 Jennifer M. Felton
Jennifer M. Felton Christopher D. Lucas
Christopher D. Lucas Adriano G. Rossi
Adriano G. Rossi Ian Dransfield
Ian Dransfield- MRC Centre for Inflammation Research, Queen’s Medical Research Institute, The University of Edinburgh, Edinburgh, UK
Due to the key role of the lung in efficient transfer of oxygen in exchange for carbon dioxide, a controlled inflammatory response is essential for restoration of tissue homeostasis following airway exposure to bacterial pathogens or environmental toxins. Unregulated or prolonged inflammatory responses in the lungs can lead to tissue damage, disrupting normal tissue architecture, and consequently compromising efficient gaseous exchange. Failure to resolve inflammation underlies the development and/or progression of a number of inflammatory lung diseases including asthma. Eosinophils, granulocytic cells of the innate immune system, are primarily involved in defense against parasitic infections. However, the propagation of the allergic inflammatory response in chronic asthma is thought to involve excessive recruitment and impaired apoptosis of eosinophils together with defective phagocytic clearance of apoptotic cells (efferocytosis). In terms of therapeutic approaches for the treatment of asthma, the widespread use of glucocorticoids is associated with a number of adverse health consequences after long-term use, while some patients suffer from steroid-resistant disease. A new approach for therapeutic intervention would be to promote the resolution of inflammation via modulation of eosinophil apoptosis and the phagocytic clearance of apoptotic cells. This review focuses on the mechanisms underpinning eosinophil-mediated lung damage, currently available treatments and therapeutic targets that might in future be harnessed to facilitate inflammation resolution by the manipulation of cell survival and clearance pathways.
Introduction
In response to tissue injury or the presence of micro-organisms, initiation of host protective mechanisms associated with the acute inflammatory response can also cause damage to the surrounding tissue. The release of proteases, glycosidases, and reactive oxygen/nitrogen species can be particularly destructive in the lung, where disruption of the normal tissue architecture compromises efficient gaseous exchange. A corollary of this close relationship between inflammation and tissue injury is that successful resolution of inflammation is crucial to optimal restoration and maintenance of lung function.
The detection of pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) via their cognate receptors leads to the production of pro-inflammatory mediators including tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1, and IL-6. Well characterized chemoattractants such as complement fragments (e.g., C3a and C5a), lipids [e.g., leukotriene B4 (LTB4) and platelet-activating factor (PAF)], and chemokines [e.g., IL-8 (CXCL8), MCP-1 (CCL2), and eotaxin (CCL11)] act to recruit and/or activate inflammatory cells. Together these mediators rapidly perpetuate inflammation via the activation of vascular endothelial cells, increased vascular permeability, and edema, concurrent with the recruitment of granulocytes at the site of injury. In this review, we will discuss the mechanisms controlling acute lung inflammation, pathological conditions where the regulation of inflammation has gone awry and discuss the current and future treatments that could promote the successful resolution of inflammation.
Polymorphonuclear Granulocytes: Critical Effectors of the Innate Immune Response
Neutrophils and eosinophils are key immune cells in the host defense against invading bacteria and parasites. Excessive recruitment, uncontrolled activation, and defective removal of these cells play a prominent role in the initiation and propagation of a number of chronic inflammatory conditions (1). Apoptosis, a major form of programed cell death, is a fundamental process regulating the tissue longevity of inflammatory cells. Apoptosis provides an efficient non-inflammatory mechanism for the removal of potentially damaging cells and cellular content from the inflamed site by resident or recruited monocyte/macrophage populations (2) or by “non-professional” phagocytes such as epithelial cells (3). The observation of failed apoptotic cell clearance seen in a number of chronic inflammatory diseases, including asthma, bronchiectasis, and chronic obstructive pulmonary disease (COPD) provides strong evidence that granulocyte apoptosis and non-inflammatory clearance has a key role in the resolution of inflammation.
Neutrophils are continuously generated from pluripotent stem cells in the bone marrow and are released into the circulation in large numbers [up to 2 × 1011 cells/day (4)]. Once appropriately triggered, circulating neutrophils or those mobilized from the large marginated pools in the lungs, liver, spleen, and bone marrow (5, 6), can be rapidly recruited to the inflammatory site and engage a number of effector mechanisms to destroy invading pathogenic organisms. This distinctive machinery includes a combination of reactive oxygen species (ROS) generation, the release of a cocktail of cytotoxic and proteolytic molecules, phagocytosis, and NETosis (the formation of extracellular chromatin traps) to destroy invading pathogenic organisms. Recruited neutrophils can undergo apoptosis which is associated with the “disabling” of secretion of their potentially harmful granule content thereby preventing damage to the surrounding tissues (7). The removal of these apoptotic cells by recruited macrophages or other local phagocytes, including airway epithelial cells (3), is believed to facilitate the resolution of inflammation. In addition to apoptotic cell death, a number of other forms of neutrophil cell death have been documented, including necrosis, NETosis, autophagocytic cell death, necroptosis, oncosis, and pyroptosis [reviewed in Ref. (8, 9)]. Although the impact of these alternative forms of cell death on the resolution of inflammatory responses is less clear, several are believed to be predominantly pro-inflammatory. As well as local cell death recent studies have provided evidence that recruited granulocytes can also undergo reverse migration away from the site of inflammation (10–12), although the consequences of this on inflammatory processes requires further investigation.
Eosinophils are also derived from granulocytic precursor populations in the bone marrow and are readily recruited from residence within hematopoietic and lymphatic organs such as the lymph nodes, thymus, spleen, and bone marrow (13) via the vasculature to the site of injury in response to parasitic or allergic inflammation (14). Historically, these cells were considered to play little role in immunoregulation, however, several lines of investigation have now shown eosinophils to be multifunctional granulocytes involved in the initiation and propagation of numerous inflammatory responses, including modulation of the adaptive immune response (14). Once at the site of injury, eosinophil degranulation contributes to both the removal of the inflammatory stimuli and also the propagation of inflammation. Eosinophil-derived granules contain a wide range of proteins including, major basic protein, eosinophil cationic protein, eosinophil peroxidase, and eosinophil-derived neurotoxin, which are known to be cytotoxic to airway epithelial cells (15, 16). Eosinophils have also been shown to undergo “traditional” extracellular trap formation (ETosis) termed EETosis (17) as well as facilitating the extracellular release of mitochondrial chromatin in a ROS-dependent manner (13). Released mitochondrial DNA and eosinophil-derived granule proteins combine to form structures, which are capable of both binding and killing invading organisms in vitro and in vivo (13), indicating that eosinophils may play a previously unrecognized role in antimicrobial defense. The fate of tissue eosinophils includes apoptosis (18) and subsequent clearance by phagocytes, although alternative fates have also been reported.
Apoptotic Pathways
There are two major pathways of apoptosis. The intrinsic pathway is characterized by a conformational change in pro-apoptotic Bcl-2 protein family members, resulting in outer mitochondrial membrane pore formation. The subsequent release of cytochrome c leads to formation of a complex with apoptotic protease-activating factor-1 (APAF-1), which then activates the downstream caspases that facilitate apoptosis. In contrast, the extrinsic pathway is triggered by cell surface death receptor trimerization resulting in the activation of Fas-associated protein with death domain (FADD) and TNF-receptor type 1-associated death domain protein (TRADD), which is responsible for the autocatalytic activation of initiator and effector caspases leading to the synchronized molecular alterations and morphological changes associated with apoptosis. Thus, the result of these two divergent pathways is the activation of intracellular caspases (a family of cysteine–aspartic proteases), which represents a hallmark event in apoptosis [reviewed in Ref. (8, 19, 20)].
Airway Inflammation
Normal Lung Structure
The lung is made of up three distinctly different anatomical areas, the proximal cartilaginous airways, distal bronchioles, and alveoli (21). The trachea and main bronchi form the proximal cartilaginous airways and are responsible for the conduction of inhaled air, during which the proximal pseudostratified epithelium provides defense against invading pathogens and environmental toxins. In contrast, the epithelium of the distal airways becomes more columnar and is populated by a large number of ciliated epithelial cells and mucus-secreting goblet cells (22) – aiding the entrapment and further removal of unwanted inhaled particles (23). Two types of cells make up the alveolar epithelium; type 1 cells, which facilitate gaseous exchange, and the type 2 cells produce numerous secretory vesicles filled with surfactant material, including surfactant-associated protein C (24). Thus, in a normal lung the architectural structure of the tissue works to provide the most efficient environment for gaseous exchange.
Due to the large surface area and constant barrage of pathogens and debris found in the air, the lungs have developed efficient mechanisms for the recognition of microbe-specific motifs. The respiratory tract is also unique in that it has both an external epithelial layer (the respiratory epithelium) and an internal endothelial layer in close apposition. Therefore, this unique structure could provide difficulties when attempting to pharmacologically target the tissue resident eosinophils rather than the airway-resident cells.
Neutrophil-Dominant Airway Inflammation
In tissue localized infection, the exposure of neutrophils to bacterial products or endogenous mediators leads to “priming” of function and facilitates chemotaxis toward the site of infection or injury. Up regulation of surface adhesion molecules (P-selectin, ICAM1, and VCAM1) on the vascular endothelial cells that interact with adhesion molecules on the neutrophil is required for the tethering, rolling, intravascular crawling, and transmigration of activated neutrophils from the circulation into the tissue to carry out their effector functions [reviewed in Ref. (4)]. Development and progression of two neutrophil-driven airway diseases; COPD, characterized by impaired airflow to the lungs as a result of an abnormal inflammatory response (25), and bronchiectasis, a chronic debilitating respiratory disease, characterized by a “vicious cycle” of permanently dilated airways, increased mucus production, and recurrent infections (26), have been linked to failed resolution of inflammation (27–29). However, despite persistent neutrophil-driven inflammation, reduced bacterial clearance is also seen (30). Thus, failure to clear bacterial pathogens from the airways leads to a prolonged inflammatory response characterizing the vicious cycle of inflammation and infection described, with both neutrophil and bacterial derived products contributing to damage of the surrounding epithelial cells.
Currently prescribed treatments for COPD and bronchiectasis include β2-adrenergic receptor agonists (e.g., salmeterol and formoterol), anticholinergic therapies (e.g., tiotropium bromide), high dose inhaled glucocorticoids, theophylline and treatments to improve mucociliary clearance, and sputum expectoration. These drugs work to reduce symptoms, improve lung function, and exercise capacity in an attempt to return to normal health status (26, 31–34). Furthermore, as well as traditional anti-inflammatory effects including inhibition of ROS release, decreased adhesion to the vascular endothelium and reduced release of pro-inflammatory cytokines from macrophages (35), salmeterol has also been shown to reduce adherence of bacteria to airway epithelial cells (36, 37), demonstrating that it may be effective at treating both the underlying infections and resultant inflammatory response.
Eosinophil Dominant Airway Inflammation
Eosinophil dominant allergic inflammation is characterized by three distinct phases (Figure 1). On initial exposure of the airway to an allergen, the sensitization stage, allergens are taken up by dendritic cells either within the airway lumen or in the submucosa after penetrating the epithelial barrier. The antigens are then presented to naïve T cells, which differentiate and activate local B cells to produce IgE. Secreted IgE then binds to Fcε receptors on the surface of submucosal tissue resident mast cells, thus priming the immune system. On second exposure to the allergen, surface bound IgE becomes cross-linked leading to the activation of the tissue resident mast cells. Inflammatory mediators are then released and initiate the propagation of inflammation characterized as the second phase of allergic inflammation, the early-phase reaction. Release of histamine, LTB4, TNF-α, IL-8, IL-13, CCL2, and VEGFA from mast cells leads to increased vascular endothelial permeability, promoting the recruitment and transmigration of granulocytes from the circulation into the tissue. IL-13, histamine and TNF-α also act directly on the goblet cells found within the airway epithelium, causing increased mucus production.
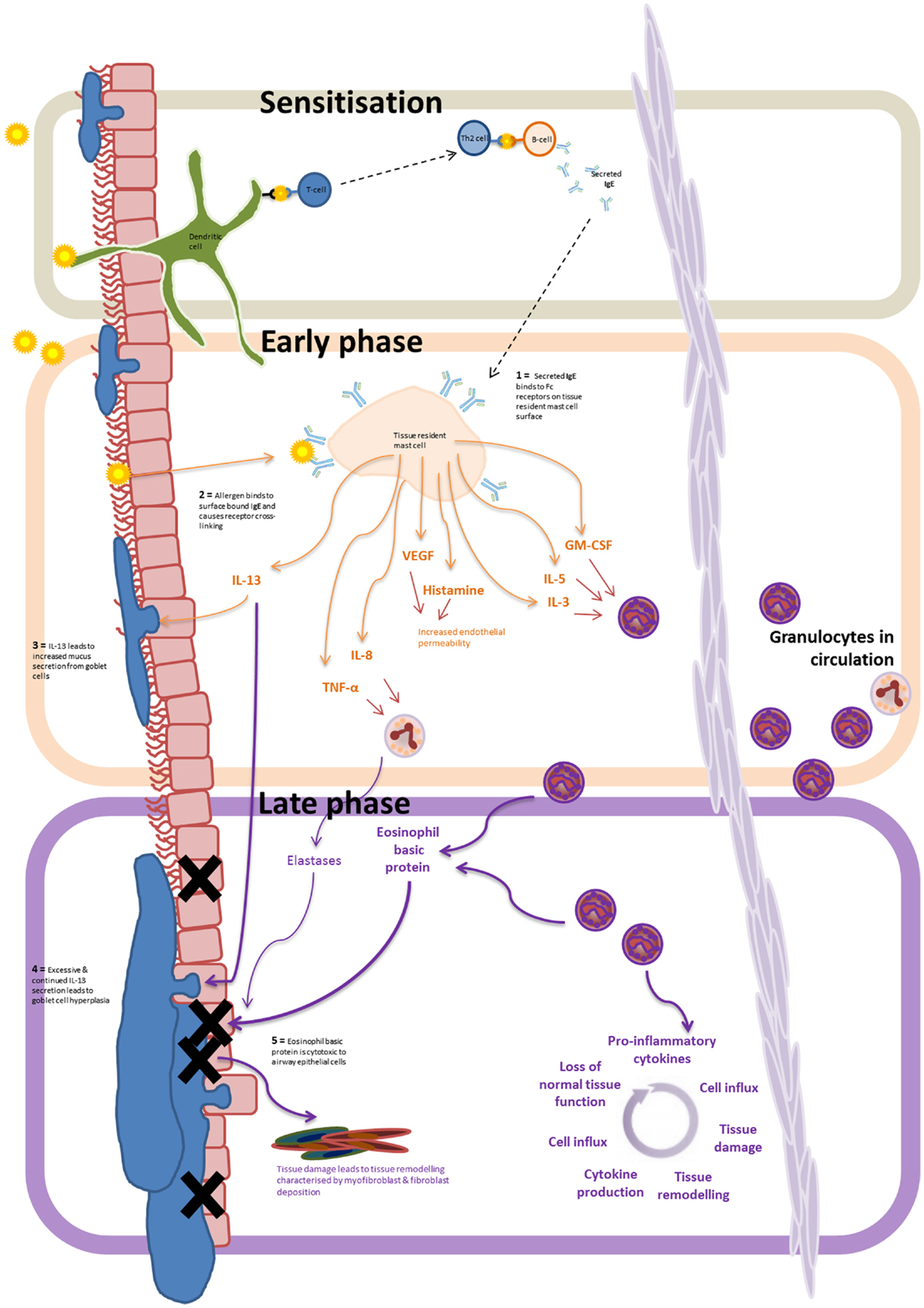
Figure 1. Allergen driven allergic inflammation: progression from the sensitization phase, to early and late inflammatory phase. Airway exposure results in the allergen being taken up by submucosal dendritic cells. Antigen presentation to B cells, via T cell–dendritic cell interactions, leads to IgE release – marking the sensitization phase. IgE binding to Fcε receptors on the tissue resident mast cells leads to allergen-induced IgE receptor crosslinking and mast cell degranulation – marking the early-phase of the inflammatory response. The release of of LTB4, TNF-α, IL-8, IL-13, CCL2, and VEGFA from mast cells results in eosinophil recruitment, increased vascular permeability, and increased mucus secretion by goblet cells. Continued exposure to the allergen, and infiltration of granulocytes to the inflamed tissue marks the progression to the late phase of the inflammatory response. Prolonged secretion of IL-13 and the release of intracellular cytotoxic granules by recruited eosinophils (and neutrophils) lead to continual tissue damage, mucus hypersecretion, and tissue remodeling resulting in the gradual loss of normal lung function.
Transendothelial migration of eosinophils to the inflamed site marks the progression into the third stage of the inflammatory response – the late-phase reaction. This usually develops 6–9 h after allergen exposure. Continued secretion of eosinophil recruiting cytokines (e.g., GM-CSF, IL-5, and IL-3) by mast cells leads to the prolonged eosinophil infiltration, representing a major contributory factor to the initiation and maintenance of eosinophilic airway inflammation in asthma (19, 38) – key cytokines involved in eosinophil recruitment are summarized in Table 1. Subsequent eosinophil degranulation and release of intracellular cytotoxic contents such as eosinophil basic protein results in damage to airway epithelial cells with increased mucus production from goblet cells and airway bronchoconstriction as a result of IL-13 secretion lead to reduced airflow, airway damage, goblet cell hyperplasia, and disrupted tissue architecture and remodeling. Mast cell production of IL-8 and TNFα also triggers the recruitment of neutrophils and elastase release causing further tissue degradation.

Table 1. Key cytokines involved in eosinophil recruitment.
Continuous exposure to allergens leads to persistent, chronic inflammation, which is associated with changes in tissue architecture and cell composition and extensive tissue remodeling. In particular, patients with chronic asthma develop increased goblet cell hyperplasia. Persistent residence of eosinophils and neutrophils within the submucosa results in the continuous production of pro-inflammatory cytokines, lack of inflammatory resolution, and a repetitive cycle of tissue injury and inflammation (Figure 1).
The most characteristic features of eosinophil dominant allergic asthma is bronchial inflammation leading to non-specific airway hyperreactivity (39), mucus plugging of airways, epithelial cells loss, mucus gland hyperplasia, epithelial basement membrane thickening, edema of the submucosa, smooth muscle hypertrophy, and inflammatory cell infiltration (40). In vitro studies have shown that eosinophil granule-derived proteins are partly responsible for the damage seen in asthma as eosinophil cationic protein and major basic protein are both cytotoxic to the bronchial epithelium (16) and have been shown to affect ciliary beat and function (41) and increase non-specific bronchial hyperreactivity (42), all of which are classical pathological findings of asthma (40). Interestingly, a number of studies using eosinophil-deficient mice (PHIL) have shown that eosinophils enhance airway mucus accumulation and hyperresponsiveness, collagen deposition, and smooth muscle hypertrophy (43, 44). However, a significantly increased mucus index was still observed in ovalbumin treated PHIL mice (43) suggesting that although eosinophils contribute substantially to airway remodeling, they are not obligatory for allergen-induced injury (44), indicating activation of both eosinophil-dependent and -independent mechanisms of injury after airway allergen challenge (43).
Current treatments for asthma include inhalable bronchodilators (β2-adrenergic agonists and anticholinergic drugs), leukotriene receptor antagonists, glucocorticoids, and theophylline (45, 46). The possible mechanisms of actions for these drugs are shown in Table 2.
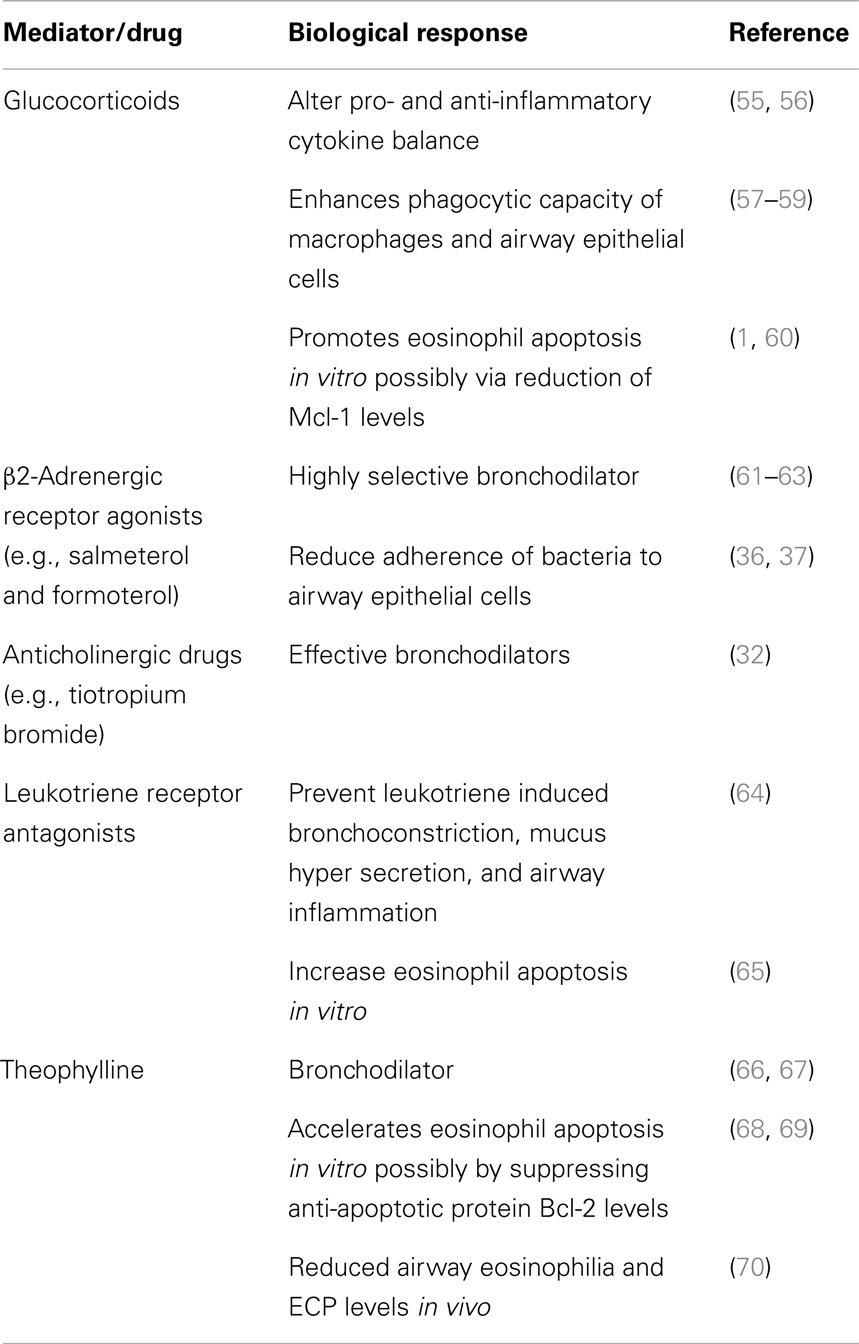
Table 2. Mechanisms of action of agents currently used for routine treatment of allergic airways disease.
Mast cells are known to play an important role in the propagation and pathogenesis of allergen-induced inflammatory disease (51, 52), due to their involvement in the sensitization stage, Figure 1. As these tissue resident cells are long lived and can survive repeated activation, the modulation of their proliferation and survival has been proposed as a potential therapeutic intervention for allergic disease (53). Recently, Wechsler et al. showed that thymol, a monocyclic phenolic plant compound with known antiseptic, antibiotic, antifungal, and antioxidant properties, was able to induce mast cell apoptosis in vitro and in vivo (54), thereby highlighting a potential pathway for modulating the allergic response through the manipulation of mast cell viability.
Failings in Current Treatments: Why New Approaches are Needed?
New approaches for the management of inflammatory airway diseases are urgently needed as current treatments are associated with a number of adverse health consequences after long-term use. For example, the management of allergic asthma is largely based around preventing exposure of the sensitized individuals to the allergen and treating with therapies which are directed toward alleviating and/or treating the symptoms of the disorder, such as inhaled glucocorticoids. However, this often poses a problem as a small subpopulation of asthma sufferers, often those with “neutrophilic” asthma, are noted to be steroid resistant (71), resulting in increased disease severity (72). Equally the undesirable side effects that arise from long-term use of glucocorticoids (the most common treatment prescribed for eosinophil dominant inflammatory conditions) include osteoporosis, hypertension, muscle atrophy, and delayed wound healing, all of which place limitations on use of glucocorticoid-based anti-inflammatory therapies.
Regulation of Granulocyte Apoptosis
As the resolution of inflammation likely depends on the apoptosis and phagocytosis of apoptotic granulocytes, research into the pharmacological manipulation of these processes is increasingly being recognized as an important area of research for the development of novel strategies to enhance the resolution of chronic inflammation (27, 73–76) and improve patient health.
The rates of granulocyte apoptosis are amenable to alteration by exogenous pharmacological compounds. Both the rates of neutrophil and eosinophil apoptosis can be accelerated by treatment with, soluble Fas ligand (Fas-L) (77), gliotoxin (78), and cyclin-dependent kinase inhibitors (CDKi) (73, 74, 79). Neutrophil apoptosis can also be delayed by pro-inflammatory cytokines (e.g., TNF-α and IL-1) (78), bacterial products [e.g., lipopolysaccharide (LPS), lipoteichoic acid, and peptidoglycan] (80, 81), growth factors [e.g., granulocyte macrophage-colony stimulating factor (GM-CSF)] (75), and pharmacological agents including dibutyryl-cAMP (82, 83) and glucocorticoids (1, 84). TNF-α has been reported to have both pro- and anti-apoptotic effects on neutrophils, with early apoptosis and late survival seen during exposure of cultured cells. It is thought that at early time points (2–8 h) during in vitro culture a subpopulation of neutrophils undergo caspase-8 dependent apoptosis, with later survival (16–24 h) dependent upon an NF-κB mediated anti-apoptotic signaling pathway. Similarly, NF-κB inhibition in eosinophils allows TNF-α mediated apoptosis to predominate (85).
One important difference between the two granulocyte populations is that in vitro treatment of human granulocytes with glucocorticoids promotes eosinophil apoptosis, whereas it delays neutrophil apoptosis (5). It is also important to note that the survival effect of glucocorticoids on neutrophil longevity may be dependent on the environmental milieu (86, 87). Marwick et al. demonstrated in vitro that the pro-survival effects of glucocorticoids on neutrophils are dependent on oxygen levels, with severe hypoxia (1% oxygen) attenuating glucocorticoid-mediated neutrophil survival (87). This observation has important implications for the therapeutic efficacy of glucocorticoids when prescribed for neutrophil-dominant inflammatory conditions, due to the relatively high oxygen concentrations found in the lung.
One way that glucocorticoids are thought to mediate their anti-inflammatory effects is through the expression and function of the downstream effector molecule Annexin A1 (AnxA1) (86, 88). AnxA1 has been shown to promote human neutrophil apoptosis via dephosphorylation of the Bcl-2-antagonist of cell death (BAD) promoting cell death via the intrinsic pathway of apoptosis (89). In vitro investigations showed that endogenous AnxA1 was released by apoptotic neutrophils and glucocorticoid-treated macrophages, which then acts in both a para- and autocrine manner to promote the phagocytic clearance of apoptotic neutrophils (90, 91). Increased production of AnxA1 by innate immune cells following glucocorticoid administration reportedly leads to decreased neutrophil endothelial transmigration, increased neutrophil apoptosis and increased phagocytosis of apoptotic cells by macrophages (88, 92). This mechanism was further supported by in vivo experiments in which administration of an anti-AnxA1 antibody prevented glucocorticoid-induced resolution of inflammation, whereas treatment with AnxA1-derived peptides promoted the resolution of inflammation (86). Further work is required to define the role of AnxA1 in the resolution of eosinophilic inflammation.
A number of classically “pro-inflammatory” eosinophil recruiting cytokines, IL-25 (93), IL-33 (94), IL-3, IL-5, and thymic stromal lymphopoietin (TSLP) (95) have also been shown to delay the rate of eosinophil apoptosis (96). Due to their key role in the recruitment and activation and of eosinophils in inflammatory sites, there is interest in developing anti-cytokine therapies, such as anti-IL-5 antibodies [reviewed in Ref. (97)], as potential therapeutic targets. Historically, there has been a number of disappointing results surrounding anti-IL-5 treatments in humans, potentially as a result of the unique architecture of the lungs localizing their effect to the airway resident, rather than the tissue resident, eosinophils.
Novel Regulators of Eosinophil Apoptosis
The regulation of cell apoptosis by pro- and anti-apoptotic Bcl-2 family members has been well documented (20). However, as granulocytes have a limited number of mitochondria (98), it was somewhat surprising when members of this protein family were found to modulate the regulation of granulocyte apoptosis. Eosinophils were found to express high levels of pro-apoptotic Bax molecules, and were also found to express a number of anti-apoptotic members of the Bcl-2 family (99). Mcl-1, an important anti-apoptotic protein in neutrophils (27), is also thought to play a predominant role in eosinophils, as previous work reported that Mcl-1 levels decreased in glucocorticoid-treated eosinophils, whereas they remained at a constant level in glucocorticoid-treated neutrophils (60), which may go some way to explain their differential effect on the two granulocyte populations.
Since granulocytes are considered to be terminally differentiated cells, the central role of active cyclin-dependent kinases (key regulators of the cell cycle) in the control of apoptosis was surprising. The structurally distinct CDKis R-roscovitine and AT7519 promoted apoptosis in a caspase-dependent manner (73, 76) by the down-regulation of intracellular Mcl-1 levels in vivo (79, 100, 101) and prevented GM-CSF-mediated up regulation of Mcl-1 (73). R-roscovitine was also shown to have pro-resolving effects in vivo in a number of models of inflammation (73) while AT7519 increased the percentage of apoptotic eosinophils as well as the percentage of macrophages containing apoptotic eosinophils in a mouse model of allergic pleurisy, indicating that AT7519 has the potential to resolve allergic inflammation by driving both eosinophil apoptosis and by increasing macrophage clearance of apoptotic cells (76). Evidence suggests that these CDKIs target CDK7 and CDK9, which are involved in transcription of key granulocyte survival proteins such as Mcl-1 (102). Further studies investigating the mechanisms underlying resolution of inflammation have also highlighted the importance of Mcl-1 in the regulation of granulocyte apoptosis. Flavones, polyphenolic plant-derived compounds, rapidly induced both eosinophil (103) and neutrophil apoptosis (101) in vitro even in the presence of powerful pro-survival mediators including LTA, GM-CSF (101), and IL-5 (103).
Another powerful driver of caspase induced eosinophil apoptosis is antibody crosslinking of sialic acid binding immunoglobulin-like lectin 8 (Siglec-8), a member of the Siglec immunoglobulin supergene family expressed only on the surface of human eosinophils, basophils, and mast cells (104). Siglec-8 cross-linking reduced eosinophil viability in a time- and concentration-dependent manner through the induction of caspase-mediated apoptosis. This was further confirmed by the use of pan (104) and selective caspase inhibitors (against caspase-8 and -9) (105), which completely inhibited Siglec-8 cross-linking induced apoptosis in vitro, while having no effect on spontaneous eosinophil apoptosis. Antibody crosslinking of the functional mouse ortholog, Siglec-F, was also shown to significantly reduce peripheral eosinophil number in a hypereosinophilic/chronic eosinophilic leukemic (HES/CEL) murine model, as well as induce eosinophil apoptosis in vivo (106). These data further highlight that regulation of eosinophil apoptosis using exogenous mediators could provide potential future therapeutic targets for eosinophilic disorders.
Pharmacological modulation of endogenous molecules involved in mediating the resolution of allergic inflammation is also a key area of research. Recently, Faustino et al. showed that tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a major player in the resolution process, with in vivo administration of anti-TRAIL markedly reducing the number of apoptotic cells in the BAL fluid of chronic allergy induced mice (107). In vivo, treatment with recombinant TRAIL, in an established mouse model of allergic airway inflammation, also significantly augmented the number of apoptotic cells found in the BAL of OVA treated mice, compared to control (PBS treated mice), as well as decreasing the overall number of eosinophils found in the BAL (107), providing further evidence that the manipulation of eosinophil apoptosis may provide avenues for the discovery of novel therapeutics.
Lipid Modulation
Lipoxins, protectins, and resolvins are bioactive lipids synthesized from arachidonic, docosahexaenoic, and eicosapentaenoic acid, respectively (108, 109). They are key pro-resolution mediators, which act to selectively prevent granulocyte migration and increase the recruitment of phagocytic cells (110). Both lipoxin A4 and B4 were reported to inhibit neutrophil recruitment to an inflammatory site and lipoxin A4 was also shown to stimulate monocyte chemotaxis and promote macrophage uptake of apoptotic neutrophils in vitro (111, 112) and in vivo (113). Similar inhibitory effects of lipoxin A4 upon the migration and chemotaxis of eosinophils in vivo and the local generation of eotaxin and IL-5 have been reported (114, 115). Resolvins also have pro-resolution effects preventing transepithelial and transendothelial migration of neutrophils in vivo (116), and stimulating the non-inflammatory phagocytosis of apoptotic neutrophils (117).
The significance of pro-resolving lipids in successful resolution of inflammation has also been noted in a number of non-allergic and allergic inflammatory conditions. Reduced levels of protectin D1 and lipoxin A4 are seen in the exhaled breath of patients after a severe and mild asthma exacerbation, respectively (118, 119). In vivo, mouse models have also provided insight into the role of pro-resolving lipids in the recruitment of granulocytes and augmentation of macrophage phagocytic capacity – resolvin E1 and lipoxin analogs were shown to reduce airway hyperresponsiveness (119), eosinophil number (118), and promote inflammation resolution in a mouse model of allergic asthma (119, 120). Given the pro-resolution roles of these lipid mediators, there is great interest in the development of them as therapeutics. However, as these endogenously produced molecules are traditionally locally active and rapidly inactivated the development of exogenously administered drugs with longer half-lives, which mimic endogenous compounds in vivo are needed to fill this pharmacological niche. Currently, the synthetic resolvin analog RX-10045, and naturally occurring small molecule lipid mediator RX-10001 and are under clinical examination for their use in a number of inflammatory diseases such as dry eye, asthma, retinal disease, and inflammatory bowel disease (109).
Manipulation of Phagocytosis
One newly emerging approach to facilitate the resolution of inflammation is the pharmacological manipulation of the phagocytosis of apoptotic granulocytes (121). As well as their effects on granulocyte apoptosis, glucocorticoids are also known to augment macrophage phagocytic function (57), which may represent an approach to drive clearance of apoptotic cells from inflamed sites (122). Glucocorticoid-treated macrophages exhibit altered cytoskeletal regulation, with increased cell motility and expression of high levels of active Rac, a key protein involved in cell motility, mitosis, wound healing, and phagocytosis (123).
Glucocorticoids induce phagocytosis of apoptotic cells via Mer, a member of the Tyro-3/Axl/Mer (TAM) receptor tyrosine kinase family (58, 124, 125). TAMs are widely expressed vertebrate-specific receptor tyrosine kinases that confer the capacity for binding and subsequent phagocytosis of apoptotic cells, together with initiation of signals that regulate cellular function. TAM-deficient mice show defective clearance of apoptotic material by retinal pigment epithelial cells of the eye, Sertoli cells of the testis and also by myeloid cells. Interestingly, a number of autoimmune conditions are associated with impaired or failed clearance of apoptotic cells (126) and the absence of TAM receptors results in progressive loss of vision, reduced fertility, and development of overt autoimmunity (122, 127, 128). Recent studies demonstrate that Mer-mediated apoptotic cell clearance has a critical importance pathophysiologically in the lung, as inflammation in an LPS-induced lung injury model was amplified following Mer blockade (129), and conversely attenuated following up regulation of Mer-signaling by use of TAPI-0 (a specific inhibitor of Mer cleavage) (130). The expression of Mer on phagocytic populations present at the inflammatory site could also be induced by treatment with glucocorticoids (57) or liver X receptor agonists (131). Alternatively, blockade of cytokines that actively suppress Mer expression (e.g., interferon-gamma) could represent an alternative strategy for promoting Mer-dependent apoptotic cell clearance (132, 133). Recent evidence suggests that Mer is down regulated by inflammatory stimuli such as LPS or bleomycin via proteolytic cleavage from the phagocyte membrane (134). Specific inhibition of ADAM17 proteolytic activity (e.g., using KD-1X-73.5 or TAPI-0) prevents Mer down-regulation and is associated with increased clearance of apoptotic cells in both LPS and bleomycin models of lung injury (135), providing a potential therapeutic approach to increase Mer-dependent clearance mechanisms in inflammation. Definition of the molecular mechanisms of phagocyte–apoptotic cell interactions and regulation by glucocorticoids will provide opportunities to identify novel targets for therapeutic gain.
Epithelial Cell Phagocytosis
In addition to the importance of therapies, which are able to modulate apoptotic cell phagocytic clearance, identification of the cell types which carry out this process in inflammatory airway conditions is of crucial importance. Induced death of airway epithelial cells as a result of exposure to environmental toxins, allergens, and pathogens has been observed and documented in patients with asthma (3). Thus, there is a need for a large population of local airway phagocytic cells to remove the apoptotic debris. There is mounting evidence that a number of “non-professional” phagocytes, including mammary epithelial (136) and microvascular endothelial cells (137) are also able to phagocytose apoptotic cells. Work published by Walsh et al. and Sexton et al. showed that bronchial epithelial cells are capable of recognizing and engulfing apoptotic eosinophils, suggesting a non-passive role of the airway epithelium in the resolution of eosinophilic inflammation in asthma (138, 139). More recently, Juncadella et al. showed that bronchial epithelial cells are also critically involved in the phagocytosis of apoptotic airway epithelial cells, which subsequently alters the production of anti-inflammatory cytokines and control of airway hyperresponsiveness in a murine model of allergic airway inflammation (3). Despite the potential for providing novel therapeutic approaches for the treatment of inflammatory diseases, little work has been done to investigate the potential for manipulation of the phagocytic ability of these cells in current models of inflammatory airway diseases.
Summary
In conclusion, recent research into the pharmacological manipulation of apoptosis and efferocytosis of apoptotic cells has provided novel insights into the treatment of inflammatory airway diseases, notably eosinophil dominant airway inflammation. This dual approach will open up new areas for therapeutic intervention, allowing the successful manipulation of inflammation resolution, as well as reducing the adverse effects associated with currently available treatments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Meagher LC, Cousin JM, Seckl JR, Haslett C. Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J Immunol (1996) 156:4422–8.
2. Savill J, Wyllie A, Henson J, Walport M, Henson P, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest (1989) 83:865. doi: 10.1172/JCI113970
3. Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature (2012) 493:547–51. doi:10.1038/nature11714
4. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13:159–75. doi:10.1038/nri3399
6. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol (2010) 31:318–24. doi:10.1016/j.it.2010.05.006
7. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol (2002) 2:965–75. doi:10.1038/nri957
8. Gabelloni ML, Trevani AS, Sabatté J, Geffner J. Mechanisms regulating neutrophil survival and cell death. Semin Immunopathol (2013) 35(4):423–37. doi:10.1007/s00281-013-0364-x
9. Galluzzi L, Vitale I, Abrams J, Alnemri E, Baehrecke E, Blagosklonny M, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ (2012) 19:107–20. doi:10.1038/cdd.2011.96
10. Woodfin A, Voisin M-B, Beyrau M, Colom B, Caille D, Diapouli F-M. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol (2011) 12:761–9. doi:10.1038/ni.2062
11. Robertson AL, Holmes GR, Bojarczuk AN, Burgon J, Loynes CA, Chimen M. A zebrafish compound screen reveals modulation of neutrophil reverse migration as an anti-inflammatory mechanism. Sci Transl Med (2014) 6:225ra29. doi:10.1126/scitranslmed.3007672
12. Lucas CD, Hoodless LJ, Rossi AG. Swimming against the tide: drugs drive neutrophil reverse migration. Sci Transl Med (2014) 6:225fs9. doi:10.1126/scitranslmed.3008666
13. Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med (2008) 14:949–53. doi:10.1038/nm.1855
14. Hogan SP, Rosenberg HF, Moqbel R, Phipps S, Foster PS, Lacy P. Eosinophils: biological properties and role in health and disease. Clin Exp Allergy (2008) 38:709–50. doi:10.1111/j.1365-2222.2008.02958.x
15. Gleich G, Flavahan N, Fujisawa T, Vanhoutte P. The eosinophil as a mediator of damage to respiratory epithelium: a model for bronchial hyperreactivity. J Allergy Clin Immunol (1988) 81:776–81. doi:10.1016/0091-6749(88)90931-1
16. Venge P, Peterson G. Epithelial injury by human eosinophils-3. Am Rev Respir Dis (1988) 138:S54–7.
17. Ueki S, Melo RC, Ghiran I, Spencer LA, Dvorak AM, Weller PF. Eosinophil extracellular DNA trap cell death mediates lytic release of free secretion-competent eosinophil granules in humans. Blood (2013) 121:2074–83. doi:10.1182/blood-2012-05-432088
18. Stern M, Meagher L, Savill J, Haslett C. Apoptosis in human eosinophils. Programmed cell death in the eosinophil leads to phagocytosis by macrophages and is modulated by IL-5. J Immunol (1992) 148:3543–9.
19. Ilmarinen P, Kankaanranta H. Eosinophil apoptosis as a therapeutic target in allergic asthma. Basic Clin Pharmacol Toxicol (2014) 114:109–17. doi:10.1111/bcpt.12163
20. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol (2007) 35:495–516. doi:10.1080/01926230701320337
21. Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology (2003) 8:432–46. doi:10.1046/j.1440-1843.2003.00493.x
22. Liu X, Driskell RR, Engelhardt JF. Airway glandular development and stem cells. Curr Top Dev Biol (2004) 64:33–56. doi:10.1016/S0070-2153(04)64003-8
23. Jeffery P. Morphologic features of airway surface epithelial cells and glands. Am Rev Respir Dis (1983) 128:S14.
24. Rawlins EL, Hogan BL. Epithelial stem cells of the lung: privileged few or opportunities for many? Development (2006) 133:2455–65. doi:10.1242/dev.02407
25. MacNee W. ABC of chronic obstructive pulmonary disease: pathology, pathogenesis, and pathophysiology. BMJ (2006) 332:1202. doi:10.1136/bmj.332.7551.1202
26. Sidhu MK, Mandal P, Hill AT. Bronchiectasis: an update on current pharmacotherapy and future perspectives. Expert Opin Pharmacother (2014) 15:505–25. doi:10.1517/14656566.2014.878330
27. Duffin R, Leitch AE, Fox S, Haslett C, Rossi AG. Targeting granulocyte apoptosis: mechanisms, models, and therapies. Immunol Rev (2010) 236:28–40. doi:10.1111/j.1600-065X.2010.00922.x
28. Parfrey H, Farahi N, Porter L, Chilvers ER. Live and let die: is neutrophil apoptosis defective in severe asthma? Thorax (2010) 65:665–7. doi:10.1136/thx.2009.134270
29. Brown V, Elborn JS, Bradley J, Ennis M. Dysregulated apoptosis and NFkappaB expression in COPD subjects. Respir Res (2009) 10:10.1186. doi:10.1186/1465-9921-10-24
30. Cole P. Host-microbe relationships in chronic respiratory infection. Respiration (1989) 55:5–8. doi:10.1159/000195745
31. Barnes PJ, Stockley R. COPD: current therapeutic interventions and future approaches. Eur Respir J (2005) 25:1084–106. doi:10.1183/09031936.05.00139104
32. Casaburi R, Mahler D, Jones P, Wanner A, San Pedro G, ZuWallack R. A long-term evaluation of once-daily inhaled tiotropium in chronic obstructive pulmonary disease. Eur Respir J (2002) 19:217–24. doi:10.1183/09031936.02.00269802
33. Kawashima K, Fujii T. The lymphocytic cholinergic system and its biological function. Life Sci (2003) 72:2101–9. doi:10.1016/S0024-3205(03)00068-7
34. ten Hacken NH, Wijkstra PJ, Kerstjens HA. Treatment of bronchiectasis in adults. BMJ (2007) 335:1089. doi:10.1136/bmj.39384.657118.80
35. Maris NA, de Vos AF, Dessing MC, Spek CA, Lutter R, Jansen HM. Antiinflammatory effects of salmeterol after inhalation of lipopolysaccharide by healthy volunteers. Am J Respir Crit Care Med (2005) 172:878–84. doi:10.1164/rccm.200503-451OC
36. Dowling RB, Rayner C, Rutman A, Jackson AD, Kanthakumar K, Dewar A. Effect of salmeterol on Pseudomonas aeruginosa infection of respiratory mucosa. Am J Respir Crit Care Med (1997) 155:327–36. doi:10.1164/ajrccm.155.1.9001332
37. Dowling R, Johnson M, Cole P, Wilson R. Effect of salmeterol on Haemophilus influenzae infection of respiratory mucosa in vitro. Eur Respir J (1998) 11:86–90. doi:10.1183/09031936.98.11010086
38. Park YM, Bochner BS. Eosinophil survival and apoptosis in health and disease. Allergy Asthma Immunol Res (2010) 2:87–101. doi:10.4168/aair.2010.2.2.87
39. Bousquet J, Chanez P, Lacoste JY, Barnéon G, Ghavanian N, Enander I, et al. Eosinophilic inflammation in asthma. N Engl J Med (1990) 323:1033–9. doi:10.1056/NEJM199010113231505
40. Beasley R, Roche WR, Roberts JA, Holgate ST. Cellular events in the bronchi in mild asthma and after bronchial provocation. Am Rev Respir Dis (1989) 139:806–17. doi:10.1164/ajrccm/139.3.806
41. Hastie A, Loegering D, Gleich G, Kueppers F. The effect of purified human eosinophil major basic protein on mammalian ciliary activity. Am Rev Respir Dis (1987) 135:848–53.
42. Flavahan N, Slifman N, Gleich G, Vanhoutte P. Human eosinophil major basic protein causes hyperreactivity of respiratory smooth muscle: role of the epithelium. Am Rev Respir Dis (1988) 138:685–8. doi:10.1164/ajrccm/138.3.685
43. Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, O’Neill KR. Defining a link with asthma in mice congenitally deficient in eosinophils. Science (2004) 305:1773–6. doi:10.1126/science.1099472
44. Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE. A critical role for eosinophils in allergic airways remodeling. Science (2004) 305:1776–9. doi:10.1126/science.1100283
45. Chung KF, Barnes PJ. Treatment of asthma. Br Med J (Clin Res Ed) (1987) 294:103. doi:10.1136/bmj.294.6564.103
46. Wood AJ, Drazen JM, Israel E, O’Byrne PM. Treatment of asthma with drugs modifying the leukotriene pathway. N Engl J Med (1999) 340:197–206. doi:10.1056/NEJM199905133401907
47. Menzies-Gow A, Ying S, Sabroe I, Stubbs VL, Soler D, Williams TJ, et al. Eotaxin (CCL11) and eotaxin-2 (CCL24) induce recruitment of eosinophils, basophils, neutrophils, and macrophages as well as features of early-and late-phase allergic reactions following cutaneous injection in human atopic and nonatopic volunteers. J Immunol (2002) 169:2712–8. doi:10.4049/jimmunol.169.5.2712
48. Gonzalo J-A, Lloyd CM, Kremer L, Finger E, Martinez-A C, Siegelman M. Eosinophil recruitment to the lung in a murine model of allergic inflammation. The role of T cells, chemokines, and adhesion receptors. J Clin Invest (1996) 98:2332. doi:10.1172/JCI119045
49. Resnick MB, Weller PF. Mechanisms of eosinophil recruitment. Am J Respir Cell Mol Biol (1993) 8:349–55. doi:10.1165/ajrcmb/8.4.349
50. Rot A, Krieger M, Brunner T, Bischoff SC, Schall T, Dahinden C. RANTES and macrophage inflammatory protein 1 alpha induce the migration and activation of normal human eosinophil granulocytes. J Exp Med (1992) 176:1489–95. doi:10.1084/jem.176.6.1489
51. Bischoff SC. Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat Rev Immunol (2007) 7:93–104. doi:10.1038/nri2018
52. Tsai M, Grimbaldeston M, Galli SJ. Mast cells and immunoregulation/immunomodulation. In: Gilfillan AM, Metcalfe DD, editors. Mast Cell Biology (Vol. 716). Springer: Bethesda, MD (2011). p. 186–211.
53. Karra L, Berent-Maoz B, Ben-Zimra M, Levi-Schaffer F. Are we ready to downregulate mast cells? Curr Opin Immunol (2009) 21:708–14. doi:10.1016/j.coi.2009.09.010
54. Wechsler JB, Hsu C-L, Bryce PJ. IgE-mediated mast cell responses are inhibited by thymol-mediated, activation-induced cell death in skin inflammation. J Allergy Clin Immunol (2014) 133(6):1735–43. doi:10.1016/j.jaci.2013.12.024
55. Richards DF, Fernandez M, Caulfield J, Hawrylowicz CM. Glucocorticoids drive human CD8+ T cell differentiation towards a phenotype with high IL-10 and reduced IL-4, IL-5 and IL-13 production. Eur J Immunol (2000) 30:2344–54. doi:10.1002/1521-4141(2000)30:8<2344::AID-IMMU2344>3.0.CO;2-7
56. Elenkov IJ. Glucocorticoids and the Th1/Th2 balance. Ann N Y Acad Sci (2004) 1024:138–46. doi:10.1196/annals.1321.010
57. Liu Y, Cousin JM, Hughes J, Van Damme J, Seckl JR, Haslett C. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol (1999) 162:3639–46.
58. McColl A, Bournazos S, Franz S, Perretti M, Morgan BP, Haslett C. Glucocorticoids induce protein S-dependent phagocytosis of apoptotic neutrophils by human macrophages. J Immunol (2009) 183:2167–75. doi:10.4049/jimmunol.0803503
59. Wang J, Wang C, Li X, Kong L, Gao K, Liu R-Y. The effects of anti-asthma drugs on the phagocytic clearance of apoptotic eosinophils by A549 cells. Respir Med (2009) 103:1693–9. doi:10.1016/j.rmed.2009.05.014
60. Sivertson KL, Seeds MC, Long DL, Peachman KK, Bass DA. The differential effect of dexamethasone on granulocyte apoptosis involves stabilization of Mcl-1L in neutrophils but not in eosinophils. Cell Immunol (2007) 246:34–45. doi:10.1016/j.cellimm.2007.05.003
61. Ullman A, Svedmyr N. Salmeterol, a new long acting inhaled beta 2 adrenoceptor agonist: comparison with salbutamol in adult asthmatic patients. Thorax (1988) 43:674–8. doi:10.1136/thx.43.9.674
62. Löfdahl CG, Svedmyr N. Formoterol fumarate, a new β2-adrenoceptor agonist. Allergy (1989) 44:264–71.
63. Waldeck B. β-Adrenoceptor agonists and asthma – 100 years of development. Eur J Pharmacol (2002) 445:1–12. doi:10.1016/S0014-2999(02)01728-4
64. Krawiec ME, Jarjour NJ. Leukotriene receptor antagonists. Semin Respir Crit Care Med (2002) 23(4):399–410. doi:10.1055/s-2002-34333
65. Lee E, Robertson T, Smith J, Kilfeather S. Leukotriene receptor antagonists and synthesis inhibitors reverse survival in eosinophils of asthmatic individuals. Am J Respir Crit Care Med (2000) 161:1881–6. doi:10.1164/ajrccm.161.6.9907054
66. Finney M, Karlsson JA, Persson C. Effects of bronchoconstrictors and bronchodilators on a novel human small airway preparation. Br J Pharmacol (1985) 85:29–36. doi:10.1111/j.1476-5381.1985.tb08827.x
67. Persson CG. Overview of effects of theophylline. J Allergy Clin Immunol (1986) 78:780–7. doi:10.1016/0091-6749(86)90061-8
68. Adachi T, Motojima S, Hirata A, Fukuda T, Kihara N, Kosaku A, et al. Eosinophil apoptosis caused by theophylline, glucocorticoids, and macrolides after stimulation with IL-5. J Allergy Clin Immunol (1996) 98:S207–15. doi:10.1016/S0091-6749(96)70068-4
69. Yasui K, Hu B, Nakazawa T, Agematsu K, Komiyama A. Theophylline accelerates human granulocyte apoptosis not via phosphodiesterase inhibition. J Clin Invest (1997) 100:1677. doi:10.1172/JCI119692
70. Lim S, Tomita K, Carramori G, Jatakanon A, Oliver B, Keller A. Low-dose theophylline reduces eosinophilic inflammation but not exhaled nitric oxide in mild asthma. Am J Respir Crit Care Med (2001) 164:273–6. doi:10.1164/ajrccm.164.2.2006043
71. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol (2013) 131:636–45. doi:10.1016/j.jaci.2012.12.1564
72. Wenzel SE. Asthma: defining of the persistent adult phenotypes. Lancet (2006) 368:804–13. doi:10.1016/S0140-6736(06)69290-8
73. Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med (2006) 12:1056–64. doi:10.1038/nm1206-1434
74. Leitch AE, Riley NA, Sheldrake TA, Festa M, Fox S, Duffin R. The cyclin-dependent kinase inhibitor R-roscovitine down-regulates Mcl-1 to override pro-inflammatory signalling and drive neutrophil apoptosis. Eur J Immunol (2010) 40:1127–38. doi:10.1002/eji.200939664
75. Leitch A, Haslett C, Rossi A. Cyclin-dependent kinase inhibitor drugs as potential novel anti-inflammatory and pro-resolution agents. Br J Pharmacol (2009) 158:1004–16. doi:10.1111/j.1476-5381.2009.00402.x
76. Alessandri AL, Duffin R, Leitch AE, Lucas CD, Sheldrake TA, Dorward DA. Induction of eosinophil apoptosis by the cyclin-dependent kinase inhibitor AT7519 promotes the resolution of eosinophil-dominant allergic inflammation. PLoS One (2011) 6:e25683. doi:10.1371/journal.pone.0025683
77. Villunger A, O’Reilly LA, Holler N, Adams J, Strasser A. FAS ligand, Bcl-2, granulocyte colony-stimulating factor, and p38 mitogen-activated protein kinase regulators of distinct cell death and survival pathways in granulocytes. J Exp Med (2000) 192:647–58. doi:10.1084/jem.192.5.647
78. Ward C, Chilvers ER, Lawson MF, Pryde JG, Fujihara S, Farrow SN. NF-κB activation is a critical regulator of human granulocyte apoptosis in vitro. J Biol Chem (1999) 274:4309–18. doi:10.1074/jbc.274.21.14956
79. Duffin R, Leitch AE, Sheldrake TA, Hallett JM, Meyer C, Fox S. The CDK inhibitor, R-roscovitine, promotes eosinophil apoptosis by down-regulation of Mcl-1. FEBS Lett (2009) 583:2540–6. doi:10.1016/j.febslet.2009.07.017
80. Lee A, Whyte M, Haslett C. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J Leukoc Biol (1993) 54:283–8.
81. Dick EP, Prince LR, Prestwich EC, Renshaw SA, Whyte MK, Sabroe I. Pathways regulating lipopolysaccharide-induced neutrophil survival revealed by lentiviral transduction of primary human neutrophils. Immunology (2009) 127:249–55. doi:10.1111/j.1365-2567.2008.02949.x
82. Gellersen B, Brosens J. Cyclic AMP and progesterone receptor cross-talk in human endometrium: a decidualizing affair. J Endocrinol (2003) 178:357–72. doi:10.1677/joe.0.1780357
83. Martin MC, Dransfield I, Haslett C, Rossi AG. Cyclic AMP regulation of neutrophil apoptosis occurs via a novel protein kinase A-independent signaling pathway. J Biol Chem (2001) 276:45041–50. doi:10.1074/jbc.M105197200
84. Cox G. Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J Immunol (1995) 154:4719–25.
85. Fujihara S, Ward C, Dransfield I, Hay RT, Uings IJ, Hayes B, et al. Inhibition of nuclear factor-κB activation un-masks the ability of TNF-α to induce human eosinophil apoptosis. Eur J Immunol (2002) 32:457–66. doi:10.1002/1521-4141(200202)32:2<457::AID-IMMU457>3.0.CO;2-1
86. Vago JP, Nogueira CR, Tavares LP, Soriani FM, Lopes F, Russo RC. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J Leukoc Biol (2012) 92:249–58. doi:10.1189/jlb.0112008
87. Marwick JA, Dorward DA, Lucas CD, Jones KO, Sheldrake TA, Fox S. Oxygen levels determine the ability of glucocorticoids to influence neutrophil survival in inflammatory environments. J Leukoc Biol (2013) 94:1285–92. doi:10.1189/jlb.0912462
88. Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol (2009) 9:62–70. doi:10.1038/nri2470
89. Solito E, Kamal A, Russo-Marie F, Buckingham JC, Marullo S, Perretti M. A novel calcium-dependent proapoptotic effect of annexin 1 on human neutrophils. FASEB J (2003) 17(11):1544–6.
90. Scannell M, Flanagan MB, Wynne KJ, Cagney G, Godson C, Maderna P. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol (2007) 178:4595–605. doi:10.4049/jimmunol.178.7.4595
91. Maderna P, Yona S, Perretti M, Godson C. Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac2–26. J Immunol (2005) 174:3727–33. doi:10.4049/jimmunol.174.6.3727
92. Perretti M, Flower RJ. Annexin 1 and the biology of the neutrophil. J Leukoc Biol (2004) 76:25–9. doi:10.1189/jlb.1103552
93. Cheung P, Wong C, Ip W, Lam C. IL-25 regulates the expression of adhesion molecules on eosinophils: mechanism of eosinophilia in allergic inflammation. Allergy (2006) 61:878–85. doi:10.1111/j.1398-9995.2006.01102.x
94. Suzukawa M, Koketsu R, Iikura M, Nakae S, Matsumoto K, Nagase H. Interleukin-33 enhances adhesion, CD11b expression and survival in human eosinophils. Lab Invest (2008) 88:1245–53. doi:10.1038/labinvest.2008.82
95. Wong CK, Hu S, Cheung PF, Lam CW. Thymic stromal lymphopoietin induces chemotactic and prosurvival effects in eosinophils: implications in allergic inflammation. Am J Respir Cell Mol Biol (2010) 43:305–15. doi:10.1165/rcmb.2009-0168OC
96. Walsh GM. Eosinophil apoptosis and clearance in asthma. J Cell Death (2013) 6:17–25. doi:10.4137/JCD.S10818
97. Garcia G, Taillé C, Laveneziana P, Bourdin A, Chanez P, Humbert M. Anti-interleukin-5 therapy in severe asthma. Eur Respir Rev (2013) 22:251–7. doi:10.1183/09059180.00004013
98. Bainton DF, Ullyot JL, Farquhar MG. The development of neutrophilic polymorphonuclear leukocytes in human bone marrow origin and content of azurophil and specific granules. J Exp Med (1971) 134:907–34. doi:10.1084/jem.134.4.907
99. Dibbert B, Daigle I, Braun D, Schranz C, Weber M, Blaser K, et al. Role for Bcl-xL in delayed eosinophil apoptosis mediated by granulocyte-macrophage colony-stimulating factor and interleukin-5. Blood (1998) 92:778–83.
100. Lucas C, Dorward D, Tait M, Fox S, Marwick J, Allen K. Downregulation of Mcl-1 has anti-inflammatory pro-resolution effects and enhances bacterial clearance from the lung. Mucosal Immunol (2014) 7(4):857–68. doi:10.1038/mi.2013.102
101. Lucas CD, Allen KC, Dorward DA, Hoodless LJ, Melrose LA, Marwick JA. Flavones induce neutrophil apoptosis by down-regulation of Mcl-1 via a proteasomal-dependent pathway. FASEB J (2013) 27:1084–94. doi:10.1096/fj.12-218990
102. Leitch A, Lucas C, Marwick J, Duffin R, Haslett C, Rossi A. Cyclin-dependent kinases 7 and 9 specifically regulate neutrophil transcription and their inhibition drives apoptosis to promote resolution of inflammation. Cell Death Differ (2012) 19:1950–61. doi:10.1038/cdd.2012.80
103. Sharma S, Dorward DA, Rossi AG, Lucas CD. Wogonin induces caspase-dependent human eosinophil apoptosis. Proceedings of the British Pharmacological Society Online (Vol 11) (2013). Available from: http://www.pa2online.org/abstracts/vol11issue3abst013p.pdf
104. Nutku E, Aizawa H, Hudson SA, Bochner BS. Ligation of Siglec-8: a selective mechanism for induction of human eosinophil apoptosis. Blood (2003) 101:5014–20. doi:10.1182/blood-2002-10-3058
105. Nutku E, Hudson SA, Bochner BS. Mechanism of Siglec-8-induced human eosinophil apoptosis: role of caspases and mitochondrial injury. Biochem Biophys Res Commun (2005) 336:918–24. doi:10.1016/j.bbrc.2005.08.202
106. Zimmermann N, McBride M, Yamada Y, Hudson S, Jones C, Cromie K. Siglec-F antibody administration to mice selectively reduces blood and tissue eosinophils. Allergy (2008) 63:1156–63. doi:10.1111/j.1398-9995.2008.01709.x
107. Faustino L, Fonseca D, Florsheim E, Resende R, Lepique A, Faquim-Mauro E. Tumor necrosis factor-related apoptosis-inducing ligand mediates the resolution of allergic airway inflammation induced by chronic allergen inhalation. Mucosal Immunol (2014). doi:10.1038/mi.2014.9
108. Dorward DA, Lucas CD, Allen KC, Rossi AG. Resolution of allergic inflammation. In: Middleton’s Allergy Principles and Practice (Vol. 1). Philadelphia: Elsevier Saunders (2013). p. 389–96.
109. Lee CH. Resolvins as new fascinating drug candidates for inflammatory diseases. Arch Pharm Res (2012) 35:3–7. doi:10.1007/s12272-012-0121-z
110. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol (2008) 8:349–61. doi:10.1038/nri2294
111. Maddox JF, Hachicha M, Takano T, Petasis NA, Fokin VV, Serhan CN. Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor. J Biol Chem (1997) 272:6972–8. doi:10.1074/jbc.272.49.30993
112. Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol (2000) 164:1663–7. doi:10.4049/jimmunol.164.4.1663
113. Mitchell S, Thomas G, Harvey K, Cottell D, Reville K, Berlasconi G. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J Am Soc Nephrol (2002) 13:2497–507. doi:10.1097/01.ASN.0000032417.73640.72
114. Lee TH, Horton CE, Kyan-Aung U, Haskard D, Crea AE, Spur BW. Lipoxin A4 and lipoxin B4 inhibit chemotactic responses of human neutrophils stimulated by leukotriene B4 and N-formyl-L-methionyl-L-leucyl-L-phenylalanine. Clin Sci (1989) 77:195–203.
115. Bandeira-Melo C, Bozza PT, Diaz BL, Cordeiro RS, Jose PJ, Martins MA. Cutting edge: lipoxin (LX) A4 and aspirin-triggered 15-epi-LXA4 block allergen-induced eosinophil trafficking. J Immunol (2000) 164:2267–71. doi:10.4049/jimmunol.164.5.2267
116. Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med (2000) 192:1197–204. doi:10.1084/jem.192.3.F5
117. Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature (2007) 447:869–74. doi:10.1038/nature05877
118. Levy BD, Kohli P, Gotlinger K, Haworth O, Hong S, Kazani S. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J Immunol (2007) 178:496–502. doi:10.4049/jimmunol.178.1.496
119. Levy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, Schmidt BA. Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A4. Nat Med (2002) 8:1018–23. doi:10.1038/nm748
120. Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-γ and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol (2008) 9:873–9. doi:10.1038/ni.1627
121. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol (2014) 14(3):166–80. doi:10.1038/nri3607
122. Rothlin CV, Lemke G. TAM receptor signaling and autoimmune disease. Curr Opin Immunol (2010) 22:740–6. doi:10.1016/j.coi.2010.10.001
123. Giles KM, Ross K, Rossi AG, Hotchin NA, Haslett C, Dransfield I. Glucocorticoid augmentation of macrophage capacity for phagocytosis of apoptotic cells is associated with reduced p130Cas expression, loss of paxillin/pyk2 phosphorylation, and high levels of active Rac. J Immunol (2001) 167:976–86. doi:10.4049/jimmunol.167.2.976
124. Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature (2001) 411:207–11. doi:10.1038/35079659
125. Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med (2002) 196:135–40. doi:10.1084/jem.20012094
126. Muñoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol (2010) 6:280–9. doi:10.1038/nrrheum.2010.46
127. Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science (2001) 293:306–11. doi:10.1126/science.1061663
128. Lauber K, Keppeler H, Munoz L, Koppe U, Schröder K, Yamaguchi H. Milk fat globule-EGF factor 8 mediates the enhancement of apoptotic cell clearance by glucocorticoids. Cell Death Differ (2013) 20:1230–40. doi:10.1038/cdd.2013.82
129. Lee Y-J, Han J-Y, Byun J, Park H-J, Park E-M, Chong YH. Inhibiting Mer receptor tyrosine kinase suppresses STAT1, SOCS1/3, and NF-κB activation and enhances inflammatory responses in lipopolysaccharide-induced acute lung injury. J Leukoc Biol (2012) 91:921–32. doi:10.1189/jlb.0611289
130. Choi J-Y, Park H-J, Lee Y-J, Byun J, Youn Y-S, Choi JH. Upregulation of Mer receptor tyrosine kinase signaling attenuated lipopolysaccharide-induced lung inflammation. J Pharmacol Exp Ther (2013) 344:447–58. doi:10.1124/jpet.112.199778
131. Gonzalez NA, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity (2009) 31:245–58. doi:10.1016/j.immuni.2009.06.018
132. Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol (2012) 189:3508–20. doi:10.4049/jimmunol.1200662
133. Heasman SJ, Giles KM, Rossi AG, Allen JE, Haslett C, Dransfield I. Interferon γ suppresses glucocorticoid augmentation of macrophage clearance of apoptotic cells. Eur J Immunol (2004) 34:1752–61. doi:10.1002/eji.200324698
134. Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, Tabas I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cδ, and p38 mitogen-activated protein kinase (MAPK). J Biol Chem (2011) 286:33335–44. doi:10.1074/jbc.M111.263020
135. Lee Y-J, Lee S-H, Youn Y-S, Choi J-Y, Song K-S, Cho M-S. Preventing cleavage of Mer promotes efferocytosis and suppresses acute lung injury in bleomycin treated mice. Toxicol Appl Pharmacol (2012) 263:61–72. doi:10.1016/j.taap.2012.05.024
136. Monks J, Rosner D, Geske FJ, Lehman L, Hanson L, Neville M. Epithelial cells as phagocytes: apoptotic epithelial cells are engulfed by mammary alveolar epithelial cells and repress inflammatory mediator release. Cell Death Differ (2005) 12:107–14. doi:10.1038/sj.cdd.4401517
137. Kirsch T1, Woywodt A, Beese M, Wyss K, Park JK, Erdbruegger U, et al. Engulfment of apoptotic cells by microvascular endothelial cells induces proinflammatory responses. Blood (2007) 109(7):2854–62.
138. Sexton DW, Blaylock MG, Walsh GM. Human alveolar epithelial cells engulf apoptotic eosinophils by means of integrin-and phosphatidylserine receptor-dependent mechanisms: a process upregulated by dexamethasone. J Allergy Clin Immunol (2001) 108:962–9. doi:10.1067/mai.2001.119414
Keywords: eosinophil, lung, inflammation, apoptosis, phagocytosis, allergy, airway, resolution
Citation: Felton JM, Lucas CD, Rossi AG and Dransfield I (2014) Eosinophils in the lung – modulating apoptosis and efferocytosis in airway inflammation. Front. Immunol. 5:302. doi: 10.3389/fimmu.2014.00302
Received: 01 May 2014; Paper pending published: 19 May 2014;
Accepted: 15 June 2014; Published online: 01 July 2014.
Edited by:
Luis Enrique Munoz, Friedrich-Alexander University, GermanyReviewed by:
Masaaki Murakami, Hokkaido University, JapanFulvio D’Acquisto, Queen Mary University of London, UK
Copyright: © 2014 Felton, Lucas, Rossi and Dransfield. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer M. Felton, MRC Centre for Inflammation Research, Queen’s Medical Research Institute, The University of Edinburgh, 47 Little France Crescent, Edinburgh, EH16 4TJ, UK e-mail:ai5tLmZlbHRvbkBzbXMuZWQuYWMudWs=