 Robert Weil
Robert Weil- Unité de Signalisation Moléculaire et Activation Cellulaire, CNRS URA 2582, Institut Pasteur, Paris, France
Autoimmune diseases are characterized by the production of antibodies against self-antigens and generally arise from a failure of central or peripheral tolerance. However, these diseases may develop when newly appearing antigens are not recognized as self by the immune system. The mechanism by which some antigens are “invisible” to the immune system is not completely understood. Apoptotic and complement system defects or autophagy imbalance can generate this antigenic autoreactivity. Under particular circumstances, cellular debris containing autoreactive antigens can be recognized by innate immune receptors or other sensors and can eventually lead to autoimmunity. Ubiquitination may be one of the mechanisms protecting autoreactive antigens from the immune system that, if disrupted, can lead to autoimmunity. Ubiquitination is an essential post-translational modification used by cells to target proteins for degradation or to regulate other intracellular processes. The level of ubiquitination is regulated during T cell tolerance and apoptosis and E3 ligases have emerged as a crucial signaling pathway for the regulation of T cell tolerance toward self-antigens. I propose here that an unrecognized role of ubiquitin and ubiquitin-like proteins could be to render intracellular or foreign antigens (present in cellular debris resulting from apoptosis, complement system, or autophagy defects) invisible to the immune system in order to prevent the development of autoimmunity.
The central tolerance occurs in the thymus, where negative selection eliminates most of the developing thymocytes that can recognize self-antigens. Peripheral T cells recognize antigens that have been processed and presented in association with the major histocompatibility complex (MHC) by the antigen presenting cells. In addition to central tolerance, several peripheral tolerance “checkpoints” act to prevent self-reactivity. Indeed, self-reactive T cells can be suppressed by regulatory T cells, eliminated by clonal deletion or inactivated by a state of unresponsiveness known as T cell anergy. In T cells, activation-induced cell death (AICD) is responsible for maintaining tolerance to self-antigen. Pathogen invasion gives rise to massive immune cell proliferation until the infection is resolved and excess of immune cells is also eliminated by activation-induced cell death (AICD) via an apoptotic Fas (CD95; APO-1) or a tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-dependent pathway. Failure of these processes (inactivation mutations or decrease expression of CD95, Fasl, or other components of the Fas signaling pathway) can lead to lymphoproliferation and autoimmunity (1–4).
The immune system can be classified in innate and adaptive immunity. Innate immunity is a non-specific defense mechanism that starts immediately or within an hour in response to pathogens. The cells of the innate system play an important role for the initiation of the adaptive immune responses, which is also known as the acquired immune system. This system is more sophisticated and specific and is composed of the humoral (production of antibodies by B lymphocytes) and the cellular immunity (clonal expansion of specific T lymphocytes such as cytotoxic T lymphocytes).
The innate functions can be divided into six steps: migration, recognition, phagocytosis, antigen processing, presentation of the antigenic peptide to lymphocytes, and cytokine secretion. There are different possibilities for a dying cell or a pathogen to be eliminated from the body. Cells that undergo programed cell death called apoptosis can set up an initiation signal (“eat me signal”) to allow their recognition and digestion by phagocytic cells (5–11). Several reports have shown that the deposition of proteins of the complement system on apoptotic cells following the activation of the complement pathways is required for their efficient digestion by macrophages (12–15).
Selective autophagy of pathogens, organelles, and protein complexes represents an important host innate mechanism that allows their removal from the body. As for apoptosis, these processes require the modification of these pathogens, organelles, and protein aggregates by an “eat me signal,” which involves a complex modification of the bacteria, organelles, or protein complexes to allow their recognition by specific cargo receptors (16–25).
We will see that improper removal of dying cells or pathogens can cause autoimmune diseases.
Role of Defective Apoptosis, Cellular Uptake by Complement, or Autophagy in Autoimmunity
Apoptotic- and Complement-Mediated Cellular Uptake Defects and Autoimmunity
In adult tissues, cell death compensates for cell division. Apoptosis (Greek for “falling off”) also known under the name of programed cell death is a form of cell death, which presents specific morphological changes. Apoptosis can be mediated by the activation of cell death receptors on their cell surface such as Fas or by intracellular inducers such as Staurosporine (26–28). The intramolecular mechanism responsible of apoptosis involves a family of proteases called Caspases that have a Cysteine at their active site and cleave their substrates at specific Aspartic acids (29–33). Caspases are synthesized as inactive precursors (pro-Caspases), which are usually activated by cleavage by other Caspases. Caspases 8, 9, and 3 play pivotal functions in apoptotic cells. Caspases 8 and 9 activate Caspase 3 that subsequently cleaves vital substrates such as nuclear lamins or DNase leading to the breakdown of nuclear lamina and fragmentation of DNA. Mitochondria play an important role in apoptosis through release of cytochrome c. Cell death can be regulated by a complex network of pro- and anti-apoptotic proteins such as p53, which act to regulate the expression of death receptors and the mitochondria outer membrane Bcl2 family of proteins, which are involved in the Caspase activation pathways. Bcl2 and Bcl-XL inhibit apoptosis by preventing the release of cytochrome c from the mitochondria (34), whereas others like Bax and Bak stimulate the release of cytochrome c from mitochondria (35). Bax and Bak are themselves activated by other proteins belonging to the Bcl2 family such as Bid (35). A third group of factors, the inhibitors of apoptosis (IAP), consists of structurally conserved proteins (XIAP, cIAP1, c-IAP2, XIAP, livin α and β, ILP2, and survivin) that can block apoptosis through their inhibitory interaction with specific Caspases (36, 37). Several IAP proteins have been shown to regulate apoptosis in a Caspase-independent manner through the JNK or NF-κB signaling pathways (38–40). Interestingly, IAPs function as E3 ubiquitin ligases and can target cellular proteins for proteasomal degradation, this process being essential for apoptosis (41). IAPs activities are regulated by second mitochondria derived activator of Caspases (smac) (42). Signals from dying cells such as expression of phosphatidylserine at cell surface can be recognized by multiple receptors of macrophages or dendritic cells and defects in the clearance of apoptotic cell debris or in the uptake of dying cells can lead to autoimmunity (43, 44).
The complement system is composed of 30 different proteins that are either circulating in the serum or attached to the cell surface. This system plays four major functions: lysis of pathogens, activation of inflammation, opsonization, and immune clearance. For example, cellular uptake can be mediated by macrophage-associated complement receptors that constitute susceptibility genes for the development of the autoimmune disease systemic lupus erythematosus (SLE) (13, 45–49). Interestingly, it has been proposed that inadequate clearance of apoptotic cells due to the reduced level of complement is responsible for these diseases (50). Once engulfed, antigens derived from dead cells are processed and presented at the cell membrane in association with the MHC. These MHC/Ag interactions subsequently stimulate T helper cells that can release cytokines such as interferon α/β (IFN) to activate macrophages, monocytes, and B cells. Defects in the clearance of apoptotic cell or in the uptake of the dying cell have been linked to autoimmune diseases (51–54). Cell debris can be recognized by innate immune receptors or other sensors to develop autoimmunity.
Autophagy Defects and Autoimmunity
Autophagy (Greek for “self-eating”) is an evolutionary conserved mechanism that was first described by Christan de Duve as a lysosome-mediated degradation process for damaged cytoplasmic constituents. In macroautophagy, a double membrane called “phagophore” forms the autophagosome that surrounds cytoplasmic proteins or organelles. Autophagosome then fuses with lysosomes to create autolysosomes in which the cytosolic cargos are degraded. Autophagosome formation requires evolutionarily conserved proteins known as Atg proteins in yeast. Microtubule-associated protein light chain 3 (LC3) is the mammalian homolog of yeast Atg8 and is a widely used marker of autophagy. LC3 is localized in autophagosomes and the amount of its phagosome-associated form, named LC3-II, is correlated with the amount of autophagosome formed. Autophagy also contributes to innate immunity by protecting host cells from invading pathogens, a process called xenophagy. Substrates for selective autophagy are recognized either directly or indirectly (through “eat me” signals) in the cell (see below). As for apoptosis, autophagy imbalance – i.e., perturbation of autophagy function or autophagy gene defects – has been involved in autoimmune diseases (55–58).
Ubiquitin as a “Camouflage Uniform” to Avoid Recognition of Antigens by the Immune System?
My reflection was guided by different publications on the biological properties of ubiquitin chains (abundance, structure, immunogenicity, and function), E3 ubiquitin ligases and deubiquitinases (Figure 1).
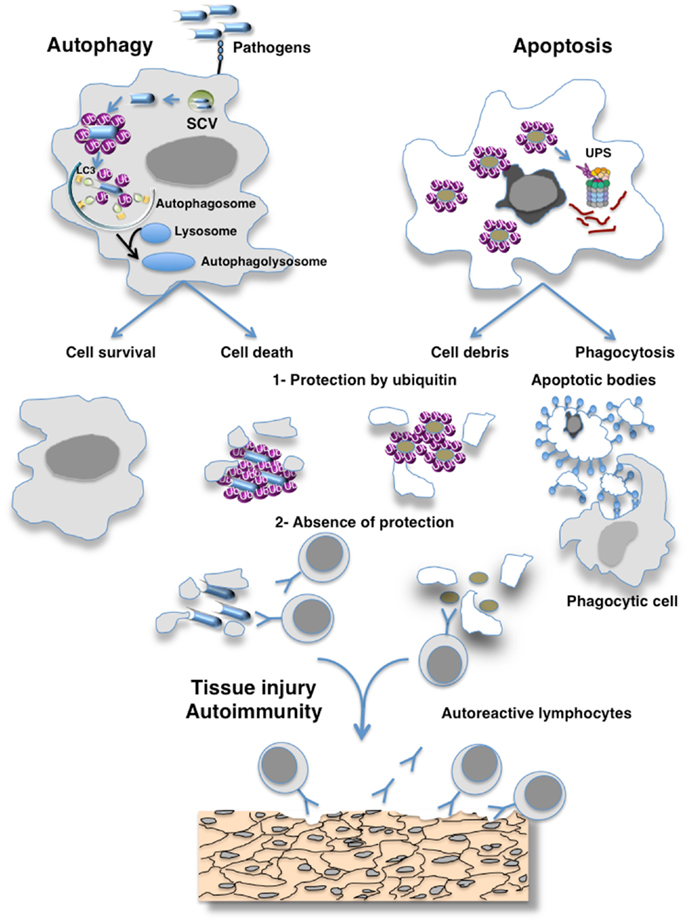
Figure 1. Hypothesis of the masking of epitopes in cell debris by ubiquitin preventing their recognition by the immune system. Eukaryotic cells use autophagy and the ubiquitin–proteasome system (UPS) as their major protein degradation pathways. Whereas the UPS is involved in the rapid degradation of proteins, autophagy pathways can selectively remove protein aggregates, damaged or excess organelles, and pathogens. Ubiquitin have been involved as a specific factor for selective autophagy as exemplified here by autophagy of pathogens. Different cellular adaptors connect pathogens to the protein light chain 3 (LC3), a key autophagy-related protein that is located at the surface of autophagosomes. Proteasome-mediated degradation also requires the ubiquitination of the cargo, which is then recognized by ubiquitin receptors allowing their degradation by the 26S proteasomes. The defective clearance of apoptotic debris by phagocytes and autophagy imbalance can result in the accumulation of cell debris that is responsible for the initiation of systemic autoimmunity. Such defect of clearance induces the release of immunogenic intracellular contents from the dying cells. I hypothesize that ubiquitination protects antigens generated by cells escaping from destruction by the immune system and that failure of ubiquitination mechanisms may induce an immune response to cross-reactive self-antigens that can lead to organ damage.
Ubiquitin, a peptide of 76 amino acids, can be covalently attached to protein substrates on lysine residues either as a monomer or polymer. Its amino acid sequence is highly conserved with little variance from insects to human. These ubiquitin chains are covalently attached to protein substrates by the concerted action of enzymes called E1, E2, and E3. Ubiquitin chains can be formed by isopeptide linkages between one of the seven internal lysine residues of an ubiquitin moiety and the carboxy-terminal residue of another ubiquitin. More recently, it has been shown that ubiquitin chains can also be formed in a head-to-tail fashion by peptidic bonds between the C-terminal glycine and the amino-terminal methionine of consecutive ubiquitin molecules. These ubiquitin modifications are reversible since deubiquitinating (DUB) enzymes can remove them.
Ubiquitin Chains Possess all of the Requirements for Masking Antigens to the Immune System
High abundance
Ubiquitin is highly abundant in all eukaryotic cells and tissues. It is the second most common post-translational protein modification after phosphorylation. Mass spectrometry analyses identified around 20,000 ubiquitination sites present on more than 5000 ubiquitin putative substrates (59–61).
Low immunogenicity
Importantly, ubiquitin is a highly conserved polypeptide and is poorly immunogenic (62). Interestingly, the presence of antibodies to ubiquitin has been found in 80% of patients exhibiting autoimmune responses associated with SLE and in those presenting a systemic sclerodermia (they are only present in 3% of normal human sera) (63–65). These reports suggest that the accumulation of ubiquitinated proteins is highly immunogenic in these autoimmune patients. However, this could not been confirmed by another study (66).
Conformation
The important variations in length and linkage architecture of these ubiquitin chains may provide a canvas surrounding the substrate that can adopt an open or more compact conformation depending on the type of ubiquitin chain and can determine the fate of conjugated proteins (67). Furthermore, polyubiquitin chains containing different linkages within the same chain (mixed chains) have also been described.
Functions
Since the discovery that ubiquitination targets proteins to proteolysis by Aaron Ciechanover, Avram Hershko, and Irwin Rose, it has been shown that ubiquitin conjugation controls different cellular processes (68).
The attachment of one or more ubiquitin provides a large interaction surface and results in a vast number of potential signals depending on the various conformations adopted by the ubiquitin chains. In addition to their role in targeting proteins for proteasomal and lysosomal degradation, signaling roles of ubiquitin have been discovered in many processes such as endocytosis, DNA repair, autophagy, and NF-κB activation (69–72).
Autophagy and the ubiquitin–proteasome system (UPS) are the two major intracellular degradation pathways. While the UPS is widely known for its role in intracellular protein degradation, autophagy is responsible for the degradation of long-lived proteins, organelles, and bacteria (71). Increased transcription of the ubiquitin gene that leads to ubiquitination of cellular proteins and degradation by the UPS is also characteristic of apoptotic cell and is required for the apoptotic process (73). The ubiquitin system is also widely used for autophagy. Various cytoplasmic bacteria are targeted for xenophagy through ubiquitin-mediated pathway. Bacteria and autophagosomes decorated with polyubiquitin chains recruit ubiquitin-binding adaptors, which in turn engage the autophagic machinery to restrict the proliferation of the bacteria (74). Two E3 ligases responsible for linking ubiquitin to the bacteria and/or bacteria-containing phagosomes have been recently identified as LRSAM1 and Parkin (75, 76). Ubiquitination of these aggregates and pathogens triggers the recruitment of different selective receptors, which in turn target these materials to the autophagosome membrane through their binding to autophagy modifiers such as LC3-like molecules that are present at the surface of autophagosomes (71). Damaged mitochondria are directly targeted to LC3-like molecules by the selective receptors Nix and FUNDC1 (16, 17). Four other autophagy receptors recognize ubiquitin chains that behave as “eat me signal” through their ubiquitin-binding domain (UBD): p62 (SQSTM1), NBR1, NDP52, and optineurin (16–25). These receptors also bind to LC3-like molecules through short LC3-interacting regions (LIRs). The sequential recruitment of DUB enzymes may negatively regulate the autophagy process. The physiological relevance of the cargo receptors is underscored by the presence of mutations in p62 and optineurin genes found in human patients with Paget’s disease of bone, primary open angle glaucoma, amyotrophic lateral sclerosis, or hepatocellular carcinoma (77–82).
In addition to phagocytosis and autophagy, neutrophil extracellular trap (NET) consisting of microbicidal molecules and extrusion of decondensed chromatin has been shown to be important strategy by which neutrophils kill microorganisms (83, 84). This process called NETosis is enhanced in SLE (85–87). A proposed hypothesis is that NETs may provide neoantigens for autoantibody formation (66, 88–91). Post-translational modifications of NET proteins such as ubiquitination and their proteolytic cleavage by NET proteases may provide the formation of neoantigens in certain circumstances. This could provide an explanation for the immunogenicity of ubiquitin in SLE and sclerodermia and in the rupture of tolerance responsible for these pathologies (see above).
It is well known that pathogens are involved in the appearance of autoimmune diseases (92), however, it is unclear whether the detection of microbial antigens by immune cells could result from a defects of masking of the antigens. Nethertheless, the detection of microbial antigens drive the clonal proliferation of specific T and B cells, and multiple mechanisms have been proposed to explain how pathogens can induce the expansion of autoreactive cells (93, 94). Among these mechanisms, it has been suggested that pathogenic antigens might react with the self and provoke tissue damage, a process known as molecular mimicry (94). Other hypotheses have been proposed to explain the emergence of autoreactive cells, including bystander activation of autoimmune cells caused by an inflammatory environment, processing and presentation of cryptic antigens, or adjuvant effects of pathogens.
I propose that, in addition to their roles in targeting proteins for degradation or cell signaling, and pathogens for destruction ubiquitin may protect antigens in cell debris resulting from apoptotic and autophagy defects and in NETs from recognition by the immune system (Figure 1).
In this hypothesis, we should expect that alteration of the ubiquitin machinery would lead to autoimmunity. Consistently, several mechanisms clearly illustrate the link between the ubiquitin system and peripheral tolerance mechanisms. These mechanisms include homeostatic regulation, T cell apoptosis, anergy, and regulatory CD4+ T cells. As described above, apoptosis is a highly regulated process involving the transcription of ubiquitin. In addition to being involved in T cell apoptosis, ubiquitin is also implicated in the molecular mechanism of T cell anergy. Clonal T cell anergy is a tolerance mechanism in which T cells are unresponsive to a second stimulation of the TCR. T cells can become anergic after encountering antigen in the absence of a CD28 co-stimulation or interleukin-2 (IL-2). The characteristic feature of clonal T cell anergy is a decrease in cytokine production and proliferation. The signaling cascade that leads to clonal T cell anergy has been the focus of many investigations and it has been shown that E3 ubiquitin ligases modulate different pathways leading to anergy. The transcription factor nuclear factor of activated T cells (NFAT) is a crucial factor for the induction of T cell anergy. In T cells, CD28 co-stimulation activates AP1-family of transcription factors, which cooperates with NFAT to induce the expression of genes encoding effectors of the T cell activation pathway. In contrast to this situation, a transcriptional program of anergy can be activated by NFAT in the absence of AP1 (95).
Interestingly, E3 ligases are over expressed in anergic T cells (96, 97) and among other anergy-associated genes, NFAT induces the transcription of E3 ligases genes (95). NFAT binds to promoter/enhancer sequences of gene related to anergy in lymphocytes (GRAIL), Cbl-b, Itch. Interestingly, the protein growth response (Egr)-2 and Egr-3 of the early Egr family of transcription factor are induced in response to NFAT and control the expression of Cbl-b and inhibition of T cell activation (98, 99). Consistently, it has been shown that Erg3−/− mice have increased susceptibility to autoimmunity (99). The upregulation of the anergy related genes were abrogated by cyclosporine A, an inhibitor of the calcium-dependent serine-threonine phosphatase calcineurin, which promotes the activation of NFAT. Since the discovery of the increased expression of E3 ligases in anergic T cells, the role of E3 ligases in peripheral T cell tolerance has been the focus of many reviews (100–104). Knocked-out mice and those carrying mutations or overexpressing ubiquitin ligases and deubiquitinases [such as autoimmune regulator (AIRE), Itch, Nedd4, Roquin, Cbl-b, TNFR-associated factor 6 (TRAF6), Act1, Peli1, NEDD4-family interacting protein 1 (Ndfip1), A20, CYLD] develop autoimmune diseases (100–102, 105–107). In particular, upregulation of the E3 ligases Cbl-b, GRAIL, Itch, and Peli1 during immune tolerance have been involved in the ubiquitination of key signaling molecules of the TCR pathway, and these studies have highlighted the key regulatory role of ubiquitin in the induction of tolerance and prevention of autoimmunity (100–102).
E3 Ubiquitin Ligases and Immune Tolerance
Cbl-b
The RING-type E3 ligase Cbl-b (Casitas B cell lymphoma b) was the first E3 ligase implicated in T cell tolerance (97, 105, 108). It belongs to a family of proteins comprising three members: c-Cbl, Cbl-b, and Cbl-3. Cbl-b is a critical regulator of T cells since Cbl-b-deficient T cells do not require CD28 co-stimulation for IL-2 production and proliferation. This observation suggests that Cbl-b regulates CD28-dependent T cell activation. Additionally, the loss of Cbl-b also results in aberrant activation of NF-κB in response to TCR stimulation. This hyperactivation of NF-κB is mediated by PKCθ, which promotes the formation of a complex formed by three signaling proteins, Carma1, Bcl10, and Malt1 as known as the CBM complex (109). Interestingly, the dysregulation of Cbl-b pathway in mice is responsible for increased susceptibility to experimental autoimmune diseases (96, 97, 108). The mechanism used by Cbl-b to downregulate T cell activation involves the GTP exchange factor Vav and the p85 subunit of phosphatidylinositol 3-kinase (PI3K). Previous studies have shown that PI3K phosphorylates phosphatidylinositol-4,5-diphosphate (PIP2) at the D3 position to form active lipid second messenger that regulate the exchange activity of Vav. It has been reported that Cbl-b induces the attachment of K48-linked ubiquitin chains to p85 and its proteasomal degradation and thus indirectly regulates Vav activation. The absence of Cbl-b increases CD28-mediated Vav1 activation and cytoskeleton reorganization (96, 97, 110).
Cbl-b constitutively interacts with the p85 subunit of the lipid kinase PI3K through its Proline rich region (111). Then, Cbl-b promotes p85 ubiquitination and affects its recruitment to the immune synapse, preventing the interaction of PI3K with CD28 (112). Finally, it has been shown that Cbl-b contributes to the disintegration of the immune synapse upon anergy induction (113).
TNFR-Associated Factor 6
TNFR-associated factor 6 is an adaptor protein that can act as an E3 ubiquitin ligase to mediate the activation of the NF-κB signaling pathway in response to TNF or interleukin-1 (IL-1)/TLR family members. Interestingly, TRAF6 expression is upregulated in activated T cells (114).
Unlike Cbl-b, TRAF6-mediated ubiquitination is associated with protein activation independently of the proteasomal degradation pathway. TRAF6 mediates the attachment of K63-linked ubiquitin chains to important signaling proteins allowing the activation of these pathways. In addition, TRAF6-deficient mice created by complementation of Rag2−/− blastocysts developed a progressive inflammation disease characterized by hyperactivation of CD4+ T cells. Moreover, T cell-specific deletion of TRAF6 (Traf6-ΔT) resulted in multiorgan inflammatory disease and resistance of T cells to the suppressor function of CD4+CD25+ regulatory T cells (114). Like Cbl-b−/− T cells, naïve Traf6-ΔT T cells hyperproliferate in response to anti-CD3 stimulation. Importantly, TRAF6-deficient cells exhibit hyperactivation of the PI3K-Akt pathway, suggesting a negative regulatory role of TRAF6 in CD28-dependent T cell activation. Consistently, as for Cbl-b, loss of TRAF6 restores the ability of CD28−/− T cells to proliferate and produce IL-2, suggesting that TRAF6 is a critical mediator of peripheral tolerance (115). TRAF6 also plays a role in thymic development since its deficiency results in disorganized distribution of medullary thymic epithelial cells (mTECs) and in the absence of mature mTECs (116). The grafting of TRAF6−/− thymic stroma tissue into athymic nude mice induced autoimmunity.
Gene Related to Anergy in Lymphocytes
Gene related to anergy in lymphocytes (GRAIL) is a RING E3 transmembrane glycoprotein that localizes to vesicular structures in the cell. GRAIL contains a single transmembrane-spanning domain that promotes its endosomal subcellular localization, a RING domain, a protease-associated (PA) domain, and a coiled-coil region (117). GRAIL was discovered using differential display to examine early changes in gene expression in anergic conditions (117). Retrovirally transduced T cell hybridoma that expresses GRAIL strongly decreases IL-2 and IL-4 production and inhibits proliferation in response to anti-CD3 and anti-CD28 co-stimulation. This function of GRAIL is dependent on endosomal trafficking, which suggests that GRAIL may target a protein of the endocytic pathway to control cytokine production. It was shown later using yeast two-hybrid system that GRAIL functionally interacts with two isoforms of the ubiquitin-specific protease Otubain 1 that belongs to the ovarian tumor (OTU) superfamily (118). Otubain 1 expressing cells contain less amount of GRAIL and secrete large amount of IL-2 following antigenic stimulation, while those expressing the alternatively spliced isoform, Otubain 1 alternative reading frame 1 (ARF-1), contain an increased amount of GRAIL and are functionally anergic. These data further demonstrate that the two isoforms of Otubain 1 have opposing effects on GRAIL and that Otubain 1 ARF-1 recruits the ubiquitin-specific protease 8 (USP-8) to promote GRAIL deubiquitination and stabilization. Using a prokaryotic system developed to screen for E3 ligase substrates, Rho guanine dissociation inhibitor (RhoGDI) was found as a potential substrate of GRAIL (119). GRAIL attaches ubiquitin-linked chains to RhoGDI and inhibits its effect on the reorganization of the cytoskeleton in T cells (120). Importantly, GRAIL-deficient mice are resistant to immune tolerance induction and exhibit a greater susceptibility to autoimmune diseases. GRAIL promotes CD3 ubiquitination and consequently, GRAIL-deficient T cells fail to regulate TCR expression in response to TCR stimulation and have an enhanced activation of NFATc1, while T cells expressing GRAIL present an enhanced TCR downregulation (107). It has been suggested that GRAIL is also responsible for the decrease cell surface expression of CD40L that occurs following anergy induction of CD4+ T cells (121). Furthermore, similarly to CD40L−/− mice, GRAIL overexpression results in reduced lymphoid follicle formation. GRAIL also targets CD83, a costimulatory signal for T cell proliferation and function, for its degradation by the proteasome 26S (122).
Itch
The E3 ligase Itch is encoded by the agouti locus, and its mutation is responsible for constant itching of the skin and development of a systemic lymphoproliferative disease characterized by enlarged secondary lymphoid organs (123). In addition, Itch−/− T cells present an activated phenotype and enhanced proliferation (124). CD4+ T cells of these mice are also resistant to Treg-dependent immunosuppression (125). Unlike Cbl-b and GRAIL, Itch contains a HECT domain that is responsible for its E3 ligase activity. Itch also includes a C-terminal C2 domain, which promotes its localization to endosomes. Itch expression is upregulated under anergizing stimuli allowing Itch to target members of the Jun family of transcription factors (i.e., c-Jun, Jun B) for ubiquitination and subsequent proteasomal degradation (126, 127). Since JunB is required for T helper 2 (Th2) differentiation, Itch downregulation in lymphocytes causes aberrant Th2 differentiation (124). Furthermore, Itch is responsible for the monoubiquitination of PLC-γ1 and PKC-θ and their lysosomal degradation (113). In these conditions, decreased PLC-γ1 expression is responsible for impaired Ca2+ signal and decreased stability of the immune synapse.
NEDD4-Family Interacting Protein 1
NEDD4-family interacting protein 1 (also known as N4WBP5) is upregulated in activated T cells. Ndfip1 interacts with NEDD4-family members and is proposed to function as an adaptor for ubiquitinated targets of the NEDD4-family such as Itch (128–130). Ndfip1 associates with Itch and promotes JunB degradation. Importantly, Ndfip1 deficiency is responsible for the failure of peripheral CD4+ T cell tolerance to self and innocuous foreign antigen, forcing them to exit cell cycle after a few divisions. This mechanism prevents CD4+ cells from differentiating into IL-4 producing cells. Ndfip1 deficiency disrupts peripheral T cell tolerance to pancreatic islets and increases the incidence of autoimmune pancreatic destruction and diabetes (131).
Roquin
Roquin was identified as a novel RING finger E3 ubiquitin ligase in a systematic screen using ethylnitrosourea (ENU)-induced mutation in the mouse and screening for autoimmunity (132). The first mutation identified was named sanroque because the accompanying lymphadenopathy exhibited all the features of SLE and small intestine inflammation: antibodies against dsDNA, proliferative glomerulonephritis with deposition of immune complexes necrotizing hepatitis, anemia, and autoimmune thrombocytomia (132, 133). Using a genetic approach, it was found that the sanroque mutation corresponded to a missense mutation of the Roquin gene. At the cellular level, these mice present increased numbers of germinal centers and follicular helper T cells. Sanroque CD4+ T cells express high level of the CD28 paralog “T cell costimulatory receptor inducible T cell costimulator” (ICOS) that was reduced upon re-expression of Roquin. Roquin was shown to localize in cytoplasmic stress granules (P bodies) (134) and to limit ICOS expression by promoting the degradation of ICOS mRNA. A conserved segment containing a region complementary to T cell expressed microRNA in ICOS 3′ untranslated mRNA was shown to be critical for the regulation by Roquin (135). However, instead of using microRNAs, a trimolecular complex containing Roquin, the RNA helicase Rck and the enhancer of decapping Edc4 was shown to promote ICOS mRNA decapping and ICOS repression (136).
To test the participation of ICOS with the sanroque phenotype, sanroque mice were crossed with ICOS−/− mice (135). Interestingly, the partial reduction of ICOS expression was accompanied by a reduction of lymphadenopathy, splenomegaly, total T- and B-cell number, and germinal center B cell number thus demonstrating that overexpression of ICOS contributes to sanroque mice autoimmune phenotype. Interestingly, it was further shown that tissue-specific ablation of Roquin in T, B cells, or in the entire hematopoietic system does not cause autoimmunity, while enforced Roquin expression in T cells exacerbates the severity of experimental arthritis (137). Finally, it was clearly shown that Roquin-1 has redundant function with Roquin-2 in the post-transcriptional repression of ICOS mRNA and that Roquin-2 compensates for the absence of Roquin-1, but not for its mutation (138, 139). It has been shown recently that Roquin-2 promotes ubiquitin-mediated proteasomal degradation of apoptosis signal-regulating kinase 1 (ASK1), a protein involved in the activation of JNK and p38 in response to stress (140). However, further investigation is required to define the ubiquitinated substrates of Roquin-1 and Roquin-2 that could explain the role of these proteins in autoimmunity.
Autoimmune Regulator
Autoimmune regulator (AIRE) gene mutation is responsible for the development of autoimmune-polyendocrinopathy-candidiasis ectodermal dystrophy (APECED), also known as autoimmune polyglandular syndrome type 1 (APS1), an organ-specific autoimmune disease. Mice carrying a defective AIRE gene also develop autoimmunity (141, 142). The clinical course in human and mice appears after a latent period. This period can be reduced in mice by the cross breeding of AIRE-deficient mice with Cbl-b KO mice (143). AIRE is predominantly expressed in medullar epithelial cells of the thymus and is considered to play important roles in the establishment of self-tolerance. mTECs have been implicated in the clonal deletion or inactivation of self-reactive thymocytes. Many ectopically expressed antigens are associated with organ-specific autoimmune diseases and it has been shown that AIRE-deficient mTECs present a decrease in the ectopic transcription of genes encoding peripheral antigens (141). Interestingly, AIRE functionally interacts with the small ubiquitin-related modifier (SUMO) ligase PIAS1 in the nuclear bodies, which cooperates to activate AIRE-known target genes (144). AIRE also functions as an E3 ligase. The AIRE gene is composed of two PHDS and SAND domains and its E3 ligase activity is mediated by the PHD1 domain and abolished by disease-causing mutations in the PHD1 (C311Y and P326Q) (145).
NF-κB Activator 1
NF-κB activator 1 (Act1) was discovered by searching for potential genes that play a role in NF-κB activation and was cloned in parallel via a yeast two-hybrid screen using NEMO as bait (146, 147). The structure of Act1 consists of two TRAF binding domains, an U-Box E3 ligase, a helix-loop-helix (HLH) and a SEF/IL-17R (SEFIR) domain. Act1 is an important negative regulator of B cell-mediated humoral immune response through its function in CD40 and BAFF signaling (two TNF receptor superfamily members). Upon CD40 stimulation, Act1 is recruited to CD40 that also interacts with TRAF3 (148). CD40 and BAFFR play critical roles in B cell survival and maturation, and dysregulation of these pathways leads to autoimmunity. In agreement with Act1 B cell function, Act1 knocked-out mice developed B cell-mediated autoimmune phenotypes including increased peripheral B cells, lymphadenopathy and splenomegaly, hypergammaglobulinemia, and autoantibodies (149). Interestingly, it was recently shown that crossing of AM14 transgenic (Tg) rheumatoid factor mice to Act1−/− mice leads to the activation of AM14 Tg B cells. AM14 Tg Act1−/− mice developed enlarged spleens and lymph nodes and presented expansions of rheumatoid factor-specific autoreactive B cells (150).
CD4+ T helper cells are divided into two lineages: T helper 1 (Th1) that secretes IFNγ and Th2 cells that secrete IL-4, IL-5, and IL-13. Recently, a third lineage that secretes IL-17 (Th17) has been found to play an important role in the defense against bacteria and fungal infections. The number of these cells is increased in autoimmune diseases. Act1 is a critical mediator of IL-17 signaling and has been involved in this pathway because its SEFIR domain is closely related to IL-17-receptor SEFIR domain. In response to IL-17, Act1 interacts with IL-17 receptor through an homotypic SEFIR–SEFIR interaction. Following this recruitment, Act1 attaches K63-linked ubiquitin chains to TRAF6 allowing its interaction with the TGFβ activated kinase 1 (TAK1) and subsequent phosphorylation of the NEMO/IKK complex followed by NF-κB activation. Because IL-17 is important in experimental autoimmune encephalomyelitis (EAE) pathogenesis, a model of multiple sclerosis, the effect of Act1 was assessed on a mouse model of EAE. Interestingly, Act1-deficient mice showed a delay in the onset of neurological impairment and had much lower severity compared to wild-type mice (151).
Pellino 1
The mammalian Peli (Pellino) family is composed of three members, Peli1, Peli2, and Peli3. The E3 ubiquitin ligase activity of Peli proteins is dependent on their C-terminal RING domain. Peli1 is essential for the TLR-mediated NF-κB activation dependent on the adaptor TRIF (152). Peli1-deficient T cells are hyper responsive to TCR and CD28 signals, they secrete more IL-2 and their naïve CD4+ T cells proliferation is not inhibited by Treg cells as opposed to wild-type CD4+ T cells (105). Consequently, Peli1−/− mice develop autoimmunity such as enlarged peripheral lymph nodes, moderate splenomegaly, and infiltration of many organs by cells from the immune response. Consistently, T cells from Peli1−/− mice show more pathogenic potential in EAE. In addition, Peli1 deficiency causes hyperactivation of late phase NF-κB and impairs ubiquitination and degradation of c-Rel, which is important for the thymic development of Tregs by directly inducing transcription of the Treg-specific transcription factor Foxp3 (153).
Deubiquitinase and Immune Tolerance
The ubiquitin-editing enzyme A20 and the deubiquitinase CYLD are important negative regulators of NF-κB signaling, and this control is important for adaptive and innate immunity. The modification of key signaling proteins such as NEMO, TRAF6, RIP1, Bcl10, MALT1 with K63-linked ubiquitin chains, or linear ubiquitin chains has emerged as an essential process for NF-κB activation. Ubiquitination can be reversed by DUB enzymes such as A20 and CYLD.
A20
A20 encoded by the TNF-α-inducible gene 3 (TNFAIP3) was identified in endothelial cells as a primary response gene induced upon treatment with TNF (154, 155). A20 was shown to be an ubiquitin-editing enzyme containing an amino-terminal DUB activity mediated by its OTU domain and a carboxy-terminal zing finger (ZnF) domain responsible for its E3 ubiquitin ligase activity (156). Further studies demonstrated a role of A20 not only in TNF signaling, but also in IL-1-, CD40-, TLR-, TCR-, and BCR-mediated NF-κB activation. A20 expression is rapidly induced upon NF-κB activation, which suggests that A20 prevents the persistent activation of NF-κB that could have negative effects on cell viability. In addition to ending NF-κB signaling, A20 exhibits NF-κB-unrelated functions. In response to pathogen invasion, A20 inhibits RIG-I-induced IRF activation and IFN responses by removing K63-linked polyubiquitin chains from the innate immune kinases TBK1 and IKKε (157–160). A20 has also been shown to control autophagy in response to TLR activation through the deubiquitination of Beclin 1, a protein essential for autophagy (161). In addition, A20 functions as an anti-apoptotic protein in several cell types and has been shown to cleave TRAIL-mediated ubiquitination of Caspase 8 in order to inhibit apoptosis (162).
Regarding autoimmunity, A20 has been reported as a disease susceptibility gene for human inflammatory and autoimmune pathology, including rheumatoid arthritis (RA) and juvenile idiopathic arthritis, SLE, inflammatory bowel disease (IBD), celiac disease, psoriasis, type 1 diabetes, Sjogren’s syndrome, coronary artery disease, rheumatic heart disease, and systemic sclerosis (163). A case-control study in African-American SLE patients with a genetic polymorphism of the A20 gene shows that it alters DUB activity and mediates risk of autoimmunity (164). As a consequence of the lack of regulation of NF-κB pathways, A20-deficient mice present severe inflammation and hypersensibility to TNF signaling and MyD88-dependent TLR signaling initiated by the commensal flora (165, 166). Cell type-specific deletion of A20 in B cells, dendritic cells, myeloid cells, intestinal epithelial cells, and keratinocytes confirmed that A20 plays a crucial role for the maintenance of tissue homeostasis and the control of systemic inflammation (167–173). Mice that specifically lack A20 in all cells of myeloid origin, develop spontaneous polyarthritis with the presence of type II collagen autoantibodies and inflammatory cytokines in serum (171). An A20-conditional KO in dendritic cells induces massive splenomegaly and lymphadenopathy. In one study, these mice developed an SLE-like phenotype, including the presence of double stranded DNA autoantibodies, glomerulonephritis, antiphospholipid syndrome, and arthritis, while, in an other study, they developed lymphocyte-dependent colitis, ankylosing arthritis, and enthesitis (172).
In B cells, A20 depletion induces enhanced B cell proliferation and survival as well as autoantibodies secretion (168, 169, 174). These mice exhibit a lupus-like autoimmune pathology characterized by increased numbers of germinal center B cells and glomerular immunoglobulin deposits. These mice also produce autoantibodies against cardiolipin, an important component of the inner mitochondrial membrane.
Cylindromatosis
Cylindromatosis (CYLD) is a tumor suppressor gene whose mutations result in a predisposition to familial cylindromatosis, a disease characterized by the development of benign tumors of the skin. CYLD exhibits deubiquitinase activity and has been identified as a critical regulator of NF-κB signaling by different approaches (175–177). As for A20, CYLD synthesis is regulated by NF-κB. CYLD KO mice confirmed that CYLD is a negative regulator of NF-κB (178, 179). The role of CYLD in B cell function is controversial. Jin and colleagues found that CYLD KO mice present several abnormalities of the immune system such as enlarged lymph nodes, B cell hyperplasia, expansion of the B cell marginal zone and B cell hyper-responsiveness in response to BCR, or LPS stimulation (178). However, in other studies, CYLD deficiency did not affect peripheral B cell numbers, but increased NF-κB activation upon stimulation (180, 181). T cells derived from CYLD KO mice displayed an hyper responsive phenotype (179). Adoptive transfer of CYLD KO T cells into RAG KO mice that lack endogenous lymphocytes induced autoimmune symptoms and intestinal inflammation (179). To explore potential overlapping functions between A20 and CYLD, the A20/CYLD double KO in B cells was generated (181). Interestingly, the lack of CYLD did not exacerbate the developmental defects and hyper-responsiveness of the A20-deficient B cell activity. The expression of CYLD must be tightly regulated since overexpression of the short spliced variant of CYLD gene (sCYLD) resulted in splenomegaly and lymphadenopathy, hyperactivation of CD4+ T cells and decrease in mTECs. When these mice were crossed onto TCR Tg background, they developed colonic inflammation associated with high production of autoantibodies.
Ubiquitin-Like Proteins and Autoimmunity
Finally, ubiquitin-like proteins (UBLs) (182), such as SUMO and ISG15, which are proteins related to ubiquitin, might also protect their substrates from recognition by the immune system. Among SUMO paralogs, SUMO4 harboring the M55V polymorphism is associated with susceptibility to autoimmune diabetes (183), although it is not clear whether SUMO4 protein is expressed. Additionally, ISGylation affects many proteins that are localized in different cellular compartments, participates in various cellular processes, and also targets a number of viral proteins. Interestingly, a large-scale microarray study of muscle samples revealed that the autoimmune disease dermatomyositis was specifically associated with enhancement of ISGylation (184).
Conclusion
Our understanding of the role played by ubiquitin in immune tolerance is still in its early stage. Although much attention focused on the function of E3 ligases and deubiquitinases in early events of T cell activation and immune tolerance, it is important to determine whether E3 ligase activity in dying cells could be involved in immune tolerance by preventing recognition by the immune system.
Another related issue is whether the UBDs that are present in a multitude of cellular proteins also protect proteins from recognition by the immune system through their interaction with ubiquitinated proteins. Intriguingly, defects in the interaction of ABIN1 with other proteins through its UBD cause autoimmunity in mice (185, 186). ABIN1 was originally identified by its interaction with the deubiquitinase A20, and mice with conditional knockout of A20 developed autoimmunity (see above) (187). Human polymorphisms were also identified in the ABIN1 coding gene, which constitutes a susceptibility gene for the development of autoimmune diseases.
In addition to changing the fate of proteins by targeting them to degradation via the proteasome pathway or to signaling complexes, ubiquitin may also mask epitopes that could lead to autoimmunity.
Finally, it is apparently difficult to conceive that the absence of ubiquitin ligase or deubiquitinases such as A20 or CYLD, which have the opposite effect leads to autoimmunity. However, it must be noted that the A20 and CYLD DUBs cleave K63-linked or linear ubiquitin chains, which are mostly important for protein–protein interactions. Consequently, their depletion increases the amount of K63-linked or linear-linked ubiquitinated proteins in signaling complexes and therefore enhances signaling. Consistently, the depletion of E3 ligases that mediate K48-linked ubiquitination of signaling proteins and proteasomal degradation also results in increased signaling. Furthermore, it is now well known that besides its DUB activity, A20 catalyzes the addition of K48-linked polyubiquitin chains to different substrates including Ubc13, UbcH5c, and RIP1 and targets them for proteasomal degradation (188) and that its depletion also impairs its E3 ubiquitin ligase activity. It is also important to consider that in lymphocytes, A20 is constitutively expressed to prevent uncontrolled activation of NF-κB and its proteasomal degradation or its cleavage by the paracaspase MALT1 impairs its inhibitory function to allow optimal NF-κB activation as exemplified by the increased antigen-mediated NF-κB signaling pathways in A20-deficient lymphocytes (189, 190). My hypothesis is that the decreased expression of E3 ubiquitin ligase induces autoimmunity by a dual mechanism: The loss of immune tolerance [absence of regulation of autoreactive lymphocytes or loss of regulatory T cell (Tregs) functions] and the absence of protection of epitopes against the immune system. However, we cannot make this assumption in the case of deubiquitinase deficiencies since epitopes are still protected by ubiquitin in these conditions.
In the past few years, it has been suggested that ubiquitin can be released from the cell to modulate the immune response (191). Ubiquitin is released from damaged erythrocytes, from damage tissues or from cells undergoing physiological turnover during prolonged blood storage. Besides its presence in the bloodstream, ubiquitin is also detectable in cerebrospinal fluid, bronchoalveolar lavage fluid, seminal plasma, and urine. Multiple diseases are known to be associated with increased concentration of extracellular ubiquitin. Its increased concentration in the serum has been reported in different pathologies including lupus erythematosus. Different properties have been attributed to extracellular ubiquitin such as antimicrobial activities. Intriguingly, ubiquitin have been originally purified from bovine thymus and characterized as a protein presenting a role in lymphocyte differentiation (192). Daily injection of ubiquitin induced T cell differentiation in the spleen and lymph nodes of athymic nu/nu mice. Interestingly, injection of ubiquitin into skeletal muscles led to a recruitment of lymphocytes (193). It has been suggested that ubiquitin presents immunosuppression activity against B and T cell functions (194). Despite the multiple effects that have been reported, little is known on the mechanism of action of extracellular ubiquitin. Recent data suggest that cellular uptake of extracellular ubiquitin is followed by its conjugaison to intracellular proteins (195, 196). Thus, the expression level of ubiquitin is not only controlled at the transcriptional level, but also by cellular uptake. Given that ubiquitin can be quickly available for protein modifications, that it lacks immunogenicity, that it attracts lymphocytes, and presents an immunosuppression activity, it is an attractive alternative hypothesis that post-translational modification of proteins with ubiquitin also neutralized their immunogenicity. Accordingly, administration of exogenous ubiquitin produces effects in various diseases including autoimmune diseases.
The experimental autoimmune EAE is a well-characterized model of the human autoimmune multiple sclerosis disease that is produced by the injection of brain extracts, which is responsible of demyelinisation. Using the same kind of approach, it would be interesting to determine whether injection of apoptotic thymocytes depleted for E1 ubiquitin enzymes to syngenic mice could induce autoimmunity, compared to injection of non-treated apoptotic thymocytes that should not induce autoimmunity (197). Another option would be to treat apoptotic extracts with non-specific DUB in vitro.
In conclusion, although it seems counterintuitive that modification of proteins by ubiquitin or UBLs may impinge on their visibility by the immune system, this unrecognized role of ubiquitin and UBL proteins in protecting from antigen-mediated detection by the immune system and its implication in immune tolerance could be a promising issue for the immunological field. Furthermore, this should highlight the therapeutic potential of manipulating E3 ligases and deubiquitinases in autoimmune diseases.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Robert Weil thanks François Rougeon, Aaron Ciechanover, Eric Vivier, Guido Kroemer, Jean-François Bach, Pierre Génin, Emmanuel Laplantine, and Andromahi Trivellas for critical reading of the manuscript and all the members of the laboratory for helpful discussions. Robert Weil is supported by CNRS. Work in the Robert Weil’s team is supported by Agence de Recherche contre le Cancer (ARC).
References
1. Linkes S, Fry C, Quinn A. Antigen-experienced CD4lo T cells are linked to deficient contraction of the immune response in autoimmune diabetes. Autoimmune Dis (2010) 2010:920148. doi:10.4061/2010/920148
2. Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature (1992) 356:314–7. doi:10.1038/356314a0
3. Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Köntgen F, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science (1999) 286:1735–8. doi:10.1126/science.286.5445.1735
4. Fortner KA, Bouillet P, Strasser A, Budd RC. Apoptosis regulators Fas and Bim synergistically control T-lymphocyte homeostatic proliferation. Eur J Immunol (2010) 40:3043–53. doi:10.1002/eji.201040577
5. Zhou Z, Hartwieg E, Horvitz HR. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell (2001) 104:43–56. doi:10.1016/S0092-8674(01)00190-8
6. Flannagan RS, Canton J, Furuya W, Glogauer M, Grinstein S. The phosphatidylserine receptor TIM4 utilizes integrins as coreceptors to effect phagocytosis. Mol Biol Cell (2014) 25(9):1511–22. doi:10.1091/mbc.E13-04-0212
7. Tyurin VA, Balasubramanian K, Winnica D, Tyurina YY, Vikulina AS, He RR, et al. Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic “eat-me” signals: cleavage and inhibition of phagocytosis by Lp-PLA2. Cell Death Differ (2014) 21:825–35. doi:10.1038/cdd.2014.1
8. Chen YZ, Mapes J, Lee ES, Skeen-Gaar RR, Xue D. Caspase-mediated activation of Caenorhabditis elegans CED-8 promotes apoptosis and phosphatidylserine externalization. Nat Commun (2013) 4:2726. doi:10.1038/ncomms3726
9. Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science (2013) 341:403–6. doi:10.1126/science.1236758
10. Shklyar B, Levy-Adam F, Mishnaevski K, Kurant E. Caspase activity is required for engulfment of apoptotic cells. Mol Cell Biol (2013) 33:3191–201. doi:10.1128/MCB.00233-13
11. Mapes J, Chen YZ, Kim A, Mitani S, Kang BH, Xue D. CED-1, CED-7, and TTR-52 regulate surface phosphatidylserine expression on apoptotic and phagocytic cells. Curr Biol (2012) 22:1267–75. doi:10.1016/j.cub.2012.05.052
12. Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol (2000) 76:227–324. doi:10.1016/S0065-2776(01)76021-X
13. Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med (1998) 188:2313–20. doi:10.1084/jem.188.12.2313
14. Quartier P, Potter PK, Ehrenstein MR, Walport MJ, Botto M. Predominant role of IgM-dependent activation of the classical pathway in the clearance of dying cells by murine bone marrow-derived macrophages in vitro. Eur J Immunol (2005) 35:252–60. doi:10.1002/eji.200425497
15. Baudino L, Sardini A, Ruseva MM, Fossati-Jimack L, Cook HT, Scott D, et al. C3 opsonization regulates endocytic handling of apoptotic cells resulting in enhanced T-cell responses to cargo-derived antigens. Proc Natl Acad Sci U S A (2013) 111:1503–8. doi:10.1073/pnas.1316877111
16. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep (2010) 11:45–51. doi:10.1038/embor.2009.256
17. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol (2012) 14:177–85. doi:10.1038/ncb2422
18. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem (2007) 282:24131–45. doi:10.1074/jbc.M702824200
19. Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell (2009) 33:505–16. doi:10.1016/j.molcel.2009.01.020
20. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science (2011) 333:228–33. doi:10.1126/science.1205405
21. Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, et al. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci (2012) 126:939–52. doi:10.1242/jcs.114819
22. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhães JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11:55–62. doi:10.1038/ni.1823
23. Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol (2009) 10:1215–21. doi:10.1038/ni.1800
24. Cemma M, Kim PK, Brumell JH. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy (2011) 7:341–5. doi:10.4161/auto.7.3.14046
25. Shvets E, Abada A, Weidberg H, Elazar Z. Dissecting the involvement of LC3B and GATE-16 in p62 recruitment into autophagosomes. Autophagy (2011) 7:683–8. doi:10.4161/auto.7.7.15279
26. Lowin B, Hahne M, Mattmann C, Tschopp J. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature (1994) 370:650–2. doi:10.1038/370650a0
27. Dive C, Hickman JA. Drug-target interactions: only the first step in the commitment to a programmed cell death? Br J Cancer (1991) 64:192–6. doi:10.1038/bjc.1991.269
28. Sen S, D’Incalci M. Apoptosis. Biochemical events and relevance to cancer chemotherapy. FEBS Lett (1992) 307:122–7. doi:10.1016/0014-5793(92)80914-3
29. Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science (1992) 256:97–100. doi:10.1126/science.1373520
30. Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature (1995) 376:37–43. doi:10.1038/376037a0
31. Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, et al. Human ICE/CED-3 protease nomenclature. Cell (1996) 87:171. doi:10.1016/S0092-8674(00)81334-3
32. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science (1998) 281:1312–6. doi:10.1126/science.281.5381.1312
33. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell (1996) 86:147–57. doi:10.1016/S0092-8674(00)80085-9
34. Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science (1997) 275:1129–32. doi:10.1126/science.275.5303.1129
35. Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ (2000) 7:1166–73. doi:10.1038/sj.cdd.4400783
36. Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature (1997) 388:300–4. doi:10.1038/40901
37. Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J (1997) 16:6914–25. doi:10.1093/emboj/16.23.6914
38. Hofer-Warbinek R, Schmid JA, Stehlik C, Binder BR, Lipp J, de Martin R. Activation of NF-kappa B by XIAP, the X chromosome-linked inhibitor of apoptosis, in endothelial cells involves TAK1. J Biol Chem (2000) 275:22064–8. doi:10.1074/jbc.M910346199
39. Sanna MG, Duckett CS, Richter BW, Thompson CB, Ulevitch RJ. Selective activation of JNK1 is necessary for the anti-apoptotic activity of hILP. Proc Natl Acad Sci U S A (1998) 95:6015–20.
40. Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J Exp Med (1998) 188:211–6. doi:10.1084/jem.188.1.211
41. Nachmias B, Ashhab Y, Ben-Yehuda D. The inhibitor of apoptosis protein family (IAPs): an emerging therapeutic target in cancer. Semin Cancer Biol (2004) 14:231–43. doi:10.1016/j.semcancer.2004.04.002
42. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell (2000) 102:33–42. doi:10.1016/S0092-8674(00)00008-8
43. Boyle KB, Randow F. The role of “eat-me” signals and autophagy cargo receptors in innate immunity. Curr Opin Microbiol (2013) 16(3):339–48. doi:10.1016/j.mib.2013.03.010
44. Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature (2001) 411:207–11. doi:10.1038/35079659
45. Moraitis E, Eleftheriou D, Cale C, Pilkington C, Brogan P. A51: c1q deficiency and autoimmunity-a single centre experience. Arthritis Rheumatol (2014) 66(Suppl 11):S76. doi:10.1002/art.38467
46. Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol (1997) 158:4525–8.
47. Nauta AJ, Daha MR, Tijsma O, van deWater B, Tedesco F, Roos A. The membrane attack complex of complement induces caspase activation and apoptosis. Eur J Immunol (2002) 32:783–92. doi:10.1002/1521-4141(200203)32:3<783::AID-IMMU783>3.0.CO;2-Q
48. Navratil JS, Watkins SC, Wisnieski JJ, Ahearn JM. The globular heads of C1q specifically recognize surface blebs of apoptotic vascular endothelial cells. J Immunol (2001) 166:3231–9. doi:10.4049/jimmunol.166.5.3231
49. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med (2001) 194:781–95. doi:10.1084/jem.194.6.781
50. Boackle SA. Complement and autoimmunity. Biomed Pharmacother (2003) 57:269–73. doi:10.1016/S0753-3322(03)00084-2
51. Pau E, Loh C, Minty GE, Chang NH, Wither JE. Identification of a lupus-susceptibility locus leading to impaired clearance of apoptotic debris on New Zealand black chromosome 13. Genes Immun (2013) 14:154–61. doi:10.1038/gene.2012.64
52. Peng Y, Elkon KB. Autoimmunity in MFG-E8-deficient mice is associated with altered trafficking and enhanced cross-presentation of apoptotic cell antigens. J Clin Invest (2011) 121:2221–41. doi:10.1172/JCI43254
53. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Möröy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet (2000) 25:177–81. doi:10.1038/76032
54. Licht R, Dieker JW, Jacobs CW, Tax WJ, Berden JH. Decreased phagocytosis of apoptotic cells in diseased SLE mice. J Autoimmun (2004) 22:139–45. doi:10.1016/j.jaut.2003.11.003
55. Bhattacharya A, Eissa NT. Autophagy and autoimmunity crosstalks. Front Immunol (2013) 4:88. doi:10.3389/fimmu.2013.00088
56. Pierdominici M, Vomero M, Barbati C, Colasanti T, Maselli A, Vacirca D, et al. Role of autophagy in immunity and autoimmunity, with a special focus on systemic lupus erythematosus. FASEB J (2012) 26:1400–12. doi:10.1096/fj.11-194175
57. Alirezaei M, Fox HS, Flynn CT, Moore CS, Hebb AL, Frausto RF, et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy (2009) 5:152–8. doi:10.4161/auto.5.2.7348
58. Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell (2007) 128:931–46. doi:10.1016/j.cell.2006.12.044
59. Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell (2011) 44:325–40. doi:10.1016/j.molcel.2011.08.025
60. Danielsen JM, Sylvestersen KB, Bekker-Jensen S, Szklarczyk D, Poulsen JW, Horn H, et al. Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol Cell Proteomics (2011) 10:M110003590. doi:10.1074/mcp.M110.003590
61. Kaiser P, Huang L. Global approaches to understanding ubiquitination. Genome Biol (2005) 6:233. doi:10.1186/gb-2005-6-10-233
62. Hershko A, Eytan E, Ciechanover A, Haas AL. Immunochemical analysis of the turnover of ubiquitin-protein conjugates in intact cells. Relationship to the breakdown of abnormal proteins. J Biol Chem (1982) 257:13964–70.
63. Elouaai F, Lulé J, Benoist H, Appolinaire-Pilipenko S, Atanassov C, Muller S, et al. Autoimmunity to histones, ubiquitin, and ubiquitinated histone H2A in NZB x NZW and MRL-lpr/lpr mice. Anti-histone antibodies are concentrated in glomerular eluates of lupus mice. Nephrol Dial Transplant (1994) 9:362–6.
64. Fujimoto M, Sato S, Ihn H, Kikuchi K, Tamaki T, Tamaki K, et al. Antiubiquitin antibody in localised and systemic scleroderma. Ann Rheum Dis (1996) 55:399–402. doi:10.1136/ard.55.6.399
65. Muller S, Briand JP, Van Regenmortel MH. Presence of antibodies to ubiquitin during the autoimmune response associated with systemic lupus erythematosus. Proc Natl Acad Sci U S A (1988) 85:8176–80. doi:10.1073/pnas.85.21.8176
66. Suzuki T, Burlingame RW, Casiano CA, Boey ML, Rubin RL. Antihistone antibodies in systemic lupus erythematosus: assay dependency and effects of ubiquitination and serum DNA. J Rheumatol (1994) 21:1081–91.
67. Ye Y, Blaser G, Horrocks MH, Ruedas-Rama MJ, Ibrahim S, Zhukov AA, et al. Ubiquitin chain conformation regulates recognition and activity of interacting proteins. Nature (2012) 492:266–70. doi:10.1038/nature11722
68. Ikeda F, Crosetto N, Dikic I. What determines the specificity and outcomes of ubiquitin signaling? Cell (2010) 143:677–81. doi:10.1016/j.cell.2010.10.026
69. Chen J, Chen ZJ. Regulation of NF-kappaB by ubiquitination. Curr Opin Immunol (2013) 25:4–12. doi:10.1016/j.coi.2012.12.005
70. Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell (2013) 49:795–807. doi:10.1016/j.molcel.2013.01.017
71. Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ (2013) 20:21–30. doi:10.1038/cdd.2012.72
72. Tanno H, Komada M. The ubiquitin code and its decoding machinery in the endocytic pathway. J Biochem (2013) 153:497–504. doi:10.1093/jb/mvt028
73. Delic J, Morange M, Magdelenat H. Ubiquitin pathway involvement in human lymphocyte gamma-irradiation-induced apoptosis. Mol Cell Biol (1993) 13:4875–83.
74. Randow F, Munz C. Autophagy in the regulation of pathogen replication and adaptive immunity. Trends Immunol (2012) 33:475–87. doi:10.1016/j.it.2012.06.003
75. Celli J. LRSAM1, an E3 ubiquitin ligase with a sense for bacteria. Cell Host Microbe (2012) 12:735–6. doi:10.1016/j.chom.2012.11.007
76. Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature (2013) 501:512–6. doi:10.1038/nature12566
77. Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science (2002) 295:1077–9. doi:10.1126/science.1066901
78. Willoughby CE, Chan LL, Herd S, Billingsley G, Noordeh N, Levin AV, et al. Defining the pathogenicity of optineurin in juvenile open-angle glaucoma. Invest Ophthalmol Vis Sci (2004) 45:3122–30. doi:10.1167/iovs.04-0107
79. Albagha OM, Visconti MR, Alonso N, Langston AL, Cundy T, Dargie R, et al. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget’s disease of bone. Nat Genet (2010) 42:520–4. doi:10.1038/ng.562
80. Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet (2002) 70:1582–8. doi:10.1086/340731
81. Hocking LJ, Lucas GJ, Daroszewska A, Mangion J, Olavesen M, Cundy T, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet (2002) 11:2735–9. doi:10.1093/hmg/11.22.2735
82. Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol (2011) 193:275–84. doi:10.1083/jcb.201102031
83. Knight JS, Carmona-Rivera C, Kaplan MJ. Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Front Immunol (2012) 3:380. doi:10.3389/fimmu.2012.00380
84. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi:10.1126/science.1092385
85. Sthoeger ZM, Bezalel S, Chapnik N, Asher I, Froy O. High alpha-defensin levels in patients with systemic lupus erythematosus. Immunology (2009) 127:116–22. doi:10.1111/j.1365-2567.2008.02997.x
86. Vordenbäumen S, Fischer-Betz R, Timm D, Sander O, Chehab G, Richter J, et al. Elevated levels of human beta-defensin 2 and human neutrophil peptides in systemic lupus erythematosus. Lupus (2010) 19:1648–53. doi:10.1177/0961203310377089
87. Ma CY, Jiao YL, Zhang J, Yang QR, Zhang ZF, Shen YJ, et al. Elevated plasma level of HMGB1 is associated with disease activity and combined alterations with IFN-alpha and TNF-alpha in systemic lupus erythematosus. Rheumatol Int (2012) 32:395–402. doi:10.1007/s00296-010-1636-6
88. Dieker JW, Fransen JH, van Bavel CC, Briand JP, Jacobs CW, Muller S, et al. Apoptosis-induced acetylation of histones is pathogenic in systemic lupus erythematosus. Arthritis Rheum (2007) 56:1921–33. doi:10.1002/art.22646
89. van Bavel CC, Dieker J, Muller S, Briand JP, Monestier M, Berden JH, et al. Apoptosis-associated acetylation on histone H2B is an epitope for lupus autoantibodies. Mol Immunol (2009) 47:511–6. doi:10.1016/j.molimm.2009.08.009
90. van Bavel CC, Dieker JW, Kroeze Y, Tamboer WP, Voll R, Muller S, et al. Apoptosis-induced histone H3 methylation is targeted by autoantibodies in systemic lupus erythematosus. Ann Rheum Dis (2011) 70:201–7. doi:10.1136/ard.2010.129320
91. Liu CL, Tangsombatvisit S, Rosenberg JM, Mandelbaum G, Gillespie EC, Gozani OP, et al. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res Ther (2012) 14:R25. doi:10.1186/ar3933
92. Bach JF. Infections and autoimmune diseases. J Autoimmun (2005) 25(Suppl):74–80. doi:10.1016/j.jaut.2005.09.024
93. Delogu LG, Deidda S, Delitala G, Manetti R. Infectious diseases and autoimmunity. J Infect Dev Ctries (2011) 5:679–87. doi:10.3855/jidc.2061
94. Oldstone MB. Molecular mimicry, microbial infection, and autoimmune disease: evolution of the concept. Curr Top Microbiol Immunol (2005) 296:1–17. doi:10.1007/3-540-30791-5_1
95. Macián F, García-Cózar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell (2002) 109:719–31. doi:10.1016/S0092-8674(02)00767-5
96. Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, et al. Cbl-b regulates the CD28 dependence of T-cell activation. Nature (2000) 403:216–20. doi:10.1038/35003235
97. Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature (2000) 403:211–6. doi:10.1038/35003228
98. Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, et al. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat Immunol (2005) 6:472–80. doi:10.1038/ni0705-737
99. Collins S, Lutz MA, Zarek PE, Anders RA, Kersh GJ, Powell JD. Opposing regulation of T cell function by Egr-1/NAB2 and Egr-2/Egr-3. Eur J Immunol (2008) 38:528–36. doi:10.1002/eji.200737157
100. Lin AE, Mak TW. The role of E3 ligases in autoimmunity and the regulation of autoreactive T cells. Curr Opin Immunol (2007) 19:665–73. doi:10.1016/j.coi.2007.10.002
101. Paolino M, Penninger JM. E3 ubiquitin ligases in T-cell tolerance. Eur J Immunol (2009) 39:2337–44. doi:10.1002/eji.200939662
102. Puga I, Rao A, Macian F. Targeted cleavage of signaling proteins by caspase 3 inhibits T cell receptor signaling in anergic T cells. Immunity (2008) 29:193–204. doi:10.1016/j.immuni.2008.06.010
103. Jiang X, Chen ZJ. The role of ubiquitylation in immune defence and pathogen evasion. Nat Rev Immunol (2012) 12:35–48. doi:10.1038/nri3111
104. Fathman CG, Lineberry NB. Molecular mechanisms of CD4+ T-cell anergy. Nat Rev Immunol (2007) 7:599–609. doi:10.1038/nri2131
105. Chang M, Jin W, Chang JH, Xiao Y, Brittain GC, Yu J, et al. The ubiquitin ligase Peli1 negatively regulates T cell activation and prevents autoimmunity. Nat Immunol (2011) 12:1002–9. doi:10.1038/ni.2090
106. Hoyne GF, Flening E, Yabas M, Teh C, Altin JA, Randall K, et al. Visualizing the role of Cbl-b in control of islet-reactive CD4 T cells and susceptibility to type 1 diabetes. J Immunol (2013) 186:2024–32. doi:10.4049/jimmunol.1002296
107. Nurieva RI, Zheng S, Jin W, Chung Y, Zhang Y, Martinez GJ, et al. The E3 ubiquitin ligase GRAIL regulates T cell tolerance and regulatory T cell function by mediating T cell receptor-CD3 degradation. Immunity (2010) 32:670–80. doi:10.1016/j.immuni.2010.05.002
108. Jeon MS, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity (2004) 21:167–77. doi:10.1016/j.immuni.2004.07.013
109. Qiao G, Li Z, Molinero L, Alegre ML, Ying H, Sun Z, et al. T-cell receptor-induced NF-kappaB activation is negatively regulated by E3 ubiquitin ligase Cbl-b. Mol Cell Biol (2008) 28:2470–80. doi:10.1128/MCB.01505-07
110. Krawczyk C, Bachmaier K, Sasaki T, Jones RG, Snapper SB, Bouchard D, et al. Cbl-b is a negative regulator of receptor clustering and raft aggregation in T cells. Immunity (2000) 13:463–73. doi:10.1016/S1074-7613(00)00046-7
111. Fang D, Wang HY, Fang N, Altman Y, Elly C, Liu YC. Cbl-b, a RING-type E3 ubiquitin ligase, targets phosphatidylinositol 3-kinase for ubiquitination in T cells. J Biol Chem (2001) 276:4872–8. doi:10.1074/jbc.M008901200
112. Fang D, Liu YC. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat Immunol (2001) 2:870–5. doi:10.1038/ni0901-870
113. Heissmeyer V, Macián F, Im SH, Varma R, Feske S, Venuprasad K, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol (2004) 5:255–65. doi:10.1038/ni1047
114. King CG, Kobayashi T, Cejas PJ, Kim T, Yoon K, Kim GK, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med (2006) 12:1088–92. doi:10.1038/nm1449
115. King CG, Buckler JL, Kobayashi T, Hannah JR, Bassett G, Kim T, et al. Cutting edge: requirement for TRAF6 in the induction of T cell anergy. J Immunol (2008) 180:34–8. doi:10.4049/jimmunol.180.1.34
116. Akiyama T, Maeda S, Yamane S, Ogino K, Kasai M, Kajiura F, et al. Dependence of self-tolerance on TRAF6-directed development of thymic stroma. Science (2005) 308:248–51. doi:10.1126/science.1105677
117. Anandasabapathy N, Ford GS, Bloom D, Holness C, Paragas V, Seroogy C, et al. GRAIL: an E3 ubiquitin ligase that inhibits cytokine gene transcription is expressed in anergic CD4+ T cells. Immunity (2003) 18:535–47. doi:10.1016/S1074-7613(03)00084-0
118. Soares L, Seroogy C, Skrenta H, Anandasabapathy N, Lovelace P, Chung CD, et al. Two isoforms of otubain 1 regulate T cell anergy via GRAIL. Nat Immunol (2004) 5:45–54. doi:10.1038/ni1017
119. Su L, Lineberry N, Huh Y, Soares L, Fathman CG. A novel E3 ubiquitin ligase substrate screen identifies Rho guanine dissociation inhibitor as a substrate of gene related to anergy in lymphocytes. J Immunol (2006) 177:7559–66. doi:10.4049/jimmunol.177.11.7559
120. Ichikawa D, Mizuno M, Yamamura T, Miyake S. GRAIL (gene related to anergy in lymphocytes) regulates cytoskeletal reorganization through ubiquitination and degradation of Arp2/3 subunit 5 and coronin 1A. J Biol Chem (2011) 286:43465–74. doi:10.1074/jbc.M111.222711
121. Lineberry NB, Su LL, Lin JT, Coffey GP, Seroogy CM, Fathman CG. Cutting edge: the transmembrane E3 ligase GRAIL ubiquitinates the costimulatory molecule CD40 ligand during the induction of T cell anergy. J Immunol (2008) 181:1622–6. doi:10.4049/jimmunol.181.3.1622
122. Su LL, Iwai H, Lin JT, Fathman CG. The transmembrane E3 ligase GRAIL ubiquitinates and degrades CD83 on CD4 T cells. J Immunol (2009) 183:438–44. doi:10.4049/jimmunol.0900204
123. Perry WL, Hustad CM, Swing DA, O’Sullivan TN, Jenkins NA, Copeland NG. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat Genet (1998) 18:143–6. doi:10.1038/ng0298-143
124. Fang D, Elly C, Gao B, Fang N, Altman Y, Joazeiro C, et al. Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat Immunol (2002) 3:281–7. doi:10.1038/ni763
125. Venuprasad K, Huang H, Harada Y, Elly C, Subramaniam M, Spelsberg T, et al. The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1. Nat Immunol (2008) 9:245–53. doi:10.1038/ni1564
126. Hartenstein B, Teurich S, Hess J, Schenkel J, Schorpp-Kistner M, Angel P. Th2 cell-specific cytokine expression and allergen-induced airway inflammation depend on JunB. EMBO J (2002) 21:6321–9. doi:10.1093/emboj/cdf648
127. Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC, et al. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science (2004) 306:271–5. doi:10.1126/science.1099414
128. Jolliffe CN, Harvey KF, Haines BP, Parasivam G, Kumar S. Identification of multiple proteins expressed in murine embryos as binding partners for the WW domains of the ubiquitin-protein ligase Nedd4. Biochem J (2000) 351(Pt 3):557–65. doi:10.1042/0264-6021:3510557
129. Harvey KF, Shearwin-Whyatt LM, Fotia A, Parton RG, Kumar S. N4WBP5, a potential target for ubiquitination by the Nedd4 family of proteins, is a novel Golgi-associated protein. J Biol Chem (2002) 277:9307–17. doi:10.1074/jbc.M110443200
130. Oliver PM, Cao X, Worthen GS, Shi P, Briones N, MacLeod M, et al. Ndfip1 protein promotes the function of itch ubiquitin ligase to prevent T cell activation and T helper 2 cell-mediated inflammation. Immunity (2006) 25:929–40. doi:10.1016/j.immuni.2006.10.012
131. Altin JA, Daley SR, Howitt J, Rickards HJ, Batkin AK, Horikawa K, et al. Ndfip1 mediates peripheral tolerance to self and exogenous antigen by inducing cell cycle exit in responding CD4+ T cells. Proc Natl Acad Sci U S A (2013) 111:2067–74. doi:10.1073/pnas.1322739111
132. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435:452–8. doi:10.1038/nature03555
133. Schaefer JS, Montufar-Solis D, Nakra N, Vigneswaran N, Klein JR. Small intestine inflammation in Roquin-mutant and Roquin-deficient mice. PLoS One (2013) 8:e56436. doi:10.1371/journal.pone.0056436
134. Athanasopoulos V, Barker A, Yu D, Tan AH, Srivastava M, Contreras N, et al. The ROQUIN family of proteins localizes to stress granules via the ROQ domain and binds target mRNAs. FEBS J (2010) 277:2109–27. doi:10.1111/j.1742-4658.2010.07628.x
135. Yu D, Tan AH, Hu X, Athanasopoulos V, Simpson N, Silva DG, et al. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature (2007) 450:299–303. doi:10.1038/nature06253
136. Glasmacher E, Hoefig KP, Vogel KU, Rath N, Du L, Wolf C, et al. Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA-independent post-transcriptional repression. Nat Immunol (2010) 11:725–33. doi:10.1038/ni.1902
137. Bertossi A, Aichinger M, Sansonetti P, Lech M, Neff F, Pal M, et al. Loss of Roquin induces early death and immune deregulation but not autoimmunity. J Exp Med (2011) 208:1749–56. doi:10.1084/jem.20110578
138. Pratama A, Ramiscal RR, Silva DG, Das SK, Athanasopoulos V, Fitch J, et al. Roquin-2 shares functions with its paralog Roquin-1 in the repression of mRNAs controlling T follicular helper cells and systemic inflammation. Immunity (2013) 38:669–80. doi:10.1016/j.immuni.2013.01.011
139. Vogel KU, Edelmann SL, Jeltsch KM, Bertossi A, Heger K, Heinz GA, et al. Roquin paralogs 1 and 2 redundantly repress the Icos and Ox40 costimulator mRNAs and control follicular helper T cell differentiation. Immunity (2013) 38:655–68. doi:10.1016/j.immuni.2012.12.004
140. Maruyama T, Araki T, Kawarazaki Y, Naguro I, Heynen S, Aza-Blanc P, et al. Roquin-2 promotes ubiquitin-mediated degradation of ASK1 to regulate stress responses. Sci Signal (2014) 7:ra8. doi:10.1126/scisignal.2004822
141. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science (2002) 298:1395–401. doi:10.1126/science.1075958
142. Ramsey C, Winqvist O, Puhakka L, Halonen M, Moro A, Kämpe O, et al. Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Hum Mol Genet (2002) 11:397–409. doi:10.1093/hmg/11.4.397
143. Teh CE, Daley SR, Enders A, Goodnow CC. T-cell regulation by casitas B-lineage lymphoma (Cblb) is a critical failsafe against autoimmune disease due to autoimmune regulator (Aire) deficiency. Proc Natl Acad Sci U S A (2010) 107:14709–14. doi:10.1073/pnas.1009209107
144. Ilmarinen T, Kangas H, Kytömaa T, Eskelin P, Saharinen J, Seeler JS, et al. Functional interaction of AIRE with PIAS1 in transcriptional regulation. Mol Immunol (2008) 45:1847–62. doi:10.1016/j.molimm.2007.10.045
145. Uchida D, Hatakeyama S, Matsushima A, Han H, Ishido S, Hotta H, et al. AIRE functions as an E3 ubiquitin ligase. J Exp Med (2004) 199:167–72. doi:10.1084/jem.20031291
146. Leonardi A, Chariot A, Claudio E, Cunningham K, Siebenlist U. CIKS, a connection to Ikappa B kinase and stress-activated protein kinase. Proc Natl Acad Sci U S A (2000) 97:10494–9. doi:10.1073/pnas.190245697
147. Li X, Commane M, Nie H, Hua X, Chatterjee-Kishore M, Wald D, et al. Act1, an NF-kappa B-activating protein. Proc Natl Acad Sci U S A (2000) 97:10489–93. doi:10.1073/pnas.160265197
148. Qian Y, Zhao Z, Jiang Z, Li X. Role of NF kappa B activator Act1 in CD40-mediated signaling in epithelial cells. Proc Natl Acad Sci U S A (2002) 99:9386–91. doi:10.1073/pnas.142294499
149. Qian Y, Qin J, Cui G, Naramura M, Snow EC, Ware CF, et al. Act1, a negative regulator in CD40- and BAFF-mediated B cell survival. Immunity (2004) 21:575–87. doi:10.1016/j.immuni.2004.09.001
150. Giltiay NV, Lu Y, Cullen JL, Jørgensen TN, Shlomchik MJ, Li X. Spontaneous loss of tolerance of autoreactive B cells in Act1-deficient rheumatoid factor transgenic mice. J Immunol (2013) 191:2155–63. doi:10.4049/jimmunol.1300152
151. Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol (2007) 8:247–56. doi:10.1038/ni1439
152. Chang M, Jin W, Sun SC. Peli1 facilitates TRIF-dependent toll-like receptor signaling and proinflammatory cytokine production. Nat Immunol (2009) 10:1089–95. doi:10.1038/ni.1777
153. Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity (2009) 31:921–31. doi:10.1016/j.immuni.2009.09.022
154. Dixit VM, Green S, Sarma V, Holzman LB, Wolf FW, O’Rourke K, et al. Tumor necrosis factor-alpha induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. J Biol Chem (1990) 265:2973–8.
155. Opipari AW Jr, Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem (1990) 265:14705–8.
156. Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature (2004) 430:694–9. doi:10.1038/nature02794
157. Lin R, Yang L, Nakhaei P, Sun Q, Sharif-Askari E, Julkunen I, et al. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J Biol Chem (2006) 281:2095–103. doi:10.1074/jbc.M510326200
158. Maelfait J, Roose K, Bogaert P, Sze M, Saelens X, Pasparakis M, et al. A20 (Tnfaip3) deficiency in myeloid cells protects against influenza A virus infection. PLoS Pathog (2012) 8:e1002570. doi:10.1371/journal.ppat.1002570
159. Parvatiyar K, Barber GN, Harhaj EW. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem (2010) 285:14999–5009. doi:10.1074/jbc.M110.109819
160. Saitoh T, Yamamoto M, Miyagishi M, Taira K, Nakanishi M, Fujita T, et al. A20 is a negative regulator of IFN regulatory factor 3 signaling. J Immunol (2005) 174:1507–12. doi:10.4049/jimmunol.174.3.1507
161. Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal (2010) 3:ra42. doi:10.1126/scisignal.2000751
162. Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell (2009) 137:721–35. doi:10.1016/j.cell.2009.03.015
163. Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends Immunol (2014) 35:22–31. doi:10.1016/j.it.2013.10.005
164. Lodolce JP, Kolodziej LE, Rhee L, Kariuki SN, Franek BS, McGreal NM, et al. African-derived genetic polymorphisms in TNFAIP3 mediate risk for autoimmunity. J Immunol (2013) 184:7001–9. doi:10.4049/jimmunol.1000324
165. Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science (2000) 289:2350–4. doi:10.1126/science.289.5488.2350
166. Turer EE, Tavares RM, Mortier E, Hitotsumatsu O, Advincula R, Lee B, et al. Homeostatic MyD88-dependent signals cause lethal inflammation in the absence of A20. J Exp Med (2008) 205:451–64. doi:10.1084/jem.20071108
167. Vereecke L, Sze M, Mc Guire C, Rogiers B, Chu Y, Schmidt-Supprian M, et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med (2010) 207:1513–23. doi:10.1084/jem.20092474
168. Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, Rhee L, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity (2010) 33:181–91. doi:10.1016/j.immuni.2010.07.017
169. Chu Y, Vahl JC, Kumar D, Heger K, Bertossi A, Wójtowicz E, et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood (2011) 117:2227–36. doi:10.1182/blood-2010-09-306019
170. Lippens S, Lefebvre S, Gilbert B, Sze M, Devos M, Verhelst K, et al. Keratinocyte-specific ablation of the NF-kappaB regulatory protein A20 (TNFAIP3) reveals a role in the control of epidermal homeostasis. Cell Death Differ (2011) 18:1845–53. doi:10.1038/cdd.2011.55
171. Matmati M, Jacques P, Maelfait J, Verheugen E, Kool M, Sze M, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet (2011) 43:908–12. doi:10.1038/ng.874
172. Kool M, van Loo G, Waelput W, De Prijck S, Muskens F, Sze M, et al. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity (2011) 35:82–96. doi:10.1016/j.immuni.2011.05.013
173. Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, Oshima S, et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol (2011) 12:1184–93. doi:10.1038/ni.2135
174. Hövelmeyer N, Reissig S, Xuan NT, Adams-Quack P, Lukas D, Nikolaev A, et al. A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies. Eur J Immunol (2011) 41:595–601. doi:10.1002/eji.201041313
175. Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature (2003) 424:797–801. doi:10.1038/nature01811
176. Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature (2003) 424:801–5. doi:10.1038/nature01802
177. Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature (2003) 424:793–6. doi:10.1038/nature01803
178. Jin W, Reiley WR, Lee AJ, Wright A, Wu X, Zhang M, et al. Deubiquitinating enzyme CYLD regulates the peripheral development and naive phenotype maintenance of B cells. J Biol Chem (2007) 282:15884–93. doi:10.1074/jbc.M609952200
179. Reiley WW, Jin W, Lee AJ, Wright A, Wu X, Tewalt EF, et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med (2007) 204:1475–85. doi:10.1084/jem.20062694
180. Hövelmeyer N, Wunderlich FT, Massoumi R, Jakobsen CG, Song J, Wörns MA, et al. Regulation of B cell homeostasis and activation by the tumor suppressor gene CYLD. J Exp Med (2007) 204:2615–27. doi:10.1084/jem.20070318
181. Chu Y, Soberon V, Glockner L, Beyaert R, Massoumi R, van Loo G, et al. A20 and CYLD do not share significant overlapping functions during B cell development and activation. J Immunol (2012) 189:4437–43. doi:10.4049/jimmunol.1200396
182. van der Veen AG, Ploegh HL. Ubiquitin-like proteins. Annu Rev Biochem (2012) 81:323–57. doi:10.1146/annurev-biochem-093010-153308
183. Sedimbi SK, Shastry A, Park Y, Rumba I, Sanjeevi CB. Association of SUMO4 M55V polymorphism with autoimmune diabetes in Latvian patients. Ann N Y Acad Sci (2006) 1079:273–7. doi:10.1196/annals.1375.041
184. Salajegheh M, Kong SW, Pinkus JL, Walsh RJ, Liao A, Nazareno R, et al. Interferon-stimulated gene 15 (ISG15) conjugates proteins in dermatomyositis muscle with perifascicular atrophy. Ann Neurol (2010) 67:53–63. doi:10.1002/ana.21805
185. Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains – from structures to functions. Nat Rev Mol Cell Biol (2009) 10:659–71. doi:10.1038/nrm2767
186. Nanda SK, Venigalla RK, Ordureau A, Patterson-Kane JC, Powell DW, Toth R, et al. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J Exp Med (2011) 208:1215–28. doi:10.1084/jem.20102177
187. Martin F, Dixit VM. A20 edits ubiquitin and autoimmune paradigms. Nat Genet (2011) 43:822–3. doi:10.1038/ng.916
188. Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science (2010) 327:1135–9. doi:10.1126/science.1182364
189. Düwel M, Welteke V, Oeckinghaus A, Baens M, Kloo B, Ferch U, et al. A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J Immunol (2009) 182:7718–28. doi:10.4049/jimmunol.0803313
190. Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol (2008) 9:263–71. doi:10.1038/ni1561
191. Majetschak M. Extracellular ubiquitin: immune modulator and endogenous opponent of damage-associated molecular pattern molecules. J Leukoc Biol (2011) 89:205–19. doi:10.1189/jlb.0510316
192. Goldstein G, Scheid M, Hammerling U, Schlesinger DH, Niall HD, Boyse EA. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc Natl Acad Sci U S A (1975) 72:11–5. doi:10.1073/pnas.72.1.11
193. Cai D, Lee KK, Li M, Tang MK, Chan KM. Ubiquitin expression is up-regulated in human and rat skeletal muscles during aging. Arch Biochem Biophys (2004) 425:42–50. doi:10.1016/j.abb.2004.02.027
194. Jaremko L, Jaremko M, Pasikowski P, Cebrat M, Stefanowicz P, Lisowski M, et al. The immunosuppressive activity and solution structures of ubiquitin fragments. Biopolymers (2009) 91:423–31. doi:10.1002/bip.21160
195. Daino H, Matsumura I, Takada K, Odajima J, Tanaka H, Ueda S, et al. Induction of apoptosis by extracellular ubiquitin in human hematopoietic cells: possible involvement of STAT3 degradation by proteasome pathway in interleukin 6-dependent hematopoietic cells. Blood (2000) 95:2577–85.
196. Majetschak M, Ponelies N, Hirsch T. Targeting the monocytic ubiquitin system with extracellular ubiquitin. Immunol Cell Biol (2006) 84:59–65. doi:10.1111/j.1440-1711.2005.01399.x
Keywords: autoimmunity, ubiquitin, UBLs, immune tolerance, apoptosis, autophagy
Citation: Weil R (2014) Does antigen masking by ubiquitin chains protect from the development of autoimmune diseases? Front. Immunol. 5:262. doi: 10.3389/fimmu.2014.00262
Received: 19 February 2014; Accepted: 19 May 2014;
Published online: 03 June 2014.
Edited by:
Daniel Hawiger, Saint Louis University, USAReviewed by:
Vigo Heissmeyer, Helmholtz Zentrum München, GermanyAntonio A. Freitas, Institut Pasteur, France
Copyright: © 2014 Weil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Weil, Unité de Signalisation Moléculaire et Activation Cellulaire, CNRS URA 2582, Institut Pasteur, 75724 Paris Cedex 15, France e-mail:cndlaWxAcGFzdGV1ci5mcg==