 Claudia Rival
Claudia Rival Yulius Setiady
Yulius Setiady Eileen T. Samy
Eileen T. Samy Jessica Harakal
Jessica Harakal Kenneth S. K. Tung
Kenneth S. K. Tung- 1Departments of Pathology and Microbiology, Beirne Carter Center for Immunology Research, University of Virginia, Charlottesville, VA, USA
- 2ImmunoGen Inc., Waltham, MA, USA
- 3EMD Serono Research Institute, Inc., Billerica, MA, USA
Human maternal autoantibodies can trigger autoimmune diseases such as congenital heart block (CHB) in the progeny of women with lupus or Sjogren’s disease. The pathogenic effect of early autoantibody (autoAb) exposure has been investigated in a murine neonatal autoimmune ovarian disease (nAOD) model triggered by a unique ZP3 antibody. Although immune complexes (IC) are formed in adult and neonatal ovaries, ZP3 antibody triggers severe nAOD only in <7-day-old neonatal mice. Propensity to nAOD is due to the uniquely hyper-responsive neonatal natural killer (NK) cells that lack the inhibitory Ly49C/I receptors. In nAOD, the neonatal NK cells directly mediate ovarian inflammation and oocyte depletion while simultaneously promoting de novo pathogenic ovarian-specific T cell responses. Resistance to nAOD in older mice results from the emergence of the Ly49C/I+ NK cells that regulate effector NK cells and from CD25+ regulatory T cell control. In preliminary studies, FcγRIII+ NK cells as well as the ovarian resident FcγRIII+ macrophages and/or dendritic cells were found to be as indispensable players. Activated by ovarian IC, they migrate to lymphoid organs where NK cell priming occurs. Remarkably, the findings in nAOD are very similar to those reported for neonatal responses to a retrovirus and its cognate antibody that lead to long-lasting immunity. Studies on nAOD therefore provide insights into maternal autoAb-mediated neonatal autoimmunity, including CHB, while simultaneously uncovering new properties of the neonatal innate and adaptive responses, lethality of premature infant infection, and novel neonatal antiviral vaccine design.
Human Neonatal Autoimmune Disease Induction by Maternal Autoantibody Exemplifies Neonatal Propensity to Autoimmunity
Increased susceptibility of premature human infants and neonatal mice to infections is well-known (1). What is less appreciated is that neonatal mice are also more susceptible to autoimmune disease. For example, spontaneous autoimmune diseases occur in mice thymectomized between days 1 and 4 but not after day 7 of life (2). Autoimmune diseases induced by tissue antigen or peptide immunization require complete Freund’s adjuvant (CFA) in adult but not in neonatal mice (3–6). Indeed, neonatal female mice immunized with an auto-peptide from the gender-specific ovarian zona pellucida 3 (pZP3) antigen, mounted peptide-specific T cell responses, and developed autoimmune ovarian disease (AOD) in 3 weeks. In contrast, the same ZP3 peptide induced tolerance in neonatal male mice as a foreign antigen (4). The opposing responses to self and foreign antigens in the neonates suggest that the neonatal tolerance paradigm (7, 8) should be revisited because it is built mainly on responses to foreign antigens or peptides including alloantigen (9–15). Indeed, in contrast to the neonatal tolerance paradigm, newborn mice responded as efficiently as adult mice to viral, nominal, and autoantigen, when the antigen dose, injection site, and adjuvant type were adjusted (16–18). Finally, neonatal propensity to autoimmunity is supported by the induction of fetal or neonatal self-tissue damage after the transplacental transfer of maternal autoantibody (autoAb) that often does not harm the adult.
Autoimmune disease occurs preferentially in women of reproductive age, and circulating autoAb is a hallmark of autoimmunity. The finding of neonatal autoimmune disease caused by maternal autoAb establishes its pathogenic potential. The impact of maternal autoAb is often transient, and neonates recover as antibody level declines. However, in some cases, maternal autoAb effects persist and induce permanent tissue damage. An example is congenital heart block (CHB) that occurs in the fetuses or infants of women with systemic lupus erythrematosus (SLE) or Sjogren’s disease. Although CHB has many possible etiologies (19), a strong candidate is the transplacental transfer of maternal autoAb against the ribonucleoproteins SSA/Ro and SSB/La (20), with SSB/La having a stronger association (21). The maternal autoAb damages the atrioventricular node of the cardiac conduction system by an unknown mechanism. Likely, CHB pathogenesis involves excessive apoptosis of cardiomyocytes, tissue inflammation and fibrosis, and the interaction of macrophages and fibroblasts (21). The clinical impact of CHB is significant. Although about 2% of neonates from mothers with autoAb to SSA/Ro and SSB/La develop CHB (20), the recurrence rate for an autoAb-positive mother with a previously affected child is 16–18% (22). Importantly, complete or third degree CHB is irreversible with a mortality rate of 12–43% (22). Although first or second degree heart block may reverse with treatment, most children require permanent pacemakers (22).
Maternal autoAb can induce neonatal autoimmune diseases other than CHB. Neonatal myasthenia gravis, associated with severe deformity and difficult deliveries, is caused by maternal autoAb to the fetal form of acetylcholine receptor and a muscle-specific kinase (23, 24). Neonatal Graves’ disease involves agonist autoAb targeting the thyroid stimulating hormone receptor (19). Neonatal pemphigus is mediated by autoAb to the desmoglein-1 or 3 antigens (25, 26). Several studies have shown a positive correlation between anti-phospholipid or lupus anticoagulant autoAb, present in patients with SLE or anti-phospholipid syndrome, and a high risk of premature onset of labor, low birth weight and early miscarriages of their progeny (19, 27). In addition, neuronal apoptosis and subsequent abortion are consequences of maternal autoAb to DNA in SLE patients (28); and autism spectrum disorders are linked to maternal autoAb against fetal brain antigens (29, 30). The mechanism of human neonatal autoimmunity is currently uncertain, and we have used the neonatal autoimmune ovarian disease (nAOD) model to address the questions of why neonates are more susceptible to autoimmune disease, and how maternal autoAb induce the tissue damage. Below, we have described some of the characteristics and advantages of nAOD that make it a very useful model.
In addition to its ability to cause tissue injury and disease, antibody has been found to elicit changes in neonatal or very young mice and promote subsequent full-blown autoimmune diseases. An example of the “pre-disease” state is the pre-diabetes that precedes clinical diabetes by months to years (31–34). In NOD mice, a very early cascade of cellular and cytokine responses to local immune complexes (IC) in pancreatic islets was found to cause subsequent development of pathogenic T cell responses and clinical juvenile diabetes (35). In addition, maternal autoAb directed to islet antigens was reported as a requirement for type I diabetes in the NOD mice (36) or to accelerate diabetes onset in a transgenic mouse model (37).
Therefore, maternal autoAb can induce either transient pathology or more persistent tissue damage in the progeny. It may also condition the fetus/infant early in life for a late onset autoimmune disease. In these situations, the design of novel preventive therapies will necessitate a thorough understanding of the pathogenic autoimmune response in the very young individuals, including their innate and adaptive neonatal responses to IC created by autoAb.
Neonatal Autoimmune Ovarian Disease Model and Its Unique Features
The aforementioned findings indicate that newborns are simultaneously more susceptible to infections and to autoimmunity. While “immaturity” of the neonatal immune system might explain the increased sensitivity to pathogens, it is not immediately apparent why newborns are more susceptible to autoimmune disease. A potential explanation is that the maturing neonatal immune system is less stringently regulated relative to the adults. Thus, an overactive neonatal response would induce both autoimmune disease and severe post-infection immunopathology, including sepsis. While other authors support the latter (38), our study focuses on the questions of why newborns are more susceptible to nAOD and how nAOD is induced. We have summarized our results in this review.
Autoimmune ovarian disease is a known cause of primary or secondary premature ovarian failure that can lead to infertility of pubertal and adult women (39, 40). Because ovarian dysfunction is not generally manifested until puberty, it cannot be certain that primary AOD, like type I diabetes, is preceded by a “pre-disease” exemplified by nAOD. While the clinical relevance of nAOD remains unresolved, the murine nAOD model itself has proven to be an excellent platform for investigating the role of the neonatal immune response to autoantigen and the role of autoAb in autoimmune disease pathogenesis.
ZP3 is a major sperm receptor in fertilization (41). The pZP3 (330–342) contains a pathogenic T cell epitope and a native B cell epitope 335–342 (42), and antibodies to this pZP3 B cell epitope inhibit sperm binding to the zona pellucida (41, 43). Adult mice immunized with pZP3 in CFA develop a pathogenic CD4+ T cell response and a non-pathogenic antibody response. The latter was confirmed in adult mice immunized with a chimeric ZP3 peptide that contains the native B cell epitope 335–342 linked to a foreign T cell peptide from the bovine RNAse (43). The mice produce ZP3 antibodies that bind to the ovarian zona pellucida without causing ovarian pathology. However, 70% of the females immunized with the chimeric ZP3 peptide were infertile because of the contraceptive effect of ZP3 antibodies. In the remnant fertile females, the maternal ZP3 autoAb transferred to the neonates induced severe nAOD (Figure 1). Remarkably, nAOD developed in the progeny only when the ZP3 antibody exposure was initiated within the first 6 days of life, as documented by feeding antibody-positive milk to normal pups of different ages, and by antibody transfer (44).
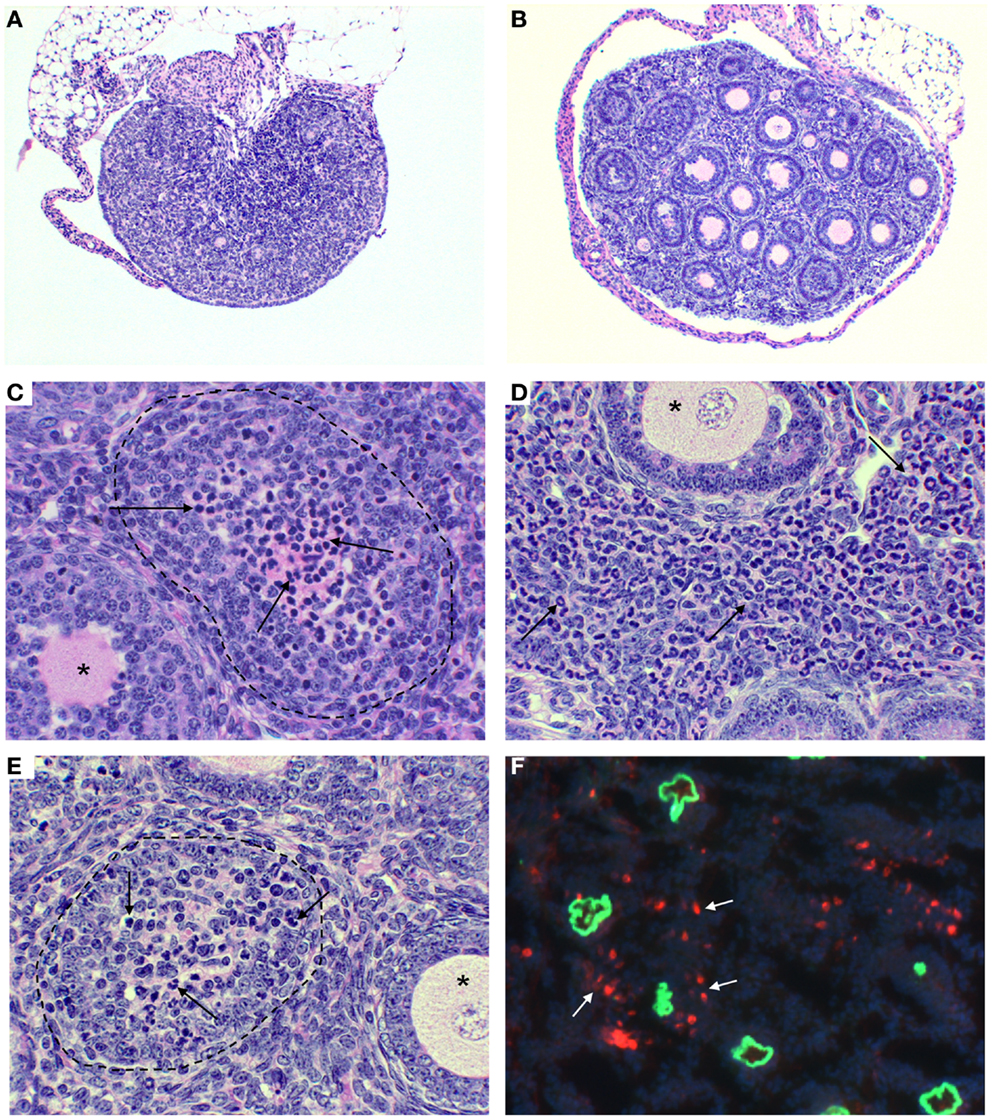
Figure 1. Ovarian immunopathology of TI-nAOD in C57BL/6 Rag1−/− mice injected with pZP3 monoclonal antibody (mAb) in the first week of life. (A) Ovarian atrophy with major oocyte depletion and disrupted ovarian architecture. (B) Normal ovarian histology in mice injected with ZP3 mAb and NK cell-depleting (NK1.1) mAb. (C–E) Mononuclear cell-dominant (C, arrows), or granulocyte-dominant (E, arrows) infiltrates inside ovarian follicles is a common feature of ovarian pathology in TI-nAOD. The dotted lines (C,E) outline ovarian follicles where oocytes are replaced by inflammatory cells while the asterisks (C–E) indicate normal ovarian follicles. (D), Inflammatory cells also infiltrate the ovarian interstitium (arrows). In (F), Immunofluorescence detection of mouse IgG (green fluorescence represents zona pellucida-bound ZP3 mAb); and NKG2D+ NK cells (red fluorescence) detectable outside and inside the zona pellucida (white arrows). DAPI (nuclear blue staining). Magnification [(A,B): 50×; (C–F): 400×]. Hematolyxin and eosin stain (A–E). [Reproduced from Rival et al. (45), J. Immunol. 191, 2865–2869].
Neonatal autoimmune ovarian disease is a unique and versatile model. First, ovaries can be safely removed to deplete ovarian autoantigens; and ovaries from donors of different ages and with different genetic modifications can be implanted under the kidney capsule can be used to monitor the effect of putative molecules in nAOD development. This approach has allowed us to show that nAOD susceptibility is not related to the intrinsic differences between adult and neonatal ovaries (44). It has also allowed us to find, unexpectedly, that nAOD induction requires resident ovarian cells that express FcγRIII. Second, nAOD induced by maternal immunization with the novel ZP3 chimeric peptide induces ZP3 antibodies without concomitant pathogenic T cell response. In addition to feeding pups with ZP3 antibody-positive mothers, nAOD is also inducible by passive transfer of a monoclonal antibody that recognizes the pZP3 335–342 (45). This allowed us to adjust the antibody dose to body weight between neonatal or adult mice and show that the nAOD resistance of adult mice is not related to insufficient antibody or ovarian IC. Third, there has been uncertainties regarding the physiological relevance of the ZP3 auto-peptide and its cognate antibody because (1) pZP3 was arbitrarily chosen as a novel auto-peptide with T and B cell epitopes; and (2) AOD induction also requires immunization with adjuvants. However, regulatory T cell (Treg) depletion from normal female mice leads to spontaneous production of autoAb that targets the same ZP3 335–342 epitope and the transfer of the sera from these animals induces nAOD in naïve pups. Thus, the pZP3 that has been studied for over 20 years is, in fact, a physiologically relevant B cell autoepitope. Fourth, nAOD develops in wild-type mice, and interestingly, nAOD also develops in mice that lack T and B cells. We call them T cell-dependent nAOD (TD-nAOD) model and T cell-independent nAOD (TI-nAOD) model, respectively. TD-nAOD induction is MHC-restricted, and it provides a useful platform for studying neonatal innate and adaptive responses and their interaction. On the other hand, the study of TI-nAOD in the recombination activation gene (Rag) knock out (KO) mice allows a reductionist approach to critically dissect the neonatal innate response. For example, we can define the novel properties of neonatal natural killer (NK) cells in vivo without the complex interaction between the innate and adaptive immune cells.
With the two nAOD models, we have addressed two fundamental questions on neonatal autoimmune disease: (1) Why are neonates more susceptible to autoimmunity; and (2) How do maternal autoAb induce an autoimmune disease in neonatal mice?
Why are Neonates More Susceptible to nAOD?
The neonatal time window of nAOD induction applies equally to TD-nAOD and TI-nAOD. Therefore, neonatal innate responses are sufficient to confer propensity to nAOD in newborn mice (44, 45). NK cells are components of the innate response that can perform antibody-dependent cellular cytotoxicity (ADCC) through FcγRIII and produce multiple cytokines. Differences in the phenotype and function of neonatal and adult NK cells have been described (46). However, while some reports show a poor neonatal NK cell function compared to adults, others demonstrate equal or enhanced effector functions in neonatal NK cells. Our recent work has convincingly demonstrated that neonatal NK cells and their unique properties are the key explanation for newborn propensity to nAOD (45). Strikingly, the age of the donors of NK cells that restore nAOD in genetically NK cell-deficient recipients that lack the rag and the common gamma chain genes, correlated precisely with the neonatal time window for nAOD induction. Thus, neonatal NK cells are critical for nAOD susceptibility (45). NK cell activation depends on the balance of signaling through stimulatory and inhibitory receptors (47). Self-tolerance by NK cells is achieved by the interaction of major histocompatibility complex class I (MHC I) with the murine Ly49 receptors or the human killer cell Ig-like receptors (KIR). However, expression of these receptors is stochastic. Those NK cells that lack receptors for self-MHC I are potentially autoreactive. However, in a process termed licensing, the NK cells that do not recognize self-MHC I become “anergic,” thus ensuring self-tolerance (48). Strikingly, it has been recently shown that the “anergic” NK cells that do not recognize self can become activated during inflammatory conditions and are more efficient in clearing infections or tumor cells than the licensed NK cells (49–51).
The expression of Ly49 receptors is known to be ontogenetically regulated; while ~5% of NK cells express Ly49C/I receptors in the first week of life, the frequency in adult mice is 10 times higher (52–54). This prompted us to investigate the role of Ly49 receptors in nAOD induction, and to address whether the delayed expression of Ly49C/I on NK cells confers adult resistance to nAOD. In a pivotal experiment, we showed that adult Ly49C/I negative NK cells could also induce nAOD only after the Ly49C/I+ NK cell subpopulation has been depleted in vivo. Our findings made three important points: (1) neonatal NK cells are documented for the first time to be functional and they are, in fact, hyper-reactive, (2) both neonatal and adult Ly49C/I negative NK cells can induce nAOD, and (3) they raise the interesting possibility that adult Ly49C/I+ NK cells can inhibit the activation of Ly49C/I negative NK cells, as shown by their capacity to block nAOD induction. Our findings are supported by recent literature on NK cells. Ivarsson et al. (55) recently showed that human fetal NK cells are hyper-responsive to cytokine stimulation, and CD16 engagement can overcome their hypo-responsiveness in killing HLA-negative targets. This study provided a clinical correlate to our findings. In addition, it has been shown that Ly49-negative NK cells can acquire full effector functions under inflammatory conditions. We should emphasize that whereas previous work (49, 51) has described intrinsic NK cell regulation, our study on nAOD provides the first evidence of extrinsic NK cell inhibition, where Ly49C/I+ NK cells may inhibit Ly49C/I negative NK cells (Figure 2). However, this possibility still requires further investigation. We currently speculate an indirect regulation that depends on the competition between the two NK cell subsets for the interaction with dendritic cells, which are required for NK cell priming (56, 57).
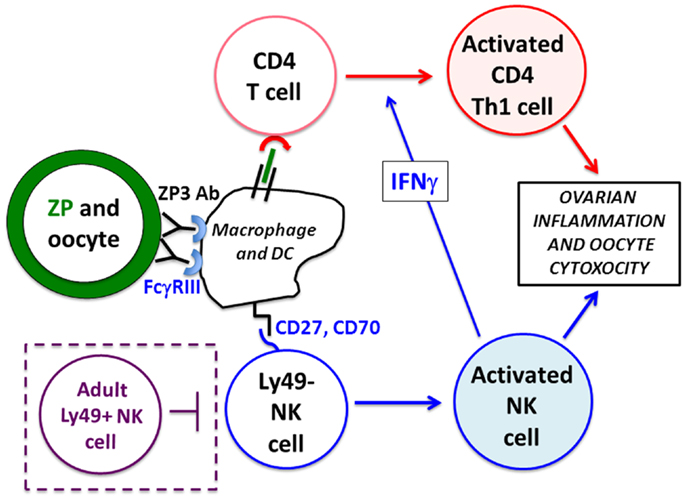
Figure 2. Mechanism of nAOD induction. Ovarian ZP3 immune complexes on the zona pellucida (ZP, green) are formed after maternal autoAb transfer and stimulate FcγRIII+ macrophage/dendritic cells that activate (1) a de novo CD4+ T cell response (in red), and (2) Ly49-negative neonatal NK cells (in blue). These NK cells produce IFNγ promoting a Th1 pathogenic CD4+ T cell response to ovarian antigens. Ovarian inflammation and oocyte depletion is, in turn, mediated by the FcγRIII+ neonatal NK cells and activated Th1 effector CD4+ T cells. Mice older than 9 days fail to develop nAOD because of the emergence of Ly49+ NK cells (in purple dotted box) and Treg function (not depicted).
As a mechanism of nAOD resistance in mice older than 7 days, Treg were documented to control susceptibility to nAOD. Thus, when Treg were depleted from 9-day-old pups with CD25 antibody, the neonatal time window was extended and the older mice became fully susceptible to nAOD (44). Therefore, at least two mechanisms acquired by adult mice confer resistance to autoimmune disease: (1) the innate system, by restraining NK cell activation with the acquisition of the inhibitory Ly49 receptors, and (2) the adaptive system, by acquiring Treg function. In contrast, these mechanisms are deficient in the neonatal mouse.
How Do the Maternal ZP3 autoAb Cause Ovarian Injury?
Dual NK Cell Requirements in nAOD: Induction of Antibody-Dependent Cytotoxicity (ADCC) and Ovarian Antigen-Specific Pathogenic CD4 T Cell Response
Maternal ZP3 autoAb transferred through the milk but not the placenta was critical for nAOD induction (44). Within 24 h, ZP3 IC was detectable in the zona pellucida of the ovary, a process that culminated in significant ovarian inflammation and loss of oocytes over the next 2 weeks [Figure 1; Ref. (44)]. The IC can cause immunopathology by: (1) activation of the complement cascade, (2) FcγRIII-dependent ADCC, and (3) T cell activation mediated by FcγRIII and/or complement receptor-bearing antigen presenting cells (APC). Complement C3b and C5b were undetectable in ovaries of mice with nAOD. However, studies with blocking antibodies and gene KO mice indicated that nAOD is dependent on FcγRIII expression (44, 58). In fact, we found that TD-nAOD pathogenesis requires both FcγRIII-dependent ADCC and a de novo T cell response. First, the neonatal NK cell must express FcγRIII to support nAOD (58). Second, the critical CD4+ T cells are required in TD-nAOD because (1) T cell depletion prevents nAOD, (2) CD4+ T cells from mice with nAOD transfer ovarian disease to naïve pups (44). Importantly, NK cells are required in both the inductive phase and the effector phase of the T cell response (58). Likely, the NK cell-derived IFNγ skews the T cell response toward IFNγ-producing Th1 cells (Figure 2). Indeed, IFNγ neutralization by antibody in the cell donors blocked the adoptive transfer of nAOD (58) and IFNγ-deficient NK cells failed to induce nAOD. Importantly, requirement of NK cells and FcγRIII expression have also been documented in the adult model of myasthenia gravis (59).
In our ongoing research, we have further clarified the pathogenesis of nAOD by making the following observations. First, in addition to NK cells, ovarian resident FcγRIII+ macrophages and/or dendritic cells are requisites in TD-nAOD. Second, NK cell activation most likely occurs in the lymph nodes where they are primed by the ovarian-derived FcγRIII+ cells. Third, NK cell homing and activation are dependent on IL-15, CD70 and CD27, and CXCR3.
Antibody to Viral Antigen also Co-Stimulates Active Immunity to Virus in Neonatal but Not Adult Mice
Maternal anti-microbial antibodies confer transient protection from infection to the progeny. However, compelling studies have shown that co-injection of a small amount of antiviral antibody with a live virus in neonatal mice can evoke effective long-term immunity against the virus. Neonatal infection with the FrCasE murine retrovirus before day 5–6 after birth leads to a virus-specific Treg response that inhibits efficient antiviral immunity (60). This results in fatality from neurodegeneration within 2 months and from erythroleukemia within 4–5 months. However, the Treg response is dramatically curtailed when the virus-infected neonatal mice receive a small dose of epitope-specific antiviral antibody within 2 days after the viral infection (61, 62). Concomitantly, the mice develop a strong antivirus CD8+ T cell response and life-long protection against the virus. As will be described below, the underlying mechanisms behind this successful neonatal antiviral vaccination schema are remarkably similar to the mechanism involved in nAOD induction. Therefore, neonatal exposure to antibody can enhance immunity against both autologous and foreign antigens.
Common Mechanisms are Shared between Autoimmunity and Viral Immunity Induction by Neonatal Immune Complexes
It is remarkable that neonatal IC formation by an epitope-specific antibody can lead to both autoimmune disease and antiviral immunity. Even more striking is the fact that the two responses deploy very similar mechanisms. They both require: (1) antibody injection and IC formation within the first week of life, (2) epitope-specific antibody that targets the functional domain of the antigen, (3) NK cells, (4) FcγRIII, (5) de novo induction of T cell responses, and (6) IFNγ. In both cases, the responses to IC beyond the neonatal time window were inhibited by Treg. Based on findings from our recent nAOD study, we can now add to this list, the critical influence of NK cell expression of the Ly49 inhibitory receptors.
General Conclusion/Summary
It is generally accepted that the higher susceptibility of newborns to infections is a consequence of the immature neonatal immune system. In support of this concept, many studies have described a weaker response by the neonatal immune system. However, this concept is still controversial, as others have found an equivalent or even stronger neonatal immune responses over the adult response. Moreover, a recent study showed that the poor protection to respiratory syncytial virus infection in newborns is due to the inhibition of the antibody production by the IFNγ produced by neonatal NK cells and T cells (63), suggesting that the neonatal immune system involves a complex cellular and molecular interplay. Experimental data derived from nAOD research have added new insight on the in vivo responsiveness of the newborn immune system. They clearly indicated that neonatal mice have the capacity to mount a robust immune response to tissue-associated IC that surpasses an adult response. The primary mechanism is the lower threshold of neonatal NK cell responses to tissue IC; and the capacity of neonatal NK cells to promote a pathogenic neonatal T cell response. It is important to emphasize that recent studies on human NK cells in support of this concept are also emerging (55). Our findings on nAOD are concordant with the observations by Michaud et al. (61) and Nasser et al. (60, 62) on the neonatal response to a foreign antigen in the context of a viral infection in the newborn. In both, the antibody exposure restricted to the first few days of life has been clearly documented to strongly enhance the neonatal immune response against its cognate antigen. The mechanisms shared between these two models should support a novel approach in effective vaccine design for the very young.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Institutes of Health Grant R01 AI51420. Claudia Rival is supported by National Institutes of Health Training Grant 5T32 DK007769-13.
References
1. PrabhuDas M, Adkins B, Gans H, King C, Levy O, Ramilo O, et al. Challenges in infant immunity: implications for responses to infection and vaccines. Nat Immunol (2011) 12:189–94. doi: 10.1038/ni0311-189
2. Nishizuka Y, Sakakura T. Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science (1969) 166:753–5. doi:10.1126/science.166.3906.753
3. Claeys D, Saraga E, Rossier BC, Kraehenbuhl JP. Neonatal injection of native proton pump antigens induces autoimmune gastritis in mice. Gastroenterology (1997) 113:1136–45. doi:10.1053/gast.1997.v113.pm9322508
4. Garza KM, Griggs ND, Tung KSK. Neonatal injection of an ovarian peptide induces autoimmune ovarian disease in female mice: requirement of endogenous neonatal ovaries. Immunity (1997) 6:89–96. doi:10.1016/S1074-7613(00)80245-9
5. Agersborg SS, Garza KM, Tung KS. Intestinal parasitism terminates self tolerance and enhances neonatal induction of autoimmune disease and memory. Eur J Immunol (2001) 31:851–9. doi:10.1002/1521-4141(200103)31:3<851::AID-IMMU851>3.0.CO;2-9
6. Ivanovska N, Yordanov M, Raykovska V. Single immunization of newborn mice with heterologous type-II collagen induces arthritic disease. Autoimmunity (2003) 36:205–10. doi:10.1080/0891693031000116057
9. Traub E. Factors influencing the persistence of choriomeningitis virus in the blood of mice after clinical recovery. J Exp Med (1938) 68:229–50. doi:10.1084/jem.68.2.229
10. Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature (1953) 172:603–6. doi:10.1038/172603a0
11. Hanan R, Oyama J. Inhibition of antibody formation in mature rabbits by contact with the antigen at an early age. J Immunol (1954) 73:49–53.
12. Dixon FJ, Maurer PH. Immunologic unresponsiveness induced by protein antigens. J Exp Med (1955) 101:245–50. doi:10.1084/jem.101.3.245
13. Nossal GJ. The immunological response of foetal mice to influenza virus. Aust J Exp Biol Med Sci (1957) 35:549–57. doi:10.1038/icb.1957.57
14. Schurmans S, Brighouse G, Kramer G, Wen L, Izui S, Merino J, et al. Transient T and B cell activation after neonatal induction of tolerance to MHC class II or Mls alloantigens. J Immunol (1991) 146:2152–60.
15. Guerau-de-Arellano M, Martinic M, Benoist C, Mathis D. Neonatal tolerance revisited: a perinatal window for Aire control of autoimmunity. J Exp Med (2009) 206:1245–52. doi:10.1084/jem.20090300
16. Ridge JP, Fuchs EJ, Matzinger P. Neonatal tolerance revisited: turning on newborn T cells with dendritic cells. Science (1996) 271:1723–6. doi:10.1126/science.271.5256.1723
17. Sarzotti M, Robbins DS, Hoffman PM. Induction of protective CTL responses in newborn mice by a murine retrovirus. Science (1996) 271:1726–8. doi:10.1126/science.271.5256.1726
18. Forsthuber T, Yip HC, Lehmann PV. Induction of TH1 and TH2 Immunity in neonatal mice. Science (1996) 271:1728–30. doi:10.1126/science.271.5256.1728
19. Chang C. Neonatal autoimmune diseases: a critical review. J Autoimmun (2012) 38:J223–38. doi:10.1016/j.jaut.2011.11.018
20. Capone C, Buyon JP, Friedman DM, Frishman WH. Cardiac manifestations of neonatal lupus: a review of autoantibody-associated congenital heart block and its impact in an adult population. Cardiol Rev (2012) 20:72–6. doi:10.1097/CRD.0b013e31823c808b
21. Gleicher N, Elkayam U. Preventing congenital neonatal heart block in offspring of mothers with anti-SSA/Ro and SSB/La antibodies: a review of published literature and registered clinical trials. Autoimmun Rev (2013) 12:1039–45. doi:10.1016/j.autrev.2013.04.006
22. De Carolis S, Salvi S, Botta A, Garofalo S, Garufi C, Ferrazzani S, et al. The impact of primary Sjogren’s syndrome on pregnancy outcome: our series and review of the literature. Autoimmun Rev (2014) 13:103–7. doi:10.1016/j.autrev.2013.09.003
23. Cavalcante P, Bernasconi P, Mantegazza R. Autoimmune mechanisms in myasthenia gravis. Curr Opin Neurol (2012) 25:621–9. doi:10.1097/WCO.0b013e328357a829
24. Verschuuren JJ, Huijbers MG, Plomp JJ, Niks EH, Molenaar PC, Martinez-Martinez P, et al. Pathophysiology of myasthenia gravis with antibodies to the acetylcholine receptor, muscle-specific kinase and low-density lipoprotein receptor-related protein 4. Autoimmun Rev (2013) 2:918–23. doi:10.1016/j.autrev.2013.03.001
25. Hertl M, Veldman C. Pemphigus – paradigm of autoantibody-mediated autoimmunity. Skin Pharmacol Appl Skin Physiol (2001) 14:408–18. doi:10.1159/000056375
26. Nishie W, Sawamura D, Natsuga K, Shinkuma S, Goto M, Shibaki A, et al. A novel humanized neonatal autoimmune blistering skin disease model induced by maternally transferred antibodies. J Immunol (2009) 183:4088–93. doi:10.4049/jimmunol.0800389
27. Carvalheiras G, Faira R, Braga J, Vasconcelos C. Fetal outcome in autoimmune diseases. Autoimmun Rev (2012) 11:A520–30. doi:10.1016/j.autrev.2011.12.002
28. Wang L, Zhou D, Lee J, Niu H, Faust TW, Frattini S, et al. Female mouse fetal loss mediated by maternal autoantibody. J Exp Med (2012) 209:1083–9. doi:10.1084/jem.20111986
29. Braunschweig D. Maternal autoantibodies in autism. Arch Neurol (2012) 69:693–9. doi:10.1001/archneurol.2011.2506
30. Fox E, Amaral D, Van de Water J. Maternal and fetal antibrain antibodies in development and disease. Dev Neurobiol (2012) 72:1327–34. doi:10.1002/dneu.22052
31. Yu LP, Robles DT, Abiru N, Kaur P, Rewers M, Kelemen K, et al. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc Natl Acad Sci U S A (2000) 97:1701–6. doi:10.1073/pnas.040556697
32. Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature (2010) 464:1293–300. doi:10.1038/nature08933
33. Fu W, Wojtkiewicz G, Weissleder R, Benoist C, Mathis D. Early window of diabetes determinism in NOD mice, dependent on the complement receptor CRIg, identified by noninvasive imaging. Nat Immunol (2012) 13:361–8. doi:10.1038/ni.2233
34. Melanitou E, Devendra D, Lin E, Miao D, Eisenbarth GS. Early and quantal (by litter) expression of insulin autoantibodies in the nonobese diabetic mice predict early diabetes onset. J Immunol (2004) 173:6603–10. doi:10.4049/jimmunol.173.11.6603
35. Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al. Crosstalk between neutrophils, B-1a cells and plasmcytoid dendritic cells initiates autoimmune diabetes. Nat Med (2012) 19:65–73. doi:10.1038/nm.3042
36. Greeley SAW, Katsumata M, Yu L, Eisenbarth GS, Moore DJ, Goodarzi H, et al. Elimination of maternally transmitted autoantibodies prevents diabetes in nonobese diabetic mice. Nat Med (2002) 8:399–402. doi:10.1038/nm0402-399
37. Silva DG, Daley SR, Hogan J, Lee SK, The CE, Hu DY, et al. Anti-islet autoantibodies trigger autoimmune diabetes in the presence of an increased frequency of islet-reactive CD4 T cells. Diabetes (2011) 60:2102–11. doi:10.2337/db10-1344
38. Zhao J, Kim KD, Yang X, Auh S, Fu YX, Tang H. Hyper innate responses in neonates lead to increased morbidity and mortality after infection. Proc Natl Acad Sci U S A (2008) 105:7528–33. doi:10.1073/pnas.0800152105
39. Kim JG, Moon SY, Chang YS, Lee JY. Autoimmune premature ovarian failure. J Obstet Gynaecol (1995) 21:59–66. doi:10.1111/j.1447-0756.1995.tb00899.x
40. Hoek A, Schoemaker J, Drexhage HA. Premature ovarian failure and ovarian autoimmunity. Endocr Rev (1997) 18:107–34. doi:10.1210/edrv.18.1.0291
41. Millar SE, Chamow SM, Baur AW, Oliver C, Robey F, Dean J. Vaccination with a synthetic zona pellucida peptide produces longterm contraception in female mice. Science (1989) 246:935–8. doi:10.1126/science.2479101
42. Tung KS, Setiady YY, Samy ET, Lewis J, Teuscher C. Autoimmune ovarian disease in day 3-thymectomized mice: the neonatal time window, antigen specificity of disease suppression, and genetic control. Curr Top Microbiol Immunol (2005) 293:209–47. doi:10.1007/3-540-27702-1_10
43. Lou Y, Ang J, Thai H, McElveen F, Tung KSK. A zona pellucida 3 peptide vaccine induces antibodies and reversible infertility without ovarian pathology. J Immunol (1995) 155:2715–20.
44. Setiady YY, Samy ET, Tung KSK. Maternal autoantibody triggers de novo T cell-mediated neonatal autoimmune disease. J Immunol (2003) 170:4656–64. doi:10.4049/jimmunol.170.9.4656
45. Rival C, Samy E, Setiady Y, Tung K. Cutting edge: Ly49C/I-neonatal NK cells predispose newborns to autoimmune ovarian disease induced by maternal autoantibody. J Immunol (2013) 191:2865–9. doi:10.4049/jimmunol.1301500
46. Lee YC, Lin SJ. Neonatal natural killer cell function: relevance to antiviral immune defense. Clin Dev Immunol (2013) 2013:427696. doi:10.1155/2013/427696
47. Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol (2011) 89:216–24. doi:10.1038/icb.2010.78
48. Elliot JM, Yokoyama WM. Unifying concepts of MHC-dependent natural killer education. Trends Immunol (2011) 32:364–72. doi:10.1016/j.it.2011.06.001
49. Orr MT, Murphy WJ, Lanier LL. “Unlicensed” natural killer cells dominate the response to cytomegalovirus infection. Nat Immunol (2010) 11:321–7. doi:10.1038/ni.1849
50. Sun JC. Re-educating natural killer cells. J Exp Med (2010) 207:2049–52. doi:10.1084/jem.20101748
51. Tarek N, Le Luduec JB, Gallagher MM, Zheng J, Venstrom JM, Chamberlain E, et al. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J Clin Invest (2012) 122:3260–70. doi:10.1172/JCI62749
52. Dorfman JR, Raulet DH. Acquisition of Ly49 receptor expression by developing natural killer cells. J Exp Med (1998) 187:609–18. doi:10.1084/jem.187.4.609
53. Kubota A, Lubota S, Lohwasser S, Mager DL, Takei F. Diversity of NK cell receptor repertoire in adult and neonatal mice. J Immunol (1999) 163:212–6.
54. Ortaldo JR, Winkler-Pickett R, Wiegand G. Activating Ly49D NK receptors: expression and function in relation to ontogeny and Ly49 inhibitor receptors. J Leukoc Biol (2000) 68:748–56. doi:10.1189/jlb.1938-3673
55. Ivarsson MA, Loh L, Marquardt N, Kekäläinen E, Berglin L, Björkström NK, et al. Differentiation and functional regulation of human fetal NK cells. J Clin Invest (2013) 123:3889–901. doi:10.1172/JCI68989
56. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime Natural Killer cells by trans-presenting Interleukin-15. Immunity (2007) 26:503–17. doi:10.1016/j.immuni.2007.03.006
57. Marcenaro E, Della Chiesa M, Pesce S, Agaugué S, Moretta A. The NK/DC complot. Adv Exp Med Biol (2009) 633:7–16. doi:10.1007/978-0-387-79311-5_2
58. Setiady YY, Pramoonjago P, Tung KSK. Requirements of NK cells and proinflammatory cytokines in T cell-dependent neonatal autoimmune ovarian disease triggered by immune complex. J Immunol (2004) 173:1051–8. doi:10.4049/jimmunol.173.2.1051
59. Shi FD, Wang HB, Li H, Hong S, Taniguchi M, Link H, et al. Natural killer cells determine the outcome of B cell-mediated autoimmunity. Nat Immunol (2000) 1:245–51. doi:10.1038/79792
60. Nasser R, Pelegrin M, Plays M, Gros L, Piechaczyk M. Control of regulatory T cells is necessary for vaccine-like effects of antiviral immunotherapy by monoclonal antibodies. Blood (2013) 121:1102–11. doi:10.1182/blood-2012-06-432153
61. Michaud HA, Gomard T, Gros L, Thiolon K, Nasser R, Jacquet C, et al. A crucial role for infected-cell/antibody immune complexes in the enhancement of endogenous antiviral immunity by short passive immunotherapy. PLoS Pathog (2010) 6:e1000948. doi:10.1371/journal.ppat.1000948
62. Nasser R, Pelegrin M, Michaud HA, Plays M, Piechaczyk M, Gros L. Long-lasting protective antiviral immunity induced by passive immunotherapies requires both neutralizing and effector functions of the administered monoclonal antibody. J Virol (2010) 84:10169–81. doi:10.1128/JVI.00568-10
Keywords: NK cells, Ly49 receptors, neonatal immunology, immune complex, autoimmune ovarian disease, regulatory T cells, congenital heart block, neonatal viral immunity
Citation: Rival C, Setiady Y, Samy ET, Harakal J and Tung KSK (2014) The unique neonatal NK cells: a critical component required for neonatal autoimmune disease induction by maternal autoantibody. Front. Immunol. 5:242. doi: 10.3389/fimmu.2014.00242
Received: 27 March 2014; Accepted: 09 May 2014;
Published online: 28 May 2014.
Edited by:
Sinuhe Hahn, University Clinics Basel, SwitzerlandReviewed by:
Daisuke Kamimura, Osaka University, JapanAna Claudia Zenclussen, Otto-von-Guericke University, Germany
Copyright: © 2014 Rival, Setiady, Samy, Harakal and Tung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Rival and Kenneth S. K. Tung, Department of Pathology, University of Virginia, Carter-Harrison Building, 345 Crispell Drive, Room 2710, Charlottesville, VA 22908, USA e-mail:Y21yM2JAdmlyZ2luaWEuZWR1;a3N0N2tAdmlyZ2luaWEuZWR1