 Christian Maueröder1
Christian Maueröder1 Luis Enrique Munoz1
Luis Enrique Munoz1 Ricardo Alfredo Chaurio1
Ricardo Alfredo Chaurio1 Martin Herrmann1Georg Schett1
Martin Herrmann1Georg Schett1 Christian Berens2*
Christian Berens2*- 1Institute of Clinical Immunology, Department of Internal Medicine 3, University of Erlangen-Nuremberg, Erlangen, Germany
- 2Department of Biology, University of Erlangen-Nuremberg, Erlangen, Germany
The sequel to the landmark article “The Hallmarks of Cancer” adds two emerging hallmarks and two enabling characteristics to the six original hallmarks (1). One emerging hallmark is the property of cancer cells to escape the immune system. Clinically apparent tumors arise as winners in a complex, hard-fought duel between cancer cell survival and eradication by the immune system.
Immunoediting, a term used for describing interactions between tumor and immune system, only occurs when, during the process of malignant transformation, cells develop features recognized by the immune system (2). The contribution of the immune system to recognition and elimination of malignant cells has been and still is being discussed controversially: some studies support the concept of immunosurveillance (3, 4), whereas others only observed small effects of the immune system in the prevention of cancer (5, 6). Recent studies suggest that, while there is evidence for immunosurveillance, not all aspects of the interaction between malignant cells and the immune system can be explained by immunoediting alone (7): some tumors never show properties making them targets of the immune system, whereas other tumors are recognized, but not eliminated due to immune suppression induced by the tumor.
However, if tumor cells are recognized as “altered cells,” their perpetual confrontation with the immune system evokes strong selection conditions favoring tumor cells that (I) lose properties making them targets of the immune system and (II) gain properties making them appear non-dangerous (8). If the tumor succeeds in decreasing its immunogenicity, it will reach a stage when the immune system does not consider those cells to be “altered-self” anymore. The tumor is now perceived as “self” and non-dangerous, with all privileges of normal healthy tissues.
When we think about therapies that elicit anti-tumor responses at this stage, we actually have to think about re-creating and enforcing tumor recognition, because, malignant tissues, although having been infiltrated by T-effector lymphocytes and, thus, being recognized by the immune system, frequently do not show remission. This correlates with reports that recruitment of T-effector lymphocytes to the site of the tumor is not necessarily sufficient for its eradication and that tumor immunity heavily depends on breaking tumor tolerance, i.e., by depletion of T-regulatory lymphocytes or by shielding T-effector lymphocytes from immune-suppressive molecules like PD-L1 (9). We propose that the need for inducing immunity and breaking of tolerance might be akin to activating some kind of tumor-specific (auto)-immunity.
The ideal tumor therapy results in local control of the primary tumor, systemic control of potential metastases and triggers an anti-tumor immune response ultimately leading to the elimination of all malignant cells. To achieve this, tumor therapy needs to deal with the problem that the immune system does not consider the tumor being dangerous anymore – it has been adopted as “self-organ.” Consequently, tumor therapy should focus on making the immune system aware of this hidden danger.
This concept was first put into practice by William Coley, who injected a cocktail of dead bacteria into tumors in the late 1800s, achieving cures in ≈30% of his patients with sarcoma and lymphoma (10, 11). The mechanism responsible for this seems to be LPS-induced IL-12 secretion triggering a robust bystander Th1-response against the tumor cells (12). Likewise, an attenuated Salmonella vaccine can induce a shift in the tumor milieu from an immune-suppressive to an immunogenic microenvironment (13). The most successful application derived from Coley’s work is treatment of bladder cancer with the Bacillus Calmette–Guerin vaccine: it has become the standard therapy for superficial bladder cancer, eradicating existing tumors, reducing the frequency of tumor recurrence, delaying stage progression, and increasing survival (14). The advantage of such strategies is their lack of specificity. The immune response is not restricted to a single and, most likely, highly specific and selectable “tumor-antigen,” but the presence of danger signals at the site of the tumor “uncloaks” the cancer cells, turning them into broad range immune targets. At this point, we can exploit a mechanism, which causes a break in self-tolerance in autoimmune diseases: transient autoimmunity accompanying any inflammatory process can, in the context of steady exposure to auto-antigens and danger signals, develop into stable autoimmunity. Following Polly Matzinger’s ideas, the key to success of danger-based tumor vaccination strategies rests on repeated administration of the vaccine (15). Repeated immunization should help overcome transient tumor immunity and establish persistent protection.
One danger-based tumor vaccination approach conducts the immunization with dying tumor cells (16, 17). Certain kinds of dying or dead cells can trigger immune responses under the right conditions. The potential of dying/dead cells to induce autoimmunity can be seen in “systemic lupus erythematosus” (SLE), a chronic inflammatory disease, in which defective clearance of apoptotic cells leads to the accumulation of secondary necrotic cells, the release of danger signals, the presentation of auto-antigens and, finally, a chronic break in self-tolerance (18–20). Based on these observations, one can assume that, under the appropriate conditions, entities once considered to be non-dangerous can become re-considered dangerous. We propose that one can learn from the processes which cause breaks of self-tolerance in patients with SLE and try to harness them to induce tumor (auto-) immunity.
In the context of tumor immunology, cell death is a double-edged sword. Tumor cells often modulate apoptotic pathways rendering them less responsive to death stimuli. Down-regulation of Fas expression or resistance to Fas-mediated apoptosis are common strategies of tumor cells to escape immunosurveillance (21) and are associated with resistance to therapy, metastatic capacity, and poor prognosis. For example, c-Jun and Stat-3 act as oncogenes by cooperatively repressing the transcription of Fas, rendering tumor cells insensitive to FasL-induced apoptosis (22). A complete loss of Fas expression is less common, possibly to low-level expression of Fas supporting tumor growth (23). Many other mechanisms to evade elimination by apoptosis, i.e., suppression of caspase-8 activity by CDK1/CYCLIN B1 dependent phosphorylation (24), bcl-2 amplification (25), and loss of pro-apoptotic proteins like BAX (26) and PUMA (27), have been reported for a large variety of cancer types (28).
These findings are hard to reconcile with the observation that a high rate of tumor cell apoptosis is accompanied by poor prognosis in some types of cancer (29–31). It is known that cancer cells show many different changes to the apoptotic machinery (28, 32); but does this mean they have lost all capability to execute apoptosis? Apoptosis is necessary for tissue homeostasis, contributes to the maintenance of peripheral tolerance and might even play a role in the induction of the latter (33, 34). The fact that most chemotherapeutics at least initially induce tumor apoptosis confirms that cancer cells frequently retain their ability to execute apoptosis (35, 36). It is reasonable to assume that those parts of the apoptotic machinery involved in the induction of extrinsic apoptosis by the immune system preferentially experience negative selection. If other parts of the apoptotic pathway would also be a potential source of harm, why do they, in defiance of the exceptional adaptability of cancer cells, still function properly? We suggest that, in contrast to the oversimplified illustration, cancer cells do not completely lose their capability to undergo apoptosis, but that their apoptotic machinery can instead be “hijacked” in a way that not only sustains their existence, but also accelerates tumor formation (37–39): an “altruistic” death of limited amounts of cancer cells is a possible way to support the survival of the tumor on the whole.
Over the years, the tumor-supportive effects of apoptotic tumor cells have received greater recognition, and it is now assumed that apoptotic tumor cells and the corresponding phagocytes participate in forming and shaping the tumor microenvironment (40). Apoptotic cells release a diverse spectrum of molecules, which act as “keep-out,” “find-me,” “eat-me,” and “tolerate-me” signals and ensure that the clearance of apoptotic cells is facilitated by defined groups of phagocytes, in particular by macrophages (41).
Of particular interest are lipid mediators, which are released from cells undergoing apoptosis: (I) lysophosphatidylcholine is a potent chemoattractant for macrophages and is released from cells executing apoptosis (42). (II) Upon proteolytic activation of sphingosine kinase 2, sphingosine-1-phosphate (S1P) is released from apoptotic cells (43). In addition to its role as a chemoattractant (44), S1P polarizes macrophages toward a non-inflammatory phenotype (M2), characterized by decreased secretion of TNF-α and IL-12-p70 and increased formation of IL-8 and Il-10 (45).
The engulfment of apoptotic cells by macrophages induces their polarization toward the M2-phenotype (Figure 1A). These alternatively activated macrophages tune down inflammation and promote angiogenesis, tissue remodeling, and repair (46, 47). Furthermore, phagocytosis of apoptotic cells by M1-macrophages also triggers a shift toward alternative activation (48). Fittingly, a large number of macrophages at the site of the tumor are associated with a poor prognosis and these tumor-associated macrophages share many characteristics with M2-macrophages (49, 50). Their presence at the site of a tumor supports Dvorak’s concept that tumors are “wounds that do not heal” (51).
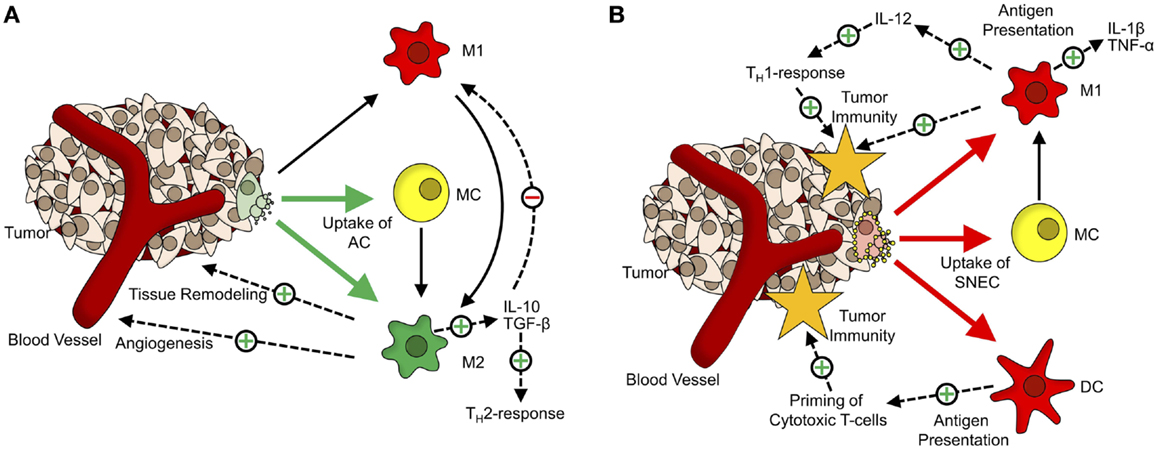
Figure 1. The dual role of cell death in tumor tolerance/immunity. (A) Role of apoptotic cells in formation of the tumor microenvironment. Apoptotic cells (AC) are mainly taken up by monocytes (MC; yellow) and alternatively activated macrophages (M2; green). Upon phagocytosis of ACs, MCs, and classically activated macrophages (M1, red) get polarized toward an M2-phenotype. M2-macrophages participate in tissue remodeling and angiogenesis and via secretion of anti-inflammatory cytokines (TGF-β, IL-10), inhibit M1-activation of macrophages and shift TH1-responses toward the TH2-phenotype. (B) Tumor-supportive effects of apoptotic cells are abrogated by Annexin-A5. Annexin-A5 (yellow circles on secondary necrotic cells) inhibits swift clearance of apoptotic cells, leading to progression of ACs into secondary necrosis. Secondary necrotic cells (SNEC) are mainly taken up by MCs, classically activated macrophages and dendritic cells (DC; red). Upon phagocytosis of SNEC, MCs get polarized toward the M1-phenotype. Phagocytosis of SNEC by DCs leads to antigen presentation and priming of T cells. Classically activated macrophages secrete inflammatory cytokines (TNF-α, IL-1β) and induce TH1-responses via IL-12.
In line with these findings is the observation that inhibiting the clearance of apoptotic tumor cells by administration of Annexin-A5 retards tumor growth in a colorectal carcinoma model and greatly enhances the effect of immunization with irradiated lymphoma cells in a lymphoma model (52, 53). The data presented suggests that this is due to the fact that the non-inflammatory clearance of apoptotic cells by macrophages is blocked so that the apoptotic cells get secondarily necrotic. The concomitant loss of membrane integrity is accompanied by the release of danger-associated molecular patterns (DAMP), which act as natural adjuvants. Phagocytosis of secondary necrotic cells by macrophages (Figure 1B) leads to an increased expression of TNF-α and IL-1β. In addition, several DAMPs released from secondary necrotic cells, like HMGB1 and HMGN1, are potent stimuli for dendritic cell maturation (54).
The close interaction between tumors, the immune system and cell death gives rise to new therapeutic approaches. Some aspects of this interaction may be exploited to support conventional cancer therapies. Systemic administration of Annexin-A5 or other phosphatidylserine ligands may help slow down tumor progression by blocking the tumor-supportive properties of apoptotic cells. In combination with radio- or chemotherapy, Annexin-A5 could be used as a natural adjuvant, which increases the immunogenicity of dying tumor cells and, thus, helps elicit an anti-tumor immune response (55). This may be especially helpful in targeting cancer cells, which have resisted therapy and would possibly lead to a relapse.
Until recently, cell death was either characterized as programed and apoptotic, or accidental and necrotic. This paradigm has been undermined by the discovery of several other forms of cell death, ranging from immunogenic apoptosis (56) or necroptosis (57) to pyroptosis (58, 59). So, in addition to manipulating cell death induced by radio- or chemotherapy in a way to increase its immunogenicity, the direct induction of immunogenic tumor cell death pathways might become a promising approach in cancer therapy (17, 54, 60), especially, since our means of controlling the manner of cell death have greatly increased during recent years (61–63).
Surgical removal of malignant tissue plays an important role in modern cancer therapy. The cancer cells obtained in this process may be used as a vaccine to establish anti-tumor immunity, if treated and administered properly. The focus must be on cancer cells dying by immunostimulatory forms of cell death leading to necrotic cell corpses, whose deployment would activate antigen-presenting-cells. This way, the specific autologous tumor cells can serve as reservoirs of tumor antigens, which, upon phagocytosis by inflammatory macrophages and dendritic cells, are effectively (cross-)presented. The impact of the vaccine could be optimized by repeated administration of the dying cells. However, we have to be very careful, since a recent study indicates that excessive immune responses against cancer can result in an increased risk of developing the autoimmune disease scleroderma (64), pointing out several parallels between the induction of autoimmunity and immunosurveillance. While this study actually supports the idea that mechanisms inducing autoimmunity can also be used to elicit tumor immunity, it also suggests that any agents used to recruit anti-tumor responses must be well-balanced. After all, nobody wants to escape cancer’s fire by jumping into the frying pan of autoimmunity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was supported by the German Research Foundation (SPP1468-IMMUNOBONE, collaborative research centers 643/TP-B5 and 796/TP-C4, GA 1507/1-1, and DFG-Graduiertenkolleg 1660: key signals of the adaptive immune response), by the Emerging Fields Initiative (EFI) of the FAU Erlangen-Nuremberg, by the German Federal Ministry of Education and Research (BMBF; m4 Cluster, 01EX1021R) and the K. und R. Wucherpfennig-Stiftung.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
2. Vesely MD, Schreiber RD. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Ann N Y Acad Sci (2013) 1284:1–5. doi:10.1111/nyas.12105
3. Engel AM, Svane IM, Rygaard J, Werdelin O. MCA sarcomas induced in scid mice are more immunogenic than MCA sarcomas induced in congenic, immunocompetent mice. Scand J Immunol (1997) 45:463–70. doi:10.1046/j.1365-3083.1997.d01-419.x
4. Street SE, Cretney E, Smyth MJ. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood (2001) 97:192–7. doi:10.1182/blood.V97.1.192
5. Ciampricotti M, Vrijland K, Hau CS, Pemovska T, Doornebal CW, Speksnijder EN, et al. Development of metastatic HER2(+) breast cancer is independent of the adaptive immune system. J Pathol (2011) 224:56–66. doi:10.1002/path.2837
6. Stutman O. Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. Science (1974) 183:534–6. doi:10.1126/science.183.4124.534
7. Manjili MH. Revisiting cancer immunoediting by understanding cancer immune complexity. J Pathol (2011) 224:5–9. doi:10.1002/path.2865
8. Riedl S, Rinner B, Asslaber M, Schaider H, Walzer S, Novak A, et al. In search of a novel target – phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim Biophys Acta (2011) 1808:2638–45. doi:10.1016/j.bbamem.2011.07.026
9. Binder DC, Engels B, Arina A, Yu P, Slauch JM, Fu YX, et al. Antigen-specific bacterial vaccine combined with anti-PD-L1 rescues dysfunctional endogenous T cells to reject long-established cancer. Cancer Immunol Res (2013) 1:123–33. doi:10.1158/2326-6066.CIR-13-0058
10. Nauts HC, Fowler GA, Bogatko FH. A review of the influence of bacterial infection and of bacterial products (Coley’s toxins) on malignant tumors in man; a critical analysis of 30 inoperable cases treated by Coley’s mixed toxins, in which diagnosis was confirmed by microscopic examination selected for special study. Acta Med Scand Suppl (1953) 276:1–103.
11. Coley WB. Further observations upon the treatment of malignant tumors with the toxins of erysipelas and Bacillus prodigious with a report of 160 cases. Bull Johns Hopkins Hosp (1896) 7:715.
12. Tsung K, Norton JA. Lessons from Coley’s toxin. Surg Oncol (2006) 15:25–8. doi:10.1016/j.suronc.2006.05.002
13. Hong EH, Chang SY, Lee BR, Pyun AR, Kim JW, Kweon MN, et al. Intratumoral injection of attenuated Salmonella vaccine can induce tumor microenvironmental shift from immune suppressive to immunogenic. Vaccine (2013) 31:1377–84. doi:10.1016/j.vaccine.2013.01.006
14. Herr HW, Morales A. History of Bacillus Calmette-Guerin and bladder cancer: an immunotherapy success story. J Urol (2008) 179:53–6. doi:10.1016/j.juro.2007.08.122
15. Matzinger P. The danger model: a renewed sense of self. Science (2002) 296:301–5. doi:10.1126/science.1071059
16. Griffith TS, Ferguson TA. Cell death in the maintenance and abrogation of tolerance: the five Ws of dying cells. Immunity (2011) 35:456–66. doi:10.1016/j.immuni.2011.08.011
17. Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol (2009) 9:353–63. doi:10.1038/nri2545
18. Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol (2010) 6:280–9. doi:10.1038/nrrheum.2010.46
19. Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum (2002) 46:191–201. doi:10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K
20. Munoz LE, Janko C, Grossmayer GE, Frey B, Voll RE, Kern P, et al. Remnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosus. Arthritis Rheum (2009) 60:1733–42. doi:10.1002/art.24535
21. Mottolese M, Buglioni S, Bracalenti C, Cardarelli MA, Ciabocco L, Giannarelli D, et al. Prognostic relevance of altered Fas (CD95)-system in human breast cancer. Int J Cancer (2000) 89:127–32. doi:10.1002/(SICI)1097-0215(20000320)89:2<127::AID-IJC5>3.0.CO;2-4
22. Ivanov VN, Bhoumik A, Krasilnikov M, Raz R, Owen-Schaub LB, Levy D, et al. Cooperation between STAT3 and c-Jun suppresses Fas transcription. Mol Cell (2001) 7:517–28. doi:10.1016/S1097-2765(01)00199-X
23. Chen L, Park SM, Tumanov AV, Hau A, Sawada K, Feig C, et al. CD95 promotes tumour growth. Nature (2010) 465:492–6. doi:10.1038/nature09075
24. Matthess Y, Raab M, Sanhaji M, Lavrik IN, Strebhardt K. Cdk1/cyclin B1 controls Fas-mediated apoptosis by regulating caspase-8 activity. Mol Cell Biol (2010) 30:5726–40. doi:10.1128/MCB.00731-10
25. Cleary ML, Sklar J. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci USA (1985) 82:7439–43. doi:10.1073/pnas.82.21.7439
26. Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science (1997) 275:967–9. doi:10.1126/science.275.5302.967
27. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature (2010) 463:899–905. doi:10.1038/nature08822
28. Fernald K, Kurokawa M. Evading apoptosis in cancer. Trends Cell Biol (2013) 23:620–33. doi:10.1016/j.tcb.2013.07.006
29. Soini Y, Raunio H, Paakko P. High-grade malignant non-Hodgkin’s lymphomas differ from low-grade lymphomas in the extent of apoptosis and their expression of bcl-2, mcl-1, bax and p53. Tumour Biol (1998) 19:176–85. doi:10.1159/000030005
30. Stammler G, Sauerbrey A, Zintl F, Volm M. Apoptotic index, Fas and bcl-2 in initial and relapsed childhood acute lymphoblastic leukaemia. Apoptosis (1997) 2:377–83. doi:10.1023/A:1026405707823
31. Bendardaf R, Ristamaki R, Kujari H, Laine J, Lamlum H, Collan Y, et al. Apoptotic index and bcl-2 expression as prognostic factors in colorectal carcinoma. Oncology (2003) 64:435–42. doi:10.1159/000070304
32. Delbridge AR, Valente LJ, Strasser A. The role of the apoptotic machinery in tumor suppression. Cold Spring Harb Perspect Biol (2012) 4:a008789. doi:10.1101/cshperspect.a008789
33. Uderhardt S, Herrmann M, Oskolkova OV, Aschermann S, Bicker W, Ipseiz N, et al. 12/15-Lipoxygenase orchestrates the clearance of apoptotic cells and maintains immunologic tolerance. Immunity (2012) 36:834–46. doi:10.1016/j.immuni.2012.03.010
34. Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med (2000) 191:411–6. doi:10.1084/jem.191.3.411
35. Jordan P, Carmo-Fonseca M. Molecular mechanisms involved in cisplatin cytotoxicity. Cell Mol Life Sci (2000) 57:1229–35. doi:10.1007/PL00000762
36. Del Gaizo Moore V, Letai A. BH3 profiling – measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer Lett (2013) 332:202–5. doi:10.1016/j.canlet.2011.12.021
37. Sarosiek KA, Ni Chonghaile T, Letai A. Mitochondria: gatekeepers of response to chemotherapy. Trends Cell Biol (2013) 23:612–9. doi:10.1016/j.tcb.2013.08.003
38. Chaurio R, Janko C, Schorn C, Maueröder C, Bilyy R, Gaipl U, et al. UVB-irradiated apoptotic cells induce accelerated growth of co-implanted viable tumor cells in immune competent mice. Autoimmunity (2013) 46:317–22. doi:10.3109/08916934.2012.754433
39. Lauber K, Munoz LE, Berens C, Jendrossek V, Belka C, Herrmann M. Apoptosis induction and tumor cell repopulation: the Yin and Yang of radiotherapy. Radiat Oncol (2011) 6:176. doi:10.1186/1748-717X-6-176
40. Gregory CD, Pound JD. Cell death in the neighbourhood: direct microenvironmental effects of apoptosis in normal and neoplastic tissues. J Pathol (2011) 223:177–94. doi:10.1002/path.2792
41. Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol (2013) 5:a008748. doi:10.1101/cshperspect.a008748
42. Lauber K, Bohn E, Kröber SM, Xiao Y-J, Blumenthal SG, Lindemann RK, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell (2003) 113:717–30. doi:10.1016/S0092-8674(03)00422-7
43. Weigert A, Cremer S, Schmidt MV, von Knethen A, Angioni C, Geisslinger G, et al. Cleavage of sphingosine kinase 2 by caspase-1 provokes its release from apoptotic cells. Blood (2010) 115:3531–40. doi:10.1182/blood-2009-10-243444
44. Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J (2008) 22:2629–38. doi:10.1096/fj.08-107169
45. Weigert A, Tzieply N, von Knethen A, Johann AM, Schmidt H, Geisslinger G, et al. Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol Biol Cell (2007) 18:3810–9. doi:10.1091/mbc.E06-12-1096
46. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol (2008) 18:349–55. doi:10.1016/j.semcancer.2008.03.004
47. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature (1997) 390:350–1. doi:10.1038/37022
48. Reiter I, Krammer B, Schwamberger G. Cutting edge: differential effect of apoptotic versus necrotic tumor cells on macrophage antitumor activities. J Immunol (1999) 163:1730–2.
49. Takanami I, Takeuchi K, Kodaira S. Tumor-associated macrophage infiltration in pulmonary adenocarcinoma: association with angiogenesis and poor prognosis. Oncology (1999) 57:138–42. doi:10.1159/000012021
50. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med (2010) 362:875–85. doi:10.1056/NEJMoa0905680
51. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med (1986) 315:1650–9. doi:10.1056/NEJM198612253152606
52. Bondanza A, Zimmermann VS, Rovere-Querini P, Turnay J, Dumitriu IE, Stach CM, et al. Inhibition of phosphatidylserine recognition heightens the immunogenicity of irradiated lymphoma cells in vivo. J Exp Med (2004) 200:1157–65. doi:10.1084/jem.20040327
53. Frey B, Schildkopf P, Rödel F, Weiss EM, Munoz LE, Herrmann M, et al. AnnexinA5 renders dead tumor cells immunogenic – implications for multimodal cancer therapies. J Immunotoxicol (2009) 6:209–16. doi:10.3109/15476910903204058
54. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer (2012) 12:860–75. doi:10.1038/nrc3380
55. Munoz LE, Franz S, Pausch F, Fürnrohr B, Sheriff A, Vogt B, et al. The influence on the immunomodulatory effects of dying and dead cells of Annexin V. J Leukoc Biol (2007) 81:6–14. doi:10.1189/jlb.0306166
56. Garg AD, Krysko DV, Vandenabeele P, Agostinis P. The emergence of phox-ER stress induced immunogenic apoptosis. Oncoimmunology (2012) 1:786–8. doi:10.4161/onci.19750
57. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol (2005) 1:112–9. doi:10.1038/nchembio0905-234a
58. Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol (2000) 38:31–40. doi:10.1046/j.1365-2958.2000.02103.x
59. Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature (2014) 505:509–14. doi:10.1038/nature12940
60. Vacchelli E, Senovilla L, Eggermont A, Fridman WH, Galon J, Zitvogel L, et al. Trial watch: chemotherapy with immunogenic cell death inducers. Oncoimmunology (2013) 2:e23510. doi:10.4161/onci.23510
61. Maueröder C, Chaurio RA, Platzer S, Munoz LE, Berens C. Model systems for rapid and slow induction of apoptosis obtained by inducible expression of pro-apoptotic proteins. Autoimmunity (2013) 46:329–35. doi:10.3109/08916934.2012.752463
62. Lohmann C, Muschaweckh A, Kirschnek S, Jennen L, Wagner H, Häcker G. Induction of tumor cell apoptosis or necrosis by conditional expression of cell death proteins: analysis of cell death pathways and in vitro immune stimulatory potential. J Immunol (2009) 182:4538–46. doi:10.4049/jimmunol.0803989
63. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell (2005) 122:927–39. doi:10.1016/j.cell.2005.07.002
Keywords: immunotherapy, tumor vaccination, cell death, tumor microenvironment, autoimmunity, danger model
Citation: Maueröder C, Munoz LE, Chaurio RA, Herrmann M, Schett G and Berens C (2014) Tumor immunotherapy: lessons from autoimmunity. Front. Immunol. 5:212. doi: 10.3389/fimmu.2014.00212
Received: 06 February 2014; Accepted: 28 April 2014;
Published online: 13 May 2014.
Edited by:
Fang-Ping Huang, Imperial College London, UKReviewed by:
Pedro Berraondo, Centro de Investigación Médica Aplicada, SpainAinhoa Arina, The University of Chicago, USA
Copyright: © 2014 Maueröder, Munoz, Chaurio, Herrmann, Schett and Berens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:Y2hyaXN0aWFuLmJlcmVuc0BmYXUuZGU=