 Yili Li1,2
Yili Li1,2 Roy A. Mariuzza1,2*
Roy A. Mariuzza1,2*- 1W. M. Keck Laboratory for Structural Biology, Institute for Bioscience and Biotechnology Research, University of Maryland, Rockville, MD, USA
- 2Department of Cell Biology and Molecular Genetics, University of Maryland, College Park, MD, USA
Natural killer (NK) cells are key components of innate immune responses to tumors and viral infections. NK cell function is regulated by NK cell receptors that recognize both cellular and viral ligands, including major histocompatibility complex (MHC), MHC-like, and non-MHC molecules. These receptors include Ly49s, killer immunoglobulin-like receptors, leukocyte immunoglobulin-like receptors, and NKG2A/CD94, which bind MHC class I (MHC-I) molecules, and NKG2D, which binds MHC-I paralogs such as the stress-induced proteins MICA and ULBP. In addition, certain viruses have evolved MHC-like immunoevasins, such as UL18 and m157 from cytomegalovirus, that act as decoy ligands for NK receptors. A growing number of NK receptor–ligand interaction pairs involving non-MHC molecules have also been identified, including NKp30–B7-H6, killer cell lectin-like receptor G1–cadherin, and NKp80–AICL. Here, we describe crystal structures determined to date of NK cell receptors bound to MHC, MHC-related, and non-MHC ligands. Collectively, these structures reveal the diverse solutions that NK receptors have developed to recognize these molecules, thereby enabling the regulation of NK cytolytic activity by both host and viral ligands.
Introduction
Natural killer (NK) cells are essential components of the innate immune response against viral infections and tumors (1–5). They not only eliminate virally infected or malignantly transformed cells by means of their cytolytic capabilities, but also produce cytokines and chemokines such as interferon-γ that modulate immune responses and help maintain tissue homeostasis. To perform these diverse functions, NK cells express a multitude of activating and inhibitory receptors that act in concert to regulate their activities (6, 7). NK receptors belong to two distinct structural families: the immunoglobulin (Ig) superfamily and the C-type lectin superfamily. In humans, NK receptors of the Ig superfamily are encoded in the leukocyte receptor complex (LRC) on chromosome 19 (7 in mouse) (8) and the NK gene complex (NKC) on chromosome 12 (6 in mouse) (9). Both superfamilies include inhibitory and activating receptors. In addition, NK receptors have been shown to recognize both cellular and viral ligands, including major histocompatibility complex (MHC), MHC-like, and non-MHC molecules.
The cytolytic activity of NK cells is regulated by positive signaling activating receptors (resulting in target cell lysis) and negative signaling inhibitory receptors (preventing lysis). It is the dynamic interplay between these signals that ultimately determines the outcome of NK cell–target cell encounters (4, 6, 7). The dominant signal received by an NK cell is inhibitory, provided by the interaction of its receptors with normal levels of MHC class I (MHC-I) molecules. If MHC-I expression is reduced by infectious or tumorigenic processes, this inhibitory signal is attenuated and the NK cell undergoes activation. As a consequence, cells with reduced MHC-I expression become subject to lysis by NK cells (1–5). The process by which NK receptors direct the cytolytic activity of NK cells against virally infected or tumor cells that have lost MHC-I expression is known as “missing-self” recognition.
Several receptor families on primate and rodent NK cells are responsible for monitoring MHC-I expression on surrounding cells (2–5, 10–13). These include the killer immunoglobulin-like receptors (KIRs) in humans, members of the Ly49 family (Ly49s) in rodents, NKG2/CD94 receptors, and leukocyte immunoglobulin-like receptors (LILRs). Although most Ly49s and KIRs inhibit NK function on binding to MHC-I ligands, some are activating (6, 7). Furthermore, the activating NK receptor NKG2D binds paralogs of MHC-I molecules, including MICA and RAE-I that are selectively upregulated in stressed tissues (14). The interaction of activating Ly49s with MHC-like proteins encoded by mouse cytomegalovirus (MCMV) has demonstrated a direct role for Ly49 receptors in anti-viral immunity (15–17).
Besides receptors specific for MHC-I or MHC-related ligands, a number of other receptors that recognize non-MHC proteins are involved in regulating NK cell cytotoxic activity (2, 7). These include CD16 (18), CD69 (19), NKR-P1 (20, 21), NTB-A (22, 23), 2B4 (22–24), DNAM-1 (25), NKp30 (26), NKp44 (27), NKp46 (28), NKp65 (21, 29), and NKp80 (21, 30, 31), which contribute to NK cell activation, and the inhibitory receptors, killer cell lectin-like receptor G1 (KLRG1) (32, 33) and LAIR-1 (34). The biological ligands for most (but not all) of these receptors have now been identified, including IgG Fc for CD16, Clr for NKR-P1, CD48 for 2B4, CD155 for DNAM-1, B7-H6 for NKp30, keratinocyte-associated C-type lectin (KACL) for NKp65, AICL for NKp80, E-cadherin for KLRG1, and collagen for LAIR-1 (2, 7).
Considerable progress has been made over the past few years in determining crystal structures of representative NK receptors, both in isolation and bound to MHC, MHC-related, or non-MHC ligands. These include both Ig-like (e.g., KIRs, LILRs, NKp30) and C-type lectin-like (e.g., Ly49s, NKG2D, NKG2/CD94, NKp65) receptors. These structures have revealed the multiplicity of solutions that NK receptors have evolved to recognize MHC, MHC-like, and non-MHC molecules, which collectively mediate crucial interactions for regulating the cytolytic activity of NK cells by host and viral ligands.
MHC-I Recognition by KIR Receptors
The highly polymorphic KIR receptor family encodes the main MHC-monitoring molecules on primate NK cells and includes both inhibitory and activating members. KIRs are transmembrane glycoproteins containing two (D1 and D2) or three (D0, D1, and D2) extracellular C2-type Ig-like domains (10, 12). KIRs with two Ig-like domains are designated KIR2D; KIRs with three Ig-like domains are designated KIR3D. Whereas KIR2D receptors bind HLA-C alleles, KIR3D receptors bind HLA-A and HLA-B alleles. Crystallographic studies of KIR2D molecules, both in free form (35–39) and bound to HLA-C ligands (40, 41), have provided a framework for understanding the specificity of KIR2D receptors for HLA-C at the atomic level. In addition, the structure of KIR3DL1 in complex with HLA-B*5701 has revealed the basis for HLA recognition by three-domain KIRs (42).
The two N-terminal domains (D1 and D2) of KIR2D receptors are linked by a short hinge segment of three to five amino acids (Figure 1A). These Ig-like domains are each formed by two anti-parallel β-sheets, such that a β-sheet of four (in D1) or three (in D2) anti-parallel strands (ABED and ABE, respectively) juxtaposes a β-sheet of four anti-parallel strands (CC′ FG). The relative disposition of D1 and D2 is similar to that found in hematopoietic receptors (43, 44), with the angle between D1 and D2 ranging from 60° to 80° in different KIRD2 receptors.
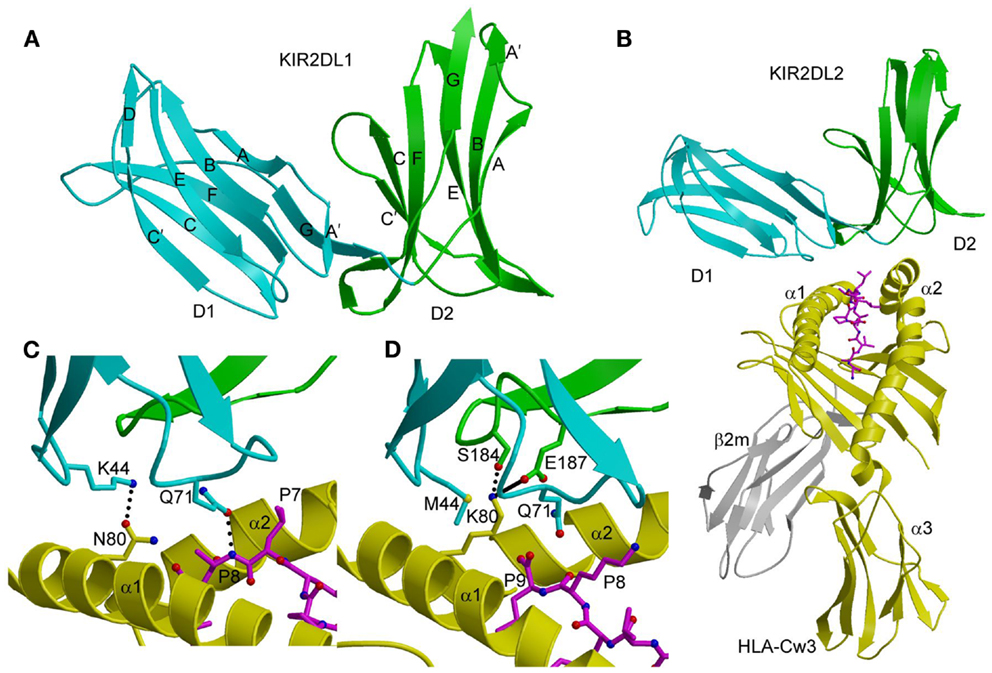
Figure 1. Three-dimensional structures of KIR2DL and KIR2DL–HLA-C complexes. (A) Ribbon diagram of KIR2DL1 (PDB accession code 1NKR). The D1 domain is cyan; D2 is green. The secondary structural elements are labeled. (B) Ribbons diagram of KIR2DL2 bound to HLA-Cw3 (1EFX). The α1, α2, and α3 domains of the HLA-Cw3 heavy chain are yellow; β2m is gray; the peptide is magenta. (C) Basis for allelic specificity and peptide selectivity of KIR2D receptors. The dotted lines represent hydrogen bonds formed by Asn80 of HLA-Cw3 with Lys44 of KIR2DL2, and by Gln71 of HLA-Cw3 with P8 of the peptide. (D) Interactions of Lys80 of HLA-Cw4 (yellow) with specificity-determining residues of KIR2DL1 (D1 domain in cyan, D2 domain in green) in the KIR2DL1–HLA-Cw4 complex (1IM9). The solid line represents a salt bridge.
In both the KIR2DL2–HLA-Cw3 (40) and KIR2DL1–HLA-Cw4 (41) complexes, the KIRs engage HLA-C through the α1 and α2 helices of the α1/α2 platform domain and the C-terminal portion of the MHC-bound peptide, with the D1–D2 axis orthogonal to the axis of the peptide (Figure 1B). This docking mode roughly resembles the way T-cell receptors (TCRs) bind MHC, but is completely distinct from the docking mode of LILR and Ly49 NK receptors (see below). Each KIR2D binds HLA-C using six loops from D1 and D2, which contact the α1 and α2 helices of the MHC-I molecule, respectively.
The structures of the KIR2DL2–HLA-Cw3 and KIR2DL1–HLA-Cw4 complexes explain the allelic specificity of KIR2DLs (40, 41). Of 12 HLA-Cw3 residues in contact with KIR2DL2, 11 are invariant in HLA-Cw4 and in all other HLA-C alleles. The only exception is Asn80, which defines the allelic specificity of KIR2DLs. Similarly, on the receptor side of the interface, 14 of 16 KIR2DL2 residues that contact HLA-Cw3 are conserved in KIR2DL1. The two exceptions are at positions 44 and 70. In the KIR2DL2–HLA-Cw3 structure, KIR2DL2 Lys44 makes a hydrogen bond with HLA-Cw3 Asn80; this hydrogen bond cannot be formed with KIR2DL1 Met44 (Figure 1C). In the KIR2DL1–HLA-Cw4 structure, the side chain of HLA-Cw4 Lys80 is situated in a negatively charge pocket of KIR2DL1 that includes Met44, which contacts HLA-Cw4 Lys80 (Figure 1D). Replacement of Met44 by lysine, as in KIR2DL2, would cause charge repulsion with HLA-Cw4 Lys80, resulting in loss of binding.
KIR2DS4 is an activating receptor that specifically recognizes HLA-A*11, as well as HLA-C allotypes bearing the C1 and C2 epitopes (39). A comparison of the unbound KIR2DS4 structure with the KIR2DL2–HLA-Cw3 and KIR2DL1–HLA-Cw4 complexes revealed two features that likely explain the binding specificity of KIR2DS4. First, a backbone displacement of one of the predicted HLA-contacting loops (L2) of KIR2DS4, relative to its position in KIR2DL2, may disrupt the interaction between Lys44 of KIR2DS4 and Asn80 of the HLA-C C1 epitope, resulting in weaker binding of KIR2DS4 to C1+ allotypes than KIR2DL2 (39). Conversely, this displacement could increase avidity for C2+ allotypes by accommodating Lys80 of the C2 epitope. Second, the Pro71–Val72 motif of KIR2DS4 that confers reactivity with HLA-A*11 is part of a loop (L3), which in KIR2DL2 contacts HLA-C using the Gln71–Asp72 motif (40). Replacement of Gln71–Asp72 by Pro71–Val72 in KIR2DS4, which is the result of gene conversion with KIR3DL2, reduces avidity for C1+ allotypes but increases avidity for HLA-A*11 and C2+ allotypes (39).
The binding of KIR2D receptors to HLA-C molecules displays preferences for certain peptides (45, 46); however, whether peptide selectivity has a role in NK receptor function is not clear. Intriguingly, KIR-associated HIV-1 sequence polymorphisms in chronically infected individuals have been found to increase the binding of inhibitory KIRs to CD4+ T cells infected with HIV-1, and to decrease the anti-viral activity of KIR-positive NK cells (47). Consistent with the observation that the KIR binding site is centered near C-terminal residues P7 and P8 of the MHC-bound peptide (40, 41), the KIR–HLA interaction is most sensitive to substitutions at these two peptide positions. By contrast, TCRs, which exhibit much greater peptide specificity than KIRs, typically focus on the central P5 position of the peptide (48). Since the peptide positions recognized by KIRs are not usually directly involved in TCR binding, MHC molecules may be able to evolve their polymorphic regions to present diverse microbial (i.e., foreign) peptides for T-cell-mediated immunity, while at the same time maintaining non-variant regions to bind self-peptides for KIR recognition and NK-cell-mediated immune defense.
In the KIR3DL1–HLA-B*5701 complex (42), KIR3DL1 engages HLA-B*5701 with its D1 and D2 domains situated over the C-terminal half of the peptide-binding groove in an overall orientation highly similar to that of the KIR2D–HLA-C complexes (Figure 2A). KIR3DL1 adopts an elongated conformation, which allows D0 to extend down toward β2-microglobulin (β2m) and engage a region of the MHC-I molecule that is nearly invariant across HLA-A and HLA-B allotypes. The D1 domain contacts the α1 helix and the self-peptide, while the D2 domain contacts the α2 helix. The D2 domain mainly interacts with HLA-B*5701 residues 142–151, which display restricted polymorphism across HLA-B alleles. At the center of the D2–HLA-B*5701 interface, KIR3DL1 residues Tyr200 and Phe276 form an aromatic cluster that converges on the α2 helix (Figure 2B). Alanine substitution of these residues abrogated binding to HLA-B*5701, demonstrating the importance of this central core to HLA recognition.
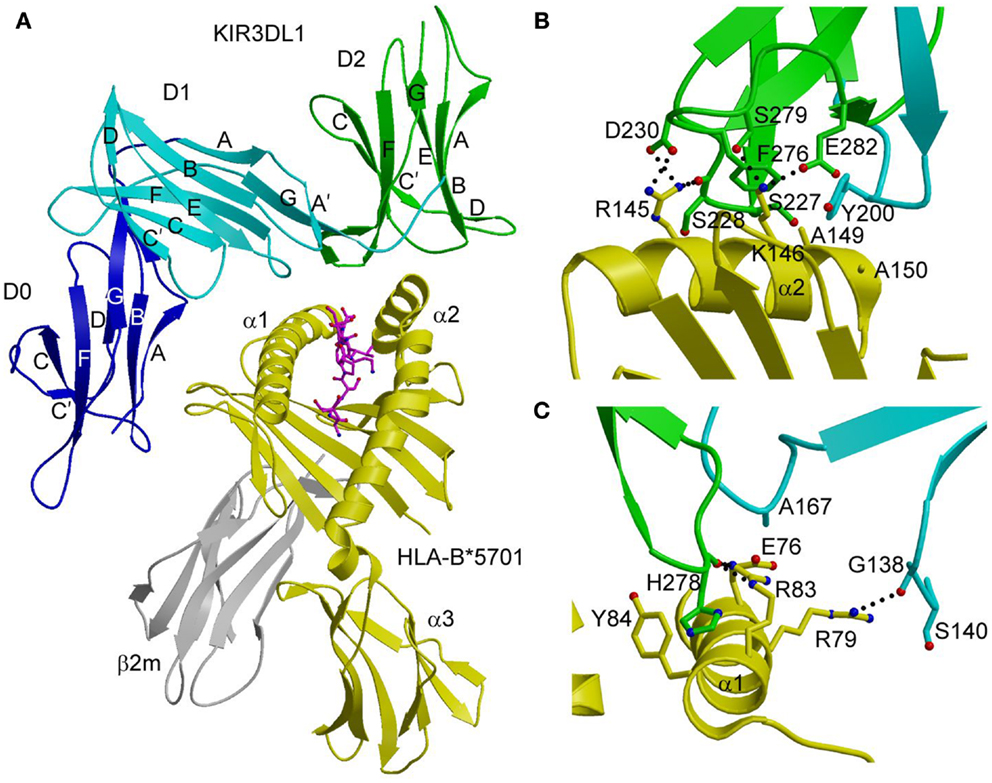
Figure 2. Structure of the KIR3DL1–HLA-B*5701 complex. (A) Ribbon diagram of KIR3DL1 bound to HLA-B*5701 (3VH8). The orientation of the MHC-I ligand is similar to that of HLA-Cw3 in the KIR2DL2–HLA-Cw3 complex (Figure 1B). The HLA-B*5701 heavy chain is yellow; β2m is gray; the peptide is magenta. The KIR3DL1 D0 domain is dark blue; D1 is cyan; D2 is green. The secondary structural elements of KIR3DL1 are labeled. (B) Contacts between KIR3DL1 and the HLA-B*5701 α2 helix. The D2 domain mainly interacts with HLA-B*5701 residues 142–151, which display limited polymorphism among HLA-B alleles. At the center of the D2–HLA-B*5701 interface, KIR3DL1 residues Tyr200 and Phe276 form an aromatic cluster that converges on the α2 helix. (C) Contacts between KIR3DL1 and the HLA-B*5701 α1 helix. KIR3DL1 recognizes HLA allotypes that contain the Bw4 epitope-defining residues 77–83 on the α1 helix, which likely accounts for the allelic specificity of KIR3DLs.
KIR3DL1 recognizes HLA allotypes that contain the Bw4 epitope, which is defined by residues 77–83 of the α1 helix. In the structure (42), KIR3DL1 contacts residues 79, 80, and 83 within the Bw4 epitope through its D1 domain (Figure 2C), which likely accounts for the allelic specificity of KIR3DLs. The D1 domain also makes limited contacts with the self-peptide at position P8, analogous to the interaction of KIR2D receptors with peptides bound to HLA-C (see above).
Unexpectedly, the extensive polymorphisms found within individual KIR3D families are located predominantly at positions not implicated in HLA binding. This implies that most KIR3D polymorphisms, a number of which are subject to positive selection (49), are unlikely to impact affinity directly, but could potentially affect HLA binding indirectly by altering the clustering or expression levels of KIR3D receptors on the NK cell surface. In this way, evolutionary pressures may drive the diversification of KIR3D sequences at sites remote from the HLA-binding site.
MHC-I Recognition by LILRs
The human LILR family of immunoreceptors (also referred to as Ig-like transcripts, or ILTs) is broadly expressed on NK cells, T cells, monocytes, B cells, and dendritic cells (50). The mouse orthologs of LILRs are known as paired immunoglobulin receptors (PIRs). Like KIRs, LILR receptors contain either two or four tandem extracellular Ig-like domains. LILRA1, LILRA2, LILRA3, LILRB1, and LILRB2 bind classical MHC-I proteins (HLA-A, -B, and -C), whereas LILRA4, LILRA5, LILRA6, LILRB3, LILRB4, and LILRB4 do not appear to recognize MHC-I. The inhibitory LILRB1 and LILRB2 receptors bind multiple MHC-I molecules, both classical and non-classical (HLA-E, -F, and -G), with comparable kinetics and affinities (51, 52). By contrast, individual KIR receptors display allelic specificity, as discussed above. In addition to their role as MHC-I sensors, LILRs may be involved in immune responses to viral infections, as suggested by the finding that LIRLB1 is a receptor for UL18 (53). This immunoevasin is an MHC-I homolog encoded by human cytomegalovirus (HCMV). The crystal structure of LILRB1 (domains D1 and D2 only) has been solved in free form (54) and bound to HLA-A2 (55) and UL18 (56). Structures have also been reported for LILRB2 (D1 and D2) in unbound form (57) and in complex with HLA-G (58).
Similar to KIR2D (Figure 1A), the two tandem Ig-like domains of both LILRB1 and LILRB2 form a bent structure characterized by an acute interdomain angle (Figure 3A). Each domain comprises two anti-parallel β-sheets arranged in a topology like that of KIRs. In the LILRB1–HLA-A2 complex (55), LILRB1 D1D2 binds the side of HLA-A2, forming two contact surfaces that include residues from β2m, which is invariant, and the HLA-A2 α3 domain, which is relatively non-polymorphic. The D1–D2 interdomain hinge region contacts β2m, while the tip of LILRB1 D1 contacts the HLA-A2 α3 domain, (Figure 3A). Similar to LILRB1, LILRB2 recognizes β2m and the HLA-G α3 domain using the interdomain hinge and D1, respectively (58). The docking mode utilized by LILRB1 and LILRB2, which differs completely from that of KIRs (Figure 1B), is consistent with MHC-I recognition in a peptide-independent manner. The focus by LILRs on conserved elements of MHC-I molecules, both classical and non-classical, accounts for the broad specificity of these NK receptors for numerous HLA alleles.
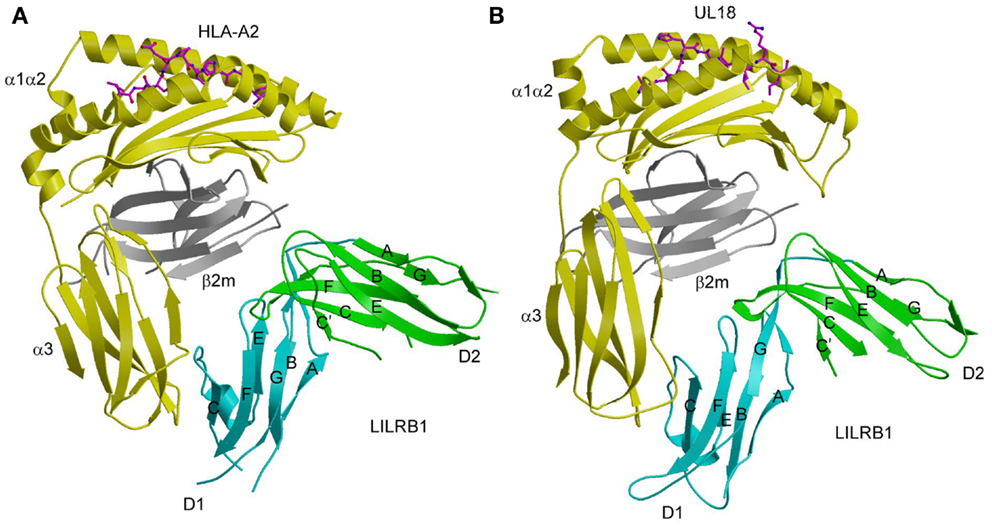
Figure 3. Interaction of LILRB1 with MHC-I and a viral MHC-I mimic. (A) Structure of LILRB1 bound to HLA-A2 (1P7Q). The α1, α2, and α3 domains of the HLA-A2 heavy chain are yellow; β2m is gray; the peptide is magenta. The D1 and D2 domains of LILRB1 are colored in cyan and green, respectively. The secondary structural elements of LILRB1 are labeled. (B) Structure of LILRB1 bound to the HCMV MHC-I mimic UL18 (3D2U).
Whereas LILRB1 undergoes an interdomain angle change of ~15° after binding MHC-I, LILRB2 maintains the same interdomain angle (55, 58). Overall, however, LILRB2 exhibits greater conformational changes than LILRB1 upon complex formation. In particular, free LILRB2 contains only one 310 helix (residues 52–55) involving binding site residues, whereas bound LILRB2 contains two such helices in the interface with HLA-G (residues 46–50 and 53–57). By contrast, LILRB1 contains two 310 helices in both free and bound states. Although affinity measurements indicate that the membrane-distal D1 and D2 domains are mainly responsible for HLA binding (54, 55), a role for the membrane-proximal D3 and D4 domains cannot be formally excluded in the absence of a structure of a four-domain LILR in complex with ligand. A complete D1–D4 LILR structure is also required to understand the apparent ability of LILR/PIR receptors to bind not only MHC-I molecules on opposing cells (trans interaction), but also ones on the same cell (cis interaction) (59, 60), as discussed below.
LILR Recognition of UL18, a Viral MHC-I Mimic
Among the microorganisms that have achieved great success in inventing strategies for immune evasion are the cytomegaloviruses, whose genomes encode proteins that interfere with both NK cell and T-cell recognition, as well as antigen processing and presentation (61–63). These include proteins that are known, or predicted to be, structural homologs of host MHC-I molecules. HCMV encodes an MHC-I homolog, UL18, that binds the inhibitory receptor LILRB1 (64). This interaction is believed to allow HCMV-infected cells to avoid NK-cell-mediated lysis (65). UL18 is a heavily glycosylated transmembrane protein that associates with β2m, and with endogenous peptides derived from host cytoplasmic proteins that resemble those bound to HLA alleles (66). Remarkably, UL18 binds LILRB1 >1000-fold more tightly than MHC-I proteins, enabling this decoy ligand to compete effectively with MHC-I for binding to LILRB1 (67).
Despite sharing only ~25% sequence with its MHC-I counterparts, the structure of UL18 bound to LILRB1 shows striking similarity to the LILRB1–HLA-A2 and LILRB2–HLA-G complexes, with the tip LILRB1 D1 domain contacting the UL18 α3 domain and the D1–D2 interdomain hinge contacting β2m (Figure 3B) (56). Variable residues in the UL18 α1 domain, which were identified by sequence analysis of laboratory and clinical HCMV strains, do not contact LILRB1, although domains D3 and D4, which are not present in the structure, could potentially engage this region of UL18. Most contacts between LILRB1 and U18 involve the UL18-specific portion of the UL18/β2m heterodimer (i.e., the heavy chain), whereas the majority of LILRB1 interactions with HLA-A2 involve the invariant β2m light chain. Additional salt bridges and better surface complementarity in the LILRB1–UL18 interface compared with the LILRB1–HLA-A2 interface likely explain the >1000-fold higher affinity of UL18.
A major difference between UL18 and MHC-I molecules is the exceptionally high carbohydrate content of UL18, which is attributable to its 13 potential N-glycosylation sites, compared to only one N-glycan attached to human MHC-I molecules. In fully glycosylated UL18 (the protein used for crystallization was minimally glycosylated), most of the surface of UL18 was predicted to be covered by carbohydrate, with the notable exceptions of the binding site for LILRB1 and the docking interface with β2m (56). This suggests that UL18 evolved a glycan shield to prevent neutralization by antibodies, while preserving the binding site for LILRs. Such a strategy for reducing immunogenicity is analogous to that employed by other viruses with heavily glycosylated envelope proteins, notably HIV and influenza (68).
Natural Cytotoxicity Receptors
Natural cytotoxicity receptors (NCRs) were discovered in a search for receptors that activated NK cells independently of MHC (69). To date, the NCR family includes NKp30 (NCR3, CD337), NKp44 (NCR2, CD336), and NKp46 (NCR1, CD335). In humans, NKp44 and NKp30 are encoded in the class III region of the MHC locus, while NKp46 is encoded in the LRC (69). Mice only possess a functional gene for NKp46. These very potent activating receptors comprise one (NKp30 and NKp44) or two (NKp46) Ig-like extracellular domains (69, 70). NCRs contain charged residues in their transmembrane regions for association with immunoreceptor tyrosine-based activation motif (ITAM)-bearing signaling polypeptides: ζ–γ for NKp30 and NKp46; and DAP12 for NKp44 (71). In humans, NCRs play a major role in NK-cell-mediated lysis of diverse tumor cells, including carcinomas, neuroblastomas, and leukemias (69, 70). In addition, NCRs have been implicated in protective responses against various viruses, including influenza (72), hepatitis C (73), West Nile (74), and Ebola (75).
Despite intensive efforts over many years, ligands for the NRC family have proven very elusive and, in some cases, controversial. NKp44 and NKp46 bind influenza and other viral hemagglutinins (HAs) mainly through recognition by the HA of terminal sialic acid moieties (the cellular receptor for HAs) on N-linked glycans of these NCRs (72, 76, 77). Although this mechanism would allow NKp44 and NKp46 to bind a wide variety of viruses, due to the ability of HAs to bind sialic acid-containing glycoproteins in general, this is probably not the full story, since recognition would not depend on the NCR ectodomain itself, but only on the fact that NKp44 and NKp46 are glycoproteins with terminal sialic acids (13). Binding of NKp46 to heparan sulfate proteoglycans has also been described (78), but the biological relevance of this interaction is unclear. Recently, a novel isoform of the mixed-lineage leukemia-5 protein (MLL5) was identified as a cellular ligand for NKp44 (79). This MLL5 isoform was not expressed on cells from healthy individuals, but was detected on a large panel of tumor and transformed cell lines. Moreover, MLL5 expression on target cells triggered NKp44-mediated NK cell cytotoxicity.
NKp30 binds the nuclear factor BAT3 (80) and the tumor cell surface protein B7-H6 (81). BAT3 (also known as BAG-6) has been implicated in the induction of apoptosis after endoplasmic reticulum stress or DNA damage (82). B7-H6 is a member of the B7-family (81), which includes ligands (B7-1 and B7-2) for the T-cell co-inhibitory receptor CTLA-4 and the co-stimulatory receptor CD28 (83). The B7-family also encodes PD-L1 and PD-L2, which are ligands for the T-cell co-inhibitory receptor PD-1. B7-H6 is not expressed in normal human tissues, but can be detected on a variety of human tumor cell lines that includes T and B lymphomas, melanomas, and carcinomas (81). Importantly, B7-H6 expression on tumor cells triggered NK cell cytotoxicity that was mediated specifically by NKp30. These results implicate B7-H6 as tumor-induced self-protein, analogous to MICA (2), which alerts NK cells to cellular transformation (81). NKp30 also recognizes the tegument pp65 protein of HCMV, indicating a role for this NCR in anti-viral immunity (84). Recently, NKp30 was shown to be responsible for the recognition and killing of the opportunistic fungi Cryptococcus and Candida (85). Although the fungal ligand recognized by NKp30 remains to be identified, possible candidates include β-1,3 glucans, which are major components of fungal cell wells and are highly conserved across fungal species. Thus, NKp30 interacts with multiple ligands, as do the activating NK receptors NKG2D and DNAM-1 (86, 87).
At present, crystal structures have been determined for NKp30, NKp44, and NKp46 in unbound form (88–90), and for NKp30 bound to B7-H6 (91). NKp44 comprises a single V-type Ig-like domain that features a prominent groove formed by two facing β-hairpin loops (CC′ and FG) projecting from the Ig fold core (Figure 4A) (88). The solvent accessibility of the groove, and its electropositive nature, suggest a possible binding site for anionic ligands, such as sialic acid, although no structure of a complex has been reported. NKp46 consists of two C2-set Ig-like domains whose overall fold and disposition are similar to those of the D1D2 domains of KIRs and LILRs (Figure 4B) (89). This structural resemblance suggests that similar receptor surfaces may be involved in ligand binding. The region of NKp46 analogous to the KIR or LILR ligand-recognition site is located at the interdomain hinge and comprises residues from both Ig-like domains. However, confirmation of this hypothesis awaits structural studies of NKp46–ligand complexes.
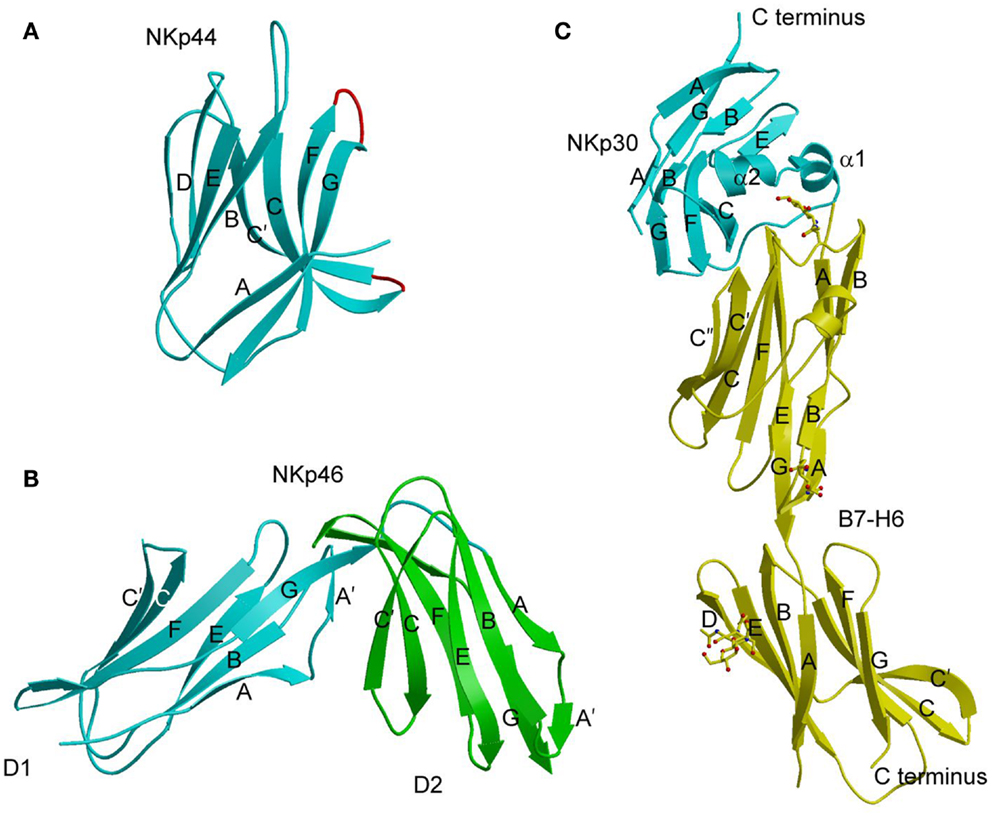
Figure 4. Natural cytotoxicity receptors. (A) Structure of NKp44 (1HKF). The β-strands are labeled. The CC′ and FG loops, drawn in red, define a positively charged surface groove that may serve as a binding site for anionic ligands. (B) Structure of NKp46 (1P6F). D1 is cyan; D2 is green. (C) Structure of NKp30 bound to its tumor cell ligand B7-H6 (3PV6). N-linked glycans at B7-H6 residues Asn43 and Asn57 in the V-like domain and Asn208 in the C-like domain are shown in ball-and-stick representation.
The Ig-like domain of NKp30 exhibits the chain topology found in C1-set domains (Figure 4C) (90, 91). The closest structural homolog of NKp30 is PD-L1, a ligand for PD-1. Like PD-1, NKp30 is a member of the CD28 family, which also includes CTLA-4, ICOS, and B and T lymphocyte attenuator (BTLA) (81). Similar to other B7-family members, the extracellular portion of B7-H6 consists of a V-like and a C-like domain, with the V-like domain distal from the membrane (91).
The structure of the NKp30–B7-H6 complex revealed a binding interface formed by the front β-sheet of the B7-H6 V-like domain and the front and back β-sheets of the NKp30 C-like domain (Figure 4C) (91). The overall architecture of the NKp30–B7-H6 complex differs considerably from those of the PD-1–PD-L1 (or PD-1–PD-L2) (92, 93) and CTLA-4–B7-1 (or CTLA-4–B7-2) complexes (94, 95), as is evident from superposing these complexes (Figures 5A,B). Relative to NKp30, PD-1 and CTLA-4 bind their ligands at angles of ~90° and ~60°, respectively. Whereas the PD-1–PD-L2 and CTLA-4–B7-1 interfaces are dominated by strand-to-strand interactions (Figures 5D,E), B7-H6 engages NKp30 in an antibody-like manner, with greater involvement by the loops of the B7-H6 V-like domain (Figure 5C). Thus, the protruding FG loop of B7-H6, which corresponds to complementarity-determining region (CDR) 3 of antibodies, fits into a deep groove on NKp30, with additional contacts provided by the BC (CDR1-like) and C′C′′(CDR2-like) loops of B7-H6.
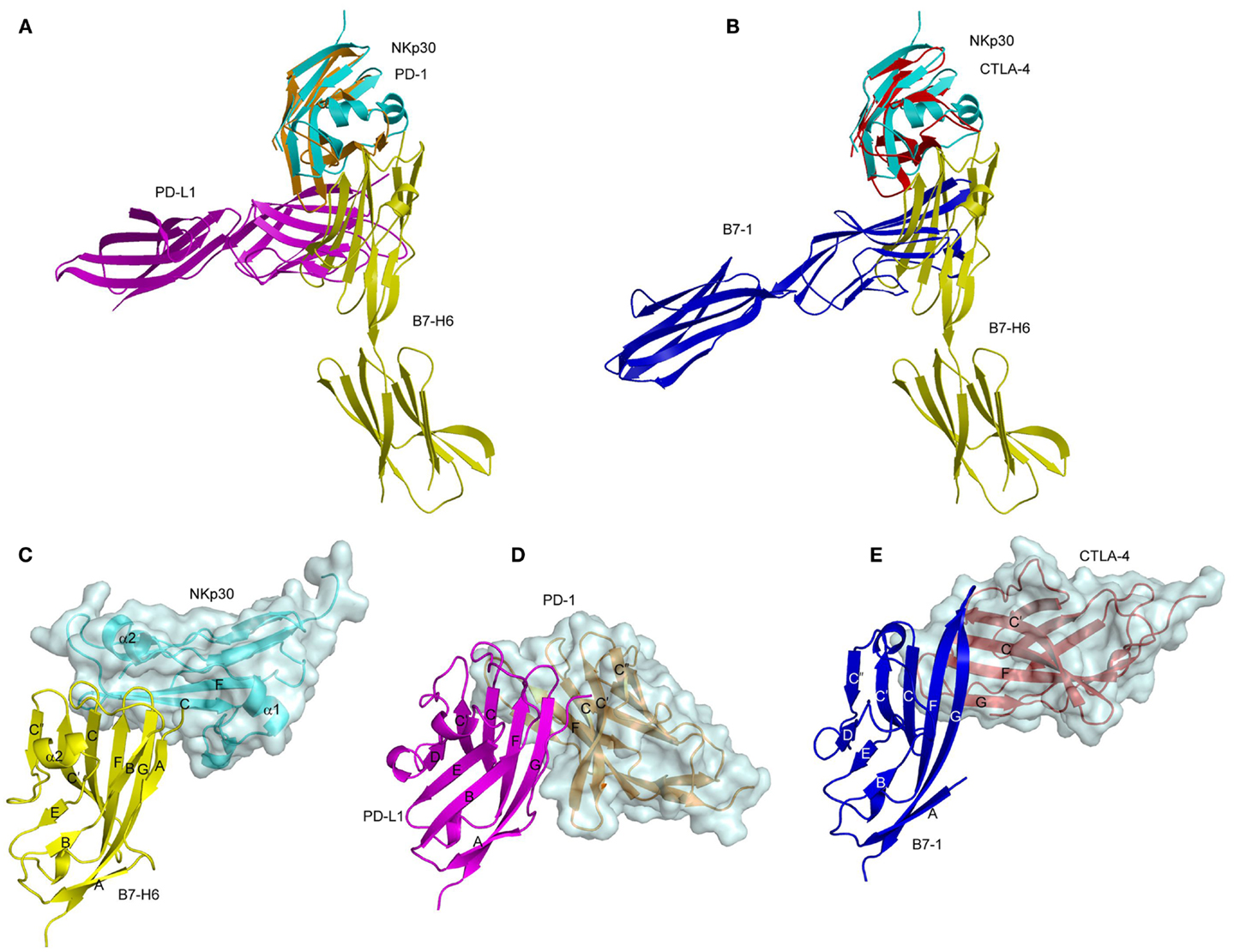
Figure 5. Comparison of the NKp30–B7-H6, PD-1–PD-L1, and CTLA-4–B7-1 complexes. (A) Overlay of the NKp30–B7-H6 (3PV6) and PD-1–PD-L1 (3BIK) complexes by superposing NKp30 (cyan) onto PD-1 (brown). B7-H6 is yellow; PD-L1 is violet. (B) Overlay of the NKp30–B7-H6 and CTLA-4–B7-1 (1I8L) complexes by superposing NKp30 (cyan) onto CTLA-4 (red). B7-H6 is yellow; B7-1 is dark blue. (C–E) Docking modes in the NKp30–B7-H6, PD-1–PD-L1, and CTLA-4–B7-1 complexes. The three complexes were overlaid by superposing the IgV domains of B7-H6 (yellow), PD-1-L1 (violet), and B7-1 (dark blue), then translated horizontally for viewing.
Besides B7-H6, NKp30 recognizes the tegument pp65 protein of HCMV (84) and the nuclear factor BAT3 (80). Remarkably, B7-H6, pp65, and BAT3 appear completely unrelated, both in terms of three-dimensional structure and origin (B7-H6 is a host surface protein; pp65 is a viral structural protein; BAT3 is a host nuclear protein). How NKp30, a relatively small receptor comprising a single Ig-like ectodomain, can bind such disparate ligands is at present a mystery.
MHC-I Recognition by Ly49 Receptors
The highly polymorphic Ly49 receptors are the principle MHC-monitoring molecules on rodent NK cells. In mice, the Ly49 family includes at least 23 members (Ly49A–W), along with multiple allelic variants (96, 97). Most Ly49s inhibit NK-cell-mediated cytolysis upon recognizing one or more H-2D or H-2K alleles (96, 98, 99). However, some Ly49s are activating. In general, Ly49s recognize MHC-I independently of the bound peptide, although Ly49C and Ly49I display considerable peptide specificity (100, 101).
Like NKG2D and NKG2/CD94, Ly49 receptors are members of the C-type lectin-like family of proteins (10, 11). However, none of these NK receptors have a functional calcium-binding site. Ly49s are homodimeric type II transmembrane proteins (N-terminus inside the NK cell), with each chain containing a C-type lectin-like domain (CTLD), known as the natural killer receptor domain (NKD). Each NKD of the Ly49 homodimer is linked by a stalk of ~70 residues to the transmembrane and cytoplasmic domains. Activating and inhibitory receptors differ with regards to their cytoplasmic domains: whereas inhibitory Ly49s transduce signals via immunoreceptor tyrosine-based inhibitory motifs (ITIM), activating Ly49s instead use the associated signaling homodimer DAP12, which possesses an ITAM (96, 98).
Extensive structural information is available for Ly49 receptors in MHC-bound and unbound forms. Crystal structures have been reported for Ly49A NKD in complex with H-2Dd (102), Ly49C NKD bound to H-2Kb (103, 104), Ly49C NKD (104), Ly49I NKD (105), Ly49G2 NKD (104), Ly49L NKD (106), Ly49L NKD with the stalk region (106), and Ly49H bound to the MCMV immunoevasin m157 (107). Together, these structures have revealed the molecular architecture of the MHC-binding site of Ly49 receptors, the basis for MHC-I engagement in trans and cis, and the means by which viral immunoevasins target Ly49s.
The Ly49 NKD is composed of two α-helices, designated α1 and α2, and two anti-parallel β-sheets formed by seven β-strands (Figure 6A). Ly49 receptors exist exclusively as dimers on the NK cell surface. In the dimers, two NKDs associate through strand β0 to create an extended anti-parallel β-sheet. Ly49 dimers may adopt two distinct conformations: “closed” and “open,” as exemplified by Ly49A (Figure 6B) and Ly49C (Figure 6C), respectively. In the closed dimer, the C-terminal ends of the α2 helices are juxtaposed, whereas in the open dimer the α2 helices do not contact each other. As explained below, this variability in Ly49 dimerization geometry serves to modulate the way NK receptors bind MHC (102–104).
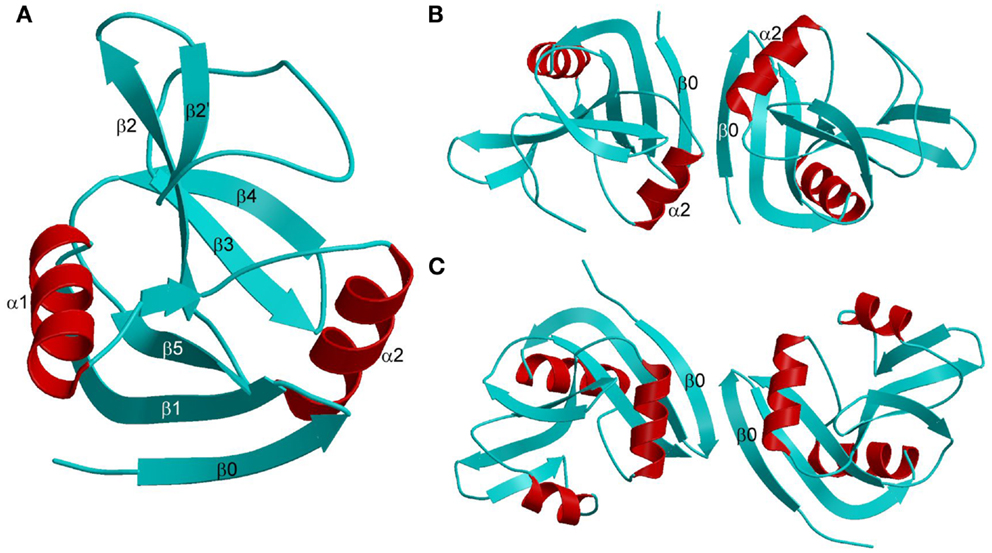
Figure 6. Structures of Ly49 NK receptors. (A) Ribbon drawing of the Ly49A C-type lectin-like domain (1QO3). Secondary structure elements are labeled. β-strands and loops are cyan; α-helices are red. (B) Structure of the “closed” Ly49A homodimer. Secondary structure elements that participate in formation of the dimer interface are labeled. The α2 helices are juxtaposed. (C) Structure of the “open” Ly49C homodimer (3C8J). The α2 helices do not make contact across the dimer interface.
Although Ly49s and other C-type lectin-like NK receptors (e.g., NKG2D, NKG2/CD94, NKp65) retain a lectin-like fold, specific structural features required for lectin activity are absent, enabling these receptors to recognize proteins as opposed to carbohydrates. Most notably, C-type lectin-like NK receptors do not contain bound calcium ions due to missing calcium-coordinating residues.
In the Ly49A–H-2Dd complex (Figure 7A), the Ly49A homodimer engages a single H-2Dd molecule using only one of its subunits, at a site beneath the peptide-binding platform of the MHC-I ligand (102). This site partially overlaps the binding sites for CD8 and LILRB1 (Figure 3A). In the Ly49C–H-2Kb complex (103, 104), by contrast, the Ly49C dimer binds H-2Kb bivalently, with each subunit making identical contacts with MHC-I at a site equivalent to Ly49A binding site on H-2Dd (Figure 7B).
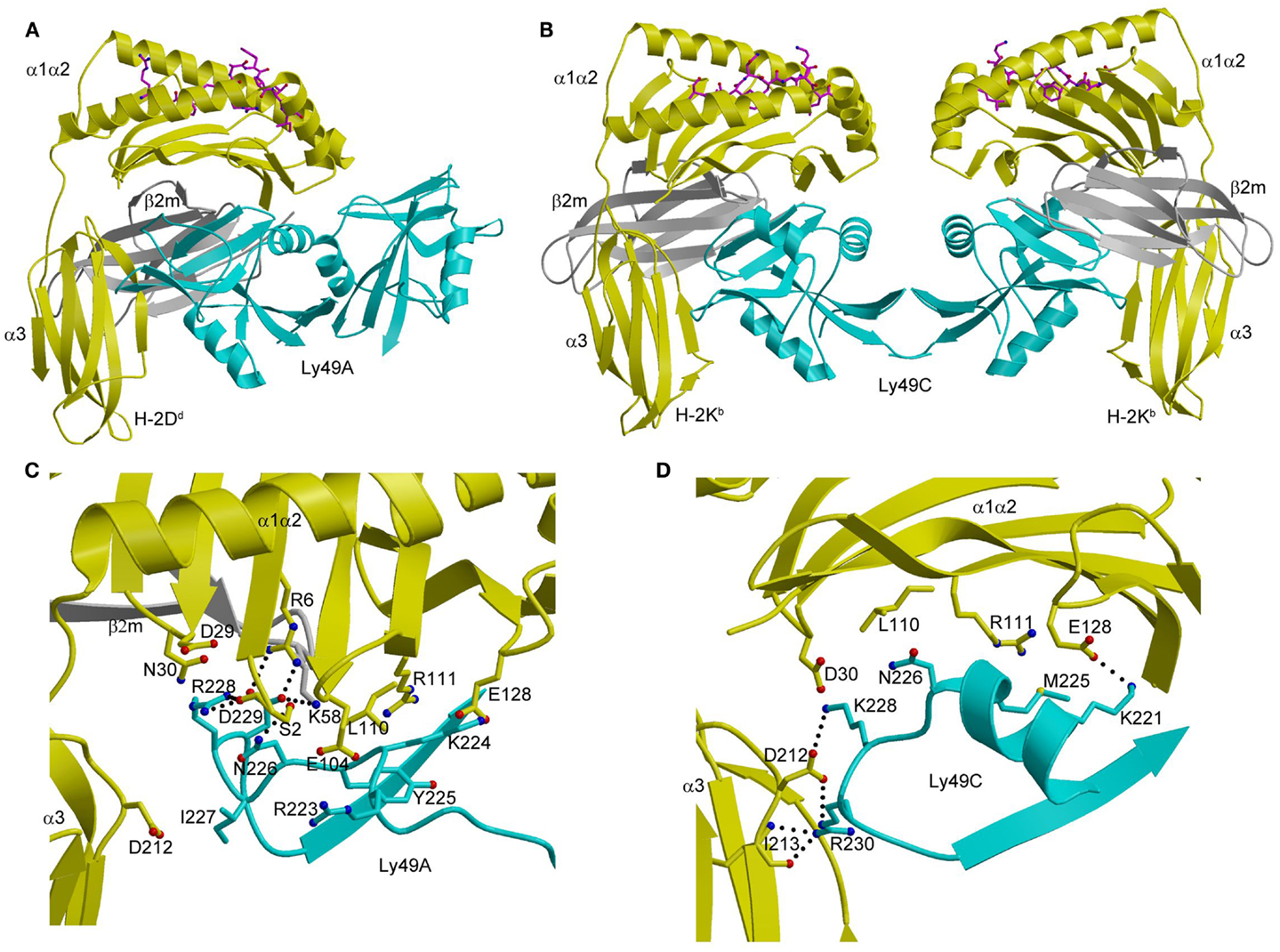
Figure 7. Structures of Ly49–MHC-I complexes. (A) Ribbon diagram of Ly49A bound to H-2Dd (1QO3). The α1, α2, and α3 domains of the MHC-I heavy chain are yellow; β2m is gray; the peptide is magenta; the Ly49A dimer is cyan. In this view, the complex is oriented with the H-2Dd molecule on the target cell at the bottom; the Ly49A homodimer reaches H-2Dd from an opposing NK cell at the top, to which it is connected via stalk regions projecting down to the N-termini of the subunits. (B) Structure of Ly49C in complex with H-2Kb (3C8K). (C) The Ly49A–H-2Dd interface, highlighting interactions made by residues 211–231 of Ly49A. (D) The Ly49C–H-2Kb interface, showing interactions made by the corresponding region of Ly49C. The side chains of interacting residues are drawn in ball-and-stick representation, with carbon atoms in cyan (Ly49A or Ly49C), yellow (H-2Dd or H-2Kb), or gray (β2m), oxygen atoms in red, and nitrogen atoms in blue. Hydrogen bonds are represented by dotted lines.
The different dimerization geometries of Ly49A and Ly49C account for the different modes of MHC engagement in the Ly49A–H-2Dd and Ly49C–H-2Kb complexes. The closed Ly49A dimer cannot simultaneously bind two MHC ligands, like the open Ly49C dimer, because this would result in major steric clashes between MHC molecules, at least in the crystal. However, an NMR study revealed that Ly49A exists predominantly as an open dimer in solution that can bind two MHC-I molecules (108). The most likely interpretation of the combined results from X-ray crystallography and NMR is that Ly49 receptors are present on the NK cell surface in dynamic equilibrium between an open form, which permits bivalent MHC binding, and a closed form, which only allows engagement of one MHC ligand.
As in the case of KIRs (see above), Ly49s display specificity for different MHC alleles. Thus, whereas the promiscuous Ly49C receptor recognizes H-2Kb, H-2Kd, H-2Db, H-2Dd, and H-2Dk, the more specific Ly49A and Ly49C receptors only bind H-2Dd and H-2Dk (97, 101). Ly49s have developed a two-tiered strategy for recognizing MHC, as deduced from the Ly49A–H-2Dd and Ly49C–H-2Kb structures (104). Primary recognition is carried out by a central region consisting of strand β3, loop L5, and strand β4 (residues 232–243). This central region has a conserved structure across Ly49s and contributes most to the binding energetics. Supplementing these primary interactions are secondary ones mediated by a region flanking the central region that comprises residues 218–231. This region, which exhibits high sequence variability across the Ly49 family, confers different MHC specificities. It adopts markedly different conformations in Ly49A (Figure 7C) and Ly49C (Figure 7D), and separates Ly49s into ones that recognize both H-2D and H-2K ligands (e.g., Ly49C) versus ones that only recognize H-2D (e.g., Ly49A) (104).
The finding that MHC recognition by Ly49A is independent of the sequence of the MHC-bound peptide in both cellular and binding assays is readily explained by the total absence of direct contacts between Ly49A and the peptide in the Ly49A–H-2Dd structure (Figure 7A) (102). By contrast, the remarkable peptide selectivity of Ly49C is difficult to understand in terms of the Ly49C–H-2Kb complex, in which there is also a complete lack of contacts between Ly49C and the peptide (Figure 7B) (103, 104). Although the potential biological role of the peptide selectivity of certain Ly49s (and KIRs) remains obscure, the description of a functional interaction between Ly49P and H-2Dk on MCMV-infected cells that confers resistance to the virus suggests an intriguing possibility (17). This interaction requires the participation of the MCMV gene product m04/gp34 (109). This virulence factor associates with MHC-I in the endoplasmic reticulum and travels to the cell surface (110). Although the molecular nature of the Ly49P–H-2Dk interaction on MCMV-infected cells remains to be defined, one possibility is that m04/gp34 provides a specific peptide recognized by Ly49P in an H-2Dk-dependent manner, which would confer on NK cells a degree of specificity for viral pathogens reminiscent of that of cytotoxic T cells (11, 17, 109).
Trans and Cis Interactions of Ly49 Receptors with MHC-I
Cell surface receptors mediate cell-to-cell communication by interacting with ligands expressed on other cells (trans interactions). In addition, some cell surface receptors have been shown to bind ligands expressed on the same cell via cis interactions (60, 111). These include the structurally unrelated NK receptors Ly49 and LILRs, which interact not only with MHC-I molecules on other cells in trans, but also with MHC-I molecules on the same cell in cis (59, 112, 113). Other examples of cell surface receptors that bind ligands in both trans and cis are: siglec-2 (CD22), a negative regulator of B cell receptor signaling that recognizes sialic acid modifications of glycoproteins (114–117); herpes virus entry mediator (HVEM), which is regulated by its ligand BTLA (118); plexin receptors that bind semaphorins (119); and notch receptors that bind Delta (120, 121). The emerging theme from these studies is that cis interactions regulate effector cell function by modulating (decreasing or increasing) the threshold at which cellular activation signals produce a biological response (60, 111).
Cis interactions between Ly49 receptors and MHC-I ligands facilitate NK cell activation (112). This effect is not the result of inhibitory signaling through constitutive ITIM phosphorylation of Ly49s. Rather, cis interactions with MHC-I sequester, or mask, Ly49s to render them physically unavailable for functional trans interactions (113). In this way, cis interactions lower the threshold at which NK cell activation exceeds inhibition, considerably increasing the sensitivity of NK cells to diseased cells (60). Remarkably, in addition to modulating inhibitory function, cis interactions of Ly49A are necessary for NK cell education (122). As in the case of Ly49, the interaction of LILRB/PIR-B receptors with MHC-I in cis is constitutive (59). However, unlike Ly49, the ITIMs of LILRB/PIR-B receptors are phosphorylated constitutively, such that cis binding generates tonic inhibitory signals that dampen NK cell activation.
Trans and cis interactions by Ly49 receptors utilize the same binding site beneath the peptide-binding platform of MHC-I (112). Therefore, Ly49s must drastically reorient their NKDs relative to the NK cell membrane in order to bind MHC-I in trans versus cis. Most likely, it is the exceptionally long stalk regions of Ly49s (~70 residues) that provide the requisite flexibility. In the crystal structure of Ly49L, which includes the C-terminal 40 residues of the stalk region (designated the α3s segment), the stalk is composed of an α-helix and a 12-residue loop linking the helix to the NKD (Figure 8A) (106). However, instead of projecting from the NKD, as is typical for a stalk region, the Ly49L stalk backfolds onto the NKD. In a modeled Ly49–MHC-I complex (Figure 8A), the N-termini of the stalk regions point in a completely opposite direction from the C-termini of the MHC-I molecules. Therefore, Ly49s likely adopt the backfolded conformation to bind MHC-I in trans. On the other hand, cis binding requires the stalks to assume an extended conformation that orients the N-termini of the NKDs toward the NK cell (Figure 8B). Unlike the trans interaction, where one Ly49 dimer engages two MHC-I ligands (Figure 3A), this model precludes cis engagement of both NKDs by MHC-I, because of the orientation that binding of one MHC-I ligand would impose on the Ly49 dimer (Figure 3B). In agreement with the model, biochemical analyses confirmed that trans binding of MHC-I by Ly49 dimers occurs in a bivalent fashion, whereas cis binding is monovalent (106). Moreover, Ly49 receptors appear able to switch between backfolded and extended conformations (108, 123).
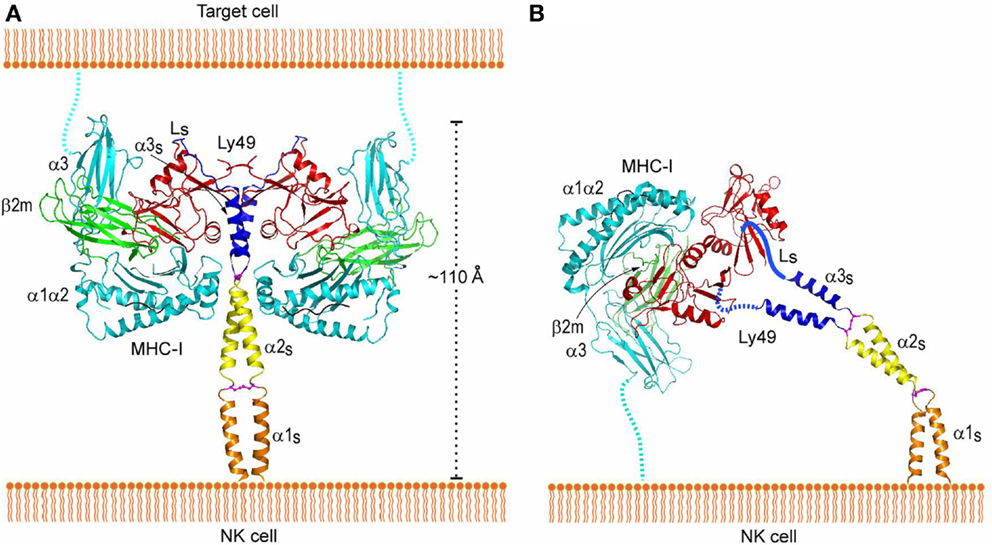
Figure 8. Interaction of Ly49 receptors with MHC-I in trans and cis. (A) Trans interaction of an Ly49 receptor with two MHC-I molecules, based on the structures of Ly49L (3G8L) and the Ly49C–H-2Kb complex (3C8K). The α1, α2, and α3 domains of the MHC-I heavy chain are cyan; β2m is green; Ly49 NKD is red; helix α3S of the Ly49 stalk and loop LS connecting α3S to the NKD are blue; the disulfide bond linking the α3S helices is magenta. The predicted α1S and α2S helices of the stalk are orange and yellow, respectively, with the disulfide bond in magenta. The Ly49 homodimer on the NK cell binds two MHC-I molecules on the target cell. To bind in trans, the stalks must adopt a backfolded conformation, as the N-termini of the Ly49 monomers point away from the NK cell membrane (Ly49s are type II transmembrane proteins). (B) Cis interaction of Ly49 with MHC-I, based on the structure of Ly49L and the Ly49A–H-2Dd complex (1QO3). The LS loops connecting the α3S helices to the NKDs are drawn arbitrarily. The Ly49 homodimer binds one MHC-I molecule on the NK cell itself. In this case, the stalks must assume an extended conformation, as the N-termini of the Ly49 monomers point toward the NK cell. Reproduced with permission from Immunity (106).
Cis–trans binding may typically require major structural changes analogous to those of Ly49. However, the stalk regions of LILRB/PIR-B receptors are much shorter than those of Ly49. Accordingly, the ligand-binding D1 and D2 domains of LILRB would need to reverse direction relative to the surface of the NK cell in order to engage MHC-I in cis. To do so, LILRB would need to bend back on itself and assume a horseshoe-shaped arrangement of the four Ig domains (D1–D4), as observed for the four N-terminal Ig-like domains of Drosophila Dscam (124). Such a large reversal implies considerable flexibility in the segment connecting D2 with D3. In the case of PIB-B, which has two additional membrane-proximal Ig-like domains compared to LILRB, the D4–D5 or D5–D6 connecting segments might provide additional flexibility.
Ly49 Recognition of a Viral Immunoevasin
Studies of the susceptibility of different mouse strains to infection by MCMV were the first to demonstrate direct recognition of a viral pathogen by NK cells that confers host protection (125). Resistance to infection in C57BL/6 mice is mediated by a single genetic locus in the NKC, which contains both inhibitory and activating Ly49 receptors (126). This locus encodes the activating receptor Ly49H, which impairs MCMV replication (127–130). By contrast, BALB/c mice do not restrict MCMV replication because they lack a gene for Ly49H. Subsequent studies revealed that Ly49H binds directly to a viral glycoprotein, the MHC-I homolog m157, which is expressed on MCMV-infected cells (15, 16). It was also discovered that m157 binds not only to the activating receptor Ly49H in resistant mouse strains, but also to the inhibitory receptor Ly49I in susceptible ones, which explains why MCMV would possess a gene that confers a selective disadvantage to its survival (15, 16, 131).
The crystal structure of m157 showed that this immunoevasin has an MHC-like fold, although it does not bind peptides or associate with β2m (132). Surprisingly, m157 binds to the stalk region of Ly49H, rather than the NKDs, which recognize MHC-I (107). Although m157 was well resolved in the Ly49H–m157 structure, only the α3s segment of Ly49H could be seen in the electron density (Figure 9A). The lack of density for the Ly49H NKD implies considerable flexibility of the NKD in the crystal lattice. In agreement with the structure, solution binding measurements using Ly49H constructs lacking the NKD or stalk region showed that binding to m157 was mediated entirely by the α3s stalk segment, and that the NKD made no appreciable contribution to the interaction (107).
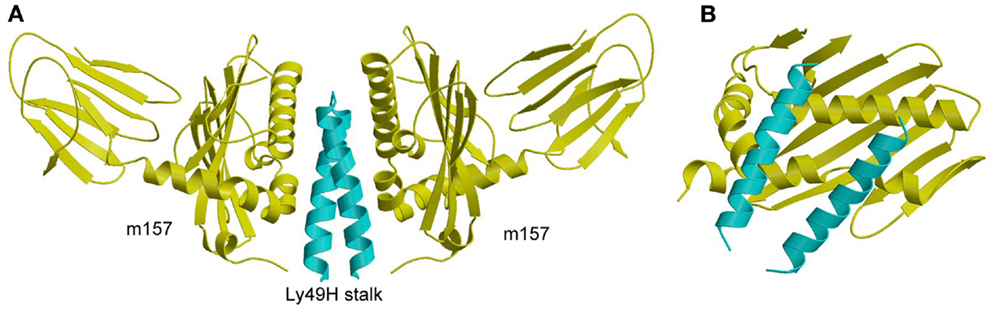
Figure 9. Structure of m157 bound to the stalk region of Ly49H. (A) Side view of the Ly49H–m157 complex (4JO8), in which two m157 monomers (yellow) engage the Ly49H stalk (cyan). Only part of the helical stalk region of Ly49H (the α3s segment) was visible in electron density. The rest of the Ly49H stalk (segments α1s and α2s) and the NKD were not resolved in the structure. (B) Top view of the Ly49H–m157 complex, in which the helical stalks of Ly49H lie across the α1/α2 platform of m157.
In the complex, two m157 monomers engage the Ly49H dimer, such that the helical stalks lie diagonally across the α1/α2 platform of m157 (Figure 9B). This binding mode is completely distinct from that used by Ly49A and Ly49C to engage MHC-I, whereby the NKDs contact MHC-I at a site beneath the α1/α2 peptide-binding platform (Figures 7A,B). Consequently, m157 does not compete with MHC-I for binding to the NKD. Central to the Ly49H–m157 interaction are two exposed aromatic residues in the Ly49H α3s stalk segment (Tyr115 and Trp123) that make extensive contacts with the immunoevasin. The ability of m157 to target some, but not all, members of the Ly49 receptor family can be correlated with sequence differences in the stalk region (107).
As discussed above, Ly49 receptors can adopt two distinct conformations, backfolded or extended (106). However, the recognition mode observed in the Ly49H–m157 complex only appears possible with Ly49 in the extended state, the conformation that recognizes MHC-I in cis (Figure 8B). In the backfolded conformation, by contrast, the Ly49 α3s stalk segment would not be accessible to m157, due to its intimate association with the NKD (Figure 8A). For both the Ly49H–m157 and Ly49I–m157 interactions, kinetic and thermodynamic measurements showed that binding involves a conformational selection mechanism where only the extended conformation of Ly49 is able to bind a first m157 ligand, followed by binding of a second m157 (123). The interaction is characterized by strong positive cooperativity, such that the second m157 binds the Ly49 homodimer 1,000-fold more tightly than the first. The rate-limiting step in the overall mechanism is a conformational transition in Ly49 from its backfolded to extended form.
Ligand Recognition by NKG2D
NKG2D is a homodimeric C-type lectin-like NK receptor that is expressed on NK cells and cytotoxic T cells. It recognizes multiple structural homologs of MHC-I, including MICA, MICB, ULBP13, and RAE-1β, which all lack a peptide-binding groove and β2m (14, 133, 134). ULBP3 and RAE-1β also lack an α3 domain, and are present on the cell surface as glycophosphatidylinositol-linked α1/α2 domains. In humans, expression of MICA and MICB is upregulated in epithelial tumors and stressed cells, compared to little or no expression in normal tissues (135, 136). In rodents, RAE-1, MULT-1, and H-60 are upregulated in tumor cells but not normal cells, similar to MICA and MICB in humans (137, 138). The differential expression pattern of these MHC-related ligands indicates that NKG2D is a key receptor for tumor surveillance by NK cells. In mice, the MCMV-encoded immunoevasins m145, m152, and m155 are involved in downregulating surface expression of the NKG2D ligands MULT-1, RAE-1, and H-60, respectively, thereby thwarting an NKG2D-mediated anti-viral response (63, 139). In humans, the HCMV-encoded immunoevasin UL16 acts as a decoy receptor by binding the NKG2D ligands MICB, ULBP1, and ULBP2 (140). Crystal structures have been reported for human and mouse NKG2D in free form (141, 142), for human NKG2D bound to MICA and ULBP3 (134, 143), and for mouse NKG2D in complex with RAE-1β (144). In addition, structures have been determined for m152 in complex with RAE-1γ (139), and for UL16 bound to MICB (140).
MICA consists of an α1/α2 platform domain, which contains the α1 and α2 helices that define the peptide-binding groove in bona fide MHC-I molecules, and a C-type Ig-like α3 domain (Figure 10A) (145). The NKG2D homodimer binds MICA orthogonally to the α1 and α2 helices of the α1/α2 platform (143). This docking mode roughly resembles that of TCR onto MHC-I, but is completely distinct from the way Ly49C binds MHC-I (Figure 7B). Recognition of the asymmetric MICA ligand by the symmetric NKG2D receptor is mediated by similar sites on the NKG2D subunits that contact distinct sites on MICA. In particular, most (7 of 11) contact residues from each NKG2D monomer are shared by both MICA binding sites.
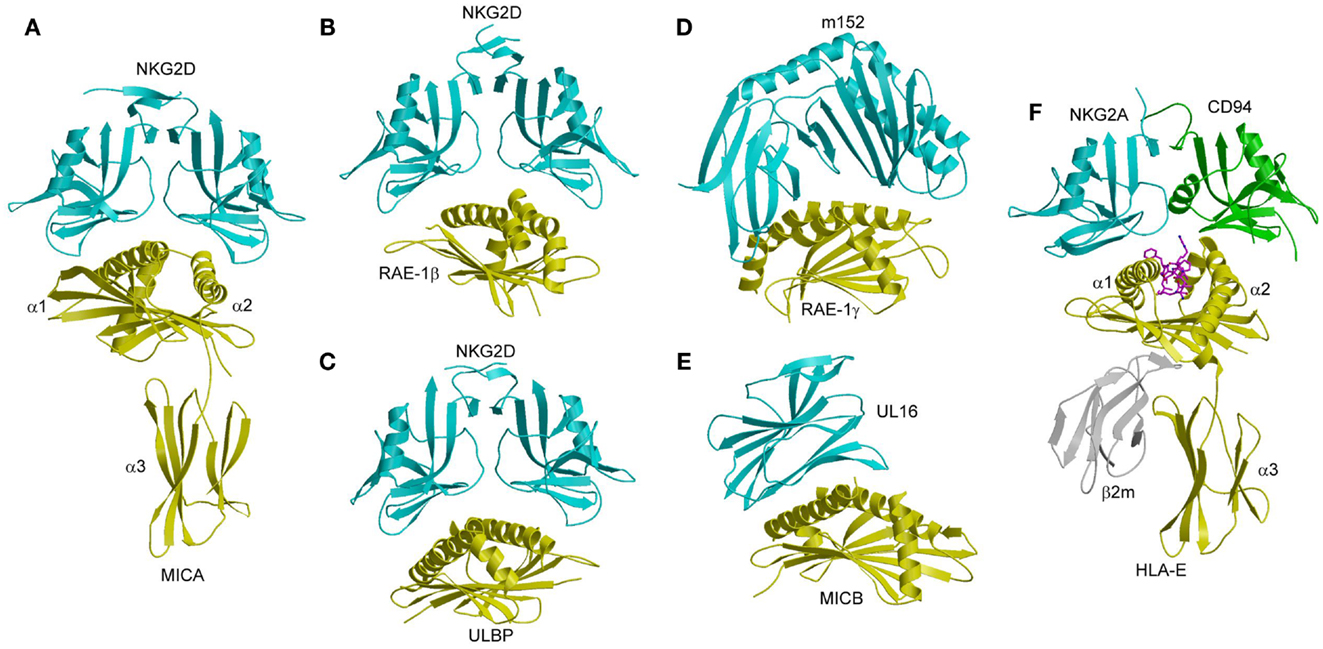
Figure 10. Structures of NKG2D and NKG2A/CD94 complexes. (A) The human NKG2D–MICA complex (1HYR). The two subunits of the NKG2D homodimer are cyan; MICA is yellow. (B) The mouse NKG2D–RAE-1β complex (1JSK). (C) The human NKG2D–ULBP3 complex (1KCG). (D) Structure of the MCMV immunoevasin m152 bound to the NKG2D ligand RAE-1γ (4G59). (E) Structure of the Ig-like HCMV immunoevasin UL16 bound to MICA (2WY3). (F) The human NKG2A/CD94–HLA-E complex (3CDG). The NKG2A and CD94 subunits of the NKG2A/CD94 heterodimer are cyan and green, respectively.
NKG2D binds ULBP3 and RAE-1β orthogonally to the α1/α2 domain of these MHC-like ligands, in a manner resembling the NKG2D–MICA complex (Figures 10B,C) (134, 144). Most notably, a shared subset of NKG2D residues mediates hydrophobic interactions with all three ligands. However, the binding interfaces also display significant differences, such that only one salt bridge and two hydrogen bonds are common to the NKG2D–ULBP3, NKG2D–RAE-1β, and NKG2D–MICA complexes (134, 143, 144).
These structural studies have demonstrated that NKG2D has a remarkable ability to recognize MICA, ULBP3, and RAE-1β using a single binding site, even though these ligands share only ~25% sequence identity. This ability is explained by a rigid adaptation recognition mechanism, rather than induced fit (146). Detailed mutational analysis of NKG2D has shown that the most energetically important residues of the receptor (“hot spots”) interact with relatively conserved residues of MICA, ULBP3, and RAE-1β, without significant conformational changes in NKG2D upon ligand binding (141, 146).
In the MCMV m152–RAE-1γ complex (139), the MHC-I-like immunoevasin binds the α1 and α2 helices of RAE-1γ in a pincer-like manner that resembles the interaction of NKG2D with RAE-1β (Figure 10D). In the HCMV UL16–MICB complex (140), by contrast, the Ig-like UL16 protein uses a three-stranded β-sheet to engage the α1 and α2 helices of MICB, such that residues at the center of the β-sheet mimic a binding motif employed by the structurally unrelated C-type lectin-like NKG2D to bind its diverse ligands (Figure 10E). By competing with NKG2D for ligand binding, m152 and UL16 prevent NKG2D-mediated NK cell activation and thus promote viral survival (147–149).
Recognition of HLA-E by NKG2/CD94 Receptors
In addition to NKG2D, which exists as a homodimer on the NK cell surface, the NKG2D family includes NKG2A, NKG2B, NKG2C, and NKG2E, all of which form obligate heterodimers with CD94 (150–152). NKG2A and NKG2B contain ITIM motifs in their cytoplasmic tails and function as inhibitory receptors; NKG2C and NKG2E associate with the ITAM-containing DAP12 molecule and are activating receptors. The ligand for NKG2/CD94 receptors is the non-classical MHC-I molecule HLA-E, which binds a restricted set of peptides derived from the leader peptides of classical and non-classical MHC-I proteins (150–152). Because HLA-E does not express on the cell surface without a bound peptide, HLA-E expression depends on the production of other MHC-I molecules. Therefore, recognition of HLA-E by NKG2/CD94 receptors enables NK cells to monitor the expression of other HLA class I proteins on cells. This double-check mechanism ensures that cells are producing MHC-I molecules in a normal manner.
The crystal structure of NKG2A/CD94 has been determined in unbound form (153), and in complex with HLA-E bound to a peptide derived from the leader sequence of HLA-G (154, 155). In the complex, NKG2A/CD94 straddles the peptide-binding cleft of HLA-E, with the NKG2A and CD94 subunits mainly interacting with the α2 and α1 helices of HLA-E, respectively (Figure 10F). No significant conformational changes in NKG2A/CD94 or HLA-E occur upon complex formation, indicating a lock-and-key binding mechanism, as in the case of NKG2D (141, 146).
Most (~70%) of the buried surface area in the NKG2A/CD94–HLA-E complex is contributed by the invariant CD94 subunit (154, 155). Thus, CD94 dominates the interaction with HLA-E, whereas NKG2A is more peripheral to the interface. The peptide accounts for ~20% of the buried surface area on the HLA-E side of interface, in which CD94 again dominates the interactions with peptide, albeit with poor shape and chemical complementarity (154). CD94 is positioned over the P8 residue of the peptide, with additional contacts to residue P5. The focus of NKG2A/CD94 on the C-terminal half of the peptide is notable, since nearly all of the limited sequence variation among HLA-E-restricted peptides is concentrated in the C-terminal residues, which are read out primarily by the invariant CD94 subunit.
In sharp contrast to the dominant role of hydrophobic interactions in ligand recognition by NKG2D (134, 144), the NKG2A/CD94–HLA-E interface is mostly electrostatic in nature (154). The interface is characterized by a large number of polar interactions, including 19 hydrogen bonds and 8 salt bridges. This helps explain the fidelity of NKG2A/CD94 for HLA-E compared to the promiscuity of NKG2D for multiple ligands, as discussed above.
Cadherin Recognition by KLRG1
Killer cell lectin-like receptor G1 is a C-type lectin-like inhibitory receptor that contains an ITIM motif in its cytoplasmic region (156, 157). KLRG1 is found on 50–80% of human NK cells, and its expression is highly upregulated following infection with viruses or parasites (158–161). The biological ligand for KLRG1 is E-cadherin (32, 33, 162). E-cadherin, whose extracellular region comprises five Ig-like domains (EC1–EC5), is localized at the basolateral membrane of epithelial cells where it establishes tight binding between neighboring cells in adherens junctions (163). Besides E-cadherin, KLRG1 recognizes N- and R-cadherins (32), which are present in analogous structures in other cell types. The binding of E-cadherin to KLRG1 prevents lysis of E-cadherin-expressing epithelial cells by KLRG1+ NK cells, thereby preventing tissue damage (32, 71, 164). In addition, KLRG1 may play a role in tumor immunosurveillance analogous to missing self-recognition by inhibitory NK receptors that bind MHC-I (Ly49s and KIRs) (164, 165). Because the malignancy of epithelial tumors is frequently associated with down-regulation of E-cadherin, the KLRG1–E-cadherin system may serve to detect potentially metastatic tumors with abnormal cadherin expression (71, 164, 166).
In the crystal structure of the complex between KLRG1 and the EC1 domain of E-cadherin, one KLRG1 CTLD binds one EC1 molecule (Figure 11A) (167). In this respect, KLRG1 recognition of its non-MHC ligand is reminiscent of Ly49 recognition of MHC-I, in which each CTLD monomer contains an entire ligand-binding site (Figure 7B). By contrast, the binding site of NKG2D for the MHC-related ligand MICA (143) (Figure 10A), as well as the binding site of NKG2A/CD94 for HLA-E (154, 155) (Figure 10F), is formed by two CTLD subunits. E-cadherin docks onto a surface of KLRG1 that roughly corresponds to the ligand-binding site of Ly49s and other C-type lectin-like NK receptors (167). This site is formed by three loops (L3, L4, and L6) and β-strand 4 (Figure 11A), which interact primarily with residues Val3–Ile7 of E-cadherin (Figure 11B). These five residues are absolutely conserved in E-, N-, and R-cadherins, which enables NK cells bearing a single KLRG1 receptor to monitor expression of multiple cadherins on target cells, resulting in MHC-independent missing self-recognition.
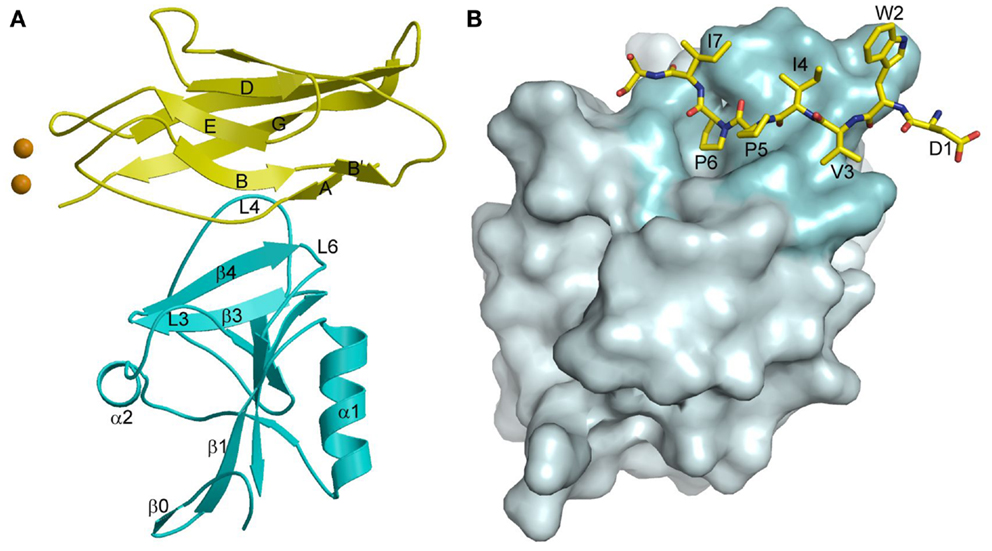
Figure 11. Cadherin recognition by KLRG1. (A) Structure of KLRG1 in complex with the membrane-distal D1 domain of E-cadherin (3FF8). KLRG1 is cyan and E-cadherin is yellow. Secondary structure elements are labeled. Bound Ca2+ ions are drawn as brown spheres. (B) The KLRG1–E-cadherin binding interface. The KLRG1 molecular surface is shown in gray with the region contacting E-cadherin colored cyan. Residues 1–8 of E-cadherin are drawn in stick format and labeled.
The KLRG1–E-cadherin complex buries a total solvent-accessible surface of only 1140 2. This exceptionally small interface is at the lower limit of the average value of 1600 (±400) Å2 for stable protein–protein complexes (168), and likely explains the relatively low affinity of the KLRG1–E-cadherin interaction (KD = 150 μM), which it is considerably weaker than for any other NK receptor–ligand pair characterized to date (167). KLRG1 may compensate for its exceptionally low monomeric affinity for cadherins through multipoint attachment to cadherin molecules on the target cell surface. Additionally, the ability of KLGR1 to form disulfide-linked dimers (169), or even multimers (170), may further increase the avidity of KLRG1–cadherin interactions. In this way, KLRG1–cadherin recognition could be achieved through the cooperativity of multiple associations, rather than by relying on the stability of individual complexes, while still allowing for dissociation of the complexes during transient NK cell–target cell encounters.
Genetically Linked C-Type Lectin-Like Receptor–Ligand Pairs
The NKC encodes approximately 30 type II transmembrane glycoproteins that are members of the C-type lectin-like superfamily (171). NKC genes are divided into killer cell lectin-like receptor (KLR) genes and C-type lectin receptor (CLEC) genes. KLR genes code for molecules expressed on NK cells. CLEC genes code for molecules expressed on other cell types, such as dendritic (CLEC9A) and myeloid (CLEC2B) cells.
The KLR family includes Ly49, NKG2D, and CD94/NKG2A receptors that bind MHC-I or MHC-I-like molecules, as discussed above. The KLR family also includes receptors that recognize non-MHC ligands. This category includes KLRG1, which binds E-cadherin (167) (Figure 11A), in addition to receptors that bind CLEC2 proteins which themselves belong to the C-type lectin-like superfamily (21). The genes encoding these KLR–CLEC2 receptor–ligand pairs are genetically linked in the NKC. For example, in mice, the inhibitory KLR family member receptor Nkrp1d binds Clrb (172, 173). Down-regulation of Clrb expression by genotoxic stress or tumorigenesis triggers NK-cell-mediated lysis, supporting the concept of MHC-independent control of NK cell function by Nkrp1 receptors (173, 174). In humans, the inhibitory NK receptor NKR-P1A binds the CLEC2 family member LLT1, reducing NK-cell-mediated cytotoxicity and interferon-γ secretion (175–177). Viral induction of LLT1 expression in B cells points to a role for the NKR-P1A–LLT1 interaction in modulating immune responses to pathogens (178). The human activating NK receptor NKp80 recognizes the CLEC2 family member AICL, which is genetically coupled to NKp80 in the NKC (21, 31). The NKp80–AICL pair promotes cross-talk between NK cells and monocytes (31). In addition to monocytes, AICL is expressed on monokine-activated human NK cells that also express NKp80, which may enable autonomous control of NK cell responses (179).
Keratinocyte-associated C-type lectin (KACL) is a newly identified member of the human CLEC2 family (180). Notably, KACL is expressed almost exclusively in the skin. KACL is a ligand for the activating receptor NKp65, which is genetically linked to KACL in the NKC (29). Upon binding KACL on keratinocytes, NKp65 triggers NK-mediated cytotoxicity and proinflammatory cytokine release. Thus, the NKp65–KACL interaction may contribute to the immunosurveillance of human skin (21, 29, 181).
The structure of NKp65 bound to KACL has revealed the basis for genetically coupled recognition in the NKC (182). KACL forms a homodimer similar to the NKG2D and Ly49 homodimers; NKp65, contrast, is monomeric (Figure 12A). KACL binds NKp65 bivalently, in a manner resembling the Ly49C–H-2Kb complex (Figure 12B) (103, 104), except that, in the NKp65–KACL complex, it is the ligand (KACL), instead of the receptor (NKp65) that is dimeric. This bivalent binding mode is completely different from those employed by the dimeric NKG2D and Ly49A receptors. Thus, the NKG2D dimer engages one MICA molecule using a single binding site formed by the association of its two subunits (Figure 12C), whereas the Ly49A dimer binds a single H-2Dd ligand using only one subunit (Figure 12D).
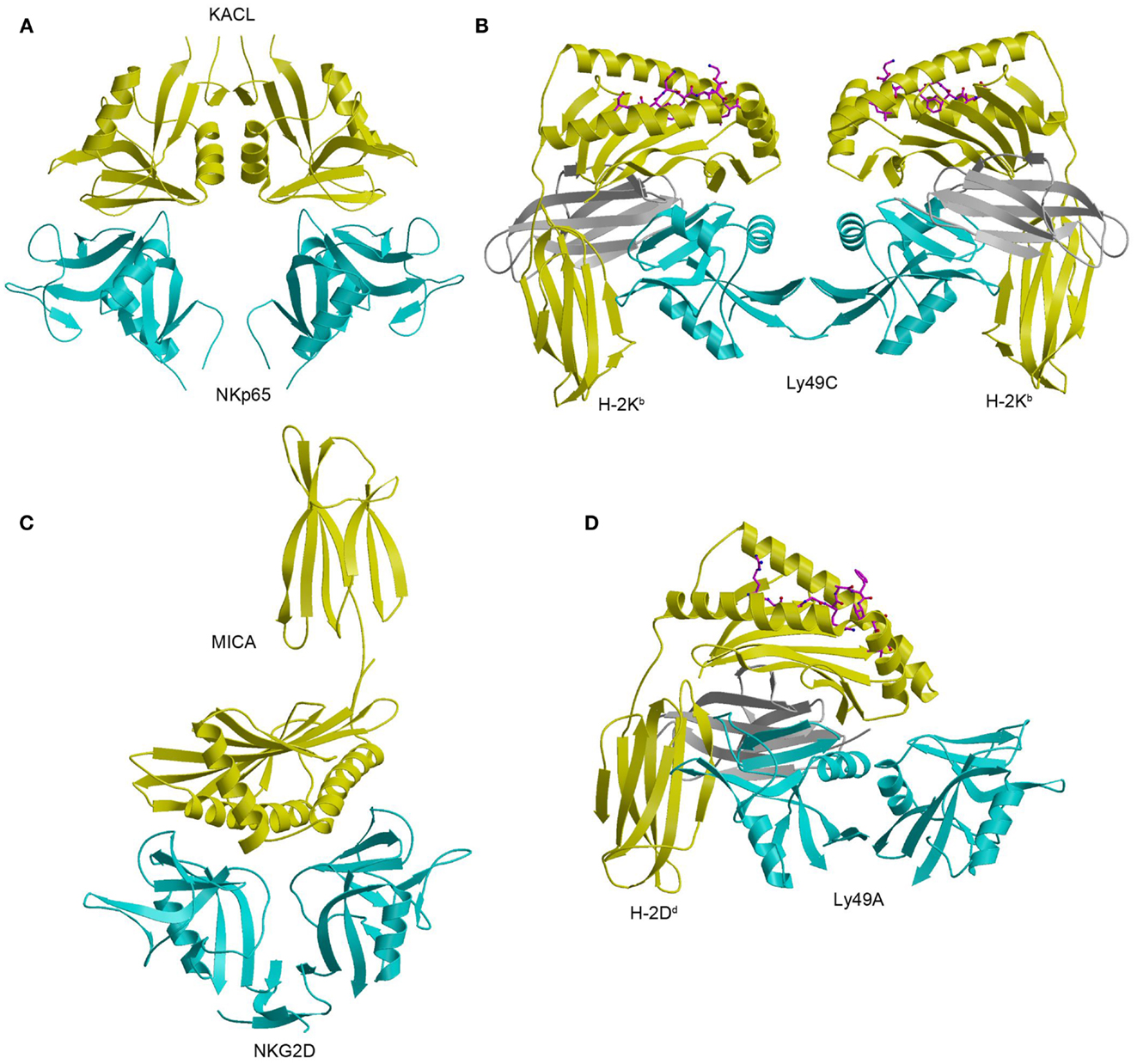
Figure 12. Structure of the NKp65–KACL complex and comparison with other NKC-encoded receptor complexes. (A) Structure of the human NKp65–KACL complex (4IOP). NKp65 is cyan and the KACL dimer is yellow. (B) Structure of the Ly49C–H-2Kb complex (3C8K). The Ly49C dimer is cyan, the H-2Kb heavy chain is yellow, and β2m is gray. (C) Structure of the NKG2D–MICA complex (1HYR). The NKG2D dimer is cyan and MICA is yellow. (D) Structure of the Ly49A–H-2Dd complex (1QO3). The Ly49A dimer is cyan, the H-2Dd is heavy chain is yellow, and β2m is gray.
In the NKp65–KACL complex (182), two C-type lectin-like proteins engage each other in a head-to-head orientation utilizing similar structural elements: NKp65 uses loops L0, L3, L5, and L6 and strands β3 and β4 to contact the analogous loops and strands of KACL (Figure 12A). A mutational analysis of KACL residues in contact with NKp65 showed that all hotspot residues of KACL are conserved or conservatively substituted in AICL and LLT1, and that these hotspot residues contact residues on NKp65, NKp80, and NKR-P1A that are themselves conserved (182). Therefore, the docking mode observed in the NKp65–KACL complex also applies to other NKC-encoded receptor–ligand pairs, including NKp80–AICL, NKR-P1A–LLT1, and Nkrp1–Clr.
NKp65 binds KACL with exceptionally high affinity (KD = 6.7 × 10−10 M), compared to other cell–cell recognition molecules, whose KDs are generally micromolar (183). Indeed, the affinity of NKp65 for KACL is 70,000-fold higher than that of NKR-P1A for LLT1 (184) and 3,000-fold higher than that of NKp80 for AICL (31). In contrast to NKR-P1A and NKp80, which exist as disulfide-linked dimers, NKp65 is not disulfide-linked on the NK cell surface. Likewise, AICL and LLT1 (21, 176, 177, 181), but not KACL (29), form disulfide-linked dimers on cells. Dimerization of NKp80 and NKR-P1A may compensate for the low (micromolar) affinities of these receptors, relative to NKp65, by increasing avidity via bivalent binding of their AICL and LLT1 ligands, which are themselves dimeric. By contrast, the high (nanomolar) affinity of the NKp65–KACL interaction may overcome the need for receptor dimerization by generating complexes of half-life comparable to those of the NKp80–AICL and NKR-P1A–LLT1 complexes, resulting in efficient signaling.
Future Directions
The structural studies described in this review have enabled us to understand how representative NK receptors recognize cellular and viral ligands at the atomic level. However, the biophysical mechanisms by which inhibitory or activating signals are transmitted to the NK cell following ligand engagement remain largely a mystery. It is also unknown how inhibitory and activating signals are integrated within the NK cell to ultimately determine the outcome of NK cell–target cell encounters.
Only recently have structural studies begun to elucidate the molecular details of the signal transduction process. Crucial for NK cell triggering is the association of the transmembrane region of activating NK receptors, such as NKG2D and NKp44, with ITAM-bearing signaling molecules, such as DAP10 and DAP12. NMR has been used to determine the structure of the heterotrimeric assembly formed by the transmembrane regions of NKG2C and DAP12 (185). The main contact site comprises an intricate electrostatic network involving five hydrophilic transmembrane residues: two aspartates and two threonines from the DAP12 dimer that together interact with a lysine from NKG2C. Such studies of membrane-embedded NK receptors, and their association with signaling proteins, promise to provide critical information for linking ligand recognition to NK cell activation or inhibition.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health Grant AI047990 to Roy A. Mariuzza.
References
1. Di Santo JP. Natural killer cells: diversity in search of a niche. Nat Immunol (2008) 9:473–5. doi: 10.1038/ni.f.201
2. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9:503–10. doi:10.1038/ni1582
3. Caliguiri MA. Human natural killer cells. Blood (2008) 112:461–9. doi:10.1182/blood-2007-09-077438
4. Bryceson YT, Chiang SC, Darmanin S, Fauriat C, Schlums H, Theorell J, et al. Molecular mechanisms of natural killer cell activation. J Innate Immun (2011) 3:216–26. doi:10.1159/000325265
5. Vivier E, Raulet DH, Moretta A, Caliguiri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science (2011) 331:44–9. doi:10.1126/science.1198687
6. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol (2008) 9:495–502. doi:10.1038/ni1581
7. Long EO, Sik KH, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol (2013) 31:227–58. doi:10.1146/annurev-immunol-020711-075005
8. Kelley J, Walter L, Trowsdale J. Comparative genomics of natural killer cell receptor gene clusters. PLoS Genet (2005) 1:129–39. doi:10.1371/journal.pgen.0010027
9. Yokoyama WM, Plougastel BFM. Immune functions encoded by the natural killer gene complex. Nat Rev Immunol (2003) 3:304–16. doi:10.1038/nri1055
10. Natarajan K, Dimasi N, Wang J, Mariuzza RA, Margulies DH. Structure and function of natural killer cell receptors: multiple molecular solutions to self, non-self discrimination. Annu Rev Immunol (2002) 20:853–85. doi:10.1146/annurev.immunol.20.100301.064812
11. Deng L, Mariuzza RA. Structural basis for recognition of MHC and MHC-like ligands by natural killer cell receptors. Semin Immunol (2006) 18:159–66. doi:10.1016/j.smim.2006.03.004
12. Joyce MG, Sun PD. The structural basis of ligand recognition by natural killer cell receptors. J Biomed Biotechnol (2011) 2011:203628. doi:10.1155/2011/203628
13. Finton KA, Strong RK. Structural insights into activation of antiviral NK cell responses. Immunol Rev (2012) 250:239–57. doi:10.1111/j.1600-065X.2012.01168.x
14. Mistry AR, O’Callaghan CA. Regulation of ligands for the activating receptor NKG2D. Immunology (2007) 121:439–47. doi:10.1111/j.1365-2567.2007.02652.x
15. Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science (2002) 296:1323–6. doi:10.1126/science.1070884
16. Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci U S A (2002) 99:8826–31. doi:10.1073/pnas.092258599
17. Derosiers M-P, Kielczewska A, Loredi-Osti J-C, Adam SG, Makrigiannis AP, Lemiex S, et al. Epistasis between mouse Klra and major histocompatibility complex class I loci is associated with a new mechanism of natural killer cell-mediated innate resistance to cytomegalovirus infection. Nat Genet (2005) 37:593–9. doi:10.1038/ng1564
18. Lanier LL, Ruitenberg JJ, Phillips JH. Functional and biochemical analysis of CD16 antigen on natural killer cells and granulocytes. J Immunol (1988) 141:3478–85.
19. Moretta A, Poggi A, Pende D, Tripodi G, Orengo AM, Pella N, et al. CD69-mediated pathway of lymphocyte activation: anti-CD69 monoclonal antibodies trigger the cytolytic activity of different lymphoid effector cells with the exception of cytolytic T lymphocytes expressing T cell receptor α/β. J Exp Med (1991) 174:1393–8. doi:10.1084/jem.174.6.1393
20. Plougastel BF, Yokoyama WM. Extending missing self? Functional interactions between lectin-like NKrp1 receptors on NK cells with lectin-like ligands. Curr Top Microbiol Immunol (2006) 298:77–89.
21. Bartel Y, Bauer B, Steinle A. Modulation of NK cell function by genetically coupled C-type lectin-like receptor/ligand pairs encoded in the human natural killer gene complex. Front Immunol (2013) 4:362. doi:10.3389/fimmu.2013.00362
22. Veillette A, Dong Z, Latour S. Consequence of the SLAM-SAP signaling pathway in innate-like and conventional lymphocytes. Immunity (2007) 27:698–710. doi:10.1016/j.immuni.2007.11.005
23. Ma CS, Nichols KE, Tangye SG. Regulation of cellular and humoral responses by the SLAM and SAP families of molecules. Annu Rev Immunol (2007) 25:337–79. doi:10.1146/annurev.immunol.25.022106.141651
24. Chlewicki LK, Velikovsky CA, Balakrishnan V, Mariuzza RA, Kumar V. Molecular basis of the dual function of 2B4 (CD244). J Immunol (2008) 180:8159–67.
25. Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity (1996) 4:573–81. doi:10.1016/S1074-7613(00)70060-4
26. Pende D, Parolini S, Pessino A, Sivori S, Augugliaro R, Morelli L, et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J Exp Med (1999) 190:1505–16. doi:10.1084/jem.190.10.1505
27. Cantoni C, Bottino C, Vitale M, Pessino A, Augugliaro R, Malaspina A, et al. NKp44, a triggering receptor involved in tumor cell lysis by activated human natural killer cells, is a novel member of the immunoglobulin superfamily. J Exp Med (1999) 189:787–96. doi:10.1084/jem.189.5.787
28. Sivori S, Vitale M, Morelli L, Sanseverino L, Augugliaro R, Bottino C, et al. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J Exp Med (1997) 186:1129–36. doi:10.1084/jem.186.7.1129
29. Spreu J, Kuttruff S, Stejfova V, Dennehy KM, Schittek B, Steinle A. Interaction of C-type lectin-like receptors NKp65 and KACL facilitates dedicated immune recognition of human keratinocytes. Proc Natl Acad Sci U S A (2010) 107:5100–5. doi:10.1073/pnas.0913108107
30. Vitale M, Falco M, Castriconi R, Parolini S, Zambello R, Semenzato G, et al. Identification of NKp80, a novel triggering molecule expressed by human NK cells. Eur J Immunol (2001) 31:233–42. doi:10.1002/1521-4141(200101)31:1<233::AID-IMMU233>3.3.CO;2-W
31. Welte S, Kuttruff S, Waldhauer I, Steinle A. Mutual activation of natural killer cells and monocytes mediated by NKp80-AICL interaction. Nat Immunol (2006) 7:1334–42. doi:10.1038/ni1402
32. Ito M, Maruyama T, Saito N, Koganei S, Yamamoto K, Matsumoto N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J Exp Med (2006) 203:289–95. doi:10.1084/jem.20051986
33. Gründemann C, Bauer M, Schweier O, von Oppen N, Lässing U, Saudan P, et al. Cutting edge: Identification of E-cadherin as a ligand for the murine killer cell lectin-like receptor G1 (KLRG1). J Immunol (2006) 176:1311–5.
34. Lebbink RJ, de Ruiter T, Adelmeijer J, Brenkman AB, van Helvoort JM, Koch M, et al. Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J Exp Med (2006) 203:1419–25. doi:10.1084/jem.20052554
35. Fan QR, Mosyak L, Winter CC, Wagtmann N, Long EO, Wiley DC. Structure of the inhibitory receptor for human natural killer cells resembles hematopoietic receptors. Nature (1997) 389:96–100. doi:10.1038/38028
36. Snyder GA, Brooks AG, Sun PD. Crystal structure of the HLA-Cw3 allotype-specific killer cell inhibitory receptor KIR2DL2. Proc Natl Acad Sci U S A (1999) 96:3864–9. doi:10.1073/pnas.96.7.3864
37. Maenaka K, Juji T, Stuart DI, Jones EY. Crystal structure of the human p58 killer cell inhibitory receptor (KIR2DL3) specific for HLA-Cw3-related MHC class I. Structure (1999) 7:391–8. doi:10.1016/S0969-2126(99)80052-5
38. Saulquin X, Gastinel LN, Vivier E. Crystal structure of the human natural killer cell activator receptor KIR2DS2 (CD158j). J Exp Med (2003) 197:933–8. doi:10.1084/jem.20021624
39. Graef T, Moesta AK, Norman PJ, Abi-Rached L, Vago L, Older Aguilar AM, et al. KIR2DS4 is a product of gene conversion with KIR3DL2 that introduced specificity for HLA-A*11 while diminishing avidity for HLA-C. J Exp Med (2009) 206:2557–72. doi:10.1084/jem.20091010
40. Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature (2000) 405:537–43. doi:10.1038/35014520
41. Fan QR, Long EO, Wiley DC. Crystal structure of the human natural killer inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat Immunol (2001) 2:452–60.
42. Vivian JP, Duncan RC, Berry R, O’Connor GM, Reid HH, Beddoe T, et al. Killer cell immunoglobulin-like receptor 3DL1-mediated recognition of human leukocyte antigen B. Nature (2011) 479:401–5. doi:10.1038/nature10517
43. de Vos AM, Ultsch M, Kossiakoff AA. Human growth hormone and extracellular domain of its receptor: crystal structure of the complex. Science (1992) 255:306–12. doi:10.1126/science.1549776
44. Somers W, Ultsch M, De Vos AM, Kossiakoff AA. The X-ray structure of a growth hormone-prolactin receptor complex. Nature (1994) 372:478–81. doi:10.1038/372478a0
45. Rajakopalan S, Long EO. The direct binding of a p58 killer cell inhibitory receptor to human histocompatibility leukocyte antigen (HLA)-Cw4 exhibits peptide selectivity. J Exp Med (1997) 185:1523–8. doi:10.1084/jem.185.8.1523
46. Zappacosta F, Borrego F, Brooks AG, Parker KC, Coligan JE. Peptides isolated from HLA-Cw*0304 confer different degrees of protection from natural killer cell-mediated lysis. Proc Natl Acad Sci U S A (1997) 94:6313–8. doi:10.1073/pnas.94.12.6313
47. Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature (2011) 476:96–100. doi:10.1038/nature10237
48. Yin Y, Li Y, Mariuzza RA. Structural basis for self-recognition by autoimmune T-cell receptors. Immunol Rev (2012) 250:32–48. doi:10.1111/imr.12002
49. Norman PJ, Abi-Rached L, Gendzekhadze K, Korbel D, Gleimer M, Rowley D, et al. Unusual selection on the KIR3DL1/S1 natural killer cell receptor in Africans. Nat Genet (2007) 39:1092–9. doi:10.1038/ng2111
50. Brown D, Trowsdale J, Allen R. The LILR family: modulators of innate and adaptive immune pathways in health and disease. Tissue Antigens (2004) 64:215–25. doi:10.1111/j.0001-2815.2004.00290.x
51. Chapman TL, Haikema AP, Bjorkmann PJ. The inhibitory receptor LIR-1 uses a common binding interaction to recognize class I MHC molecules and the viral MHC homolog UL18. Immunity (1999) 11:603–11. doi:10.1016/S1074-7613(00)80135-1
52. Shiroishi M, Tsumoto K, Amano K, Shirakihara Y, Colonna M, Braud VM, et al. Human inhibitory receptors ILT2 and ILT4 compete with CD8 for MHC class I binding and bind preferentially to HLA-G. Proc Natl Acad Sci U S A (2003) 100:8856–61. doi:10.1073/pnas.1431057100
53. Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, et al. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity (1997) 7:273–82. doi:10.1016/S1074-7613(00)80529-4
54. Chapman TL, Heikema AP, West AP Jr., Bjorkman PJ. Crystal structure and ligand binding properties of the D1D2 region of the inhibitory receptor LIR-1 (ILT-2). Immunity (2000) 13:727–36. doi:10.1016/S1074-7613(00)00071-6
55. Willcox BE, Thomas LM, Bjorkman PJ. Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat Immunol (2003) 4:913–9. doi:10.1038/ni961
56. Yang Z, Bjorkman PJ. Structure of UL18, a peptide-binding viral MHC mimic, bound to a host inhibitory receptor. Proc Natl Acad Sci U S A (2008) 105:10095–100. doi:10.1073/pnas.0804551105
57. Willcox BE, Thomas LM, Chapman TL, Heikema AP, West AP Jr., Bjorkman PJ. Crystal structure of LIR-2 (ILT4) at 1.8 Å: differences from LIR-1 (ILT2) in regions implicated in the binding of the human cytomegalovirus class I MHC homolog UL18. BMC Struct Biol (2002) 2:6. doi:10.1186/1472-6807-2-6
58. Shiroishi M, Kuroki K, Rasubala L, Tsumoto K, Kumagai I, Kurimoto E, et al. Structural basis for recognition of the nonclassical MHC molecule HLA-G by the leukocyte Ig-like receptor B2 (LILRB2/LIR2/ILT4/CD85d). Proc Natl Acad Sci U S A (2006) 103:16412–7. doi:10.1073/pnas.0605228103
59. Masuda A, Nakamura A, Maeda T, Sakamoto Y, Takai T. Cis binding between inhibitory receptors and MHC class I can regulate mast cell activation. J Exp Med (2007) 204:907–20. doi:10.1084/jem.20060631
60. Held W, Mariuzza RA. Cis interactions of immunoreceptors with MHC and non-MHC ligands. Nat Rev Immunol (2008) 8:269–78. doi:10.1038/nri2278
61. Farrell H, Degli-Esposti M, Densley E, Cretney E, Smyth M, Davis-Poynter N. Cytomegalovirus MHC class I homologues and natural killer cells: an overview. Microbes Infect (2000) 2:521–32. doi:10.1016/S1286-4579(00)00315-4
62. Basta S, Bennink JR. A survival game of hide and seek: cytomegaloviruses and MHC class I antigen presentation pathways. Viral Immunol (2003) 16:231–42.
63. Revilleza MJ, Wang R, Mans J, Hong M, Natarajan K, Margulies DH. How the virus outsmarts the host: function and structure of cytomegalovirus MHC-I-like molecules in the evasion of natural killer cell surveillance. J Biomed Biotechnol (2011) 2011:724607. doi:10.1155/2011/724607
64. Beck S, Barrell BG. Human cytomegalovirus encodes a glycoprotein homologous to MHC class I antigens. Nature (1988) 331:269–72. doi:10.1038/331269a0
65. Wagner CS, Ljunggren HG, Achour A. Immune modulation by the human cytomegalovirus-encoded molecule UL18, a mystery yet to be solved. J Immunol (2008) 180:19–24.
66. Fahnestock ML, Johnson JL, Feldman RM, Neveu JM, Lane WS, Bjorkman PJ. The MHC class I homolog encoded by human cytomegalovirus binds endogenous peptides. Immunity (1995) 3:583–90. doi:10.1016/1074-7613(95)90129-9
67. Wagner CS, Rölle A, Cosman D, Ljunggren HG, Berndt KD, Achour A. Structural elements underlying the high binding affinity of human cytomegalovirus UL18 to leukocyte immunoglobulin-like receptor-1. J Mol Biol (2007) 373:695–705. doi:10.1016/j.jmb.2007.08.020
68. Julien J-P, Lee PS, Wilson IA. Structural insights into key sites of vulnerability on HIV-1 Env and influenza HA. Immunol Rev (2012) 250:180–98. doi:10.1111/imr.12005
69. Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol (2001) 19:197–223. doi:10.1146/annurev.immunol.19.1.197
70. Moretta L, Bottino C, Pende D, Castriconi R, Mingari MC, Moretta A. Surface NK receptors and their ligands on tumor cells. Semin Immunol (2006) 18:151–8. doi:10.1016/j.smim.2006.03.002
71. Bryceson YT, Long EO. Line of attack: NK cell specificity and integration of signals. Curr Opin Immunol (2008) 20:344–52. doi:10.1016/j.coi.2008.03.005
72. Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature (2001) 409:1055–60. doi:10.1038/35059110
73. De Maria A, Fogli M, Mazza S, Basso M, Picciotto A, Costa P, et al. Increased natural cytotoxicity receptor expression and relevant IL-10 production in NK cells from chronically infected viremic HCV patients. Eur J Immunol (2007) 37:445–55. doi:10.1002/eji.200635989
74. Hershkovitz O, Rosental B, Rosenberg LA, Navarro-Sanchez ME, Jivov S, Zilka A, et al. NKp44 receptor mediates interaction of the envelope glycoproteins from the West Nile and dengue viruses with NK cells. J Immunol (2009) 183:2610–21. doi:10.4049/jimmunol.0802806
75. Fuller CL, Ruthel G, Warfield KL, Swenson DL, Bosio CM, Aman MJ, et al. NKp30-dependent cytolysis of filovirus-infected human dendritic cells. Cell Microbiol (2007) 9:962–76. doi:10.1111/j.1462-5822.2006.00844.x
76. Ho JW, Hershkovitz O, Peiris M, Zilka A, Bar-Ilan A, Nal B, et al. H5-type influenza virus hemagglutinin is functionally recognized by the natural killer-activating receptor NKp44. J Virol (2008) 82:2028–32. doi:10.1128/JVI.02065-07
77. Jarahian M, Fiedler M, Cohnen A, Djandji D, Hämmerling GJ, Gati C, et al. Modulation of NKp30- and NKp46-mediated natural killer cell responses by poxviral hemagglutinin. PLoS Pathog (2011) 7:e1002195. doi:10.1371/journal.ppat.1002195
78. Hecht ML, Rosental B, Horlacher T, Hershkovitz O, De Paz JL, Noti C, et al. Natural cytotoxicity receptors NKp30, NKp44 and NKp46 bind to different heparan sulfate/heparin sequences. J Proteome Res (2009) 8:712–20. doi:10.1021/pr800747c
79. Baychelier F, Sennepin A, Ermonval M, Dorgham K, Debré P, Vieillard V. Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood (2013) 122:2935–42. doi:10.1182/blood-2013-03-489054
80. Pogge von Strandmann E, Simhadri VR, von Tresckow B, Sasse S, Reiners KS, Hansen HP, et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity (2007) 27:965–74. doi:10.1016/j.immuni.2007.10.010
81. Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med (2009) 206:1495–503. doi:10.1084/jem.20090681
82. Sasaki T, Gan EC, Wakeham A, Kornbluth S, Mak TW, Okada H. HLA-B-associated transcript 3 (Bat3)/Scythe is essential for p300-mediated acetylation of p53. Genes Dev (2007) 21:848–61. doi:10.1101/gad.1534107
83. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol (2008) 8:467–77. doi:10.1038/nri2326
84. Arnon TI, Achdout H, Levi O, Markel G, Saleh N, Katz G, et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat Immunol (2005) 6:515–23. doi:10.1038/ni1190
85. Li SS, Kyei SK, Timm-McCann M, Ogbomo H, Jones GJ, Shi M, et al. The NK receptor NKp30 mediates direct fungal recognition and killing and is diminished in NK cells from HIV-infected patients. Cell Host Microbe (2013) 14:387–97. doi:10.1016/j.chom.2013.09.007
86. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol (2003) 3:781–90. doi:10.1038/nri1199
87. Soriani A, Zingoni A, Cereboni C, Iannitto ML, Ricciardi MR, DiGialleonardo V, et al. ATM-ATR dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK cell susceptibility and is associated with a senescent phenotype. Blood (2009) 113:3503–11. doi:10.1182/blood-2008-08-173914
88. Cantoni C, Ponassi M, Biossoni R, Conte R, Spallarossa A, Moretta A, et al. The three-dimensional structure of the human NK cell receptor NKp44, a triggering partner in natural cytotoxicity. Structure (2003) 11:725–34. doi:10.1016/S0969-2126(03)00095-9
89. Foster CE, Colonna M, Sun PD. Crystal structure of the human natural killer (NK) cell activating receptor NKp46 reveals structural relationship to other leukocyte receptor complex immunoreceptors. J Biol Chem (2003) 278:46081–6. doi:10.1074/jbc.M308491200
90. Joyce MG, Tran P, Zhuravleva MA, Jaw J, Colonna M, Sun PD. Crystal structure of human natural cytotoxicity receptor NKp30 and identification of its ligand binding site. Proc Natl Acad Sci U S A (2011) 108:6223–8. doi:10.1073/pnas.1100622108
91. Li Y, Wang Q, Mariuzza RA. Structure of the human activating natural cytotoxicity receptor NKp30 bound to its tumor cell ligand B7-H6. J Exp Med (2011) 208:703–14. doi:10.1084/jem.20102548
92. Lin DY, Tanaka Y, Iwasaki M, Gittis AG, Su H-P, Mikami B, et al. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc Natl Acad Sci U S A (2008) 105:3011–6. doi:10.1073/pnas.0712278105
93. Lazar-Molnar E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc Natl Acad Sci U S A (2008) 105:10483–8. doi:10.1073/pnas.0804453105
94. Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, et al. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature (2001) 410:608–11. doi:10.1038/35069118
95. Schwartz J-CD, Zhang X, Federov AA, Nathenson SG, Almo SC. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature (2001) 410:604–7. doi:10.1038/35069112
96. Plougastel BFM, Yokoyama WM. Immune functions encoded by the natural killer gene complex. Nat Rev Immunol (2003) 3:304–16. doi:10.1038/nri1055
97. Anderson SK, Ortaldo JR, McVicar DW. The ever-expanding Ly49 gene family: repertoire and signaling. Immunol Rev (2001) 181:79–89. doi:10.1034/j.1600-065X.2001.1810106.x
98. Lanier LL. NK cell recognition. Annu Rev Immunol (2005) 23:225–74. doi:10.1146/annurev.immunol.23.021704.115526
99. McFall E, Tu MM, Al-Khattabi N, Tai L-H, St-Laurent AS, Tzankova V, et al. Optimized tetramer analysis reveals Ly49 promiscuity for MHC ligands. J Immunol (2013) 191:5722–9. doi:10.4049/jimmunol.1300726
100. Franksson L, Sundbäck J, Achour A, Bernlind J, Glas R, Kärre K. Peptide dependency and selectivity of the NK inhibitory receptor Ly-49C. Eur J Immunol (1999) 29:2748–58. doi:10.1002/(SICI)1521-4141(199909)29:09<2748::AID-IMMU2748>3.3.CO;2-3
101. Hanke T, Takizawa H, McMahon CW, Busch DH, Pamer EG, Miller JD, et al. Direct assessment of MHC class I binding by seven Ly49 inhibitory NK cell receptors. Immunity (1999) 11:67–77. doi:10.1016/S1074-7613(00)80082-5
102. Tormo J, Natarajan K, Margulies DH, Mariuzza RA. Crystal structure of a lectin-like natural killer cell receptor bound to its MHC class I ligand. Nature (1999) 402:623–31. doi:10.1038/45170
103. Dam J, Guan R, Natarajan K, Dimasi N, Chlewicki LK, Kranz DM, et al. Variable MHC class I engagement by Ly49 NK receptors revealed by the crystal structure of Ly49C bound to H-2Kb. Nat Immunol (2003) 4:1213–22. doi:10.1038/ni1006
104. Deng L, Cho S, Malchiodi EL, Kerzic MC, Dam J, Mariuzza RA. Molecular architecture of the major histocompatibility complex class I-binding site of Ly49 natural killer cell receptors. J Biol Chem (2008) 283:16840–9. doi:10.1074/jbc.M801526200
105. Dimasi N, Sawicki MW, Reineck LA, Li Y, Natarajan K, Margulies DH, et al. Crystal structure of the Ly49I natural killer cell receptor reveals variability in dimerization mode within the Ly49 family. J Mol Biol (2002) 320:573–85. doi:10.1016/S0022-2836(02)00498-9
106. Back J, Malchiodi EL, Cho S, Scarpellino L, Schneider P, Kerzic MC, et al. Distinct conformations of Ly49 natural killer cell receptors mediate MHC class I recognition in trans and cis. Immunity (2009) 31:598–608. doi:10.1016/j.immuni.2009.07.007
107. Berry R, Ng N, Saunders PM, Vivian JP, Lin J, Deuss FA, et al. Targeting of a natural killer cell receptor family by a viral immunoevasin. Nat Immunol (2013) 14:699–705. doi:10.1038/ni.2605
108. Dam J, Baber J, Grishaev A, Malchiodi EL, Schuck P, Bax A, et al. Variable dimerization of the Ly49A natural killer cell receptor results in differential engagement of its MHC class I ligand. J Mol Biol (2006) 362:102–13. doi:10.1016/j.jmb.2006.07.005
109. Kielczewska A, Pyzik M, Sun T, Krmpotic A, Lodoen MB, Munks MW, et al. Ly49P recognition of cytomegalovirus-infected cells expressing H2-Dk and CMV-encoded m04 correlates with the NK cell antiviral response. J Exp Med (2009) 206:515–23. doi:10.1084/jem.20080954
110. Kavanagh DG, Gold MC, Wagner M, Koszinowski UH, Hill AB. The multiple immune-evasion genes of murine cytomegalovirus are not redundant: m4 and m152 inhibit antigen presentation in a complementary and cooperative fashion. J Exp Med (2001) 194:967–78. doi:10.1084/jem.194.7.967
111. Held W, Mariuzza RA. Cis-trans interactions of cell surface receptors: biological roles and structural basis. Cell Mol Life Sci (2011) 68:3469–78. doi:10.1007/s00018-011-0798-z
112. Doucey M-A, Scarpellino L, Zimmer J, Guillaume P, Luescher IF, Bron C, et al. Cis association of Ly49A with MHC class I restricts natural killer cell inhibition. Nat Immunol (2004) 5:328–36. doi:10.1038/ni1043
113. Back J, Chalifour A, Scarpellino L, Held W. Stable masking by H-2Dd cis ligand limits Ly49A relocalization to the site of NK cell/target cell contact. Proc Natl Acad Sci U S A (2007) 104:3978–83. doi:10.1073/pnas.0607418104
114. Razi N, Varki A. Masking and unmasking of the sialic acid-binding lectin activity of CD22 (Siglec-2) on B lymphocytes. Proc Natl Acad Sci U S A (1998) 95:7469–74. doi:10.1073/pnas.95.13.7469
115. Collins BE, Blixt O, Bovin NV, Danzer CP, Chui D, Marth JD, et al. Constitutively unmasked CD22 on B cells of ST6Gal I knockout mice: novel sialoside probe for murine CD22. Glycobiology (2002) 12:563–71. doi:10.1093/glycob/cwf067
116. Poe JC, Fujimoto Y, Hasegawa M, Haas KM, Miller AS, Sanford IG, et al. CD22 regulates B lymphocyte function in vivo through both ligand-dependent and ligand-independent mechanisms. Nat Immunol (2004) 5:1078–87. doi:10.1038/ni1121
117. Collins BE, Blixt O, DeSieno AR, Bovin N, Marth JD, Paulson JC. Masking of CD22 by cis ligands does not prevent redistribution of CD22 to sites of cell contact. Proc Natl Acad Sci U S A (2004) 101:6104–9. doi:10.1073/pnas.0400851101
118. Cheung TC, Oborne LM, Steinberg MW, Macauley MG, Fukuyama S, Sanjo H, et al. T cell intrinsic heterodimeric complexes between HVEM and BTLA determine receptivity to the surrounding microenvironment. J Immunol (2009) 183:7286–96. doi:10.4049/jimmunol.0902490
119. Haklai-Topper L, Mlechkovich G, Savariego D, Gokhman I, Yaron A. Cis interaction between semaphorin-6A and plexin-A4 modulates the repulsive response to Sema6A. EMBO J (2010) 29:2635–45. doi:10.1038/emboj.2010.147
120. Cordle J, Johnson S, Tay JZ, Roversi P, Wilkin MB, de Madrid BH, et al. A conserved face of the jagged/serrate DSL domain is involved in notch trans-activation and cis-inhibition. Nat Struct Mol Biol (2008) 15:849–57. doi:10.1038/nsmb.1457
121. Sprinzak D, Lakhanpal A, Lebon L, Santat LA, Fontes ME, Anderson GA, et al. Cis-interactions between notch and Delta generate mutually exclusive signalling states. Nature (2010) 465:86–90. doi:10.1038/nature08959
122. Chalifour A, Scarpellino L, Back J, Brodin P, Devevre E, Gros F, et al. A role for cis interaction between the inhibitory Ly49A receptor and MHC class I for NK cell education. Immunity (2009) 30:337–47. doi:10.1016/j.immuni.2008.12.019
123. Romasanta PN, Curto LM, Urtasun N, Sarratea MB, Chiappini S, Miranda MV, et al. A positive cooperativity binding model between Ly49 natural killer cell receptors and the viral immunoevasin m157: kinetic and thermodynamic studies. J Biol Chem (2014) 289:5083–96. doi:10.1074/jbc.M113.532929
124. Meijers R, Puettmann-Holgado R, Skiniotis G, Liu JH, Walz T, Wang JH, et al. Structural basis of Dscam isoform specificity. Nature (2007) 449:487–91. doi:10.1038/nature06147
125. Lanier LL. Evolutionary struggles between NK cells and viruses. Nat Rev Immunol (2008) 8:259–68. doi:10.1038/nri2276
126. Daniels KA, Devora G, Lai WC, O’Donnell CL, Bennett M, Walsh RM. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J Exp Med (2001) 194:29–44. doi:10.1084/jem.194.1.29
127. Lee S-H, Girard S, Macina D, Busa M, Zafer A, Belouchi A, et al. Susceptibility to mouse cytomegalovirus is associated with depletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nat Genet (2001) 28:42–5. doi:10.1038/88247
128. Lee S-H, Zafer A, de Repentigny Y, Kothary R, Tremblay ML, Gros P, et al. Transgenic expression of the activating natural killer cell receptor Ly49H confers resistance to cytomegalovirus in genetically susceptible mice. J Exp Med (2003) 197:515–26. doi:10.1084/jem.20021713
129. Voigt V, Forbes CA, Tonkin JN, Degli-Esposti MA, Smith HR, Yokoyama WM, et al. Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc Natl Acad Sci U S A (2003) 100:13483–8. doi:10.1073/pnas.2233572100
130. French AR, Pingel JT, Wagner M, Bubic I, Yang L, Kim S, et al. Escape of mutant double-stranded DNA virus from innate immune control. Immunity (2004) 20:747–56. doi:10.1016/j.immuni.2004.05.006
131. Corbett AJ, Coudert JD, Forbes CA, Scalzo AA. Functional consequences of natural sequence variation of murine cytomegalovirus m157 for Ly49 receptor specificity and NK cell activation. J Immunol (2011) 186:1713–22. doi:10.4049/jimmunol.1003308
132. Adams EJ, Juo ZS, Venook RT, Boulanger MJ, Arase H, Lanier LL, et al. Structural elucidation of the m157 mouse cytomegalovirus ligand for Ly49 natural killer cell receptors. Proc Natl Acad Sci U S A (2007) 104:10128–33. doi:10.1073/pnas.0703735104
133. Vivier E, Tomasello E, Paul P. Lymphocyte activation via NKG2D: towards a new paradigm in immune recognition? Curr Opin Immunol (2002) 14:306–11. doi:10.1016/S0952-7915(02)00337-0
134. Radaev S, Rostro B, Brooks AG, Colonna M, Sun P. Conformational plasticity revealed by the co-crystal structure of the activating NK receptor NKG2D and its MHC-like ligand ULBP. Immunity (2001) 15:1039–49. doi:10.1016/S1074-7613(01)00241-2
135. Groh V, Steinle A, Bauer S, Spies T. Recognition of stress-induced MHC molecules by intestinal epithelial γδ T cells. Science (1998) 279:1737–40. doi:10.1126/science.279.5357.1737
136. Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived γδ T cells of MICA and MICB. Proc Natl Acad Sci U S A (1999) 96:6879–84. doi:10.1073/pnas.96.12.6879
137. Cerwenka A, Bakker AB, McClanahan T, Wagner J, Wu J, Phillips JH, et al. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity (2000) 12:721–7. doi:10.1016/S1074-7613(00)80222-8
138. Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol (2000) 1:119–26. doi:10.1038/77793
139. Wang R, Natarajan K, Revilleza MJ, Boyd LF, Zhi L, Zhao H, et al. Structural basis of mouse cytomegalovirus m152/gp40 interaction with RAE1γ reveals a paradigm for MHC/MHC interaction in immune evasion. Proc Natl Acad Sci U S A (2012) 109:E3578–87. doi:10.1073/pnas.1214088109
140. Müller S, Zocher G, Steinle A, Stehle T. Structure of the HCMV UL16- MICB complex elucidates select binding of a viral immunoevasin to diverse NKG2D ligands. PLoS Pathog (2010) 6:e1000723. doi:10.1371/journal.ppat.1000723
141. McFarland BJ, Kortemme T, Yu SF, Baker D, Strong RK. Symmetry recognizing asymmetry: analysis of the interactions between the C-type lectin-like immunoreceptor NKG2D and MHC class I-like ligands. Structure (2003) 11:411–22. doi:10.1016/S0969-2126(03)00047-9
142. Wolan DW, Teyton L, Rudolph MG, Villmow B, Bauer S, Busch DH, et al. Crystal structure of the murine NK cell-activating receptor NKG2D at 1.95 Å. Nat Immunol (2001) 2:248–54. doi:10.1038/85311
143. Li P, Morris DL, Willcox BE, Steinle A, Spies T, Strong RK. Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat Immunol (2001) 2:443–51.
144. Li P, McDermott G, Strong RK. Crystal structure of RAE-1β and its complex with the activating immunoreceptor NKG2D. Immunity (2002) 16:77–86. doi:10.1016/S1074-7613(02)00258-3
145. Li P, Willie ST, Bauer S, Morris DL, Spies T, Strong RK. Crystal structure of the MHC class I homolog MICA, a γδ T cell ligand. Immunity (1999) 10:577–84. doi:10.1016/S1074-7613(00)80057-6
146. McFarland BJ, Strong RK. Thermodynamic analysis of degenerate recognition by the NKG2D immunoreceptor: not induced fit but rigid adaptation. Immunity (2003) 19:803–12. doi:10.1016/S1074-7613(03)00320-0
147. Lodoen M, Ogasawara K, Hamerman JA, Arase A, Houchins JP, Mocarski ES, et al. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med (2003) 197:1245–53. doi:10.1084/jem.20021973
148. Lodoen MB, Abenes G, Umamoto S, Houchins JP, Liu F, Lanier LL. The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J Exp Med (2004) 200:1075–81. doi:10.1084/jem.20040583
149. Krmpotic A, Hasan M, Loewendorf A, Saulig T, Halenius A, Lenac T, et al. NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by mouse cytomegalovirus gene m145. J Exp Med (2005) 201:211–20. doi:10.1084/jem.20041617
150. Borrego F, Ulbrecht M, Weiss EH, Coligan JE, Brooks AG. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J Exp Med (1998) 187:813–8. doi:10.1084/jem.187.5.813
151. Braud VM, Allan DS, O’Callaghan CA, Söderström K, D’Andrea A, Ogg GS, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature (1998) 391:795–9.
152. Lee N, Llano M, Carretero M, Ishitani A, Navarro F, López-Botet M, et al. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A (1998) 95:5199–204. doi:10.1073/pnas.95.9.5199
153. Sullivan LC, Clements CS, Beddoe T, Johnson D, Hoare HL, Lin J, et al. The heterodimeric assembly of the CD94-NKG2 receptor family and implications for human leukocyte antigen-E recognition. J Exp Med (2008) 205:725–35.
154. Petrie EJ, Clements CS, Lin J, Sullivan LC, Johnson D, Huyton T, et al. CD94-NKG2A recognition of human leukocyte antigen (HLA)-E bound to an HLA class I leader sequence. J Exp Med (2008) 205:725–35. doi:10.1084/jem.20072525
155. Kaiser BK, Pizarro JC, Kerns J, Strong RK. Structural basis for NKG2A/CD94 recognition of HLA-E. Proc Natl Acad Sci U S A (2008) 105:6696–701. doi:10.1073/pnas.0802736105
156. Guthman MD, Tal M, Pecht I. A secretion inhibitory signal transduction molecule on mast cells is another C-type lectin. Proc Natl Acad Sci U S A (1995) 92:9397–401. doi:10.1073/pnas.92.20.9397
157. Hanke T, Corral L, Vance RE, Raulet DH. 2F1 antigen, the mouse homolog of the rat “mast cell function-associated antigen”, is a lectin-like type II transmembrane receptor expressed by natural killer cells. Eur J Immunol (1998) 28:4409–17. doi:10.1002/(SICI)1521-4141(199812)28:12<4409::AID-IMMU4409>3.0.CO;2-3
158. Voehringer D, Blaser P, Brawand P, Raulet DH, Hanke T, Pircher H. Viral infections induce abundant numbers of senescent CD8 T cells. J Immunol (2001) 167:4838–43.
159. Robbins SH, Terrizzi SC, Sydora BC, Mikayama T, Brossay L. Differential regulation of killer cell lectin-like receptor G1 expression on T cells. J Immunol (2003) 170:5876–85.
160. Thimme R, Appay V, Koschella M, Panther E, Roth E, Hislop AD, et al. Increased expression of the NK cell receptor KLRG1 by virus-specific CD8 T cells during persistent antigen stimulation. J Virol (2005) 79:12112–6. doi:10.1128/JVI.79.18.12112-12116.2005
161. Ibegbu CC, Xu YX, Harris W, Maggio D, Miller JD, Koutis AP. Expression of killer cell lectin-like receptor G1 on antigen-specific human CD8+ T lymphocytes during active, latent, and resolved infection and its relation with CD57. J Immunol (2005) 174:6088–94.
162. Tessmer MS, Fugere C, Stevenaert F, Naidenko OV, Chong HJ, Leclercq G, et al. KLRG1 binds cadherins and preferentially associates with SHIP-1. Int Immunol (2007) 4:391–400. doi:10.1093/intimm/dxm004
163. Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol (2005) 6:622–34. doi:10.1038/nrm1699
164. Colonna M. Cytolytic responses: cadherins put out the fire. J Exp Med (2006) 203:261–4. doi:10.1084/jem.20052559
165. Schwartzkopff S, Gründemann C, Schweier O, Rosshart S, Karjalainen KE, Becker K-F, et al. Tumor-associated E-cadherin mutations affect binding to the killer cell lectin-like receptor G1 in humans. J Immunol (2007) 179:1022–9.
166. Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene (2008) 24:6920–9. doi:10.1038/onc.2008.343
167. Li Y, Hofmann M, Wang Q, Teng L, Chlewicki LK, Pircher H, et al. Structure of natural killer cell receptor KLRG1 bound to E-cadherin reveals basis for MHC-independent missing self recognition. Immunity (2009) 31:35–46. doi:10.1016/j.immuni.2009.04.019
168. Lo Conte LL, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol (1999) 285:2177–98. doi:10.1006/jmbi.1998.2439
169. Corral L, Hanke T, Vance RE, Cado D, Raulet DH. NK cell expression of the killer cell lectin-like receptor G1 (KLRG1), the mouse homolog of MAFA, is modulated by MHC class I molecules. Eur J Immunol (2000) 30:920–30. doi:10.1002/1521-4141(200003)30:3<920::AID-IMMU920>3.3.CO;2-G
170. Rosshart S, Hofmann M, Schweier O, Pfaff A-K, Yoshimoto K, Takeuchi T, et al. Interaction of KLRG1 with E-cadherin: new functional and structural insights. Eur J Immunol (2008) 38:3354–64. doi:10.1002/eji.200838690
171. Hao L, Klein J, Nei M. Heterogeneous but conserved natural killer receptor gene complexes in four major orders of mammals. Proc Natl Acad Sci U S A (2006) 103:3192–7. doi:10.1073/pnas.0511280103
172. Iizuka K, Naidenko OV, Plougastel BF, Fremont DH, Yokoyama WM. Genetically linked C-type lectin-related ligands for the NKRP1 family of natural killer cell receptors. Nat Immunol (2003) 4:801–7. doi:10.1038/ni954
173. Carlyle JR, Jamieson AM, Gasser S, Clingan CS, Arase H, Raulet DH. Missing self recognition of Ocil/Clr-b by inhibitory NKR-P1 natural killer cell receptors. Proc Natl Acad Sci U S A (2004) 101:3527–32. doi:10.1073/pnas.0308304101
174. Fine JH, Chen P, Mesci A, Allan DS, Gasser S, Raulet DH, et al. Chemotherapy-induced genotoxic stress promotes sensitivity to natural killer cell cytotoxicity by enabling missing self recognition. Cancer Res (2010) 70:7102–13. doi:10.1158/0008-5472.CAN-10-1316
175. Aldemir H, Prod’homme V, Dumaurier MJ, Retiere C, Poupon G, Cazareth J, et al. Cutting edge: lectin-like transcript 1 is a ligand for the CD161 receptor. J Immunol (2005) 175:7791–5.
176. Rosen DB, Bettadapura J, Alsharifi M, Mathew PA, Warren HS, Lanier LL. Cutting edge: lectin-like transcript-1 is a ligand for the inhibitory human NKR-P1A receptor. J Immunol (2005) 175:7796–9.
177. Rosen DB, Cao W, Avery DT, Tangye SG, Liu YJ, Houchins JP, et al. Functional consequences of interactions between human NKR-P1A and its ligand LLT1 expressed on activated dendritic cells and B cells. J Immunol (2008) 180:6508–17.
178. Germain C, Meier A, Jensen T, Knapnougel P, Poupon G, Lazzari A, et al. Induction of lectin-like transcript 1 (LLT1) protein cell surface expression by pathogens and interferon-γ contributes to modulate immune responses. J Biol Chem (2011) 286:37964–75. doi:10.1074/jbc.M111.285312
179. Klimosch SN, Bartel Y, Wiemann S, Steinle A. Genetically coupled receptor-ligand pair NKp80-AICL enables autonomous control of human NK cell responses. Blood (2013) 122:2380–99. doi:10.1182/blood-2013-01-479790
180. Spreu J, Kienle EC, Schrage B, Steinle A. CLEC2A: a novel, alternatively spliced and skin-associated member of the NKC-encoded AICL-CD69-LLT1 family. Immunogenetics (2007) 59:903–12. doi:10.1007/s00251-007-0263-1
181. Vogler I, Steinle A. Vis-à-vis in the NKC: genetically linked natural killer cell receptor/ligand pairs in the natural killer gene complex (NKC). J Innate Immun (2011) 3:227–35. doi:10.1159/000324112
182. Li Y, Wang Q, Chen S, Brown PH, Mariuzza RA. Structure of NKp65 bound to its keratinocyte ligand reveals basis for genetically linked recognition in natural killer gene complex. Proc Natl Acad Sci U S A (2013) 110:11505–10. doi:10.1073/pnas.1303300110
183. Davis SJ, Ikemizu S, Evans EJ, Fugger L, Bakker TR, van der Merwe PA. The nature of molecular recognition by T cells. Nat Immunol (2003) 4:217–24. doi:10.1038/ni0303-217
184. Kamishikiryo J, Fukuhara H, Okabe Y, Kuroki K, Maenaka K. Molecular basis for LLT1 protein recognition by human CD161 protein (NKRP1A/KLRB1). J Biol Chem (2011) 286:23823–30. doi:10.1074/jbc.M110.214254
Keywords: NK receptor, MHC, virus, KIR, Ly49, NKG2, structure
Citation: Li Y and Mariuzza RA (2014) Structural basis for recognition of cellular and viral ligands by NK cell receptors. Front. Immunol. 5:123. doi: 10.3389/fimmu.2014.00123
Received: 02 February 2014; Accepted: 10 March 2014;
Published online: 26 March 2014.
Edited by:
Simona Sivori, University of Genoa, ItalyReviewed by:
Hugh Thomson Reyburn, Spanish National Research Council, SpainRoberto Biassoni, Istituto Giannina Gaslini, Italy
Copyright: © 2014 Li and Mariuzza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roy A. Mariuzza, Institute for Bioscience and Biotechnology Research, University of Maryland, 9600 Gudelsky Drive, Rockville, MD 20850, USA e-mail:cm1hcml1enpAdW1kLmVkdQ==