 Stephen J. McSorley
Stephen J. McSorley- Department of Anatomy, Physiology and Cell Biology, Center for Comparative Medicine, School of Veterinary Medicine, University of California Davis, Davis, CA, USA
Intra-macrophage bacterial infections cause significant morbidity and mortality in both the developed and developing world. Protective host immune responses to these infections initially requires the activation and expansion of pathogen-specific CD4 Th1 cells within lymphoid tissues and subsequent relocation of these effector cells to sites of infection. After entering infected tissues, the elicitation of Th1 bactericidal activity can be triggered by cognate or non-cognate signals that are delivered by locally infected antigen-presenting cells and innate cells. However, the contribution of non-cognate stimulation to the resolution of bacterial infection remains poorly understood, especially in the context of a Th1 response. Here, we review the current data on Th1 cell activation and expansion in mouse models of Salmonella and Chlamydia infection and discuss the potential role of non-cognate Th1 cell stimulation in these disease models. Greater understanding of this pathway of T cell activation may lead to the design of therapeutics or vaccines to combat intra-macrophage pathogens.
Introduction
The mammalian immune system contains a variety of cell types that respond in a highly coordinated fashion to eradicate microbial pathogens. The different cell populations that mediate this host defense capability are conveniently assigned to innate or adaptive immune compartments depending a variety of factors, including the tempo of the effector function produced, the use of certain pathogen recognition receptors, and whether these cells have an inherent capacity to confer immune memory. Innate immune responses typically invoke an immediate effector response, make use of germ-line encoded receptors with a restricted capacity for pathogen recognition, and lack the ability to confer a stronger response to secondary infection (1). In contrast, adaptive immune responses require a period of maturation before effector functions are elaborated, utilize complex, rearranged receptors that allow a wider range of specificities, and confer a modified host response to re-infection (2). This general compartmentalization of cells into innate and adaptive arms of the immune system is useful since it provides a conceptual framework that reduces complexity in understanding the dynamics of host–pathogen interactions. However, as might be expected, this model is an oversimplification and some cells of the innate immune system can display characteristics of the adaptive response, and vice versa (3–6). In this review, we will discuss the capacity of adaptive Th1 cells to elaborate effector functions in response to innate stimuli and thus under these conditions appear to function as a component of the innate immune response. The ability of these expanded effector lymphocytes to blur the lines between innate and adaptive immunity may be a critical component of protective immunity to Salmonella, Chlamydia, and other intracellular bacteria.
Global Impact of Salmonella and Chlamydia Infections
Salmonella can cause different clinical diseases in a human host, depending upon the genome of the infecting Salmonella serovar and the immune competence of the infected host (7, 8). Typhoid fever is caused by human transmission of Salmonella enterica serovar Typhi or serovar Paratyphi and this disease remains prevalent in parts of Africa and Asia (9). Current estimates suggest that typhoid causes 217,000 deaths globally every year, the impact of which is felt predominantly in geographical regions with limited access to clean water or basic sanitation infrastructure (10). Although typhoidal serovars enter the human host via the intestine, much of the in vivo bacterial replication occurs in the systemic tissues of the liver, spleen, and bone marrow. In contrast, many other Salmonella serovars can cause local gastro-intestinal infections that are often self-limiting but are a major cause of food-borne infection in the US and other developed nations (11, 12). Thus, Salmonella infection has a global footprint and largely affects developed and developing nations with different patterns of systemic or localized disease. A third disease caused by Salmonella has emerged in sub-Saharan Africa and primarily affects patients with an immature or compromised immune system, either due to age, co-infection, or nutritional status (13, 14). These Salmonella infections can be systemic and are caused by non-typhoidal serovars and therefore this disease is collectively referred to as invasive non-typhoidal Salmonellosis (NTS). While vaccines are currently available for typhoid, these are not widely used in typhoid endemic areas due to concerns about efficacy, safety, or cost (8). The development of improved vaccines for typhoid and NTS therefore remains a priority. Greater understanding of host protective immune mechanisms during Salmonella infection will be required in order to meet this important goal.
While Salmonella is a facultative intracellular pathogen that can grow inside and outside host cells, Chlamydia is an obligate intracellular organism and is only metabolically active within host cells (15). Chlamydia trachomatis causes a sexually transmitted infection in humans that is now the most common notifiable disease in the US (16). The 1.4 million Chlamydia cases reported in 2011 represent an 8% increase over 2010 and is the largest number of cases ever reported to the Centers for Disease Control (CDC) for any single condition (16). Overall, the CDC reports an 8.3% positivity rate among young women screened at family planning clinics, making Chlamydia one of the most prevalent bacterial infections in the US (17). Although most Chlamydia infections are initially asymptomatic, they cause serious pelvic inflammatory disease (PID) in 5–15% of untreated female patients (18, 19). Approximately one in six women who develop PID become infertile, while many others develop chronic pelvic pain, ectopic pregnancy, and if exposed to HIV, Chlamydia-infected women are five times more likely to acquire the virus (18–20). Thus, Chlamydia infection represents a growing healthcare problem in the US and greater understanding of protective immunity in the female reproductive tract will be required to develop an effective vaccine.
Role of CD4 Th1 Cells in Protective Immunity to Salmonella and Chlamydia
Given the location of Chlamydia infection in the reproductive tract and Salmonella infection in the intestine, the immune response to these infections will undoubtedly contain unique tissue-specific components. However, in both mouse models of Salmonella and Chlamydia infection, pathogen-specific CD4 Th1 cells have been found to be essential for successful resolution of primary infection (21, 22). In the Salmonella model, oral infection of C57BL/6 mice with attenuated bacteria generates a systemic infection that eventually resolves over a period of several weeks (23). The ability to resolve this infection is absent in mice lacking MHC class-II-restricted T cells, IFN-γ, or the Th1 transcription factor T-bet (24, 25). Furthermore, successful resolution of Salmonella infection correlates with the expansion of Salmonella-specific Th1 cells in systemic tissues (23, 26).
Genital inoculation of C57BL/6 mice with Chlamydia muridarum generates a self-limiting ascending infection of the upper reproductive tract (21). Similar to Salmonella infection, the resolution of primary C. muridarum infection requires the presence of MHC class-II restricted T cells and IFN-γ (27). The Chlamydia-specific T cell response has been visualized using antigen-specific reagents and the predominant T helper subset detected in draining lymph nodes and spleen consists of a Th1 population that expresses T-bet and secretes IFN-γ (28, 29). In both infection models, the contribution of CD8 T cells and B cells in resolving primary infection is thought to be limited (27, 30–33), although recent data suggest a requirement for B cells in preventing bacterial dissemination to systemic tissues following Chlamydia genital challenge (28). It is not yet clear whether this implies a requirement for B cells in antigen presentation to CD4 T cells or simply a requirement for early antibody production.
Secondary responses to Salmonella and Chlamydia infection have also been examined and the data suggest a wider range of lymphocyte responses that can contribute to bacterial clearance (21, 34). Despite the fact that Salmonella and Chlamydia replicate intracellularly in an infected host, B cells and antibody can contribute to the resolution of secondary infection (30–32, 35, 36). A role for B cells is evident in experiments examining acquired immunity in B cell-deficient mice or by examining the protective immunity mediated by the transfer of immune serum (31, 32, 36–38). Similarly, CD8 T cells have been reported to contribute to secondary protection against both Salmonella and Chlamydia (24, 27, 39), although a recent report examining Salmonella infection of MHC class-I, perforin-, and granzyme-deficient mice did not detect an impaired protective response to secondary infection (33). Despite the expanded contribution of antibody and CD8 T cells in secondary protective immunity, CD4 Th1 cells are still thought to be the primary cell type involved in the resolution of secondary infection (21, 22). Thus, the development of pathogen-specific CD4 Th1 cells is essential for the development of protective immunity in mouse models of Salmonella and Chlamydia infection.
Cognate Signals Driving T Cell Activation and Reactivation
Naïve pathogen-specific CD4 T cells are activated in secondary lymphoid tissues by dendritic cells expressing CD80/86 and displaying microbial peptides on surface MHC class-II (40). In both Salmonella and Chlamydia infection models, TCR transgenic mice and MHC class-II tetramers have been used to visualize naïve T cell activation, expansion, and acquisition of effector functions in vivo (28, 41–43). Initial T cell expansion occurs in the Peyer’s patch and mesenteric lymph nodes after oral infection with Salmonella (41, 44). However, systemic expansion of CD4 T cells can also occur in the spleen and recent evidence suggests that these mucosal and systemic responses are functionally and antigenically distinct (43). Thus, while flagellin-specific CD4 Th17 cells expand in the intestine of Salmonella-infected mice, CD4 Th1 cells specific for components of the Salmonella Pathogenicity Island 2 (SPI2) Type III Secretion System were expanded in the spleen (43). Genital infection of mice with C. muridarum initially drives Chlamydia-specific T cell expansion in the draining ileac lymph, before systemic expansion occurs in the spleen (28). Unlike Salmonella infection, the antigenic targets of the CD4 response appear to be similar in mucosal and systemic locations and Th1 cells were primarily detected both locally and systemically. The most prominent feature of the immune response in both Salmonella and Chlamydia infection models is that a large pool of expanded pathogen-specific Th1 cells is generated. The activation and clonal expansion of Salmonella-specific T cells is strictly dependent on cognate stimulation since flagellin-specific T cells remain unactivated after infection of mice with flagellin-deficient Salmonella (41, 44). Thus, Th1 cells arise from a relatively infrequent pool of naïve pathogen-specific T cells in response to cognate (TCR-dependent) signals that are delivered in lymphoid tissues and these signals eventually lead to clonal expansion and effector development.
Non-Cognate Activation of Effector T Cells
When Th1 cells relocate to an infected non-lymphoid tissue, they can produce IFN-γ locally in order to restrain intracellular bacterial replication (45). The stimulatory signals required to elicit local IFN-γ from effector T cells in tissues could potentially involve cognate stimulation via peptide/MHC complexes on the surface of infected cells or resident dendritic cells (Figure 1). However, many intracellular pathogens have evolved strategies that prevent MHC presentation of microbial peptides or down-regulate surface MHC expression on infected cells (46, 47). While down-regulation of MHC class-I is often discussed as a viral evasion strategy (48), Salmonella have also been reported to reduce expression of MHC class-II of antigen-presenting cells (49). Thus, in the absence of cognate ligands, Th1 cells may simply recognize inflammatory cues such as cytokines and TLR ligands in infected tissues to secrete IFN-γ (Figure 1). However, the relative contribution of cognate versus non-cognate signals in the eradication of intracellular pathogens is not fully understood.
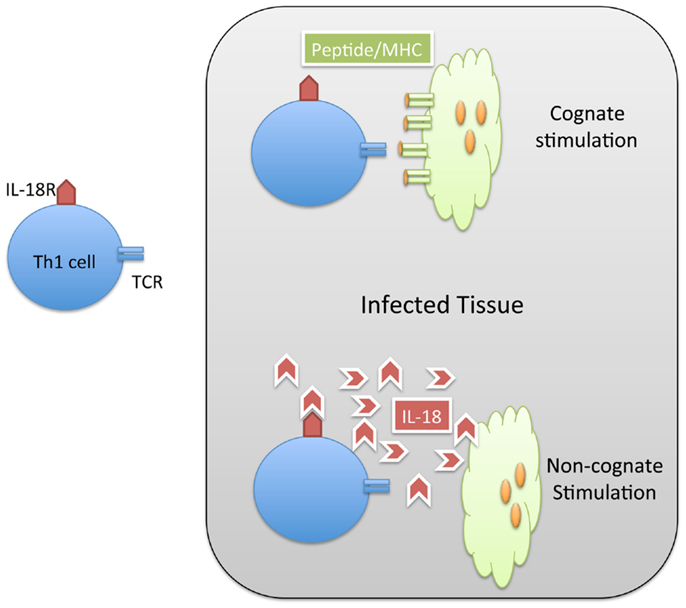
Figure 1. Th1 cells can be activated by cognate and non-cognate stimuli in infected tissue. Naïve CD4 T cells are activated in lymphoid tissues to generate Th1 cells specific for Salmonella. These Salmonella-specific T cells can enter infected tissue and be stimulated by cognate (MHC/peptide) stimuli (TOP) or non-cognate (IL-18) stimuli. In both cases, the result of this stimulation is the production of IFN-gamma and resolution of the infection.
Effector CD4 T cells that have relocated to non-lymphoid tissues retain the ability to respond to cognate signals in that location. Indeed, in a non-infectious model system, antibody that effectively blocked the peptide/MHC complex reduced the ability of CD4 T cells to produce effector cytokines (50). Similarly, recent experiments with bone marrow chimeras containing MHC class-II-deficient and MHC class-II sufficient myeloid cells demonstrated an increased burden of M. tuberculosis in host cells lacking MHC class-II (51). These data support the idea that Th1 cells scan infected tissues and can respond to local cognate signals to produce cytokines. However, the ability to respond to cognate signals may not always be required for the elaboration of effector functions. Studies of CD8 T cell effector function have demonstrated that expanded pathogen-specific T cells can secrete IFN-γ in response to a variety of inflammatory cytokines including IL-12, IL-18, and IL-15 (52–54). In a similar manner, CD4 cells have been shown to produce cytokines after direct ligation of surface TLRs by microbial products (3). Thus, non-cognate stimulation of Th1 cells could potentially be a major contributing factor to bacterial clearance from tissues during intracellular infections.
In a mouse model of Salmonella infection, a large proportion of CD4 T cells can be rapidly induced to secrete IFN-γ following intravenous injection of heat-killed bacteria (23). It was initially assumed that this complex mixture of bacterial antigens was able to efficiently activate Salmonella-specific Th1 cells via cognate signals delivered after antigen presentation of heat-killed bacteria. However, it was subsequently demonstrated that this large response from Th1 cells could also be induced following the injection of TLR ligands and more importantly also occurred in the absence of host MHC class-II (55, 56). Recently, this response was shown to be due to the induction of IL-18 and IL-33 in response to both TLR and inflammasome stimulation (57). The primary inflammasome components involved in recognition of Salmonella infection are NLRC4 and NLRP3 (58). Although NLRC4 can be activated in response to flagellin, bacteria that lacked flagellin expression were still able to induce non-cognate T cell activation suggesting that other components also participate in this response. Overall, these data suggest that inflammasome activation combines with TLR ligation to induce IL-18 and IL-33 production and that these cytokines drive T cell stimulation. Indeed, optimal IFN-γ production required T cell expression of IL-18R and IL-33R and mice containing a T cell-specific deficiency in Myd88 were less able to control the growth of Salmonella (57). A very similar pathway of non-cognate T cell activation has been reported following the injection of bacterial flagellin, although activation of CD8 T cells in this case was thought to require direct flagellin recognition by NLRC4 expressed by dendritic cells (59). Together, these data suggest that, during Salmonella infection, non-cognate signals may be vitally important for driving CD4 Th1 and CD8 T cells to produce IFN-γ and that mice lacking these particular pathways may be unable to generate an effective adaptive response. Interestingly, a similar non-cognate response was detected from Th1 cells in Chlamydia-infected mice (57), suggesting that non-cognate activation of CD4 T cells may be a common feature of the host immunity to intracellular bacteria. Future experiments examining other intracellular pathogens will be important to determine how ubiquitous this pathway is for eliciting protective Th1 responses to microbial pathogens. However, the finding that clearance of M. tuberculosis from individual myeloid cells requires direct cognate stimulation implies that an appropriate balance of cognate and non-cognate signals in infected tissues will be important for Th1 responses to different intracellular pathogens (51). Indeed, it is possible that cognate and non-cognate signals are each responsible for Th1 cytokine production at different stages of the host response, in different anatomical locations, or simply depending on the overall bacterial load within an infected tissue.
Contribution of Non-Cognate T Cell Activation to Pathogen Clearance
The non-cognate elicitation of an effector response from expanded T cells may be required to specifically deal with pathogens that are able to alter host MHC expression or affect the presentation of microbial peptides in infected tissues. Any Th1 cell that enters an infected tissue would therefore retain some capacity to produce IFN-γ in response to local inflammation. Indeed, it has been shown that IFN-γ produced locally can induce iNOS expression from locally infected macrophages, even an individual macrophage happens to lack expression of MHC class-II (45). Thus, there is a degree of non-specificity in the function of Th1 cells within infected tissues. The ability of these same T cells to respond to non-cognate signals may simply further decrease the activation threshold for eliciting bactericidal response. Although it has not been directly examined in vivo, the contribution of non-cognate Th1 cell stimulation may be directly related to the overall pathogen burden in the infected tissue. Thus, if the overall tissue burden is low, then PAMP-elicited cytokines such as IL-18 and IL-33 would also be expected to be at low concentrations, leaving Th1 cells to seek out cognate stimulation and thus constraining T cell activation to a very localized radius around the few infected cells in the tissue. In contrast, if a Th1 cell encounters high concentrations of inflammatory cytokines, the threshold for T cell stimulation would effectively be lowered, allowing immediate and widespread production of IFN-γ. Such a lower threshold of activation may be particularly important when an infected host is combating a rapidly dividing or rapidly spreading pathogen such as Salmonella, but conversely may be less important for immunity to a slow growing pathogen such as M. tuberculosis.
Another potential role for non-cognate T cell activation could occur in situations of bacterial co-infection. Indeed, a role for non-cognate T cell activation in driving pathology has been examined in the context of influenza and bacterial co-infections (60). In this case, an expanded pool of virus-specific CD8 T cells could be rapidly activated to produce harmful pathology in response to inflammatory cytokines elicited by bacterial infection. Conversely, persistent viral stimulation of macrophages can sometimes provide protection against some intracellular bacterial infections (61). In the case of Th1 cells, a pathway of non-cognate activation could be a primary driver of protective immunity during a co-infection. For example, if an individual is infected with an intracellular pathogen and therefore has invested in the expansion and functional maturation of a pool of Th1 cells, the simultaneous encounter with an unrelated secondary infection may well recruit and activate these Th1 cells in a non-cognate manner. Indeed, the original discovery of macrophage activation was surprising because the efferent phase of the adaptive response involved a relatively non-specific mechanism and was demonstrated using a co-infection model where Brucella infection prevented productive infection with Listeria. However, a role for non-cognate T cell activation in the elicitation of protective immunity during co-infections has not yet been described. Overall, it seems most likely that non-cognate mechanisms of Th1 cell activation could have evolved to help the host combat bacterial evasions of host immunity, superior bacterial cell division, or co-infections. Future research in this area is required to examine each of these possibilities.
Conclusion
Naïve CD4 T cells are activated by cognate signals leading to the expansion of an effector pool of pathogen-specific T cells that can migrate to infected tissues and deliver local anti-microbial effects. Recent data have demonstrated that Th1 cells can be activated within infected tissues in response to cognate and/or non-cognate signals that arise from TLR and inflammasome activation. Thus, although the adaptive response is regulated by highly specific antigen-specific surface receptors, an expanded pool of effector cells retains the ability to respond immediately to inflammatory cues that are normally associated with the innate arm of the immune system. This functional capability reinforces our growing understanding that innate and adaptive immune systems are not completely separate entities but instead work in a coordinated fashion to resolve infection with microbial pathogens. This ability of expanded effector lymphocytes to blur the lines between innate and adaptive immunity may be a critical component of protective immunity to Salmonella, Chlamydia, and other intracellular bacteria.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Institutes of Health to Stephen J. McSorley (AI055743, AI076278, and AI056172).
References
1. Broz P, Ohlson MB, Monack DM. Innate immune response to Salmonella typhimurium, a model enteric pathogen. Gut Microbes (2012) 3:62–70. doi: 10.4161/gmic.19141
2. Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol (2013) 13:309–20. doi:10.1038/nri3442
3. Reynolds JM, Dong C. Toll-like receptor regulation of effector T lymphocyte function. Trends Immunol (2013) 34:511–9. doi:10.1016/j.it.2013.06.003
4. Dostert C, Ludigs K, Guarda G. Innate and adaptive effects of inflammasomes on T cell responses. Curr Opin Immunol (2013) 25:359–65. doi:10.1016/j.coi.2013.02.008
5. Quintin J, Cheng SC, van der Meer JW, Netea MG. Innate immune memory: towards a better understanding of host defense mechanisms. Curr Opin Immunol (2014) 29C:1–7. doi:10.1016/j.coi.2014.02.006
6. Sheridan BS, Romagnoli PA, Pham QM, Fu HH, Alonzo F III, Schubert WD, et al. Gammadelta T cells exhibit multifunctional and protective memory in intestinal tissues. Immunity (2013) 39:184–95. doi:10.1016/j.immuni.2013.06.015
7. Dougan G, John V, Palmer S, Mastroeni P. Immunity to salmonellosis. Immunol Rev (2011) 240:196–210. doi:10.1111/j.1600-065X.2010.00999.x
8. McGregor AC, Waddington CS, Pollard AJ. Prospects for prevention of Salmonella infection in children through vaccination. Curr Opin Infect Dis (2013) 26:254–62. doi:10.1097/QCO.0b013e32835fb829
9. Parry CM, Hien TT, Dougan G, White NJ, Farrar JJ. Typhoid fever. N Engl J Med (2002) 347:1770–82. doi:10.1056/NEJMra020201
10. Crump JA, Mintz ED. Global trends in typhoid and paratyphoid fever. Clin Infect Dis (2010) 50:241–6. doi:10.1086/649541
11. Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, et al. Food-related illness and death in the United States. Emerg Infect Dis (1999) 5:607–25. doi:10.3201/eid0506.990625
12. Costa LF, Paixao TA, Tsolis RM, Baumler AJ, Santos RL. Salmonellosis in cattle: advantages of being an experimental model. Res Vet Sci (2012) 93:1–6. doi:10.1016/j.rvsc.2012.03.002
13. Gordon MA. Salmonella infections in immunocompromised adults. J Infect (2008) 56:413–22. doi:10.1016/j.jinf.2008.03.012
14. Feasey NA, Dougan G, Kingsley RA, Heyderman RS, Gordon MA. Invasive non-typhoidal Salmonella disease: an emerging and neglected tropical disease in Africa. Lancet (2012) 379:2489–99. doi:10.1016/S0140-6736(11)61752-2
15. Brunham RC, Rey-Ladino J. Immunology of Chlamydia infection: implications for a Chlamydia trachomatis vaccine. Nat Rev Immunol (2005) 5:149–61. doi:10.1038/nri1551
16. Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance, 2008. Atlanta, GA: Department of Health and Human Services (2009).
17. Miller WC, Ford CA, Morris M, Handcock MS, Schmitz JL, Hobbs MM, et al. Prevalence of chlamydial and gonococcal infections among young adults in the United States. JAMA (2004) 291:2229–36. doi:10.1001/jama.291.18.2229
18. Gottlieb SL, Brunham RC, Byrne GI, Martin DH, Xu F, Berman SM. Introduction: the natural history and immunobiology of Chlamydia trachomatis genital infection and implications for Chlamydia control. J Infect Dis (2010) 201(Suppl 2):S85–7. doi:10.1086/652401
19. Gottlieb SL, Martin DH, Xu F, Byrne GI, Brunham RC. Summary: the natural history and immunobiology of Chlamydia trachomatis genital infection and implications for Chlamydia control. J Infect Dis (2010) 201(Suppl 2):S190–204. doi:10.1086/652401
20. Fleming DT, Wasserheit JN. From epidemiological synergy to public health policy and practice: the contribution of other sexually transmitted diseases to sexual transmission of HIV infection. Sex Transm Infect (1999) 75:3–17. doi:10.1136/sti.75.1.3
21. Farris CM, Morrison RP. Vaccination against Chlamydia genital infection utilizing the murine C. muridarum model. Infect Immun (2011) 79:986–96. doi:10.1128/IAI.00881-10
22. Griffin AJ, McSorley SJ. Development of protective immunity to Salmonella, a mucosal pathogen with a systemic agenda. Mucosal Immunol (2011) 4:371–82. doi:10.1038/mi.2011.2
23. Srinivasan A, Foley J, McSorley SJ. Massive number of antigen-specific CD4 T cells during vaccination with live attenuated Salmonella causes interclonal competition. J Immunol (2004) 172:6884–93. doi:10.4049/jimmunol.172.11.6884
24. Hess J, Ladel C, Miko D, Kaufmann SH. Salmonella typhimurium aroA-infection in gene-targeted immunodeficient mice: major role of CD4+ TCR-alpha beta cells and IFN-gamma in bacterial clearance independent of intracellular location. J Immunol (1996) 156:3321–6.
25. Ravindran R, Foley J, Stoklasek T, Glimcher LH, McSorley SJ. Expression of T-bet by CD4 T cells is essential for resistance to Salmonella infection. J Immunol (2005) 175:4603–10. doi:10.4049/jimmunol.175.7.4603
26. Griffin AJ, McSorley SJ. Generation of Salmonella-specific Th1 cells requires sustained antigen stimulation. Vaccine (2011) 29:2697–704. doi:10.1016/j.vaccine.2011.01.078
27. Morrison RP, Feilzer K, Tumas DB. Gene knockout mice establish a primary protective role for major histocompatibility complex class II-restricted responses in Chlamydia trachomatis genital tract infection. Infect Immun (1995) 63:4661–8.
28. Li LX, McSorley SJ. B cells enhance antigen-specific CD4 T cell priming and prevent bacteria dissemination following Chlamydia muridarum genital tract infection. PLoS Pathog (2013) 9:e1003707. doi:10.1371/journal.ppat.1003707
29. Gondek DC, Roan NR, Starnbach MN. T cell responses in the absence of IFN-gamma exacerbate uterine infection with Chlamydia trachomatis. J Immunol (2009) 183:1313–9. doi:10.4049/jimmunol.0900295
30. Su H, Feilzer K, Caldwell HD, Morrison RP. Chlamydia trachomatis genital tract infection of antibody-deficient gene knockout mice. Infect Immun (1997) 65:1993–9.
31. McSorley SJ, Jenkins MK. Antibody is required for protection against virulent but not attenuated Salmonella enterica serovar typhimurium. Infect Immun (2000) 68:3344–8. doi:10.1128/IAI.68.6.3344-3348.2000
32. Mastroeni P, Simmons C, Fowler R, Hormaeche CE, Dougan G. Igh-6(-/-) (B-cell-deficient) mice fail to mount solid acquired resistance to oral challenge with virulent Salmonella enterica serovar typhimurium and show impaired Th1 T-cell responses to Salmonella antigens. Infect Immun (2000) 68:46–53. doi:10.1128/IAI.68.1.46-53.2000
33. Lee SJ, Dunmire S, McSorley SJ. MHC class-I-restricted CD8 T cells play a protective role during primary Salmonella infection. Immunol Lett (2012) 148:138–43. doi:10.1016/j.imlet.2012.10.009
34. Moon JJ, McSorley SJ. Tracking the dynamics of Salmonella specific T cell responses. Curr Top Microbiol Immunol (2009) 334:179–98. doi:10.1007/978-3-540-93864-4_8
35. Williams DM, Grubbs BG, Pack E, Kelly K, Rank RG. Humoral and cellular immunity in secondary infection due to murine Chlamydia trachomatis. Infect Immun (1997) 65:2876–82.
36. Nanton MR, Way SS, Shlomchik MJ, McSorley SJ. Cutting edge: B cells are essential for protective immunity against Salmonella independent of antibody secretion. J Immunol (2012) 189:5503–7. doi:10.4049/jimmunol.1201413
37. Mastroeni P, Villarreal-Ramos B, Hormaeche CE. Adoptive transfer of immunity to oral challenge with virulent Salmonella in innately susceptible BALB/c mice requires both immune serum and T cells. Infect Immun (1993) 61:3981–4.
38. Morrison SG, Morrison RP. A predominant role for antibody in acquired immunity to chlamydial genital tract reinfection. J Immunol (2005) 175:7536–42. doi:10.4049/jimmunol.175.11.7536
39. Lo WF, Ong H, Metcalf ES, Soloski MJ. T cell responses to gram-negative intracellular bacterial pathogens: a role for CD8+ T cells in immunity to Salmonella infection and the involvement of MHC class Ib molecules. J Immunol (1999) 162:5398–406.
40. Jenkins MK, Khoruts A, Ingulli E, Mueller DL, McSorley SJ, Reinhardt RL, et al. In vivo activation of antigen-specific CD4 T cells. Annu Rev Immunol (2001) 19:23–45. doi:10.1146/annurev.immunol.19.1.23
41. McSorley SJ, Asch S, Costalonga M, Rieinhardt RL, Jenkins MK. Tracking Salmonella-specific CD4 T cells in vivo reveals a local mucosal response to a disseminated infection. Immunity (2002) 16:365–77. doi:10.1016/S1074-7613(02)00289-3
42. Roan NR, Gierahn TM, Higgins DE, Starnbach MN. Monitoring the T cell response to genital tract infection. Proc Natl Acad Sci U S A (2006) 103:12069–74. doi:10.1073/pnas.0603866103
43. Lee SJ, McLachlan JB, Kurtz JR, Fan D, Winter SE, Baumler AJ, et al. Temporal expression of bacterial proteins instructs host CD4 T cell expansion and Th17 development. PLoS Pathog (2012) 8:e1002499. doi:10.1371/journal.ppat.1002499
44. Salazar-Gonzalez RM, Niess JH, Zammit DJ, Ravindran R, Srinivasan A, Maxwell JR, et al. CCR6-mediated dendritic cell activation of pathogen-specific T cells in Peyer’s patches. Immunity (2006) 24:623–32. doi:10.1016/j.immuni.2006.02.015
45. Muller AJ, Filipe-Santos O, Eberl G, Aebischer T, Spath GF, Bousso P. CD4+ T cells rely on a cytokine gradient to control intracellular pathogens beyond sites of antigen presentation. Immunity (2012) 37:147–57. doi:10.1016/j.immuni.2012.05.015
46. Bueno SM, Gonzalez PA, Schwebach JR, Kalergis AM. T cell immunity evasion by virulent Salmonella enterica. Immunol Lett (2007) 111:14–20. doi:10.1016/j.imlet.2007.05.003
47. Bedoui S, Kupz A, Wijburg OL, Walduck AK, Rescigno M, Strugnell RA. Different bacterial pathogens, different strategies, yet the aim is the same: evasion of intestinal dendritic cell recognition. J Immunol (2010) 184:2237–42. doi:10.4049/jimmunol.0902871
48. Horst D, Verweij MC, Davison AJ, Ressing ME, Wiertz EJ. Viral evasion of T cell immunity: ancient mechanisms offering new applications. Curr Opin Immunol (2011) 23:96–103. doi:10.1016/j.coi.2010.11.005
49. Lapaque N, Hutchinson JL, Jones DC, Meresse S, Holden DW, Trowsdale J, et al. Salmonella regulates polyubiquitination and surface expression of MHC class II antigens. Proc Natl Acad Sci U S A (2009) 106:14052–7. doi:10.1073/pnas.0906735106
50. McLachlan JB, Catron DM, Moon JJ, Jenkins MK. Dendritic cell antigen presentation drives simultaneous cytokine production by effector and regulatory T cells in inflamed skin. Immunity (2009) 30:277–88. doi:10.1016/j.immuni.2008.11.013
51. Srivastava S, Ernst JD. Cutting edge: direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. J Immunol (2013) 191:1016–20. doi:10.4049/jimmunol.1301236
52. Berg RE, Cordes CJ, Forman J. Contribution of CD8+ T cells to innate immunity: IFN-gamma secretion induced by IL-12 and IL-18. Eur J Immunol (2002) 32:2807–16. doi:10.1002/1521-4141(2002010)32:10<2807::AID-IMMU2807>3.0.CO;2-0
53. Freeman BE, Hammarlund E, Raue HP, Slifka MK. Regulation of innate CD8+ T-cell activation mediated by cytokines. Proc Natl Acad Sci U S A (2012) 109:9971–6. doi:10.1073/pnas.1203543109
54. Lertmemongkolchai G, Cai G, Hunter CA, Bancroft GJ. Bystander activation of CD8+ T cells contributes to the rapid production of IFN-gamma in response to bacterial pathogens. J Immunol (2001) 166:1097–105. doi:10.4049/jimmunol.166.2.1097
55. Srinivasan A, Salazar-Gonzalez RM, Jarcho M, Sandau MM, Lefrancois L, McSorley SJ. Innate immune activation of CD4 T cells in Salmonella-infected mice is dependent on IL-18. J Immunol (2007) 178:6342–9. doi:10.4049/jimmunol.178.10.6342
56. Srinivasan A, McSorley SJ. Pivotal advance: exposure to LPS suppresses CD4+ T cell cytokine production in Salmonella-infected mice and exacerbates murine typhoid. J Leukoc Biol (2007) 81:403–11. doi:10.1189/jlb.0306194
57. O’Donnell H, Pham OH, Li LX, Atif SM, Lee SJ, Ravesloot MM, et al. Toll-like receptor and inflammasome signals converge to amplify the innate bactericidal capacity of T helper 1 cells. Immunity (2014) 40:213–24. doi:10.1016/j.immuni.2013.12.013
58. Broz P, Monack DM. Molecular mechanisms of inflammasome activation during microbial infections. Immunol Rev (2011) 243:174–90. doi:10.1111/j.1600-065X.2011.01041.x
59. Kupz A, Guarda G, Gebhardt T, Sander LE, Short KR, Diavatopoulos DA, et al. NLRC4 inflammasomes in dendritic cells regulate noncognate effector function by memory CD8 T cells. Nat Immunol (2012) 13:162–9. doi:10.1038/ni.2195
60. Beadling C, Slifka MK. How do viral infections predispose patients to bacterial infections? Curr Opin Infect Dis (2004) 17:185–91. doi:10.1097/00001432-200406000-00003
Keywords: T cell, bacterial infections, protective immunity, IFN-gamma, Salmonella
Citation: McSorley SJ (2014) The role of non-cognate T cell stimulation during intracellular bacterial infection. Front. Immunol. 5:319. doi: 10.3389/fimmu.2014.00319
Received: 21 May 2014; Accepted: 24 June 2014;
Published online: 09 July 2014.
Edited by:
Kimberly Sue Schluns, University of Texas MD Anderson Cancer Center, USAReviewed by:
Hao Shen, University of Pennsylvania School of Medicine, USAKamal Khanna, University of Connecticut Health Center, USA
Copyright: © 2014 McSorley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen J. McSorley, Department of Anatomy, Physiology and Cell Biology, Center for Comparative Medicine, School of Veterinary Medicine, University of California Davis, County Road 98 and Hutchison Drive, Davis, CA 95616, USA e-mail:c2ptY3NvcmxleUB1Y2RhdmlzLmVkdQ==