 Tianlei Ying1*
Tianlei Ying1* Tina W. Ju
Tina W. Ju Ponraj Prabakaran
Ponraj Prabakaran Dimiter S. Dimitrov
Dimiter S. Dimitrov- 1Protein Interactions Group, Cancer and Inflammation Program, National Cancer Institute, National Institutes of Health, Frederick, MD, USA
- 2Basic Science Program, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, MD, USA
Antibody fragments are emerging as promising biopharmaceuticals because of their relatively small-size and other unique properties. However, when compared to full-size antibodies, most of the current antibody fragments of VH or VL display greatly reduced half-lives. A promising approach to overcome this problem is through the development of novel antibody fragments based on IgG Fc region, which contributes to the long half-life of IgG through its unique pH-dependent association with the neonatal Fc receptor (FcRn). The IgG Fc region comprises two CH2 and two CH3 domains. In this report, we present a comparative study of the FcRn binding capability of the CH2 and CH3 domains. The stability and aggregation resistance of these domains were also investigated and compared. We found that monomeric CH2 and CH3 domains exhibited the pH-dependent FcRn binding while the dimeric forms of CH2 and CH3 domains did not. Although all of these domains had high serum stability, they had aggregation tendencies as measured by dynamic light scattering. By providing a better understanding of the structure–activity relationship of the Fc fragment, these results guide further approaches to generate novel Fc-based small-size antibody fragments that possess pH-dependent FcRn binding capability, desired in vivo half-lives, and other favorable biophysical properties for their druggability.
Introduction
Therapeutic antibodies are the fastest growing class of protein biopharmaceuticals, accounting for over half of therapeutic protein sales in 2010. Monoclonal antibodies (mAbs), in particular, are enjoying significant success in the clinic and have been used for the effective treatment of a number of diseases such as cancer and immune disorders (1). Currently, the vast majority of Food and Drug Administration (FDA)-approved therapeutic antibodies and those in clinical trials are full-size antibodies in IgG1 format (2). Due to their large size (~150 kDa), they exhibit poor penetration in tissues and limited binding to surface targets that could be accessible by molecules of smaller size. Therefore, antibody fragments – with a reduced size of 12–50 kDa – have the potential to overcome the fundamental limitations of full-size antibodies. Due to their smaller size, they could achieve enhanced tissue penetration and a wider range of possible targets, as well as require lower production costs (3–5). However, compared to IgG format antibodies, they display greatly reduced half-lives and as such have limited clinical potential.
IgG1 antibodies exhibit extremely long half-lives (up to 3 weeks in circulation) and broad biodistribution due to their unique pH-dependent association with the neonatal Fc receptor (FcRn). The FcRn is an intracellular receptor localized in the acidic compartments of endosomes (6). By fluid-phase endocytosis, circulating IgGs enter endosomes, the first step of the proteolytic degradation pathway. However, at the low pH of the endosome (pH 6.0), IgGs form a complex with FcRn and are then recycled to the cell surface. The higher physiological pH of the circulation (pH 7.4) triggers the release of IgGs from their complex with FcRn back into circulation (7). Therefore, a promising approach to improve half-life is the development of antibody fragments based on the IgG Fc region, which comprises the unit necessary for binding to FcRn. Fc-based fragments can be used to create Fc-fusion proteins and novel antibody scaffolds.
Fc-based fragments have proven to be successful in enhancing the half-life of many drug candidates that alone would have been in circulation for a scale of minutes to hours rather than weeks. These Fc fusions are composed of the effector domain of a macromolecule linked to the IgG Fc region, which confers a prolonged half-life and better production due to its interactions with FcRn and its relative stability, respectively (8). The first Fc-fusion etanercept (Enbrel™) was introduced in 1998 for the treatment of rheumatoid arthritis (9) and later a broad range of autoimmune diseases. As a tumor necrosis factor (TNF) inhibitor, Enbrel™ is composed of the TNF receptor fused to the Fc region of IgG1. Since its introduction, this class of therapeutic has grown rapidly, as the FDA has approved several Fc fusions for clinical use.
Fc-derived antibody fragments can also serve as novel antibody scaffolds that exhibit both effector functions and binding to FcRn (10–14). Moreover, the loop regions at the C-terminal of its CH3 regions can be engineered to form new antigen-binding sites (15). The Fc antigen-binding (Fcab) small-size antibody format developed by F-star has been shown to possess not only properties similar to that of full-size antibodies but also the added benefit of lower production costs. However, to further reduce the size of possible scaffolds, the IgG Fc region can be divided into two CH2 regions and two CH3 domains. These domains have the potential to be engineered into even smaller antibody fragments.
We have previously engineered monomeric CH2 (16), CH3 (11), and Fc-based fragments (12). Here, we present a comparative study of the FcRn binding capability of the monomeric and dimeric CH2 and CH3 domains. Both monomeric and dimeric CH2 and CH3 domains had high serum stability but also aggregation tendencies at different levels. Among these, we found that monomeric CH2 and CH3 domains exhibited the pH-dependent FcRn binding. Further, structural modeling studies of these domains in complex with FcRn were performed to understand the molecular basis of the FcRn binding. There results could be helpful to generate novel Fc region-based small-size antibody fragments that possess pH-dependent FcRn binding capability, desired in vivo half-lives, and other biophysical properties.
Materials and Methods
Antibody Expression and Purification
All antibody fragments, including Fc, mFc, dCH2, CH2, dCH3, mCH3, were expressed in E. coli HB2151, as described previously (11, 12, 17). The bacterial pellet was collected after centrifugation at 6,000 × g for 15 min, and resuspended in PBS (pH 7.4) containing 0.5 M NaCl and 0.5 milliunits/mL polymyxin B (Sigma-Aldrich). After 30 min incubation at room temperature, the bacterial pellet was subsequently clarified by centrifugation at 25,000 × g for 30 min at 4°C. The clarified supernatant was used for purification of antibody fragments by using HisTrap HP column (GE Healthcare) according to manufacturer’s protocols. Protein purity was determined by SDS-PAGE, and protein concentration was measured spectrophotometrically (NanoVue, GE Healthcare).
Dynamic Light Scattering
For comparison of aggregation tendency, the antibody fragments were filtered through a 0.22-μm filter (Millipore, Bedford, MA, USA) and adjusted to the same concentration (0.25 mg/mL). Measurements were performed using a Zetasizer Nano ZS ZEN3600 (Malvern Instruments Limited, Westborough, MA, USA) to determine the size of protein particles. The samples (0.5 mL) were analyzed in polystyrene cuvettes with a path length of 10 mm at 25°C. Each sample was recorded three times with seven subruns of 10 s. The same samples were also used to run size exclusion chromatography (SEC) using FPLC AKTA BASIC pH/C system (GE Healthcare) with a Superdex 75 10/300 GL column. PBS (pH 7.4) was used as running buffer (flow rate 0.5 mL/min), and eluting proteins were monitored at 280 nm. The molecular mass standards used were ribonuclease A (13.7 kDa), chymotrypsinogen A (25 kDa), ovalbumin (44 kDa), bovine serum albumin (67 kDa), and aldolase (158 kDa), as described previously.
Serum Stability
The stabilities of dCH2, CH2, dCH3, and mCH3 in serum were evaluated for up to 12 days. Normal human serum was collected from healthy human donors approved by the NCI-Frederick Research Donor Program. Aliquots (0.1 mL) of each antibody fragment solution (12 μM) were mixed with 0.5 mL of normal human serum to give a final concentration of 2 μM. The mixture was passed through a 0.22-μm filter (Millipore, Bedford, MA, USA), and then incubated at 37°C. An aliquot was taken out at each time point and immediately stored at −80°C. After 10 days incubation, Western blots were performed to check the serum stability. Samples were electrophoresed through SDS-PAGE and transferred onto 0.2 μm nitrocellulose membranes (Bio-Rad). After transfer, membranes were blocked with 3% milk in PBS for 1 h and then incubated with anti-His Tag monoclonal antibody (ABM, Vancouver, BC, Canada). Membranes were washed and then incubated with anti-mouse IgG–alkaline phosphatase antibody (Sigma-Aldrich). After washing, immune complexes were detected by reaction with BCIP/NBT substrate solution (Sigma-Aldrich).
FcRn Binding Measured by ELISA
Recombinant human single-chain soluble FcRn (sFcRn), containing both β and α chains in a 1:1 molar ratio, was expressed in mammalian cells and purified as described previously (18). ELISA wells were coated with sFcRn at 50 ng per well in PBS (pH 7.4) overnight at 4°C, and blocked with protein-free blocking buffer (Thermo Scientific) at 37°C for 2 h. Twofold serially diluted protein, prepared with PBS containing 0.2% BSA, pH 6.0 or PBS containing 0.2% BSA, pH 7.4, was added and incubated at 37°C for 1.5 h. The plates were washed with PBST (PBS plus 0.05% Tween-20), pH 6.0 or pH 7.4, and horseradish peroxidase (HRP)-conjugated anti-FLAG tag antibody (Sigma-Aldrich) in PBS (pH 6.0 or 7.4) was incubated with wells for 45 min at 37°C. After extensive washes with PBST (pH 6.0 or 7.4), the binding was detected by the addition of ABTS substrate (Roche, Indianapolis, IN, USA), and monitored at 405 nm.
Homology-Modeling and Energy Minimization
The three dimensional (3D) models of human FcRn, mFc, mCH2, and mCH3 were predicted by using the structural components from the crystal structure of rat FcRn–Fc complex (19) (PBD entry: 1I1A) as templates. Homology-modeling was carried out using SWISS-MODEL workspace (20), a fully automated protein structure homology-modeling server accessible via the ExPASy web server. After the generation of initial coordinates, each of the resulting models was refined by energy minimization using “Not just A Molecular Dynamics” (NAMD) program (21). The structures were solvated in a spherical box of water molecules using VMD program, with a minimal distance of 3 Å from the protein to the box boundary of sphere, and all the histidine residues were assigned as protonated. Systems were first minimized 5,000 steps, followed by a molecular dynamics simulation with the temperature gradually dropped from 350 to 100 K. The system was then re-minimized for 50,000 steps with conjugate gradient method. The non-bonded interaction cutoff distance was set to be 9 Å. The final minimized structures were taken for molecular docking.
Molecular Docking and Interface Analysis
Docking of FcRn and Fc-based fragments were performed using the ZDOCK module (22) on Discovery Studio 2.5 (Accelrys Inc.), which uses a grid-based representation of two proteins and a 3D fast Fourier transform (FFT) to effectively explore the rigid body search space of docking positions. The top ranked complex prediction, formed by the receptor FcRn and the ligand mFc, CH2, or mCH3, were solvated and subjected to further dynamic simulations run for 250 ps, with other parameters the same as above, to allow adjustment of the positions of side-chains and helices. The final minimized complex structures were analyzed by using VMD software (23) and PDBePISA (24), an interactive tool for the exploration of macromolecular interfaces at the European Bioinformatics Institute.
Results and Discussion
Aggregation Propensities of Fc, mFc, dCH2, CH2, dCH3, and mCH3 Fragments
We used dynamic light scattering (DLS) to measure the aggregation tendencies of Fc-based fragments consisting of Fc, CH2, and CH3 in monomeric and dimeric forms at 37°C. We observed that both dimeric Fc and dCH3 had a major peak at a smaller size followed by a minor peak at a larger size indicating the predominance of monomeric species with the lesser extent of soluble oligomers (Figure 1). In contrast, the peaks at the larger size were increased for mFc, mCH3, dCH2, and CH2 reflecting the formation of large soluble oligomers of these fragments. Interestingly, the supernatants from most of the fragments were still monomeric according to the results obtained by using SEC (data not shown). Such findings indicate that the formation of the soluble oligomers may be reversible and the oligomers disassociate to monomers in diluted condition when running SEC.
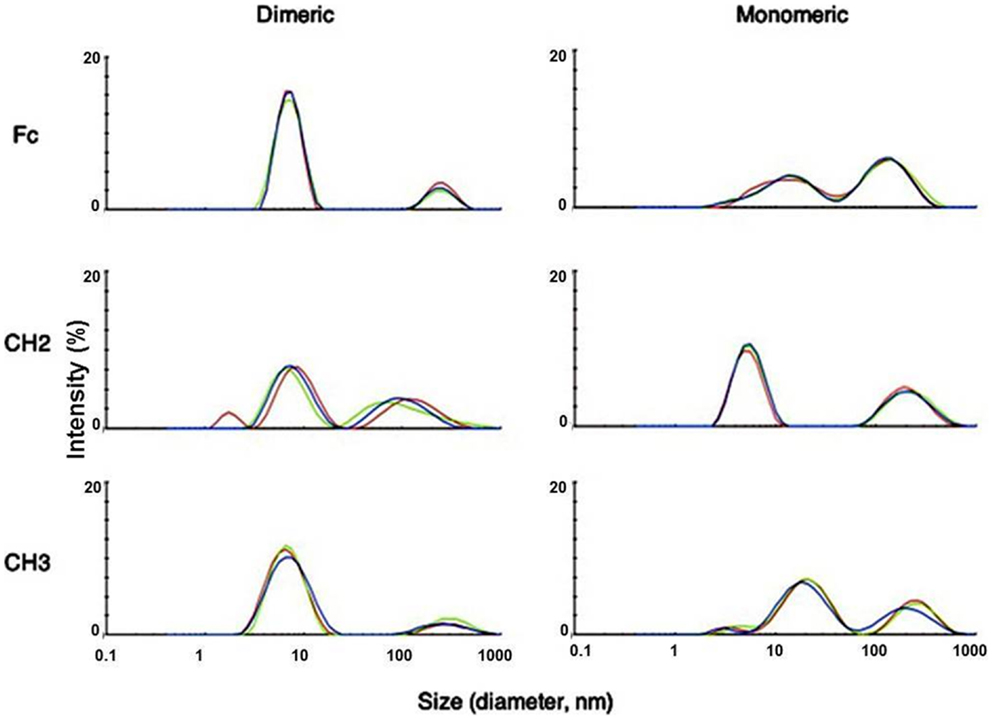
Figure 1. Measurement of aggregation for IgG1 Fc fragments at 37°C by DLS with the X-axis denoting protein particle size (diameter, nanometer) and the Y-axis intensity (%). Three different colors depict three different measurements of the same fragments. Major peaks observed at different sizes indicated possible oligomer formation in these fragments after purification.
Serum Stability of dCH2, MCH2, dCH3, and CH3 Fragments
We evaluated the stability of dCH2, mCH2, dCH3, and CH3 fragments in serum. Samples were incubated with human serum at 37°C, and an aliquot was taken out at each time point, for every other day, and stored at −80°C before Western blot (Figure 2) analyses for up to 12 days. These data suggest that dimeric and monomeric fragments of CH2 and CH3 domains have high serum stability.
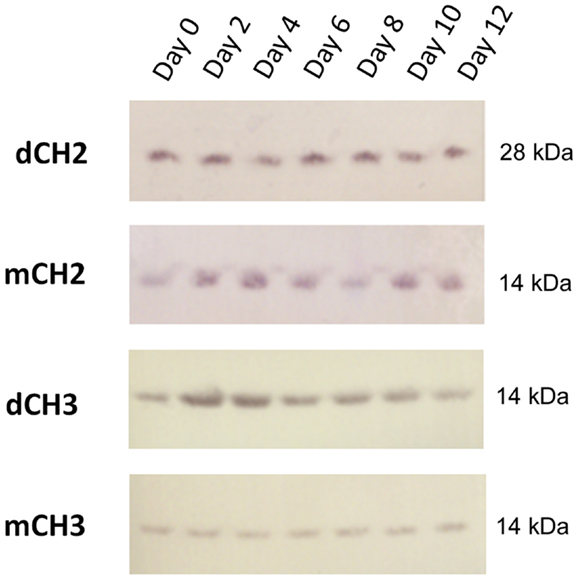
Figure 2. Western blot–direct serum analysis of dCH2, CH2, dCH3, and mCH3 measured up to 12 days.
Monomeric CH2 and CH3 Fragments Bind to FcRn
To test whether the dCH2, mCH2, dCH3, and mCH3 fragments are functional and behave in a manner similar to wild-type or mFc, ELISA binding experiments were carried out to validate their pH-dependent binding to FcRn. The binding was performed under pH 6.0 and pH 7.4, respectively, as described in the experimental method section. As shown in Figure 3, the mCH2 and mCH3 fragments displayed a behavior similar to that of the wild-type or mFc. At pH 6.0, the mCH2 and mCH3 fragments showed detectable binding to FcRn (Figure 3A) while dCH2 and dCH3 did not show any binding to FcRn. At pH 7.4, neither wild-type Fc nor the fragments showed detectable binding to FcRn (Figure 3B). These results suggested that the mCH2 and mCH3 maintained characteristic pH-dependent FcRn binding while the dimeric forms of CH2 and CH3 fragments did not.
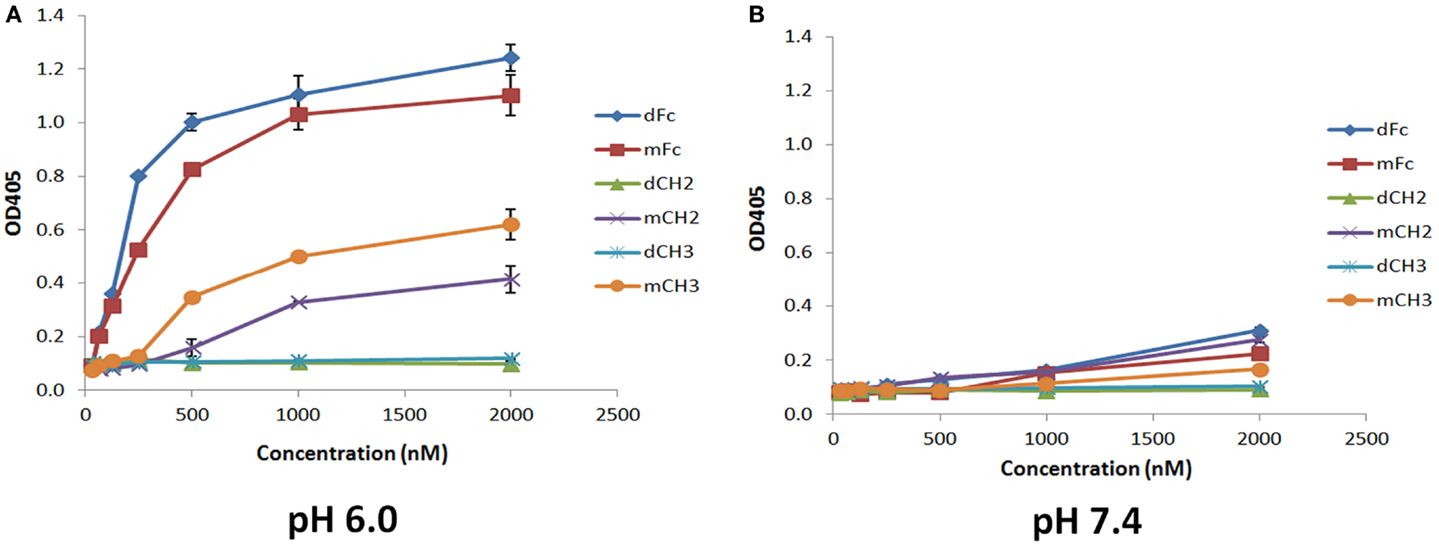
Figure 3. Binding of IgG1 Fc-dimeric/monomeric fragments, dFc/mFc, mFc, dCH2/mCH2, and dCH3/mCH3, to hFcRn at pH 6.0 (A) and pH 7.4 (B) measured by ELISA. The experimental points are represented as mean ± SD (n = 3).
Molecular Modeling and Docking Studies of FcRn Complexes with Monomeric Fc, CH2, and CH3 Domains
To understand the details of potential molecular interactions underlying FcRn binding with the Fc fragments mFc, mCH2, and mCH3, homology-based molecular modeling of hFcRn and the Fc fragments was carried out. We then used these models for docking of hFcRn complexes with mFc, mCH2, and mCH3 fragments to compare the possible difference between the complexes in hFcRn binding and properties. Figure 4 depicts the docked model of hFcRn–mFc/Fc complex as superimposed with the known rat FcRn–Fc complex structure. The structure of rat FcRn was used as a template for building the structure of its human counterpart. The root-mean-square deviation (RMSD) values for Cα atoms were calculated as 1.39 Å between the aligned human and rat FcRn components of modeled and known template structures. The inset of Figure 4 shows a close-up view highlighting the molecular interface of the hFcRn (yellow surface) and mFc (orange ribbons) complex, along with the amino acid residues of mFc (in sticks) that could potentially make contact with the hFcRn. The FcRn–mFc interface accounts for a total of 1,261 Å2 buried surface area and involves specific interactions such as hydrogen bonds, salt bridges, and hydrophobic interactions. These interactions occur mostly at the hinge region between CH2 and CH3 domains of mFc, which contains several histidine and charged residues (Table 1) that resemble the rat FcRn/heterodimeric Fc complex (19).
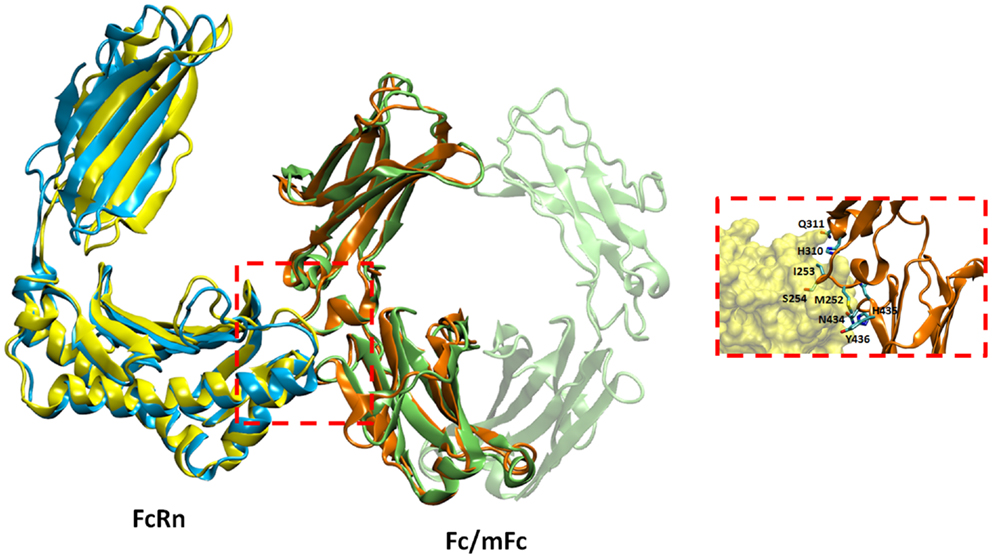
Figure 4. Docked model of hFcRn–mFc complex as superimposed with the known rat FcRn–Fc complex structure. The inset shows a close-up view of the molecular interface of hFcRn (yellow surface) and mFc (orange ribbons) with potential binding residues (as sticks).
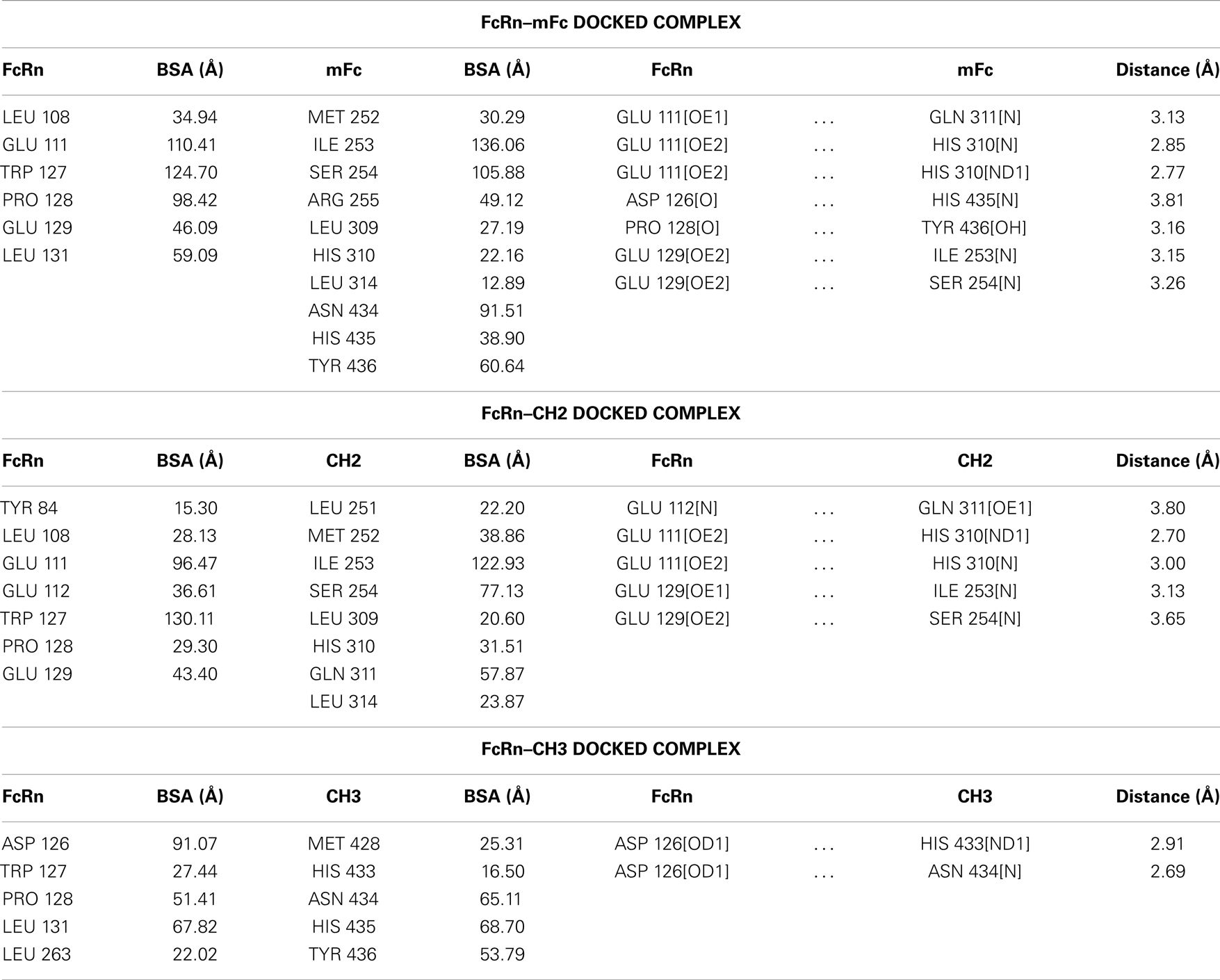
Table 1. Buried surface areas (BSA, Å2) and hydrogen bonds in human FcRn docked complexes with mFc, CH2, and CH3 fragments.
The docked models of hFcRn complexes with mCH2 and mCH3 were illustrated in Figure 5. The overall contribution to the binding interface areas of these model complexes are much smaller, with 807 and 584 Å2 of buried surface areas at the interface of hFcRn–mCH2 and hFcRn–mCH3, respectively, compared to the FcRn–mFc model complex. However, some of the critical intermolecular salt bridges were found between hFcRn and Fc fragment – mFc, mCH2, and mCH3 – docking model complexes. In particular, His310 in CH2 and His433 in CH3 could interact with the acidic residues Glu112 and Asp126 of FcRn, respectively. Similar salt bridges between Fc histidine residues, His310 and His435, and FcRn acidic residues, Glu111/Asp126, respectively, were observed in the hFcRn–mFc model complex. The putative titrating salt bridges that involve acidic–histidine residue pairs at the interfaces of hFcRn–mFc, hFcRn–mCH2, and hFcRn–mCH3 complexes might be ascribed to the mechanism of pH-dependent FcRn binding. The different intermolecular interactions predicted in the docked model hFcRn complexes of mFc, mCH2, and mCH3 fragments may have resulted in their differential binding properties (Figure 3).
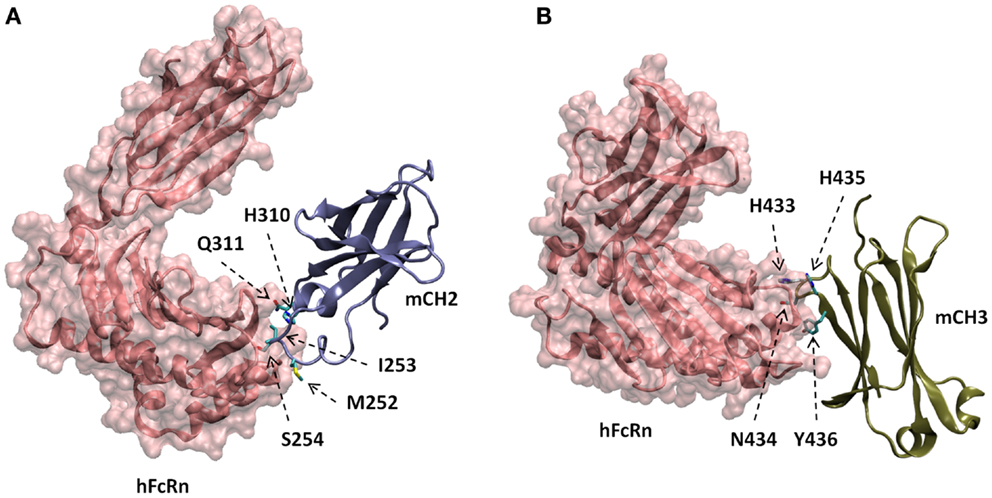
Figure 5. Docked models of hFcRn complexes with mCH2 (A) and mCH3 (B), showing with putative amino acids in mCH2 and mCH3 domains involved in the interactions with hFcRn.
One last consideration is that the preferential binding of hFcRn to mCH2/mCH3 over their dimeric counterparts, dCH2/dCH3, as seen in Figure 3, may be due to the exposure of additional residues after the monomerization of the dimers. It may not be due to the size-dependent effect alone since the hFcRn binding to dFc and mFc were much stronger. However, it should be noted that these data were obtained only using an ELISA assay where the protein conformation may not completely mimic the conformation in their native state as expressed at the cell surface. Further studies using natively expressed proteins are needed to validate these findings.
Conclusion
In summary, this study suggests that mCH2 and mCH3 could be potentially applicable in the engineering and development of novel biopharmaceuticals due to their high serum stability and pH-dependent FcRn binding. We found that while both monomeric and dimeric CH2 and CH3 domains had high serum stability and exhibited varying aggregation tendencies, only mCH2 and mCH3 bound to FcRn at pH 6.0. None of the antibody fragments bound to FcRn at pH 7.4. Our molecular modeling and docking studies suggest that this preferential binding of mCH2 and mCH3 to FcRn may be due to additional exposed residues from their monomerization. The smaller size and other described properties of mCH2 and mCH3 would overcome the fundamental problem of full-size antibodies, resulting in enhanced tissue penetration and providing access to a wide range of targets that are sterically occluded.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Sergey G. Tarasov and Marzena A. Dyba of the Biophysics Resource in the Structural Biophysics Laboratory, NCI-Frederick for helpful discussions and technical support. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by Federal funds from the NIH, National Cancer Institute, under contract numbers NO1-CO-12400 and HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
1. Dimitrov DS. Therapeutic proteins. Methods Mol Biol (2012) 899:1–26. doi:10.1007/978-1-61779-921-1_1
2. Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol (2006) 6(5):343–57. doi:10.1038/nri1837
3. Saerens D, Ghassabeh GH, Muyldermans S. Single-domain antibodies as building blocks for novel therapeutics. Curr Opin Pharmacol (2008) 8(5):600–8. doi:10.1016/j.coph.2008.07.006
4. Chen W, Gong R, Ying T, Prabakaran P, Zhu Z, Feng Y, et al. Discovery of novel candidate therapeutics and diagnostics based on engineered human antibody domains. Curr Drug Discov Technol (2014) 11(1):28–40.
5. Strohl WR, Strohl LM. Antibody fragments as therapeutics. Therapeutic Antibody Engineering: Current and Future Advances Driving the Strongest Growth Area in the Pharmaceutical Industry. Cambridge: Woodhead Publishing Series in Biomedicine (2013) p. 265–99.
6. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol (2007) 7(9):715–25. doi:10.1038/nri2155
7. Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I-related receptor FcRn. Annu Rev Immunol (2000) 18:739–66. doi:10.1146/annurev.immunol.18.1.739
8. Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. MAbs (2011) 3(5):422–30. doi:10.4161/mabs.3.5.16983
9. Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med (1997) 337(3):141–7. doi:10.1056/NEJM199707173370301
10. Gehlsen K, Gong R, Bramhill D, Wiersma D, Kirkpatrick S, Wang Y, et al. Pharmacokinetics of engineered human monomeric and dimeric CH2 domains. MAbs (2012) 4(4):466–74. doi:10.4161/mabs.20652
11. Ying T, Chen W, Feng Y, Wang Y, Gong R, Dimitrov DS. Engineered soluble monomeric IgG1 CH3 domain: generation, mechanisms of function, and implications for design of biological therapeutics. J Biol Chem (2013) 288(35):25154–64. doi:10.1074/jbc.M113.484154
12. Ying T, Chen W, Gong R, Feng Y, Dimitrov DS. Soluble monomeric IgG1 Fc. J Biol Chem (2012) 287(23):19399–408. doi:10.1074/jbc.M112.368647
13. Gong R, Wang Y, Feng Y, Zhao Q, Dimitrov DS. Shortened engineered human antibody CH2 domains: increased stability and binding to the human neonatal Fc receptor. J Biol Chem (2011) 286(31):27288–93. doi:10.1074/jbc.M111.254219
14. Gong R, Wang Y, Ying T, Dimitrov DS. Bispecific engineered antibody domains (nanoantibodies) that interact noncompetitively with an HIV-1 neutralizing epitope and FcRn. PLoS One (2012) 7(8):e42288. doi:10.1371/journal.pone.0042288
15. Wozniak-Knopp G, Bartl S, Bauer A, Mostageer M, Woisetschlager M, Antes B, et al. Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu-binding sites and antibody properties. Protein Eng Des Sel (2010) 23(4):289–97. doi:10.1093/protein/gzq005
16. Gong R, Vu BK, Feng Y, Prieto DA, Dyba MA, Walsh JD, et al. Engineered human antibody constant domains with increased stability. J Biol Chem (2009) 284(21):14203–10. doi:10.1074/jbc.M900769200
17. Gong R, Chen W, Dimitrov DS. Expression, purification, and characterization of engineered antibody CH2 and VH domains. Methods Mol Biol (2012) 899:85–102. doi:10.1007/978-1-61779-921-1_6
18. Feng Y, Gong R, Dimitrov DS. Design, expression and characterization of a soluble single-chain functional human neonatal Fc receptor. Protein Expr Purif (2011) 79(1):66–71. doi:10.1016/j.pep.2011.03.012
19. Martin WL, West AP Jr, Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell (2001) 7(4):867–77. doi:10.1016/S1097-2765(01)00230-1
20. Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc (2009) 4(1):1–13. doi:10.1038/nprot.2008.197
21. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem (2005) 26(16):1781–802. doi:10.1002/jcc.20289
22. Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins (2003) 52(1):80–7. doi:10.1002/prot.10389
23. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph (1996) 14(1):33–8. doi:10.1016/0263-7855(96)00018-5
Keywords: FcRn, antibody fragments, CH2 domain, CH3 domain, small-size
Citation: Ying T, Ju TW, Wang Y, Prabakaran P and Dimitrov DS (2014) Interactions of IgG1 CH2 and CH3 domains with FcRn. Front. Immunol. 5:146. doi: 10.3389/fimmu.2014.00146
Received: 22 January 2014; Paper pending published: 19 February 2014;
Accepted: 20 March 2014; Published online: 02 April 2014.
Edited by:
Jan Terje Andersen, Oslo University Hospital, NorwayReviewed by:
Julia Ponomarenko, University of California San Diego, USADaniel Olive, INSERM UMR 891 Institut Paoli Calmettes, France
Copyright: © 2014 Ying, Ju, Wang, Prabakaran and Dimitrov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tianlei Ying, Protein Interactions Group, Cancer and Inflammation Program, National Cancer Institute, National Institutes of Health, Building 567, Room 185, Frederick, MD 21702, USA e-mail:eWluZ3RAbWFpbC5uaWguZ292