. Update on the role of Toll-like receptors during bacterial infections and sepsis. Wien Med Wochenschr (2010) 160:107–11. doi:10.1007/s10354-010-0765-6
52. Li CH, Wang JH, Redmond HP. Bacterial lipoprotein-induced self-tolerance and cross-tolerance to LPS are associated with reduced IRAK-1 expression and MyD88-IRAK complex formation. J Leukoc Biol (2006) 79:867–75. doi:10.1189/jlb.0905505
53. Alyanakian MA, Grela F, Aumeunier A, Chiavaroli C, Gouarin C, Bardel E, et al. Transforming growth factor-beta and natural killer T-cells are involved in the protective effect of a bacterial extract on type 1 diabetes. Diabetes (2006) 55:179–85. doi:10.2337/diabetes.55.01.06.db05-0189
Keywords: Toll-like receptors, type 1 diabetes, immunity, tolerance, regulatory T-cells
Citation: Gülden E and Wen L (2014) Toll-like receptor activation in immunity vs. tolerance in autoimmune diabetes. Front. Immunol. 5:119. doi: 10.3389/fimmu.2014.00119
Received: 17 December 2013; Accepted: 07 March 2014;
Published online: 24 March 2014.
Edited by:
Christophe M. Filippi, Genomics Institute of the Novartis Research Foundation, USACopyright: © 2014 Gülden and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:bGkud2VuQHlhbGUuZWR1