 Anne-Sophie Chretien1
Anne-Sophie Chretien1 Aude Le Roy2Norbert Vey1,3Thomas Prebet3
Aude Le Roy2Norbert Vey1,3Thomas Prebet3 Didier Blaise1,4
Didier Blaise1,4 Cyril Fauriat1
Cyril Fauriat1 Daniel Olive1,2*
Daniel Olive1,2*- 1Centre de Cancérologie de Marseille, INSERM, U1068, Institut Paoli-Calmettes, Aix-Marseille Université, UM 105, CNRS, UMR7258, Marseille, France
- 2Centre de Cancérologie de Marseille, Plateforme d’Immunomonitoring en Cancérologie, INSERM, U1068, Institut Paoli-Calmettes, Aix-Marseille Université, UM 105, CNRS, UMR7258, Marseille, France
- 3Département d’Hématologie, Institut Paoli-Calmettes, Marseille, France
- 4Unité de Transplantation et de Thérapie Cellulaire, Institut Paoli-Calmettes, Marseille, France
Despite evidence of cancer immune-surveillance, which plays a key role in tumor rejection, cancer cells can escape immune recognition through different mechanisms. Thus, evasion to Natural killer (NK) cell-mediated anti-tumor activity is commonly described and is mediated by various mechanisms, mainly cancer cell-induced down-regulation of NK-activating receptors (NCRs, NKG2D, DNAM-1, and CD16) as well as up-regulation of inhibitory receptors (killer-cell immunoglobulin-like receptors, KIRs, NKG2A). Alterations of NK cells lead to an impaired recognition of tumor cells as well as a decreased ability to interact with immune cells. Alternatively, cancer cells downregulate expression of ligands for NK cell-activating receptors and up-regulate expression of the ligands for inhibitory receptors. A better knowledge of the extent and the mechanisms of these defects will allow developing pharmacological strategies to restore NK cell ability to recognize and lyse tumor cells. Combining conventional chemotherapy and immune modulation is a promising approach likely to improve clinical outcome in diverse neoplastic malignancies. Here, we overview experimental approaches as well as strategies already available in the clinics that restore NK cell functionality. Yet successful cancer therapies based on the manipulation of NK cell already have shown efficacy in the context of hematologic malignancies. Additionally, the ability of cytotoxic agents to increase susceptibility of tumors to NK cell lysis has been studied and may require improvement to maximize this effect. More recently, new strategies were developed to specifically restore NK cell phenotype or to stimulate NK cell functions. Overall, pharmacological immune modulation trends to be integrated in therapeutic strategies and should improve anti-tumor effects of conventional cancer therapy.
Introduction
Natural killer (NK) cells are key components of the innate immunity and substantially contribute to anti-tumor immune responses (1–3). The role of NK cells in immune surveillance is linked to many aspects of the NK cell biology. First, NK cells directly recognize and lyse cancer cells. Besides this direct effect, NK cells are also able to initiate anti-tumor immune responses via the secretion of various cytokines such as IFN-γ and TNF-α (1, 4).
Triggering of effector functions of NK cells is the result of a balance between activating and inhibitory signals provided by a large set of activating or inhibitory receptors. The most commonly described activating receptors involved in anti-tumor immunity are NKG2D, DNAM-1, and the natural cytotoxic receptors (NCR), NKp30, NKp44, and NKp46. Hence, NCR are NK-activating receptors of primary importance in immune surveillance and response in the context of cancer (5–7). NKp30, NKp46 are expressed by all NK cells, whereas NKp44 is only expressed by activated NK cells (8–11). The acquisition of NCR during NK cell maturation correlates with the acquisition of cytolytic activity against tumor target cells (12). NKG2D is an activating receptor also expressed by, but not restricted to, all NK cells. Ligands for NKG2D include proteins related to non-classical HLA-I such as MICA, MICB, or the structurally related ULBP1–6 (13, 14). Inhibitory receptors belong to the killer-cell immunoglobulin-like receptors (KIRs) or to the C-type lectin CD94/NKG2A heterodimer (15). These receptors recognize HLA-I and the non-classical HLA-E and inhibit NK cell activation.
The fundamental role of NK cells in oncology has been widely demonstrated in both hematologic and solid neoplasms. The relevance of this concept is illustrated by many examples in clinical practice, such as the success of hematopoietic stem cell transplantation in hematologic malignancies (16–19), poor NK cell functions associated with increased incidence of cancer (20), the importance of NK cells for the response to chemotherapy and radiotherapy (21, 22), or the use of parameters related to NK cell functions as prognostic biomarkers (23–25). Thus, NK cells can be used as prognostic biomarkers, as well as therapeutic targets or therapeutic agents.
However, although NK cells can kill target cells spontaneously without prior stimulation, a delicate balance between inhibitory and activating signals tightly regulates their activation (1, 26). In the context of cancer, this balance is often deregulated through various mechanisms (27). First of all, cancer cells are able to induce a down-regulation of activating receptors (notably NCR and NKG2D,) as well as an up-regulation of the NK cell inhibitory receptors (23, 24, 28, 29). Then, tumor cells usually poorly express ligands for activating receptors, and/or overexpress ligands for inhibitory receptors (30–32). Finally, the release of various factors such as cytokines or reactive oxygen species (ROS) within the tumor microenvironment impairs the crosstalk between NK cells and dendritic cells (DCs), enhancing the phenomenon of tumor escape (33–35).
Many efforts have been developed in the past few years to restore NK cell functionality in cancer patients. In this review, we focus on NK cells as a cornerstone to restore or improve anti-tumor immunity. We overview different pharmacological strategies aiming at counteracting the effect of tumor cells on NK cell functionality (Figure 1). Taking into account the crucial importance of NK cells for maintenance of a prolonged response to treatment, therapeutic strategies improving or restoring NK cell functions in combination with standard treatment regimens are expected to broadly impact patients’ clinical outcome.
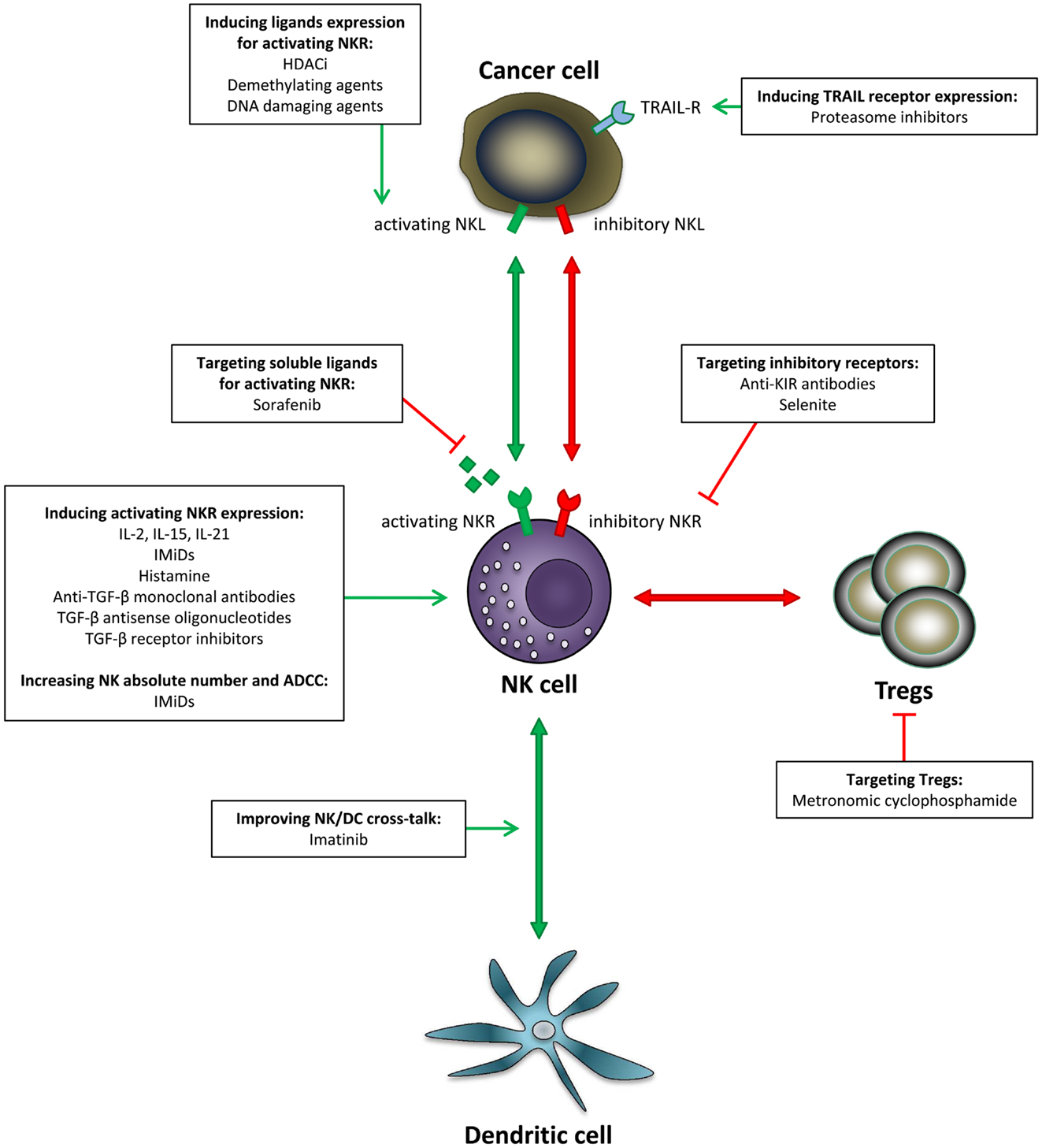
Figure 1. Pharmacological strategies aiming at improving NK anti-tumor functions. Various options have been developed to restore NK cell functionality in cancer: induction of NK triggering receptors, induction of NK ligands expression on the target cells, blockade of inhibitory signals, as well as stimulation of NK/DC crosstalk. In addition, increasing NK number and improving ADCC can enhance this effect. NKL, natural killer ligand; NKR, natural killer receptor.
Inducing Natural Cytotoxic Receptors Expression
Natural cytotoxic receptors expression is classically downregulated during cancer progression, regardless of the type of cancer (23, 24, 28, 29). The mechanisms involved in NCR down-regulation still need to be further defined. Restoring NCR expression may render NK cells more efficient against tumor cells. So far, clinical strategies aiming at restoring NCR expression remain to be proposed. However, taking into account the strong prognostic value of NCR expression, therapeutic strategies aiming at inducing their expression is expected to improve clinical outcome. Therefore, targeting events interfering with the expression of these receptors is certainly a relevant therapeutic option (23, 25). Among possible mechanisms, Transforming Growth Factor beta 1 (TGF-β1) downregulates NKp30 and NKG2D expression on NK cells, leading to a decreased ability of NK cells to kill target cells (23, 36–38). The release of TGF-β1 is done either by the tumor cell or by regulatory T cells (Tregs). Other tumor-released soluble factors are involved in NCR down-regulation, such as Activin-A, indoleamine dioxygenase (IDO), or prostaglandin E2 (PGE2) (34, 39, 40). Similarly to other activating receptors defect, the down modulation of NCR is somehow dependent on the pressure exerted by tumor cells, which reflects a pathway for tumor evasion. Hence, in acute myeloid leukemia (AML) patients, the low NCR expression acquired during leukemia development is restored in patients achieving complete remission (23). Some recently published data suggest that NCR down-regulation is consecutive to NK activation in the tumor, leading to an exhaustion of the NK cells and a subsequent down-regulation of the NCRs (41).
Cytokines
Amongst the efficient ways to improve NCR expression on NK cells, the use of cytokines, mainly IL-2, IL-15, and IL-21, may be promising. NK cell differentiation is cytokine-dependent (29). High baseline levels of circulating IL-2 constitute an independent prognostic factor for head and neck cancer patients (42).
IL-2
IL-2 is FDA-approved for cancer indications, which is not the case for IL-15 and IL-21. Most clinical trials using cytokines alone or in combination with chemotherapy or radiotherapy are set with IL-2. Conclusions of clinical trials report modest anti-tumor activity when used in monotherapy. Among its diverse immunostimulatory potentials, IL-2 is able to induce expression of NKG2D and NKp46 on NK cells (43, 44). However, following IL-2 stimulation, the NK cytolytic functions do not seem to reach normal cytolytic activity when compared to healthy volunteers (44). Moreover, IL-2 fails to induce NK cell proliferation compared to healthy volunteers, and increases the rate of apoptotic NK cells (44). Some authors evidenced the critical role of IL-2 for the development and peripheral expansion of regulatory T cells (45), which is not the case for IL-15 and IL-21. Noteworthy, the use of IL-2, especially at high doses, might be limited to ex vivo expansion of NK cells for problems of in vivo toxicity (46).
IL-15
IL-15 plays a major role in the proliferation, differentiation, survival, and functions of T and NK cells (29, 47). Exposure of NK cells to low doses of IL-15 significantly improved NKp30, NKp46, NKG2D, and NKG2C surface expression. Accordingly, this increase of receptor expression was correlated with an increase of natural cytotoxicity against autologous AML blasts (29, 48). In addition, in hematologic malignancies, low levels of circulating IL-15 after bone marrow transplantation were predictive of risk of relapse (49). In line, NK cell recovery in stem cell transplantation is strongly correlated with plasmatic concentrations of IL-15 (48).
IL-15 serum concentration increases dramatically following administration of cytotoxic agents (29, 49). For some authors, this elevation of serum IL-15 could be related to the depletion of lymphoid populations that normally consume circulating IL-15 or to inflammation induced by chemotherapy (48). In vivo, injections of the IL-15/IL-15Rα heterodimer result in significant expansion of γδ, CD8+ T, and NK cells (47). Recently, this cytokine has become available for use in early phase clinical trials as an alternative to IL-2 (29, 47). IL-15 is currently assessed as a therapy for various solid tumors including refractory metastatic melanoma, metastatic renal cell cancer. IL-15 is also assessed as an adjuvant of chemotherapy and vaccines strategies or prior to stem cell therapy and NK cells infusion.
IL-21
IL-21 shares significant structural homology with IL-2 and IL-15 (50). In phase I trials, this cytokine shows a favorable safety profile and signs of clinical activity (51). Although some reports demonstrated a deleterious effect of IL-21 by reducing activating receptor expression (NKG2D, NKp44), its main effect is to enhance NK cell functions. Hence, IL-21 is capable of inducing NK cell maturation and NKp46 and NKp30 expression (12, 52, 53). Ex vivo, IL-21 stimulates the production of IFN-γ and cytotoxic properties of NK cells (53). Several clinical trials reported the effect of IL-21 therapy on immune system after administration in patients with metastatic melanoma and renal cell carcinoma (51). Although NK and T-cell numbers were temporarily decreased during administration of IL-21, the cells had higher expression of CXCR3, HMMR, IFN-γ, perforin, and granzymes at the mRNA level. Evidence of NK cell activation was further confirmed by enhanced ability of NK cells from patients to lyse K562 target cells (51). These results were confirmed in a phase II trial for metastatic melanoma (54).
Immunomodulatory Drugs
Immunomodulatory drugs (IMiDs) present another therapeutic option to increase activating receptors expression. Two molecules are currently developed in oncology: lenalidomide, FDA-approved in hematologic malignancies, and pomalidomide. These drugs present anti-angiogenic and anti-proliferative activity, and their effect on the immune system, particularly on NK cells, is probably part of their mechanism of action. For instance, immunomonitoring of patients treated with immunomodulatory drugs, IMiDs have been associated with an increased expression of NKp44 and NKp46, in multiple myeloma (MM), myelodysplastic syndrome but also in solid tumors (55, 56). Interestingly, this effect of lenalidomide may not be a direct effect on NK cells because this effect was not observed in vitro on purified NK cells (57). In this study, IMiDs-treated NK cells displayed a lower NKp46 expression, although this had no functional consequences on cytolytic functions of NK cells.
Histamine
Blocking phenomenon responsible for NCR down-regulation is another potential strategy to induce indirect NCR expression. Thus, ROS, PGE2, and IDO, which are present in the tumor microenvironment, appear to be relevant targets (33–35). Romero et al. demonstrated that histamine was able to prevent NKp46 and NKG2D down-regulation mediated by mononuclear and polymorphonuclear phagocytes ROS production (35). Moreover, histamine maintains the cytolytic activity of NK cells toward leukemic cells despite the presence of phagocytes. A phase III clinical trial assessed the efficacy of post-consolidation immunotherapy with IL-2 and histamine dihydrochloride for patients with AML in complete remission. This treatment was shown to significantly improve leukemia-free survival, with mild to moderate side effects (33).
Inducing NKG2D Expression
NKG2D down-regulation on circulating NK cells in cancer patients compared to healthy volunteers was described in various cancer types, including breast cancer, glioma, melanoma, and lung cancer (58–62).
Cytokines
Few pharmacological agents are able to directly increase the expression of NK-activating receptors. Until now, the only described possibility to directly induce NKG2D expression on NK cells is the use of immunostimulatory cytokines. Ex vivo, IL-15 was shown to be able to induce a dramatic increase of NKG2D expression (63, 64). Although the use of IL-15 is still restricted to phase I and II clinical trials, conventional chemotherapies are able to induce a huge increase of the circulating IL-15 (29).
TGF-β Pathway
A second strategy allowing NKG2D restoration in the cancer context is indirect up-regulation by blocking the agents responsible for NKG2D down-regulation. For instance, stroma-derived factors in the tumor microenvironment, in particular TGF-β, display an immunosuppressive activity on most anti-tumor immune effectors, and an indirect immunosuppressive effect via the inhibition of MICA transcription (38, 65). Besides immune suppression, stroma-derived factors also present direct effects on the tumor cell since TGF-β promotes tumorigenesis and epithelial–mesenchymal transition (66). In vitro, TGF-β inhibits the expression of NKp30 and NKG2D (37) and blood concentration of TGF-β1 was shown to inversely correlate with NKG2D expression at the surface of NK cells of cancer patients and has been linked with impaired NK cytotoxicity (58, 60). TGF-β antagonizes the IL-15-induced proliferation and gene expression associated with NK cell activation, inhibiting the expression of NK cell activation receptor molecules (67). Moreover, ex vivo addition of neutralizing anti-TGF-β monoclonal antibodies completely restores surface NKG2D expression at the surface of NK cells and partially restores NKp30 expression (60, 67). In addition, blocking TGF-β completely restores IFN-γ production by tumor-associated NK cells (67).
Some approaches aiming at decreasing circulating TGF-β in patients are currently under investigation (68). These early stage clinical trials currently assess several approaches, mainly the use of anti-TGF-β monoclonal antibodies and antisense oligonucleotides. For example, fresolimumab (GC-1008), a fully humanized pan-neutralizing antibody directed against all the three isoforms of TGF-β, has been assessed in renal cell carcinoma and in metastatic melanoma (68, 69). In this phase I/II trial, fresolimumab was safe and well-tolerated with no dose-limiting toxicities and displayed encouraging results.
The impact of TGF-β blockade on immune parameters was recently assessed in patients with malignant pleural mesothelioma treated with fresolimumab (70). Fresolimumab had no effect in the expression of NK, CD4+, or CD8+ T-cell-activating and inhibitory markers, other than a decrease in the expression of 2B4 and DNAM-1 on NK cells, although TGF-β serum concentrations were markedly decreased. The authors conclude that acute changes in serum TGF-β concentration are not associated with the set of biomarker changes that were predicted based on animal models. No effect was detected on the expression of NKG2D nor NKp30, and the effect on DNAM-1 expression, although significant, was minor (70).
Another possibility to decrease TGF-β in the tumor milieu is the use of antisense oligonucleotides. Some of these compounds are currently in clinical evaluation. Belagenpumatucel-L, a therapeutic vaccine comprised of four TGF-β2 antisense gene-modified allogeneic NSCLC cell lines was assessed in grade III/IV NSCLC patients. In a phase II study, positive clinical outcomes were correlated with immune response to the vaccine and induction of immune enhancement of tumor antigen, but the effect on NK cells was not assessed (65). This compound is still currently investigated in non-small cell lung carcinoma in phases II and III trials.
Alternatively, SD-208, a TGF-β receptor I kinase inhibitors, restores the lytic activity of polyclonal NK cells against glioma cells in the presence of recombinant TGF-β or of TGF-β-containing glioma cell supernatant (71). This molecule is able to restore NKG2D expression on NK cells, whose expression was altered in vitro by cancer cell lines supernatants or direct inhibition with recombinant TGF-β (72).
To conclude, NKG2D expression has never been shown to present a prognostic value unlike NKG2D ligands expression, thus suggesting that the best strategy to target the NKG2D/NKG2D ligand system might be to induce ligands expression rather than the receptor itself.
Inducing Ligands Expression for NK-Activating Receptors
The main ligands for NKG2D are the MHC class I chain-related molecules MICA and MICB and the ULBP1–4. These ligands have been extensively studied in various malignancies. Ligands of DNAM-1 are CD112 (Nectin-2) and CD155 (Poliovirus receptor, PVR). Ligands of NCRs have been elusive for many years and although pathogen-related ligands have been suggested (hemagglutinins, heparate sulfates), only ligands for NKp30 have been identified. B7-H6, an Ig molecule from the family of B7 molecules has been identified as NKp30 ligand (73). B7-H6 is expressed by several cell lines and by primary tumors (74). Mechanisms of induction of B7-H6 expression have been described in non-transformed cells with TLR agonists as well as the pro-inflammatory cytokines TNFα and IL-1β (75). In primary tumors, recent experimental data suggest that B7-H6 expression is regulated by HDACs, in particular HDAC3 (74). In addition, BAG6/BAT3, a nuclear protein localized at the plasma membrane or on exosomes of tumor cells, has also been assigned as an NKp30 ligand (76). The importance of ligands expression for tumor cell recognition by NK cells is a key factor for anti-tumor immune response, as illustrated by the strong prognostic value of MICA/MICB, RAET1G, and ULBP2 expression in colorectal cancer and breast cancer (30–32). Tumor cells poorly express ligands for NK-activating receptors, and tumor ligands expression is inversely correlated with clinical stage (77).
Histone Deacetylase Inhibitors
Histone deacetylase inhibitors were successfully introduced as anti-cancer agents for their ability to block gene transcription and promote cell differentiation. These molecules induce cell cycle arrest and induce apoptosis of tumor cells, with minimal effects on normal tissue (78). Unexpectedly, their effect on anti-tumor immunity is part of their mechanism of action.
The main impact of these molecules on immunity is mediated through up-regulation of tumor antigens, in particular NKG2D ligands (79). HDACi-mediated immune modulation is also linked to the ability of these molecules to enhance immune recognition and lysis of the tumor cells by T cells and NK cells (79). To date, two molecules, romidepsin and vorinostat, have received approval from the FDA for the treatment of cutaneous T-cell lymphoma. In vitro, romidepsin, vorinostat, and sodium valproate were shown to increase MICA/B and ULBPs expression on various cancer cell lines and primary tumor cells, and render the target cells more sensitive to NK cell lysis (80–84). Depending on the authors, this mechanism was found to be GSK3- or ERK-dependent (81, 83).
Induction of MICA and MICB expression was associated with a shedding of the soluble forms of these NKG2D ligands, sMICA and sMICB (82). This raises the question of the potential counterbalancing of the clinical benefits in this particular case, since increase of the serum concentrations of sMICA and sMICB are responsible for NKG2D endocytosis and degradation, and represents a mode of T-cell silencing and immune escape (62, 82). Thus, Poggi et al. monitored NKG2D ligands shedding following treatment of AML patients treated with valproic acid. In this study, MICA, ULBP2, and ULBP3 expression on blasts was significantly increased after treatment with valproic acid. No ligand shedding was detected despite a strong up-regulation of the ligands on leukemic cells. Consequently, leukemic cells from patients treated with valproic acid, become able to trigger lytic granule exocytosis by autologous CD8+ T and NK cells (85).
However, some studies evidenced that HDACi down-regulate ligands for other NK cells-activating receptors, such as B7-H6, a ligand for NKp30, and impair tumor cell recognition by NK cells. These results were obtained with first and second generation HDACi (vorinostat, trichostatin A, valproic acid, and apicidin) on various cancer cell lines (74). Moreover, treatment of human NK cells with trichostatin A, valproic acid, or sodium butyrate affects the functional response of human NK cells, evidenced by a strong inhibition of IFN-γ secretion and a decreased ability to lyse target cells (86). Furthermore, the authors evidenced a down-regulation of activating receptors NKG2D and NCRs on resting and cytokine-stimulated NK cells.
Another study assessed the effect of vorinostat and valproic acid on NK cells. At therapeutic concentration, these drugs induced the down-regulation of NKp30 and NKp46, and inhibited IL-2 activation of NK cells, thus suppressing their cytolytic activity toward leukemic cell lines. This effect seems to be mediated by the inhibition of NFκB. In addition, the authors showed that vorinostat was toxic to NK cells in the range of therapeutic concentrations (87).
Demethylating Agents
The hypomethylating drugs decitabine and azacytidine are epigenetic drugs that are currently used in treatment of hematological malignancies (88). Besides their direct effect on the tumor cell, these drugs probably act through their impact on innate immunity. In vitro, both drugs induce ULBP1 and MICB on cell lines and primary tumor cells when incubated with either decitabine or 5-azacytidine (89, 90). This effect was related to promoter DNA methylation and DNA damage and correlates with enhanced NK cytotoxicity (90, 91).
However, DNA methylation is an important regulator of KIR expression by NK cells, potentially impacting on NK cell functions (92, 93). Hence, 5-azacytidine induces an increase in the percentage of KIR+ NK cells upon treatment with clinically relevant concentrations of 5-azacytidine, which correlated with an impaired granzyme B and perforin release, IFN-γ production, and decreased cytotoxicity (91, 94). However, this effect seems to be restricted to 5-azacytidine, since decitabine increases NK cell cytotoxicity and enhances IFN-γ production, in a dose-dependent manner (91). These results were confirmed in recent studies in different settings. Recently, Cerdeira et al. tested the effect of 5-azacytidine in hypoxic conditions with addition of TGF-β. Although the authors confirmed the impact of this drug on KIR expression, however, the cytotoxicity of NK cells cultured in these specific conditions was not affected (92).
For some authors, the results obtained in vitro in such settings are debatable. Indeed, since 5-azacytidine and decitabine are nucleoside analogs, these molecules require DNA replication to be incorporated into the DNA strand. In vitro studies using resting NK cells are therefore more likely to reflect the direct mRNA effect of such drugs than the effect of hypomethylation (88). Thus, Kopp et al. studied the effect of decitabine on proliferating NK cells. The authors show that decitabine negatively affects NK cell viability and proliferation in a dose-dependent manner. Simultaneous increase in KIR and NKp44 expression and decrease in NKG2D expression was evidenced. However, the impact on NK functionality in terms of toxicity was biphasic, with decreased toxicity at low doses and increased toxicity at high doses. Since the target cells used in these experiments lack class I HLA, this effect is independent of KIR up-regulation. Whether this increased cytotoxicity is maintained in the presence of HLA-positive targets remains to be determinate (88).
To conclude, further investigation is required to determine whether epigenetic drugs adversely affect NK cell survival, proliferation, or functions when administrated to patients.
DNA-Damaging Agents
Some conventional chemotherapeutic agents can induce immunogenic cell death, e.g., tumor cell apoptosis and stress signals that lead to the surface expression of ligands for NKG2D and DNAM-1 (95, 96). This DNA damage pathway can be activated by several mechanisms, during the course of chemotherapy with DNA-damaging agents such as doxorubicin, mitoxantrone, cisplatin, and oxaliplatin (8, 95–99). This particular mode of cell death displays damage-associated molecular patterns, e.g., exposure of calreticulin endoplasmic reticulum proteins at the surface of the pre-apoptotic cell, as well as secretion of ATP (100).
The oncogenic stress induced by these DNA-damaging agents stimulates various aspects of anti-cancer immunity, including activation of NK cells via ULBP1, MICA/B, and PVR expression at the surface of the cancer cell in an ATM (ataxia telangiectasia, mutated), ATR (ATM- and Rad3-related) protein kinases, and/or P53-dependent manner (8, 96–99). Other agents are able to induce stress conditions, leading to the expression of ligands for NKG2D and DNAM-1, such as IMiDs and proteasome inhibitors (22). These results await clinical confirmation with immunomonitoring studies of patients undergoing DNA-damaging agent therapy.
Targeting Soluble Ligands for Activating Receptors
The expression of NKR ligands at the surface of cancer cells appears to be a good prognostic factor. However, the shedding of soluble ligands in the circulation strongly impairs NK cell functions and has been linked with tumorigenesis and tumor progression (101) and high serum concentration of ULBP2 presents a strong prognostic value in breast cancer, colorectal cancer, and melanoma (30–32). Noteworthy, the discovery of B7-H6 and BAG6, ligands for NKp30, included the detection of soluble forms, which may compete for cell–cell interaction with membrane-bound ligands, although only soluble/exosome-bound BAG6 has been detected in a cancer situation (75, 102).
The prototypical example of ligand shedding is the release of soluble MICA/MICB (sMICA/sMICB), typically by A disintegrin and metalloproteases (ADAMs) (103, 104). These proteases are overexpressed in malignant tissues compared to normal tissues (105, 106). As a consequence, serum concentrations of soluble ligands for NKG2D are elevated in various malignant conditions (103). The ligation of these soluble ligands induces internalization of NKG2D and its subsequent degradation, leading to an overall down-regulation of the receptor at the surface of NK cells. In various cancers, high levels of circulating ligands for NK-activating receptors correlated with a poor prognosis. Direct pharmacologic inhibition of these metalloproteases is still in preclinical evaluation.
Sorafenib
Sorafenib is a multi-target tyrosine kinase inhibitor targeting RAS/RAF/MAPK as well as VEGFR and PDGFR signaling pathways, implicated in cell proliferation and angiogenesis. Sorafenib is indicated in renal cell carcinoma, hepatocellular carcinoma, thyroid cancer and melanoma. In vitro, this molecule presents interesting off-target effects on ADAM9 expression as evidenced by a recent study on the human hepatocellular carcinoma cell line HepG2. In this study, sorafenib was able to strongly decrease ADAM9 expression at the proteic and transcriptional level, which correlated with a decrease of sMICA concentration in the culture supernatant and enhanced sensitivity to NK cell lysis. In addition, ADAM9 inhibition increases the expression of membrane-bound MICA on the tumor cell, enhancing the NK sensitivity of hepatocellular carcinoma cells (105).
Controversial data were published about effects of sorafenib on NK cells. NK cell function is inhibited by sorafenib as a consequence of impaired phosphorylation of PI3K and ERK, which directly control NK cell reactivity (107). Immunomonitoring of patients with renal cell carcinoma and melanoma treated with sorafenib failed to evidence modification of pERK1/2 expression in peripheral-blood NK cells after short-term or long-term administration (108). In addition, sorafenib may also positively (Th1) or negatively (DCs) impact other aspects of anti-tumor immunity (61, 109, 110). Whether this action is positive or negative remains to be determinated, as well as the overall “immune benefit” of such antagonistic effects on anti-tumor immunity, besides their direct pro-apoptotic effect on the tumor cell.
Targeting Inhibitory Receptors
Although activating NK receptors are crucial, triggering of NK cell effector functions is prevented by the expression of the inhibitory receptors KIR and NKG2A. Although in some examples of solid cancer, KIR and NKG2A expression is altered, generally expression is maintained and tumor cells may maintain sufficient amounts of HLA molecules to ensure inhibition of NK cells and evade killing. Moreover, some tumors display decreased expression of TCR-dependent HLA molecules while maintaining a normal expression of KIR-dependent HLA molecules (111). High HLA-E expression has been observed in several solid cancers (112, 113) and leukemias (114). Consequently, as 20–70% of NK cells express NKG2A, HLA-E expression by tumor cells impairs the anti-tumor activity of a predominant proportion of NK cells.
Anti-KIR Monoclonal Antibodies
Among the strategies to improve the recognition of tumor cells by NK cells, blocking the inhibitory interactions is appealing. The most advanced therapeutic compound as for today is the anti-KIR monoclonal antibody, IPH2101. This fully humanized antibody blocks the interaction of the major KIR expressed by NK cells with their cognate ligands, i.e., HLA-C. This reagent has been tested in early phase clinical trials and was shown to be well-tolerated in patients suffering from AML (115). In some instance, NK cells from treated patients expressed the activation marker CD69 and IFN-γ or MIP-1β was detected in the sera of patients. Another clinical trial in patients with MM has also shown that IPH2101 is safe and also enhances ex vivo NK cell cytotoxicity against MM cells (116). IPH2101 (and its replacement IPH2102) is therefore a novel immune-therapeutic agent that may improve anti-tumor activity of patients. More trials are programed and already necessary but yet this reagent has reached the promises for clinical use against cancer cells.
Selenite
As mentioned above, control of NK cell activation is either achieved by KIR/HLA interactions but also NKG2A/HLA-E interaction. In healthy individuals, at steady state, the two systems compensate for each other to ensure a total control of NK cell reactivity. Regarding NKG2A-mediated inhibition of NK cells by HLA-E expressing tumor cells, very few data are available. Interestingly, an FDA-approved reagent, selenium, may be a promising tool. Supplementation with selenium has been associated with reduced risk of solid cancer (117). The mechanism of action of selenium is not entirely known, but it induces apoptosis of tumor cells by generating an oxidative stress, which may be more effective on tumor cells compared to healthy cells (118, 119). Alternatively, selenium blocks the synthesis of HLA-E and consequently increases cytotoxicity mediated by NKG2A-positive NK cells (120). This effect, combined to the direct toxicity on tumor cells may result in reduced disease progression and improved survival. Sodium selenite is currently under investigation in several clinical trials for the treatment of different cancers.
Altogether, targeting inhibitory NK receptors reflects a novel orientation taken for innovative therapeutic approaches, as it represents another way to counteract the immune escape via ligands for inhibitory receptors. Of note, this strategy relies on the expression of activating ligands by leukemic cells. Hence, removing of inhibition will allow NK cells killing their targets provided that they express the ligands for activating NK receptors.
Alternative Pathways to Improve NK Activity
Increasing NK Cell Lysis Capacity with IMiDs
Immunomodulatory drugs are capable to enhance monoclonal antibodies anti-tumor activity. First in vitro, Wu et al. have shown an enhancement of NK cell-mediated tumor cell ADCC by lenalidomide for a variety of rituximab-treated NHL (non-Hodgkin lymphoma), cetuximab-coated CRC (colorectal cancer), and trastuzumab-coating breast cancer cell lines (121, 122). Another team highlighted the enhancement of ADCC by lenalidomide in vitro. They have shown an increase of Raji cell apoptosis mediated by PBMC combination with rituximab by lenalidomide (123). In the first case, the effect was observed on purified NK cell but Wu et al. have explained that this mechanism is dependent on the presence of antibody and either interleukin-2 or interleukin-12. In the second case, Zhu et al. have observed this effect on PBMC. Finally, the researches of Hayashi et al. have shown that IMiDs-enhanced NK cell ADCC by triggering IL-2 production from T cells (124). All these works suggest that in vitro IMiDs-positive effect on NK cell ADCC could be dependent on IL-2.
In animal models, lenalidomide or pomalidomide in combination with rituximab improves severe combined immunodeficient (SCID) lymphoma-bearing mouse survival compared to rituximab in monotherapy (125). Three years later, the same team explained this enhancement of anti-tumor activity by an expanding, activating, and trafficking of NK cells into the tumor bed, which facilitate a more efficient ADCC. The IMiDs effect on NK cells in this model is also associated with DC activation and production of chemokines and pro-inflammatory cytokines (126).
In the same way, IMiDs are also capable to enhance natural cytotoxicity of NK cell against cancer cells. First, Davies et al. highlighted the potency of thalidomide, lenalidomide, and pomalidomide to increase PBMC cytotoxicity toward MM tumor cells (cell lines and patient cells) in vitro. They presented this effect as an NK-dependent effect (127). Then, Zhu et al. have shown the similar effect with lenalidomide and pomalidomide on K562 and PC-3 cell lines (i.e., enhanced PBMC-mediated tumor cell apoptosis). They have also shown that NK cells are essential in inducing cancer cell apoptosis (123). In the same manner as ADCC, Hayashi et al. have explained this IMiDs enhancement of NK cell cytotoxicity via induction of IL-2 production in T cells (124).
In line with in vitro studies, IMiDs also increased NK cell natural cytotoxicity in patients suffering from MDS or solid tumors (56). At last, IMiDs have an important property toward NK cell numbers. Hence, the number and the localization of NK cells in cancer patients is often correlated with prognosis (24, 25, 128–130).
Davies et al. observed that thalidomide treatment for MM patients resulted in an increase of absolute NK cell numbers (127). This observation was confirmed with lenalidomide in some metastatic malignant melanoma patients and other advanced cancers (131), and in children with solid tumors or MDS (56). This effect was also highlighted in lenalidomide and pomalidomide treated mice (lymphoma-bearing SCID mice) at the tumor site. Reddy et al. have shown in their study an increase of tumor central infiltration by NK cells in mice treated by lenalidomide or pomalidomide compared to DMSO-treated mice. They could explain that by the IMiDs effects on DCs stimulation and modification of the cytokine microenvironment (126).
Inducing TRAIL Receptor Expression on Target Cells
Proteasome inhibitors are a class of anti-cancer drugs that are used in first line of treatment of MM, and that are currently evaluated in hematologic and solid malignancies. These molecules disrupt proteasome activity, resulting in cell growth arrest, apoptosis, angiogenesis inhibition, and decreased binding of tumor cells to stromal cells (132). In vitro, bortezomib was shown to sensitize tumor cell lines as well as primary tumor cells to perforin/granzyme-mediated NK-tumor cytotoxicity. This effect was found to be dependent on augmentation of tumor caspase-8 activity as well as on up-regulation of Fas and TNF related apoptosis-inducing ligand (TRAIL) receptor on tumor cells, thus inducing target apoptosis by NK cells through Fas/FasL and TRAIL/DR5 interactions (133–135). Other proteasome inhibitors such as the b-A15 share this property (136). In addition, proteasome inhibitors up-regulate ULBP1 and ULBP2 expression (137–139). This effect is accompanied by a down-regulation of HLA class I molecules (140).
In vivo, bortezomib sensitizes tumors to killing by NK cells. This anti-tumor effect is enhanced upon depletion of Tregs (134, 141). Based on these results, a non-randomized phase I study is currently ongoing in order to evaluate the safety and the anti-tumor effects of adoptively infused ex vivo expanded autologous NK cells against metastatic cancers or hematological malignancies sensitized to NK TRAIL cytotoxicity with bortezomib (134). However, bortezomib paradoxically renders tumor cells resistant to killing by tumor-specific T cells, thus potentially counterbalancing the benefits obtained through the sensitization to killing by NK cells (136, 142). In addition, in vitro assays evidenced that bortezomib presents pro-apoptotic effects on NK cells, and induces a down-regulation of NKp46 expression with subsequent decrease in NKp46-mediated activity (143). b-AP15, a new proteasome inhibitor, appears to overcome this deleterious effect on T cells: in vitro evaluation of this molecule was shown to sensitize tumor cell lines to both NK and T cell-mediated killing (136). However, at equipotent doses, this molecule seems to be more toxic to NK cells than bortezomib (144).
Improving NK/DC Crosstalk
The relevance of the NK/DC crosstalk has been demonstrated in various physiopathological settings and alterations of these interactions have been shown to contribute to tumor progression (145). Imatinib mesylate is a tyrosine kinase inhibitor that inhibits the tyrosine kinase encoded by the bcr-abl oncogene and tyrosine kinases encoded by the c-kit and the PDGFR oncogenes. Targeting these tyrosine kinases directly induces apoptosis of the cancer cell, which constitutes the main mechanism of action of imatinib. Besides this direct anti-proliferative effect, an “off-target” effect, inducing DC-mediated NK activation was described by Borg et al. (145). In this study, patients with GIST were assessed for NK cell functions during the course of treatment with imatinib. Anti-tumor response correlated with enhanced NK-mediated anti-tumor response, thus bringing out a new mechanism of action of this molecule. The authors then defined immunologic responder patients with increased RFS. In a more recent study conducted in GIST patients, the authors validated the concept, showing a correlation between clinical outcome and NK cell activation induced by therapy with imatinib (21). Immunomonitoring of NK cell functions included IFN-γ production and NKG2D expression. Although IFN-γ production was associated with clinical outcome, enhanced NKG2D-dependent lysis observed at 1 year of imatinib therapy did not impact survival (21). Interestingly, this DC-mediated NK activation seems to occur in lymph nodes where imatinib promotes the formation of immunologic synapses with resting or preactivated NK cells as a consequence of the blocking of KIT signaling in DCs (21, 97).
Depleting Tregs
Tregs inhibit antigen-specific immune response both in a cytokine-dependent and cell contact-dependent manner (146–148). Tregs alter both T cells and NK cells proliferation and activity through the down-regulation of NKG2D (147–149). Increased frequency of Treg cells and low T effector (Teff)–Treg ratios are associated with a poor clinical outcome and a lack of treatment response (147, 150–153). Impairment of Treg activity by either specific blockade or depletion can enhance immune response against tumor-associated antigens (147, 148). To date, drugs that specifically target Tregs are not available (153).
Although cyclophosphamide is immunosuppressive at high doses, this molecule displays particularly interesting immunostimulatory properties in metronomic scheduling (iterative administration of low doses) mainly by its ability to suppress FOXP3+ regulatory T cells (95, 149, 154) and to induce TH2/TH1 to TH17 shifts in cytokine production, induction of TH17, and resetting of dendritic cell homeostasis (153, 155). In murine models, metronomic cyclophosphamide strongly induces NKp46 expression as well as perforin and granzymes (156). Importantly, immunomonitoring studies evidenced that low-dose cyclophosphamide regimen restores patients’ T cells and NK cells functions as evidenced by killing assays (149, 157). Metronomic cyclophosphamide is currently tested in combination with anti-cancer vaccines, for its ability to suppress Tregs in order to facilitate vaccine-induced tumor rejection (153). Despite metronomic cyclophosphamide provides promising clinical results, some authors point the absence of randomization in these trials (158).
Concluding Remarks
Accumulating evidence based on immunomonitoring analyses highlights immune parameters as strong prognostic factors, both in hematopoietic and solid neoplasms. These conclusions provide a strong rationale for developing therapeutic strategies aiming at restoring key immune parameters. Among the major mechanisms used by tumor cells to escape immunity, the evasion from receptor–ligand-mediated anti-tumor activity by NK cells represents the most prevalent pathway. Hence, the recognition of tumor cells by NK cells via NCR or NKG2D-activating receptors is often impaired in various cancers and enhancing NK cell functions appears as one of the most promising approaches. One important question remains the ability of a cancer cell to overcome immune suppression upon exposure to immunostimulating drugs. Recent studies suggest that NK cells on tumor site exhibit a phenotype of exhaustion and terminal differentiation. Restoring NK functionality in this context could be of limited interest since these cells may hardly become highly anti-tumoral. This parameter should be considered to maximize the effects of such approaches.
To conclude, targeting immune evasion mechanisms, in association with conventional chemotherapy, may improve clinical outcome and is clinically feasible with limited side effects. To date, clinical application of this concept is mainly limited to drugs designed to target cancer cells, with off-target effects on the immune system. The problem of these strategies is that the overall benefit on the different immune effectors is sometimes hard to predict, and can be deleterious on crucial immune effectors, although restoring other cells. New strategies aiming at specifically restored immune functions will be potentially more efficient, and are currently in preclinical and clinical development. Further development of these immune therapies urges to associate clinical trials with translational immunology and immunomonitoring. A better knowledge regarding immune evasion mechanisms will definitely provide the absolutely required bases for the next-generation immune cancer therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science (2011) 331(6013):44–9. doi:10.1126/science.1198687
2. Moretta L, Bottino C, Pende D, Vitale M, Mingari MC, Moretta A. Human natural killer cells: molecular mechanisms controlling NK cell activation and tumor cell lysis. Immunol Lett (2005) 100(1):7–13. doi:10.1016/j.imlet.2005.07.004
3. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol (2012) 12(4):239–52. doi:10.1038/nri3174
4. Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, et al. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med (1999) 5(4):405–11. doi:10.1038/7403
5. Pende D, Parolini S, Pessino A, Sivori S, Augugliaro R, Morelli L, et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J Exp Med (1999) 190(10):1505–16. doi:10.1084/jem.190.10.1505
6. Kaifu T, Escaliere B, Gastinel LN, Vivier E, Baratin M. B7-H6/NKp30 interaction: a mechanism of alerting NK cells against tumors. Cell Mol Life Sci (2011) 68(21):3531–9. doi:10.1007/s00018-011-0802-7
7. Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol (2013) 34(4):182–91. doi:10.1016/j.it.2013.01.003
8. Raulet DH, Guerra N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nat Rev Immunol (2009) 9(8):568–80. doi:10.1038/nri2604
9. Sivori S, Vitale M, Morelli L, Sanseverino L, Augugliaro R, Bottino C, et al. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J Exp Med (1997) 186(7):1129–36. doi:10.1084/jem.186.7.1129
10. Vitale M, Bottino C, Sivori S, Sanseverino L, Castriconi R, Marcenaro E, et al. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J Exp Med (1998) 187(12):2065–72. doi:10.1084/jem.187.12.2065
11. Hudspeth K, Silva-Santos B, Mavilio D. Natural cytotoxicity receptors: broader expression patterns and functions in innate and adaptive immune cells. Front Immunol (2013) 4:69. doi:10.3389/fimmu.2013.00069
12. Sivori S, Cantoni C, Parolini S, Marcenaro E, Conte R, Moretta L, et al. IL-21 induces both rapid maturation of human CD34+ cell precursors towards NK cells and acquisition of surface killer Ig-like receptors. Eur J Immunol (2003) 33(12):3439–47. doi:10.1002/eji.200324533
13. Mistry AR, O’Callaghan CA. Regulation of ligands for the activating receptor NKG2D. Immunology (2007) 121(4):439–47. doi:10.1111/j.1365-2567.2007.02652.x
14. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol (2013) 31:413–41. doi:10.1146/annurev-immunol-032712-095951
15. Lanier LL. NK cell recognition. Annu Rev Immunol (2005) 23:225–74. doi:10.1146/annurev.immunol.23.021704.115526
16. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood (2010) 116(14):2411–9. doi:10.1182/blood-2010-05-283051
17. Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A, Aloisi T, et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood (2007) 110(1):433–40. doi:10.1182/blood-2006-07-038687
18. Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood (1990) 75(3):555–62.
19. Norell H, Moretta A, Silva-Santos B, Moretta L. At the bench: preclinical rationale for exploiting NK cells and gammadelta T lymphocytes for the treatment of high-risk leukemias. J Leukoc Biol (2013) 94(6):1123–39. doi:10.1189/jlb.0613312
20. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet (2000) 356(9244):1795–9. doi:10.1016/S0140-6736(00)03231-1
21. Menard C, Blay JY, Borg C, Michiels S, Ghiringhelli F, Robert C, et al. Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res (2009) 69(8):3563–9. doi:10.1158/0008-5472.CAN-08-3807
22. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity (2013) 39(1):74–88. doi:10.1016/j.immuni.2013.06.014
23. Fauriat C, Just-Landi S, Mallet F, Arnoulet C, Sainty D, Olive D, et al. Deficient expression of NCR in NK cells from acute myeloid leukemia: evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood (2007) 109(1):323–30. doi:10.1182/blood-2005-08-027979
24. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest (2011) 121(9):3609–22. doi:10.1172/JCI45816
25. Delahaye NF, Rusakiewicz S, Martins I, Menard C, Roux S, Lyonnet L, et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med (2011) 17(6):700–7. doi:10.1038/nm.2366
26. Long EO, Sik Kim H, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol (2013) 31:227–58. doi:10.1146/annurev-immunol-020711-075005
27. Baier C, Fino A, Sanchez C, Farnault L, Rihet P, Kahn-Perles B, et al. Natural killer cells modulation in hematological malignancies. Front Immunol (2013) 4:459. doi:10.3389/fimmu.2013.00459
28. Markel G, Seidman R, Besser MJ, Zabari N, Ortenberg R, Shapira R, et al. Natural killer lysis receptor (NKLR)/NKLR-ligand matching as a novel approach for enhancing anti-tumor activity of allogeneic NK cells. PLoS One (2009) 4(5):e5597. doi:10.1371/journal.pone.0005597
29. Szczepanski MJ, Szajnik M, Welsh A, Foon KA, Whiteside TL, Boyiadzis M. Interleukin-15 enhances natural killer cell cytotoxicity in patients with acute myeloid leukemia by upregulating the activating NK cell receptors. Cancer Immunol Immunother (2010) 59(1):73–9. doi:10.1007/s00262-009-0724-5
30. McGilvray RW, Eagle RA, Watson NF, Al-Attar A, Ball G, Jafferji I, et al. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin Cancer Res (2009) 15(22):6993–7002. doi:10.1158/1078-0432.CCR-09-0991
31. de Kruijf EM, Sajet A, van Nes JG, Putter H, Smit VT, Eagle RA, et al. NKG2D ligand tumor expression and association with clinical outcome in early breast cancer patients: an observational study. BMC Cancer (2012) 12(1):24. doi:10.1186/1471-2407-12-24
32. Paschen A, Sucker A, Hill B, Moll I, Zapatka M, Nguyen XD, et al. Differential clinical significance of individual NKG2D ligands in melanoma: soluble ULBP2 as an indicator of poor prognosis superior to S100B. Clin Cancer Res (2009) 15(16):5208–15. doi:10.1158/1078-0432.CCR-09-0886
33. Brune M, Castaigne S, Catalano J, Gehlsen K, Ho AD, Hofmann WK, et al. Improved leukemia-free survival after postconsolidation immunotherapy with histamine dihydrochloride and interleukin-2 in acute myeloid leukemia: results of a randomized phase 3 trial. Blood (2006) 108(1):88–96. doi:10.1182/blood-2005-10-4073
34. Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res (2012) 72(6):1407–15. doi:10.1158/0008-5472.CAN-11-2544
35. Romero AI, Thoren FB, Brune M, Hellstrand K. NKp46 and NKG2D receptor expression in NK cells with CD56dim and CD56bright phenotype: regulation by histamine and reactive oxygen species. Br J Haematol (2006) 132(1):91–8. doi:10.1111/j.1365-2141.2005.05842.x
36. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol (2010) 10(8):554–67. doi:10.1038/nri2808
37. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor β inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A (2003) 100(7):4120–5. doi:10.1073/pnas.0730640100
38. Friese MA, Wischhusen J, Wick W, Weiler M, Eisele G, Steinle A, et al. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res (2004) 64(20):7596–603. doi:10.1158/0008-5472.CAN-04-1627
39. Robson NC, Wei H, McAlpine T, Kirkpatrick N, Cebon J, Maraskovsky E. Activin-A attenuates several human natural killer cell functions. Blood (2009) 113(14):3218–25. doi:10.1182/blood-2008-07-166926
40. Cremer I, Fridman WH, Sautes-Fridman C. Tumor microenvironment in NSCLC suppresses NK cells function. Oncoimmunology (2012) 1(2):244–6. doi:10.4161/onci.1.2.18309
41. Gillard-Bocquet M, Caer C, Cagnard N, Crozet L, Perez M, Fridman WH, et al. Lung tumor microenvironment induces specific gene expression signature in intratumoral NK cells. Front Immunol (2013) 4:19. doi:10.3389/fimmu.2013.00019
42. Vacchelli E, Eggermont A, Fridman WH, Galon J, Zitvogel L, Kroemer G, et al. Trial watch: immunostimulatory cytokines. Oncoimmunology (2013) 2(7):e24850. doi:10.4161/onci.24850
43. El-Sherbiny YM, Meade JL, Holmes TD, McGonagle D, Mackie SL, Morgan AW, et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res (2007) 67(18):8444–9. doi:10.1158/0008-5472.CAN-06-4230
44. Kiladjian JJ, Bourgeois E, Lobe I, Braun T, Visentin G, Bourhis JH, et al. Cytolytic function and survival of natural killer cells are severely altered in myelodysplastic syndromes. Leukemia (2006) 20(3):463–70. doi:10.1038/sj.leu.2404080
46. Farag SS, Caligiuri MA. Cytokine modulation of the innate immune system in the treatment of leukemia and lymphoma. Adv Pharmacol (2004) 51:295–318. doi:10.1016/S1054-3589(04)51013-X
47. Stroncek DF, Melief CJ, Castiello L, Cesano A, Cheever MA, Civini S, et al. Highlights of the society for immunotherapy of cancer (SITC) 27th annual meeting. J Immunother Cancer (2013) 1(1):4. doi:10.1186/2051-1426-1-4
48. Boyiadzis M, Memon S, Carson J, Allen K, Szczepanski MJ, Vance BA, et al. Up-regulation of NK cell activating receptors following allogeneic hematopoietic stem cell transplantation under a lymphodepleting reduced intensity regimen is associated with elevated IL-15 levels. Biol Blood Marrow Transplant (2008) 14(3):290–300. doi:10.1016/j.bbmt.2007.12.490
49. Thiant S, Yakoub-Agha I, Magro L, Trauet J, Coiteux V, Jouet JP, et al. Plasma levels of IL-7 and IL-15 in the first month after myeloablative BMT are predictive biomarkers of both acute GVHD and relapse. Bone Marrow Transplant (2010) 45(10):1546–52. doi:10.1038/bmt.2010.13
50. Mehta DS, Wurster AL, Grusby MJ. Biology of IL-21 and the IL-21 receptor. Immunol Rev (2004) 202:84–95. doi:10.1111/j.0105-2896.2004.00201.x
51. Frederiksen KS, Lundsgaard D, Freeman JA, Hughes SD, Holm TL, Skrumsager BK, et al. IL-21 induces in vivo immune activation of NK cells and CD8(+) T cells in patients with metastatic melanoma and renal cell carcinoma. Cancer Immunol Immunother (2008) 57(10):1439–49. doi:10.1007/s00262-008-0479-4
52. Burgess SJ, Marusina AI, Pathmanathan I, Borrego F, Coligan JE. IL-21 down-regulates NKG2D/DAP10 expression on human NK and CD8+ T cells. J Immunol (2006) 176(3):1490–7.
53. de Rham C, Ferrari-Lacraz S, Jendly S, Schneiter G, Dayer JM, Villard J. The proinflammatory cytokines IL-2, IL-15 and IL-21 modulate the repertoire of mature human natural killer cell receptors. Arthritis Res Ther (2007) 9(6):R125. doi:10.1186/ar2336
54. Davis ID, Brady B, Kefford RF, Millward M, Cebon J, Skrumsager BK, et al. Clinical and biological efficacy of recombinant human interleukin-21 in patients with stage IV malignant melanoma without prior treatment: a phase IIa trial. Clin Cancer Res (2009) 15(6):2123–9. doi:10.1158/1078-0432.CCR-08-2663
55. Lioznov M, El-Cheikh J, Hoffmann F, Hildebrandt Y, Ayuk F, Wolschke C, et al. Lenalidomide as salvage therapy after allo-SCT for multiple myeloma is effective and leads to an increase of activated NK (NKp44(+)) and T (HLA-DR(+)) cells. Bone Marrow Transplant (2009) 45(2):349–53. doi:10.1038/bmt.2009.155
56. Berg SL, Cairo MS, Russell H, Ayello J, Ingle AM, Lau H, et al. Safety, pharmacokinetics, and immunomodulatory effects of lenalidomide in children and adolescents with relapsed/refractory solid tumors or myelodysplastic syndrome: a Children’s Oncology Group Phase I Consortium report. J Clin Oncol (2011) 29(3):316–23. doi:10.1200/JCO.2010.30.8387
57. Dauguet N, Fournié J-J, Poupot R, Poupot M. Lenalidomide down regulates the production of interferon-gamma and the expression of inhibitory cytotoxic receptors of human natural killer cells. Cell Immunol (2010) 264(2):163–70. doi:10.1016/j.cellimm.2010.06.003
58. Mamessier E, Sylvain A, Bertucci FO, Castellano RM, Finetti P, Houvenaeghel G, et al. Human breast tumor cells induce self-tolerance mechanisms to avoid NKG2D-mediated and DNAM-mediated NK cell recognition. Cancer Res (2011) 71(21):6621–32. doi:10.1158/0008-5472.CAN-11-0792
59. Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol (2009) 12(1):7–13. doi:10.1093/neuonc/nop009
60. Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol (2004) 172(12):7335–40.
61. Romero AI, Chaput N, Poirier-Colame V, Rusakiewicz S, Jacquelot N, Chaba K, et al. Regulation of CD4+NKG2D+ Th1 cells in patients with metastatic melanoma treated with sorafenib: role of IL-15Rα and NKG2D triggering. Cancer Res (2013) 74(1):68–80. doi:10.1158/0008-5472.CAN-13-1186
62. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature (2002) 419(6908):734–8. doi:10.1038/nature01112
63. Szczepanski MJ, Szajnik M, Welsh A, Whiteside TL, Boyiadzis M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica (2011) 96(9):1302–9. doi:10.3324/haematol.2010.039743
64. Roberts AI, Lee L, Schwarz E, Groh V, Spies T, Ebert EC, et al. Cutting edge: NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J Immunol (2001) 167(10):5527–30.
65. Nemunaitis J, Dillman RO, Schwarzenberger PO, Senzer N, Cunningham C, Cutler J, et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J Clin Oncol (2006) 24(29):4721–30. doi:10.1200/JCO.2005.05.5335
66. Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov (2012) 11(10):790–811. doi:10.1038/nrd3810
67. Wilson EB, El-Jawhari JJ, Neilson AL, Hall GD, Melcher AA, Meade JL, et al. Human tumour immune evasion via TGF-beta blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLoS One (2011) 6(9):e22842. doi:10.1371/journal.pone.0022842
68. Buijs JT, Stayrook KR, Guise TA. The role of TGF-beta in bone metastasis: novel therapeutic perspectives. Bonekey Rep (2012) 1:96. doi:10.1038/bonekey.2012.96
69. Morris J, Shapiro G, Tan A, Lawrence D, Olencki T, Dezube B, et al. Phase I/II study of GC1008: a human anti-transforming growth factor-beta (TGFβ) monoclonal antibody (MAb) in patients with advanced malignant melanoma (MM) or renal cell carcinoma (RCC). J Clin Oncol (2008) 26(20 Suppl):9028.
70. Stevenson JP, Kindler HL, Papasavvas E, Sun J, Jacobs-Small M, Hull J, et al. Immunological effects of the TGFbeta-blocking antibody GC1008 in malignant pleural mesothelioma patients. Oncoimmunology (2013) 2(8):e26218. doi:10.4161/onci.26218
71. Uhl M, Aulwurm S, Wischhusen J, Weiler M, Ma JY, Almirez R, et al. SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res (2004) 64(21):7954–61. doi:10.1158/0008-5472.CAN-04-1013
72. Krockenberger M, Dombrowski Y, Weidler C, Ossadnik M, Honig A, Hausler S, et al. Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D. J Immunol (2008) 180(11):7338–48.
73. Brandt CS, Baratin M, Eugene CY, Kennedy J, Gao Z, Fox B, et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med (2009) 206(7):1495–503. doi:10.1084/jem.20090681
74. Fiegler N, Textor S, Arnold A, Rolle A, Oehme I, Breuhahn K, et al. Downregulation of the activating NKp30 ligand B7-H6 by HDAC inhibitors impairs tumor cell recognition by NK cells. Blood (2013) 122(5):684–93. doi:10.1182/blood-2013-02-482513
75. Matta J, Baratin M, Chiche L, Forel JM, Cognet C, Thomas G, et al. Induction of B7-H6, a ligand for the natural killer cell-activating receptor NKp30, in inflammatory conditions. Blood (2013) 122(3):394–404. doi:10.1182/blood-2013-01-481705
76. Pogge von Strandmann E, Simhadri VR, von Tresckow B, Sasse S, Reiners KS, Hansen HP, et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity (2007) 27(6):965–74. doi:10.1016/j.immuni.2007.10.010
77. Carbone E, Neri P, Mesuraca M, Fulciniti MT, Otsuki T, Pende D, et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood (2005) 105(1):251–8. doi:10.1182/blood-2004-04-1422
78. Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol (2009) 27(32):5459–68. doi:10.1200/JCO.2009.22.1291
79. Schrump DS. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: mechanisms and potential clinical implications. Clin Cancer Res (2009) 15(12):3947–57. doi:10.1158/1078-0432.CCR-08-2787
80. Diermayr S, Himmelreich H, Durovic B, Mathys-Schneeberger A, Siegler U, Langenkamp U, et al. NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood (2008) 111(3):1428–36. doi:10.1182/blood-2007-07-101311
81. Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res (2005) 65(23):11136–45. doi:10.1158/0008-5472.CAN-05-0599
82. Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res (2005) 65(14):6321–9. doi:10.1158/0008-5472.CAN-04-4252
83. Wu X, Tao Y, Hou J, Meng X, Shi J. Valproic acid upregulates NKG2D ligand expression through an ERK-dependent mechanism and potentially enhances NK cell-mediated lysis of myeloma. Neoplasia (2012) 14(12):1178–89. doi:10.1593/neo.121236
84. Schmudde M, Braun A, Pende D, Sonnemann J, Klier U, Beck JF, et al. Histone deacetylase inhibitors sensitize tumour cells for cytotoxic effects of natural killer cells. Cancer Lett (2008) 272(1):110–21. doi:10.1016/j.canlet.2008.06.027
85. Poggi A, Catellani S, Garuti A, Pierri I, Gobbi M, Zocchi MR. Effective in vivo induction of NKG2D ligands in acute myeloid leukaemias by all-trans-retinoic acid or sodium valproate. Leukemia (2009) 23(4):641–8. doi:10.1038/leu.2008.354
86. Rossi LE, Avila DE, Spallanzani RG, Ziblat A, Fuertes MB, Lapyckyj L, et al. Histone deacetylase inhibitors impair NK cell viability and effector functions through inhibition of activation and receptor expression. J Leukoc Biol (2012) 91(2):321–31. doi:10.1189/jlb.0711339
87. Ogbomo H, Michaelis M, Kreuter J, Doerr HW, Cinatl J Jr. Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett (2007) 581(7):1317–22. doi:10.1016/j.febslet.2007.02.045
88. Kopp LM, Ray A, Denman CJ, Senyukov VS, Somanchi SS, Zhu S, et al. Decitabine has a biphasic effect on natural killer cell viability, phenotype, and function under proliferative conditions. Mol Immunol (2013) 54(3–4):296–301. doi:10.1016/j.molimm.2012.12.012
89. Rohner A, Langenkamp U, Siegler U, Kalberer CP, Wodnar-Filipowicz A. Differentiation-promoting drugs up-regulate NKG2D ligand expression and enhance the susceptibility of acute myeloid leukemia cells to natural killer cell-mediated lysis. Leuk Res (2007) 31(10):1393–402. doi:10.1016/j.leukres.2007.02.020
90. Tang K-F, He C-X, Zeng G-L, Wu J, Song G-B, Shi Y-S, et al. Induction of MHC class I-related chain B (MICB) by 5-aza-2′-deoxycytidine. Biochem Biophys Res Commun (2008) 370(4):578–83. doi:10.1016/j.bbrc.2008.03.131
91. Schmiedel BJ, Arélin V, Gruenebach F, Krusch M, Schmidt SM, Salih HR. Azacytidine impairs NK cell reactivity while decitabine augments NK cell responsiveness toward stimulation. Int J Cancer (2011) 128(12):2911–22. doi:10.1002/ijc.25635
92. Cerdeira AS, Rajakumar A, Royle CM, Lo A, Husain Z, Thadhani RI, et al. Conversion of peripheral blood NK cells to a decidual NK-like phenotype by a cocktail of defined factors. J Immunol (2013) 190(8):3939–48. doi:10.4049/jimmunol.1202582
93. Santourlidis S, Trompeter H-I, Weinhold S, Eisermann B, Meyer KL, Wernet P, et al. Crucial role of DNA methylation in determination of clonally distributed killer cell Ig-like receptor expression patterns in NK cells. J Immunol (2002) 169(8):4253–61.
94. Gao X-N, Lin J, Wang L-L, Yu L. Demethylating treatment suppresses natural killer cell cytolytic activity. Mol Immunol (2009) 46(10):2064–70. doi:10.1016/j.molimm.2009.02.033
95. Vacchelli E, Galluzzi L, Fridman WH, Galon J, Sautes-Fridman C, Tartour E, et al. Trial watch: chemotherapy with immunogenic cell death inducers. Oncoimmunology (2012) 1(2):179–88. doi:10.4161/onci.1.2.19026
96. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi:10.1146/annurev-immunol-032712-100008
97. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol (2008) 8(1):59–73. doi:10.1038/nri2216
98. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature (2005) 436(7054):1186–90. doi:10.1038/nature03884
99. Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood (2009) 113(15):3503–11. doi:10.1182/blood-2008-08-173914
100. Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J (2012) 31(5):1062–79. doi:10.1038/emboj.2011.497
101. Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nat Rev Cancer (2008) 8(12):932–41. doi:10.1038/nrc2459
102. Reiners KS, Topolar D, Henke A, Simhadri VR, Kessler J, Sauer M, et al. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood (2013) 121(18):3658–65. doi:10.1182/blood-2013-01-476606
103. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor-associated MICA is shed by ADAM proteases. Cancer Res (2008) 68(15):6368–76. doi:10.1158/0008-5472.CAN-07-6768
104. Salih HR, Rammensee H-G, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol (2002) 169(8):4098–102.
105. Kohga K, Takehara T, Tatsumi T, Ishida H, Miyagi T, Hosui A, et al. Sorafenib inhibits the shedding of major histocompatibility complex class I-related chain A on hepatocellular carcinoma cells by down-regulating a disintegrin and metalloproteinase 9. Hepatology (2010) 51(4):1264–73. doi:10.1002/hep.23456
106. Mochizuki S, Okada Y. ADAMs in cancer cell proliferation and progression. Cancer Sci (2007) 98(5):621–8. doi:10.1111/j.1349-7006.2007.00434.x
107. Krusch M, Salih J, Schlicke M, Baessler T, Kampa KM, Mayer F, et al. The kinase inhibitors sunitinib and sorafenib differentially affect NK cell antitumor reactivity in vitro. J Immunol (2009) 183(12):8286–94. doi:10.4049/jimmunol.0902404
108. Escudier B, Lassau N, Angevin E, Soria JC, Chami L, Lamuraglia M, et al. Phase I trial of sorafenib in combination with IFN alpha-2a in patients with unresectable and/or metastatic renal cell carcinoma or malignant melanoma. Clin Cancer Res (2007) 13(6):1801–9. doi:10.1158/1078-0432.CCR-06-1432
109. Nagai H, Mukozu T, Matsui D, Kanekawa T, Kanayama M, Wakui N, et al. Sorafenib prevents escape from host immunity in liver cirrhosis patients with advanced hepatocellular carcinoma. Clin Dev Immunol (2012) 2012:607851. doi:10.1155/2012/607851
110. Hipp MM, Hilf N, Walter S, Werth D, Brauer KM, Radsak MP, et al. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood (2008) 111(12):5610–20. doi:10.1182/blood-2007-02-075945
111. Demanet C, Mulder A, Deneys V, Worsham MJ, Maes P, Claas FH, et al. Down-regulation of HLA-A and HLA-Bw6, but not HLA-Bw4, allospecificities in leukemic cells: an escape mechanism from CTL and NK attack? Blood (2004) 103(8):3122–30. doi:10.1182/blood-2003-07-2500
112. Wischhusen J, Friese MA, Mittelbronn M, Meyermann R, Weller M. HLA-E protects glioma cells from NKG2D-mediated immune responses in vitro: implications for immune escape in vivo. J Neuropathol Exp Neurol (2005) 64(6):523–8.
113. Malmberg KJ, Levitsky V, Norell H, de Matos CT, Carlsten M, Schedvins K, et al. IFN-gamma protects short-term ovarian carcinoma cell lines from CTL lysis via a CD94/NKG2A-dependent mechanism. J Clin Invest (2002) 110(10):1515–23. doi:10.1172/JCI200215564
114. Nguyen S, Beziat V, Dhedin N, Kuentz M, Vernant JP, Debre P, et al. HLA-E upregulation on IFN-gamma-activated AML blasts impairs CD94/NKG2A-dependent NK cytolysis after haplo-mismatched hematopoietic SCT. Bone Marrow Transplant (2009) 43(9):693–9. doi:10.1038/bmt.2008.380
115. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood (2012) 120(22):4317–23. doi:10.1182/blood-2012-06-437558
116. Benson DM Jr, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood (2012) 120(22):4324–33. doi:10.1182/blood-2012-06-438028
117. Clark LC, Combs GF Jr, Turnbull BW, Slate EH, Chalker DK, Chow J, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA (1996) 276(24):1957–63. doi:10.1001/jama.1996.03540240035027
118. Guan L, Han B, Li J, Li Z, Huang F, Yang Y, et al. Exposure of human leukemia NB4 cells to increasing concentrations of selenite switches the signaling from pro-survival to pro-apoptosis. Ann Hematol (2009) 88(8):733–42. doi:10.1007/s00277-008-0676-4
119. Olm E, Jonsson-Videsater K, Ribera-Cortada I, Fernandes AP, Eriksson LC, Lehmann S, et al. Selenite is a potent cytotoxic agent for human primary AML cells. Cancer Lett (2009) 282(1):116–23. doi:10.1016/j.canlet.2009.03.010
120. Enqvist M, Nilsonne G, Hammarfjord O, Wallin RP, Bjorkstrom NK, Bjornstedt M, et al. Selenite induces posttranscriptional blockade of HLA-E expression and sensitizes tumor cells to CD94/NKG2A-positive NK cells. J Immunol (2011) 187(7):3546–54. doi:10.4049/jimmunol.1100610
121. Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, et al. Lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res (2008) 14(14):4650–7. doi:10.1158/1078-0432.CCR-07-4405
122. Wu L, Parton A, Lu L, Adams M, Schafer P, Bartlett JB. Lenalidomide enhances antibody-dependent cellular cytotoxicity of solid tumor cells in vitro: influence of host immune and tumor markers. Cancer Immunol Immunother (2011) 60(1):61–73. doi:10.1007/s00262-010-0919-9
123. Zhu D, Corral LG, Fleming YW, Stein B. Immunomodulatory drugs revlimid (lenalidomide) and CC-4047 induce apoptosis of both hematological and solid tumor cells through NK cell activation. Cancer Immunol Immunother (2008) 57(12):1849–59. doi:10.1007/s00262-008-0512-7
124. Hayashi T, Hideshima T, Akiyama M, Podar K, Yasui H, Raje N, et al. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: clinical application. Br J Haematol (2005) 128(2):192–203. doi:10.1111/j.1365-2141.2004.05286.x
125. Hernandez-Ilizaliturri FJ, Reddy N, Holkova B, Ottman E, Czuczman MS. Immunomodulatory drug CC-5013 or CC-4047 and rituximab enhance antitumor activity in a severe combined immunodeficient mouse lymphoma model. Clin Cancer Res (2005) 11(16):5984–92. doi:10.1158/1078-0432.CCR-05-0577
126. Reddy N, Hernandez-Ilizaliturri FJ, Deeb G, Roth M, Vaughn M, Knight J, et al. Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br J Haematol (2008) 140(1):36–45. doi:10.1111/j.1365-2141.2007.06841.x
127. Davies FE, Raje N, Hideshima T, Lentzsch S, Young G, Tai YT, et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood (2001) 98(1):210–6. doi:10.1182/blood.V98.1.210
128. Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, et al. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer (2000) 88(3):577–83. doi:10.1002/(SICI)1097-0142(20000201)88:3<577::AID-CNCR13>3.0.CO;2-V
129. Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer (1997) 79(12):2320–8. doi:10.1002/(SICI)1097-0142(19970615)79:12<2320::AID-CNCR5>3.0.CO;2-P
130. Esendagli G, Bruderek K, Goldmann T, Busche A, Branscheid D, Vollmer E, et al. Malignant and non-malignant lung tissue areas are differentially populated by natural killer cells and regulatory T cells in non-small cell lung cancer. Lung Cancer (2008) 59(1):32–40. doi:10.1016/j.lungcan.2007.07.022
131. Bartlett J, Michael A, Clarke I, Dredge K, Nicholson S, Kristeleit H, et al. Phase I study to determine the safety, tolerability and immunostimulatory activity of thalidomide analogue CC-5013 in patients with metastatic malignant melanoma and other advanced cancers. Br J Cancer (2004) 90(5):955–61. doi:10.1038/sj.bjc.6601579
132. Mahindra A, Laubach J, Raje N, Munshi N, Richardson PG, Anderson K. Latest advances and current challenges in the treatment of multiple myeloma. Nat Rev Clin Oncol (2012) 9(3):135–43. doi:10.1038/nrclinonc.2012.15
133. Lundqvist A, Abrams SI, Schrump DS, Alvarez G, Suffredini D, Berg M, et al. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res (2006) 66(14):7317–25. doi:10.1158/0008-5472.CAN-06-0680
134. Lundqvist A, Berg M, Smith A, Childs RW. Bortezomib treatment to potentiate the anti-tumor immunity of ex-vivo expanded adoptively infused autologous natural killer cells. J Cancer (2011) 2:383. doi:10.7150/jca.2.383
135. Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, et al. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol (2008) 180(1):163–70.
136. Sarhan D, Wennerberg E, D’Arcy P, Gurajada D, Linder S, Lundqvist A. A novel inhibitor of proteasome deubiquitinating activity renders tumor cells sensitive to TRAIL-mediated apoptosis by natural killer cells and T cells. Cancer Immunol Immunother (2013) 62(8):1359–68. doi:10.1007/s00262-013-1439-1
137. Butler JE, Moore MB, Presnell SR, Chan HW, Chalupny NJ, Lutz CT. Proteasome regulation of ULBP1 transcription. J Immunol (2009) 182(10):6600–9. doi:10.4049/jimmunol.0801214
138. Kamimura H, Yamagiwa S, Tsuchiya A, Takamura M, Matsuda Y, Ohkoshi S, et al. Reduced NKG2D ligand expression in hepatocellular carcinoma correlates with early recurrence. J Hepatol (2012) 56(2):381–8. doi:10.1016/j.jhep.2011.06.017
139. Valés-Gómez M, Chisholm SE, Cassady-Cain RL, Roda-Navarro P, Reyburn HT. Selective induction of expression of a ligand for the NKG2D receptor by proteasome inhibitors. Cancer Res (2008) 68(5):1546–54. doi:10.1158/0008-5472.CAN-07-2973
140. Shi J, Tricot GJ, Garg TK, Malaviarachchi PA, Szmania SM, Kellum RE, et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood (2008) 111(3):1309–17. doi:10.1182/blood-2007-03-078535
141. Lundqvist A, Yokoyama H, Smith A, Berg M, Childs R. Bortezomib treatment and regulatory T-cell depletion enhance the antitumor effects of adoptively infused NK cells. Blood (2009) 113(24):6120–7. doi:10.1182/blood-2008-11-190421
142. Lundqvist A, Su S, Rao S, Childs R. Cutting edge: bortezomib-treated tumors sensitized to NK cell apoptosis paradoxically acquire resistance to antigen-specific T cells. J Immunol (2010) 184(3):1139–42. doi:10.4049/jimmunol.0902856
143. Wang X, Ottosson A, Ji C, Feng X, Nordenskjöld M, Henter J-I, et al. Proteasome inhibition induces apoptosis in primary human natural killer cells and suppresses NKp46-mediated cytotoxicity. Haematologica (2009) 94(4):470–8. doi:10.3324/haematol.13783
144. Feng X, Holmlund T, Zheng C, Fadeel B. Pro-apoptotic effects of the novel proteasome inhibitor b-AP15 on multiple myeloma cells and natural killer cells. Exp Hematol (2014) 42:172–82. doi:10.1016/j.exphem.2013.11.010
145. Borg C, Terme M, Taieb J, Menard C, Flament C, Robert C, et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J Clin Invest (2004) 114(3):379–88. doi:10.1172/JCI21102
146. Gasteiger G, Hemmers S, Firth MA, Le Floc’h A, Huse M, Sun JC, et al. IL-2 dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J Exp Med (2013) 210(6):1167–78. doi:10.1084/jem.20122462
147. Pasquier E, Kavallaris M, André N. Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol (2010) 7(8):455–65. doi:10.1038/nrclinonc.2010.82
148. Kosmaczewska A, Ciszak L, Potoczek S, Frydecka I. The significance of Treg cells in defective tumor immunity. Arch Immunol Ther Exp (Warsz) (2008) 56(3):181–91. doi:10.1007/s00005-008-0018-1
149. Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother (2007) 56(5):641–8. doi:10.1007/s00262-006-0225-8
150. Kono K, Kawaida H, Takahashi A, Sugai H, Mimura K, Miyagawa N, et al. CD4 (+) CD25high regulatory T cells increase with tumor stage in patients with gastric and esophageal cancers. Cancer Immunol Immunother (2006) 55(9):1064–71. doi:10.1007/s00262-005-0092-8
151. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10(9):942–9. doi:10.1038/nm1093
152. Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med (2005) 202(8):1075–85. doi:10.1084/jem.20051511
153. Le DT, Jaffee EM. Regulatory T-cell modulation using cyclophosphamide in vaccine approaches: a current perspective. Cancer Res (2012) 72(14):3439–44. doi:10.1158/0008-5472.CAN-11-3912
154. Nakahara T, Uchi H, Lesokhin AM, Avogadri F, Rizzuto GA, Hirschhorn-Cymerman D, et al. Cyclophosphamide enhances immunity by modulating the balance of dendritic cell subsets in lymphoid organs. Blood (2010) 115(22):4384–92. doi:10.1182/blood-2009-11-251231
155. Orlandi P, Fontana A, Fioravanti A, Di Desidero T, Galli L, Derosa L, et al. VEGF-A polymorphisms predict progression-free survival among advanced castration-resistant prostate cancer patients treated with metronomic cyclophosphamide. Br J Cancer (2013) 109(4):957–64. doi:10.1038/bjc.2013.398
156. Doloff JC, Waxman DJ. VEGF receptor inhibitors block the ability of metronomically dosed cyclophosphamide to activate innate immunity-induced tumor regression. Cancer Res (2012) 72(5):1103–15. doi:10.1158/0008-5472.CAN-11-3380
157. Greten TF, Ormandy LA, Fikuart A, Höchst B, Henschen S, Hörning M, et al. Low-dose cyclophosphamide treatment impairs regulatory T cells and unmasks AFP-specific CD4+ T-cell responses in patients with advanced HCC. J Immunother (2012) 2:33. doi:10.1097/CJI.0b013e3181bb499f
Keywords: cancer, immune escape, NK cell, NCR, NKG2D, KIR, immunotherapy
Citation: Chretien A-S, Le Roy A, Vey N, Prebet T, Blaise D, Fauriat C and Olive D (2014) Cancer-induced alterations of NK-mediated target recognition: current and investigational pharmacological strategies aiming at restoring NK-mediated anti-tumor activity. Front. Immunol. 5:122. doi: 10.3389/fimmu.2014.00122
Received: 17 January 2014; Accepted: 10 March 2014;
Published online: 24 March 2014.
Edited by:
Simona Sivori, University of Genoa, ItalyReviewed by:
Alexander Steinle, Goethe University Frankfurt am Main, GermanyClaudia Cantoni, University of Genoa, Italy
Copyright: © 2014 Chretien, Le Roy, Vey, Prebet, Blaise, Fauriat and Olive. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Olive, 232 Boulevard de Sainte-Marguerite, Marseille, France e-mail:ZGFuaWVsLm9saXZlQGluc2VybS5mcg==