 Hillary L. Shane
Hillary L. Shane Kimberly D. Klonowski
Kimberly D. Klonowski- Department of Cellular Biology, University of Georgia, Athens, GA, USA
Resident memory T cells (TRM) are broadly defined as a population of T cells, which persist in non-lymphoid sites long-term, do not re-enter the circulation, and are distinct from central memory T cells (TCM) and circulating effector memory T cells (TEM). Recent studies have described populations of TRM cells in the skin, gut, lungs, and nervous tissue. However, it is becoming increasingly clear that the specific environment in which the TRM reside can further refine their phenotypical and functional properties. Here, we focus on the TRM cells that develop following respiratory infection and reside in the lungs and the lung airways. Specifically, we will review recent studies that have described some of the requirements for establishment of TRM cells in these tissues, and the defining characteristics of TRM in the lungs and lung airways. With continual bombardment of the respiratory tract by both pathogenic and environmental antigens, dynamic fluctuations in the local milieu including homeostatic resources and niche restrictions can impact TRM longevity. Beyond a comprehensive characterization of lung TRM cells, special attention will be placed on studies, which have defined how the microenvironment of the lung influences memory T cell survival at this site. As memory T cell populations in the lung airways are requisite for protection yet wane numerically over time, developing a comprehensive picture of factors which may influence TRM development and persistence at these sites is important for improving T cell-based vaccine design.
Introduction
The adaptive immune system is defined by its ability to mount an antigen-specific immune response and generate long-lived memory cells. CD8+ memory T cells (Tmem) respond rapidly upon secondary encounter with the same antigen and can provide protection against the development of severe disease or chronic infection in the absence of neutralizing antibodies (1). This attribute of Tmem is particularly attractive in the context of vaccine design for viral infections such as HIV or influenza, which rapidly modify antibody targets as a result of high mutagenic rates and immune pressure.
The efficiency of Tmem-mediated protection is in part a direct result of activated T cells initiating divergent developmental and migratory programs, which provide the host with a multifaceted immune response following challenge. This Tmem diversity is acquired as a result of different levels of co-stimulation, inflammation, or T cell help, which not only vary throughout the course of a single infection but are also impacted by infection route. Initially, memory T cells were broadly categorized into two populations based on homing preferences, circulating between secondary lymphoid organs as central memory T cells (TCM) or less discretely throughout the periphery, including non-lymphoid tissues, defined as effector memory T cells (TEM) (2). These memory pools are distinguished from one another by their differential expression of the lymph node homing molecules L-selectin (CD62L) and CCR7, with TCM expressing high levels of these molecules for lymph node entry and retention (3) and TEM cells expressing low levels. While this simplified TCM/TEM paradigm predominated Tmem classification for several years, subsequent studies using parabiotic mice (4) and adoptive transfer systems (5) demonstrated that at least one additional Tmem pool exists with tissue-specific residency and little migratory potential. Additional studies confirmed the existence of these tissue-locked Tmem at portals of pathogen entry and led to the T resident memory cells (TRM) nomenclature.
As relative newcomers to the T cell memory scene, TRM cells have not been characterized to the same extent as TCM and TEM cells, and our definition of this memory population, as well our understanding of its origin is still evolving. Nonetheless, specific CD8+ TRM populations have been identified in many peripheral sites including the gut (6), skin (7), brain (8), female reproductive mucosa (9, 10), and the lung (11). Despite some similarities with TEM, lack of equilibration of Tmem between specific tissues of parabiotic mice as well as general “hallmarks” of TRM have been identified as defining characteristics. These distinguishing features include the expression of CD103 (αE integrin) and CD69, molecules traditionally associated with adhesion within epithelial layers and recent activation, respectively (12, 13). A recent paper by Mackay et al. defined a common transcriptional signature shared by CD103+ TRM cells isolated from the skin, gut, and lung consisting of 37 genes differentially expressed compared to TEM or TCM cells, demonstrating that TRM cells are a distinct Tmem lineage (14). Additionally, this study determined that TRM cells from distinct anatomical sites also possessed unique gene transcription patterns, with 127 being unique to the gut, 86 unique to the skin, and 25 unique to the lung, indicating additional diversification within the TRM pool, likely environmentally driven.
Despite the relative juvenescence of the TRM field, the importance of this cell population has been alluded to for some time. TRM cells are positioned at the site of pathogen encounter as a front line of defense, and several studies have highlighted their role in defense against pathogenic challenges (7, 15–17). Indeed, in the case of influenza virus infection, the number of antigen-specific CD8+ T cells located within the respiratory tract correlates with the highest degree of heterosubtypic immunity (18, 19), and recently it has been shown that TRM specifically are responsible for this protection (20). Defining the characteristics that lead to TRM development, and determining how they persist at sites of infection may lead to novel ways to enhance vaccine efficacy. This review will focus on the development, characteristics, and maintenance of CD8+ TRM cells in the respiratory tract, which develop after acute respiratory infection, primarily with influenza and Sendai viruses. How the lung environment affects the developmental transition and tissue residency of CD8+ TRM cells will be discussed, from the primary activation of the antigen-specific cell through the return to homeostasis and during resting conditions.
Part I: Factors Influencing TRM Development
There is great interest in deciphering the TRM developmental pathway, as understanding this mechanism could lead to modulation of the responses in ways, which could enhance the establishment of this Tmem pool. The development of TRM cells will have two main requirements: (1) the ability to survive through contraction (become a Tmem) and (2) the ability to differentiate into the appropriate memory lineage (become a TRM cell as opposed to a TEM or TCM cell). In this section, we will discuss factors that may influence TRM development in the early priming environment of the lymph node, and subsequently in the inflamed lung. Recent evidence demonstrates that T cell differentiation into distinct TEM and TCM subsets occurs soon after T cell priming (21), which begs the question: does a population of cells that is destined to become TRM cells also develop during or soon after initial activation in the lymph node? Or, do TRM arise only after tissue-specific entry based on specific cues within the microenvironment of tissues, like the lung? These scenarios are not necessarily mutually exclusive and full commitment to the TRM lineage is likely due to a combination of these two possibilities as will be further discussed and as described in Figure 1.
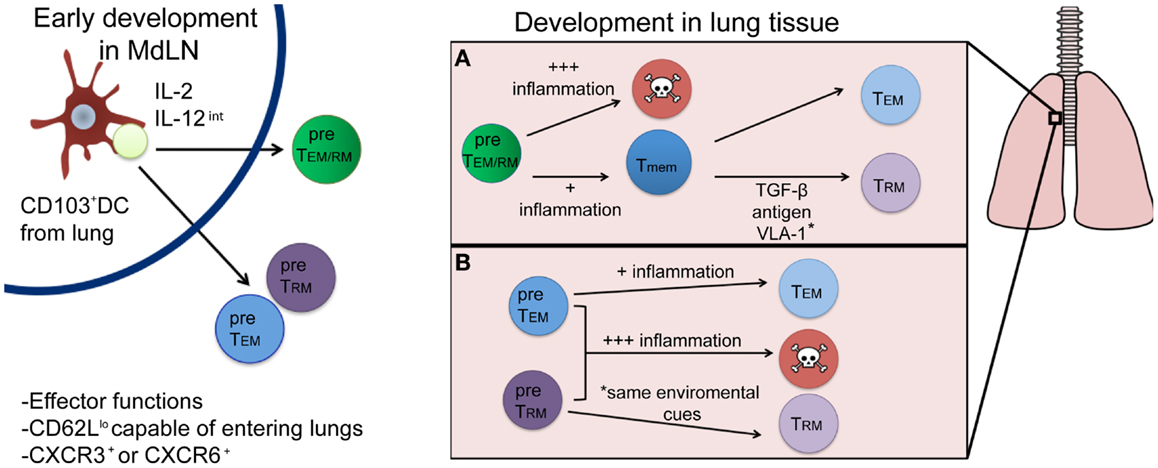
Figure 1. Proposed developmental pathways for TRM cells following respiratory infection. Priming by a CD103+ DC and appropriate cytokine signals (left) results in the generation of either a common TEM/RM precursor cell (green) or individual pre-TEM or TRM cells (blue and purple, respectively). Once in the lung (right), environmental factors will drive subsequent cell fate decisions, resulting in either terminal differentiation (and death) or the generation of memory cells. While most current evidence supports the differentiation route depicted in (A), where a common precursor differentiates first into memory, followed by environmentally driven lineage differentiation into TRM or TEM cells, it is plausible that differentiation into a TRM fate occurs immediately following priming in the lymph node (B) and is distinguished by yet to be discovered phenotypic or genetic markers.
Early Differentiation Signals in the Lymph Node: Developing TRM Potential
Activation of CD8+ T cell requires three signals: detection of cognate peptide/MHCI complex, co-stimulation, and a cytokine signal (22). The combination of these three signals, which may vary in intensity and type, results not only in clonal expansion and acquisition of effector function, but also influences long-term cellular fate (21). In many cases, the overall Tmem potential of the antigen-specific CD8+ T cell is driven by lineage-associated transcription factors and acquired epigenetic changes (23), which can be experimentally monitored. These programing signals are influenced by the type of (priming) APC, antigen availability, and inflammatory properties of the pathogen, which can vary based on the individual pathogen and the route, which infection is acquired. While an early TRM lineage-specific transcriptional program has not been identified, specific migratory signals facilitating peripheral tissue entry, with subsequent acquisition of TRM characteristics implies that at least some early signals help polarize cells toward a TRM fate. Here, we will discuss the possible early signals encountered in the lung draining mediastinal lymph nodes (MdLN), which may promote the development of respiratory TRM cells.
During influenza infection, activated, antigen-laden respiratory DCs migrate to the MdLN to interact with naïve CD8+ T cells. The majority of these migratory DCs fall into two subsets, airway localized CD103+ DCs and lung parenchyma CDllbhi DCs (24). In addition to their localization in the lung during a resting state, these DC populations differ in their induction of CD8+ T cell effector functions, with CD103+ DCs requisite for complete effector differentiation, defined by expression of standard effector markers and their potential to enter inflamed tissues (CD25hi, T-bethi, and Blimp-1hi and CD62Llo CCR5hi). In contrast, CD11bhi DCs are more likely to prime CD8+ T cells, which largely remained in the lymph nodes, expressing molecules associated with the development of TCM (CD62Lhi, T-betlo, Blimp-1lo CD25lo, and CD127hi (25). Thus, as entry into peripheral tissues is a defining characteristic of TRM cells, it is likely that TRM precursors are activated in the draining MdLN by activated respiratory CD103+ DCs, where they not only acquire effector function, but more importantly, the ability to accumulate in lung tissue, which is requisite for TRM development. Priming by CD103+ DCs may also be one of the reasons that TRM have a propensity to develop following induction of the responses in mucosal tissues, as similar CD103+ epidermal associated DCs are found predominately in these sites (26) and may be a common method promoting CD8+ T cell migration into peripheral tissues. In support of this, intranasal vaccination gives rise to populations of long lasting Tmem in the female reproductive tract, a phenomenon that is not observed following systemic infection (27–29). This indicates that a common priming requirement (possibly CD103+ DCs) can induce CD8+ T cell migration into more restrictive sites, and vaccination at certain mucosal surfaces may broadly confer protection at expanded peripheral sites (30). However, it should be noted that certain systemic infections, such as lymphocytic choriomeningitis virus (LCMV), can produce populations of TRM cells in a wide variety of tissues, including the intestinal tract, brain, and female reproductive tract, as well as organs such as the kidney, heart, and pancreas, although in this study respiratory TRM were not assessed (31). LCMV, a true systemic pathogen, can replicate in multiple cell types and locations, suggesting that pathogen promiscuity could result in activation of CD103+ DC and induction of TRM independent of mucosal infection. For the lung, it seems that priming via the respiratory route [intranasally (i.n.)] is necessary for TRM formation, as priming with influenza virus intraperitoneally (i.p.) fails to generate TRM cells (20). The difference here is that influenza will not produce a productive infection when given by the i.p. route, limiting presentation to CD103− DCs (32). It will be important for future studies to distinguish whether the lung is truly a restrictive site, limiting TRM generation only after infection via the i.n. route, or if a systemic or mucosal challenge at a divergent site can induce lung TRM populations under the right conditions.
Another important factor which can be highly variable during infection is the presence of particular cytokines, which influence both memory cell potential (33, 34), and the specific pool of Tmem that develops (35). The potential for an effector T cell (Teff) to become a Tmem cell has been defined based on the expression of CD127 and KLRG1 (36). Teffs largely fall into one of three categories: terminally differentiated short-lived effector cells (SLECs, KLRG1hi/CD127lo), early effector cells (EECs, KLRG1lo/CD127lo) or memory precursor effector cells (MPECs, KLRG1lo, CD127hi). It is the latter population, which develops into long-lived, bonafide Tmem of various phenotypes, including TCM, TEM, and TRM. Therefore, the formation of MPECs is a necessary step in TRM development, although the timing in which a cell begins to express these markers may differentially impact its memory phenotype. MPECs can form early in the lymph node, or once at the site of infection they can arise from EECs, which have the potential to differentiate into both SLECs and MPECs (37). The inflammatory cytokine IL-12 is detectable at 48 h following influenza infection, and is important for the development of IFN-γ producing cells early in the immune response (38). In regard to memory development, IL-12 promotes the development of terminally differentiated SLECs in a dose dependent manner via induction of the transcription factor T-bet (33). Interestingly, graded induction of IL-12 is observed after systemic infection with two different pathogens: L. monocytogenes (LM) induces a high concentration of IL-12, whereas vesicular stomatitis virus (VSV) induces much lower IL-12 levels. High concentrations of IL-12 during LM infection promote a skewed development favoring SLECs while VSV infection (lower IL-12) favored EECs (37). Since TRM cells arise from KLRG1lo precursors (14), high levels of IL-12 would likely negatively impact TRM development. Nonetheless, a minimum threshold of IL-12 (and T-bet) expression is required to not only promote the requisite development of Teff but promote migration into peripheral sites. In support of this, it has been shown that CD103+ DCs isolated from the small intestine are capable of producing IL-12 following TLR stimulation (39). However, high levels of IL-12 signaling had a direct effect on CD8+ T cells, leading to the down-regulation of CXCR3, a molecule necessary for the accumulation of antigen-specific CD8+ T cells in the airways following influenza infection (40). These data would suggest that CD8+ T cells at the site of priming need just the right amount of IL-12 to reach their full TRM potential. In terms of cytokines important for parsing Tmem into defined subsets, the common gamma chain cytokines IL-2 and IL-15 have been shown to play a role in CD8+ T cell differentiation into TCM and TEM cells. TCM cells can be identified as a distinct population arising from MPECs as early as 5 dpi, and are formed through IL-15 signaling (when IL-2 is limited), whereas IL-2 signaling leads to TEM phenotypes (41). As previously mentioned TCM cells develop early after infection from the MPEC population in the lymph node, and these cells may never enter peripheral tissues. Thus, TRM cells may arise from Teffs, which do not receive early TCM biasing signals in the lymph node, and retain the ability to enter peripheral sites.
While the evidence we have presented thus far suggests that specific cellular interactions and cytokines present in the lymph node at the time of priming could form a population of cells with the potential to become TRM cells, an early development pathway completely unique to TRM remains unlikely. Traditional cell surface markers and functional characteristics associated with Teff cells or TEM, such as low levels of CD62L expression, are indistinguishable from TRM early after infection. Moreover, the prototypical TRM cell surface marker, CD103, does not appear until after a certain period of tissue residency in the epidermis (14). Interestingly, TRM populations in the skin require the expression of CXCR3 for entry into the epithelium and subsequent TRM differentiation as cells lacking CXCR3 remained largely outside of the epidermis and TRM recovered from the skin were numerically reduced. Conversely, mice lacking CCR7 expression have CD8+ Teffs, which fail to leave the skin via the lymphatics and harbor larger numbers of TRM cells, suggesting environmental factors are required for complete TRM development (14). Signals encountered in the MdLN after respiratory infection likely generate a population of Teff cells with the potential to enter the lung and fully develop into true TRM cells. However, CD8+ T cells isolated from the respiratory tract phenotypically resemble TEM cells, TRM cells, and terminally differentiated SLECs (our unpublished observations), suggesting not all Teff that enter the lung become TRM. More likely, cells immigrating to the respiratory tract enter as a common TEM/RM precursor (Figure 1A). This common TEM/RM precursor population is likely primed by CD103+ DCs, expresses high levels of CD25, and encounters intermediate levels of IL-12, akin to the development of TEM cells. Therefore, TRM and TEM cells may share similar early developmental pathways, with later signals in the lung further differentiating and diverting true TRM from a common TEM/RM precursor. Indeed, evidence supports the development of a common TEM/TRM precursor. As previously noted, the development of TEM cells is dependent on IL-2 (and not IL-15) (41) and IL-15 is also dispensable for CD8+ Tmem that develops following a respiratory infection (42), which generates substantial TRM compared to systemic infection (20). In contrast, systemic infections produce large amounts of TCM cells, and Tmem in these infections require IL-15 for maintenance over time (43). Although the evidence suggests a common developmental pathway for TEM and TRM cells after initial activation in the lymph node, the possibility remains that they are distinct lineages by the time of lymph node egress (Figure 1B), identifiable by phenotypic markers or gene expression patterns yet to be discovered. Nonetheless, the full commitment to the TRM lineage will continue in the specific peripheral tissue, where these cells will be retained.
Environmental Signals Commit TEM/TRM Precursor Cells to a TRM Lineage
If T cell priming in the MdLN results in the migration of a common TEM/RM precursor population of cells to the lung, what factors in the lung facilitate the development of “full-fledged” TRM cells? At the site of infection multiple factors will continue to influence emigrating TEM/RM precursors. Evidence in cutaneous infection models suggests that commitment to the TRM lineage is a two-step process characterized by the sequential up-regulation of Bcl-2 and CD69, followed by CD103 (14). This suggests that T cells first acquire a memory phenotype, or an increased chance of survival, prior to differentiating into TRM cells based on the current TRM phenotypic markers. This section will discuss the respiratory factors that influence the transition to a memory phenotype and specific environmental components present in the lung that polarize these anti-viral CD8+ T cells toward a TRM lineage.
The inflammatory environment of the lung
The pioneer Teff cells immigrating to the lung arrive ~5–6 days after initial respiratory infection. Prior to their arrival, innate immune cells have accumulated, keeping viral titers low, and as a result, some local tissue damage has occurred via cytolysis of infected epithelial cells, affecting barrier function. The inflammatory effects of this local immune response in the lung are still very present at the time of T cell entry, and can influence the development of TRM cells. However, since anti-influenza Teff migrate to the lung asynchronously over several days (peaking at ~10 days post viral infection), all T cells do not encounter equivalent levels of inflammation which will likely affect the fate of individual Teff clones.
The first CD8+ Teffs to arrive at the site of infection will encounter the greatest level of inflammation, as infectious virus is still present (at least until ~d8 post influenza infection) and innate effectors such as NK cells are producing local IFN-γ (38). Inflammatory monocyte-derived DCs arrive in the inflamed lung at the same time as Teff and function as lung APCs, amplifying the inflammatory milieu and locally expanding the emigrating Teff (44). Additionally, CD8+ T cell proliferation continues in the lung, a process requisite for viral control after influenza infection (45). This additional expansion, however, is not without a cost. Increased levels of cellular division is not only associated with increased levels of apoptosis within the highly dividing populations (46), the aforementioned cytokines also promote terminal differentiation of the T cells and the formation of KLRG1+ SLECs (47, 48). Therefore, this early inflammatory environment skews cells away from becoming memory cells, yet may paradoxically pave the way for resolution from infection and inflammation so that later immigrants may develop into Tmem.
CD8+ Teff themselves produce cytokines in the lung, including IL-2, IFN-γ and TNF-α which enhance the overall inflammatory response (49). Interestingly, while CD8+ T cells activated in lymph nodes rapidly gain the ability to produce the inflammatory cytokine IFN-γ, entry into the lung tissue imparts IL-10 production (50, 51) in a manner seemingly dependent on the inflammatory lung environment (52), indicating that an enhanced activation status resulting from high levels of inflammation induces the CD8+ T cells to produce regulatory cytokines. IL-10 is also produced at high levels by regulatory T cells (Tregs) activated in the lung following influenza infection (53). The production of regulatory cytokines by Tregs and CD8+ T cells is important to initiate “dampening” the immune responses in the lung to prevent excessive damage and loss of function of this essential organ. Importantly, the production of IL-10 can directly impact the development of memory cells by inducing MPEC populations in a STAT3 dependent manner (54), however, it is unclear whether IL-10 has any direct consequences on the development of TRM.
A variety of other cytokines produced after influenza infection is known to modulate anti-viral CD8+ T cell responses. Thymic stromal lymphopoietin (TSLP), an epithelial derived cytokine that can be produced in the infected lung (55, 56), promotes expansion of the CD8+ T cells at the site of infection directly (56) and indirectly via CD11b+ inflammatory DCs (57). Additionally, transpresentation of IL-15 by pulmonary DCs has been shown to increase the survival of Teffs (58) and is an important component of TRM development in the skin (14). However, IL-15 does not seem necessary for the overall development of memory in the lungs or the airways following influenza infection (42), although this study as well as the TSLP studies did not address TRM populations specifically.
Localization as an important step in development of TRM cells
As residence at the peripheral site is a requisite for TRM development, cells destined to become TRM cells must first gain access into peripheral tissues, often into physically restricted areas such as within an epithelial layer or closely associated with the underlying basil lamina (within the parenchyma). The route of migration used by T cells trafficking to the lung, however, is not well understood. Cells can enter the lung via two circulatory systems: the bronchial system, which provides oxygenated blood to the lung tissue, and the pulmonary circulation, which includes vessels that bring deoxygenated blood to alveoli and subsequently drain oxygenated blood back to the heart (59). The lung epithelium surrounding the airway spaces share a fused basal lamina with the adjacent capillary endothelium to allow gas exchange and could facilitate direct blood to airway traffic. Because pulmonary vessels are small in diameter and thin walled, blood pressure in these vessels is relatively low, thus allowing lymphocytes to traverse the endothelium independent of the multistep paradigm described for lymphocyte migration through larger vessels, which are dependent on selectins, integrins, and chemokines (60). However, histological sections of lung tissues depict memory cells localized close to the airways, but within the lung parenchyma, evoking a blood → lung → airway route (19, 61).
Broadly speaking, activated CD8+ Teffs cells can gain access to peripheral sites by virtue of their expression of CD11a and CD44 with concomitant loss of CD62L expression on their cell surface (60, 62).While access into distinct anatomical sites within other mucosal tissues such as the skin and gut is highly correlated with expression of tissue-specific homing receptors (63–65), analogous molecules have not yet been identified for lung homing CD8+ T cells. Nonetheless, some chemotactic signals are associated with Teff migration into inflamed lung tissues including CXCR3 (66) and CXCR6 (67). CXCR6 is specifically up-regulated on CD8+ T cells isolated from the lung and lung airways following intranasal immunization and mice lacking CXCR6 have reduced protection against tuberculosis challenge (67), indicating that CXCR6 expression may be important for the establishment of CD8+ T cells at sites of protection. The expression of CXCR3 is important to establish migration of CD8+ T cells specifically to the airways (68). While TRM populations were not assessed in this study, CXCR3− antigen-specific CD8+ T cells isolated from the lung expressed lower levels of CD69 than WT cells occupying the airways where antigen is present. CD69 expression is upregulated on TRM populations, and contact with antigen has been suggested to be necessary for TRM formation (8). Therefore, expression of CXCR3 may be a requirement for the development of TRM cells in the lungs, akin to the requirement for CXCR3 in the skin (14).
As influenza virus replicates primarily in epithelial tissue, the localization of CD8+ T cells adjacent to antigen may expose them to unique cytokines available in and near the epithelium such as TGF-β. TGF-β plays a role in both the contraction of effector T cells (69) and the establishment of TRM cells by inducing the expression of CD103 (70). The role of TGF-β in the development of TRM cells has been well described in the intestinal mucosa and the skin, and has also been implicated in the development of TRM in the lung (71). Although TGF-β can be transiently activated by influenza virus (72, 73), it likely has lower constitutive production in the lung than other barrier sites as over-expression of TGF-β can promote pulmonary fibrosis and lung disease (74). Due to the localization of TGF-β production, CD103 expression may be specific to only those cells, which are found within epithelial layers and not necessary for TRM in the lung parenchyma, a concept discussed later in more detail. Interestingly, following influenza infection a large majority of antigen-specific CD8+ T cells begin to express the α1β1 integrin VLA-1 (61). Teffs localized cells to the collagen-rich areas near the airways and basement membranes that are VLA-1+ have a survival advantage over those that do not express VLA-1 at the peak of the CD8+ T cell response (61). The localization and retention of cells within the lung parenchyma, as well as the survival advantage may make VLA-1 expression a unique marker for cells destined to become lung TRM cells. However, this possibility has yet to be explored.
Part II: Characteristics and Maintenance of Committed CD8+ TRM in the Lung
Following the resolution from infection, antigen-specific CD8+ T cells will persist at the site of infection (19). As previously noted, these Tmem cells exist in the lung in two basic compartments, the airways and the lung parenchyma. Airway CD8+ T cells exist outside of the body, within the lumen of the respiratory tract, or they can exist much like they do in the intestinal epithelium as intraepithelial cells. Cells within the airways, and very likely some intraepithelial cells, can be isolated by performing a bronchoalveolar lavage (BAL), while the remaining parenchyma cells can be isolated through a process involving the enzymatic digestion of collagen. Additionally the localization and characterization of these cell populations can be defined by microscopic analysis of lung tissue sections, although phenotyping cells by this method is limited. It is important to distinguish between these two populations of cells in the discussion of TRM, as airway cells are likely comprised of both true TRM cells and circulating Tmem, which migrate to the airways following the resolution of infection.
Cells in the airways are subject to the external environment of the lung, where mucous and pulmonary surfactants decrease the potential for their long-term persistence. Therefore, it is thought that memory CD8+ T cells in the lung airways, at least for some period of time, are partially maintained by the continual recruitment to the airways. In support of this, Slutter et al. showed that CXCR3 is required for the continual recruitment of cells into the airways, and that loss of CXCR3 expression results in the accelerated loss of antigen-specific CD8+ T cells specifically from the airways (40). Tracking the entry of Tmem from the circulation is also possible by monitoring CD11a expression, which is lost ~40 h after CD8+ T cell emigration into the airways (75). Indeed, when Tmem are extracted from the airways (up until at least 13 months post infection), portions of the antigen-specific CD8+ T cells express high levels of CD11a. Together, these data confirm that at least a proportion of airway CD8+ cells may not be bona fide TRM based on presence within this site alone. In support of this argument, CD103 expression is reduced on antigen-specific Tmem isolated from the airway when compared to Tmem isolated from the lung parenchyma both in terms of frequency (11) and on a per cell basis (76). Finally, while evidence suggests that a circulating population of cells is actively recruited into the lung airways during steady state conditions (40, 75) it is clear that these recruits are not sufficient (either in number or function) to provide protection against heterosubtypic influenza challenge, as protection wanes while recruitment continues. Perhaps the limited migration and supplementation of competent Tmem cells from within the lung parenchyma may augment this pool and maintain heterosubtypic immunity, at least temporarily. However, cell tracking studies have not confirmed this possibility.
Tmem also exist in the respiratory tract within the lung tissue or parenchyma. As the lung is a highly vascularized organ, it can be difficult to discern at time of tissue harvest, which antigen-specific cells are trafficking through the vasculature of the lung (trapped within small capillaries) and which are truly within the parenchyma. Experiments using intravascular staining whereby antibodies are injected directly into the blood stream immediately before the lungs are examined to “tag” circulating cells demonstrated a large number of cells isolated from the lung tissue are circulating cells (naïve or TEM) despite perfusion. This method has been useful in characterizing both CD4+ (77, 78) and CD8+ (11) TRM cells in direct contrast to the circulating pool. Using this method to distinguish circulating vs. resident cells has, and will, continue to provide a clearer picture of what TRM cells look like in the resting lung.
Microscopic analysis of lung tissue sections has also been useful in determining the precise localization of TRM in the respiratory tract to gain better insight regarding the cellular associations and tissue microarchitecture, which may be important for supporting TRM development and/or survival. Turner et al. showed that CD4+ TRM cells established following influenza infection were clustered together in the lungs, in regions both close to the airways and to the pulmonary blood vessels (78). This would position the cells in an ideal place to encounter antigen entering the body. The clustering of cells in this location is not a new observation, nor is it exclusive for the CD4+ T cell population. In 2004, Ray et al. showed that influenza specific CD8+ T cells persisted in the highly collagenized area between the airways and the blood vessels, and that this retention was dependent on the expression of VLA-1 (61). VLA-1 binds to type IV and type I collagen (79, 80), which are important structural components of the lung interstitium, specifically between the bronchi and the vasculature, and the basement membranes of both the pulmonary vasculature and the epithelium of the airway, respectively (81, 82). The co-localization of TRM and collagen below the epidermal cell layer of the airways shows that TRM cells also exist within the lung parenchyma. The collagen-rich environment of the lung may provide a framework or scaffold in which TRM cells can persist close to the site of antigen acquisition, yet not actually within the epithelial layer of the lung where they may be subject to the harsh environment of the airways. Additionally, it is quite possible that this collagen matrix could also trap or capture soluble growth factors important for TRM maintenance.
The Persistence of TRM Cells in the Respiratory Tract: Role of the Lung Environment
Like other mucosal barrier sites, the resting lung is engaged in a constant balancing act regarding immunity and tolerance. It is estimated that we breathe in 10,000 l of air per day, with each breath containing a plethora of allergens, environmental pollutants, and pathogens. Inappropriate response to non-harmful antigens could lead to persistent inflammation and pulmonary disease. To prevent this, multiple layers of innate protection exist in the lung to preclude any inappropriate initiation of an immune response. The most basic of these is the mucosal barrier itself. The lining of the upper respiratory tract is composed of ciliated epithelial cells and mucus-secreting goblet cells, which together function as a “mucociliary escalator” facilitating expulsion of these innocuous agents, as well as some commensal organisms, out of the respiratory tract without activation of the adaptive immune response. However, the mucus would also prevent TRM cells from persisting in the airways of the upper respiratory tract, leading to the accumulation of TRM either within the epithelium, the parenchyma, or in the airways of the lower respiratory tract. While the lower respiratory tract does not contain mucous, it is characterized by numerous “pockets” where gas exchange occurs termed alveoli. The cells lining the alveoli are specialized epithelial cells known as type I and type II alveolar epithelial cells, which form the structural architecture of the alveoli and secrete immunosuppressive pulmonary surfactants, respectively (83). The role that these lung derived factors may play on CD8+ T cells at the site is further complicated by conditions of an inflamed lung, such as asthma and allergy. Allergens can induce the upregulation of pulmonary surfactants, which in turn can protect against allergic disease via local IL-13 inhibition (84). Due to the proximity of surfactants and TRM cells in the lower respiratory tract, and the essential role for surfactants in regulating respiratory inflammation, it is possible that TRM persistence could be dynamically regulated by perturbation in surfactant (and mucus) activity. However, this has not been analyzed.
TRM persist long-term in many non-lymphoid tissues, albeit with different kinetics. For example, VSV-specific TRM cells exist as long as 120 dpi in the brain (8) while cutaneous herpes simplex virus TRM cells persist for the lifetime of a mouse (85). This is shown to occur independently of increased proliferation (8, 15) and maintained populations are not dependent on replenishment from lymphoid organs (6, 78). Perhaps somewhat unique to the respiratory tract is that Tmem cells within this site appear to have a limited life-span, steadily decreasing over time (19). The lack of long-term survival of Tmem cells lung airways, and perhaps certain populations in the lung itself, has functional consequences since heterosubtypic immunity against influenza viruses is lost ~4–6 months post infection (18). Moreover, this loss of anti-influenza immunity is coordinate with substantial loss in CD8+ Tmem cells of the airways, despite stable numbers in the spleen (19) and the continual recruitment of cells from the circulation into the airways (40, 75). While these former studies did not directly assess the role of TRM cells, recent evidence suggests that protective heterosubtypic immunity against influenza infection is mediated solely by TRM, as the ability to control viral titers and protect from severe disease is gradually lost along with TRM cells in the airways (20). Yet, the question of why TRM cells do not persist in the lung and lung airways to the extent that they do in other tissues remains unanswered. Interestingly, following influenza infection lung TRM cells retain expression of interferon-induced transmembrane protein IFITM3, which imparts cells with a survival advantage in the face of viral infection (76). This increased survival mechanism may be particularly important at this site, due to the regularity at which respiratory infections are acquired. The unique properties of respiratory TRM cells have provided some insight into why their persistence in the lungs is limited (Table 1).
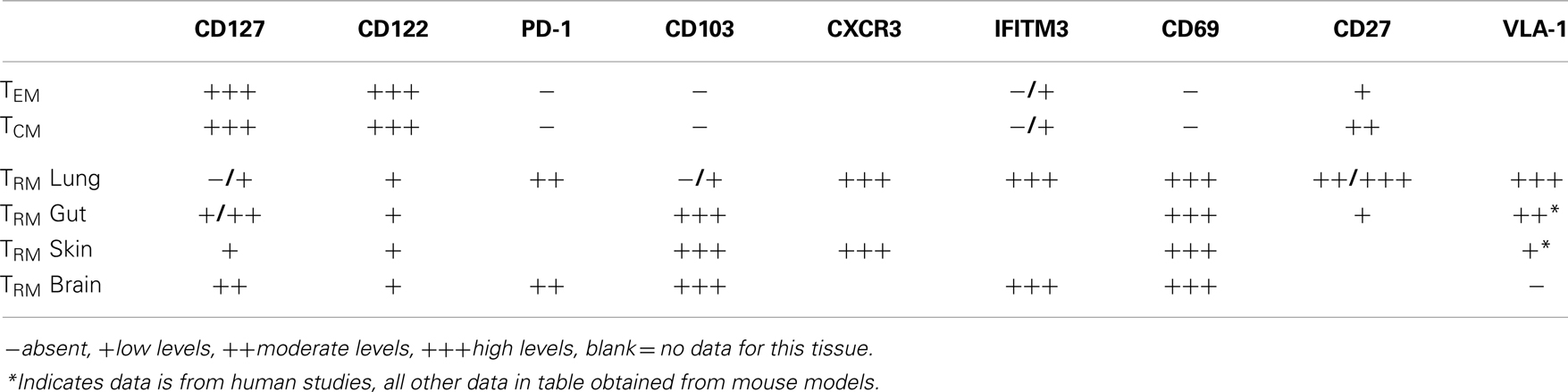
Table 1. Factors associated with the positioning and survival of defined pools of memory CD8+ T cells in specific anatomical sites.
The cytokines IL-7 and IL-15 are requisite for the development and maintenance of memory CD8+ T cells after systemic infection (35, 86). However, what role, if any, these cytokines play in the maintenance of TRM cells in the lung has not been defined. In most sites assessed to date, TRM cells express reduced levels of CD127, as compared to TCM and TEM cells. Concurrently, CD8+ T cells in the lung airways express reduced levels of CD127 (11, 87, 88) as do cells in the lung parenchyma, although to a lesser extent (56). Like CD11a, it is possible that CD127 is cleaved from CD8+ T cells in the airways, leaving these cells incapable of receiving proliferative or survival signals, either from IL-7 or from TSLP, which has been shown to be produced constitutively in the gut (89), and in the lung during both resting conditions and after inflammatory stimuli (56). IL-15 has been shown to be dispensable for the development and maintenance of memory cells that develop from respiratory infections and CD122 is lost from CD8+ T cells within the respiratory tract (87). Furthermore, CD122 or the beta chain of the IL-15R, which signals to memory CD8+ T cells is expressed at lower levels on TRM isolated from the epithelium of the small intestine (90). A recently described pool of TRM isolated from secondary lymphoid organs are maintained independently of IL-15 and even found in increased numbers in mice lacking IL-15 (91). Therefore, IL-15 appears to be uniformly dispensable for the maintenance of TRM cells, and while levels of CD127 on TRM cells is more variable, the near complete loss of this receptor in the respiratory tract may provide one mechanism in which CD8+ T cells at this site have decreased sustainability. However, it should be noted that TRM cells from the brain do not respond to IL-7 or IL-15 ex vivo, unlike splenic memory cells, which show increased survival upon exposure to these cytokines (16), indicating that perhaps the survival of TRM cells is completely independent of classical cytokine memory signals.
The maintenance of CD8+ T cells in the lungs has also been attributed to residual antigen found in the MdLN for ~2 months post influenza infection (92). Influenza antigens have also been detected in the lung tissue itself for 30 days within focal inflammatory structures (93), reminiscent of inducible bronchus associated lymphoid tissue (iBALT). iBALT develops following influenza infection and has similar structure to lymph node tissue, such as defined B cell follicles and the formation of germinal centers surrounding DCs; this structure contributes to the proliferation of B and T cells during primary influenza infection and can be protective in mice where other lymphoid organs are lacking (94). As the timing of loss of residual antigen coincides with the loss of protective heterosubtypic immunity, it has been hypothesized that antigen is necessary for the persistence of Tmem in the lung and lung airways. In support of this possibility, TRM cells in the lung express PD-1 (20), which may indicate continued exposure to antigen. While certain TRM populations have been shown to persist in the absence of antigen (8, 31) definitive studies have not been carried out for TRM cells in the lung to rule this out as a mechanism for maintenance.
It is likely in humans that the maintenance and survival of TRM cells may be much different than what is observed in mice. As previously mentioned, constant antigenic stimulation, allergic inflammation, and relatively common airway disorders such as asthma will influence the lung environment in ways that will affect many indigenous respiratory cells. In addition, the regularity of respiratory infections in humans will result in the accumulation of many pools of clonally diverse antigen-specific cells, recognizing a plethora of pathogens. de Bree et al. showed that influenza and respiratory syncytial virus-specific CD8+ T cells were enriched in the human lung compared to the circulation (95). In direct contrast, antigen-specific CD8+ T cells that developed from the blood-borne pathogens cytomegalovirus and Epstein-Barr virus equilibrated between the blood and lung of these patients (95). The accumulation of CD8+ T cells in the lung due to respiratory infection would certainly lead to large numbers of TRM cells populating the human lung during steady state conditions. Indeed, studies have determined that CD103+αβ TCR CD8+ T cells comprise about 1/3 of the total CD8+ T cell population in the human lung (96), or over 10 billion total cells (97). However, the history of human lung TRM (when developed/how long maintained) and how the history of individual clones correlates with acquisition of specific infections is difficult to determine. Furthermore, in humans, the survival of these pools may be affected by attrition resulting from heterologous infections. In these scenarios, either competition for resources in distinct environmental niches or by bystander apoptosis via cytotoxic factors present at the time of the new viral infection may deplete previously existent TRM pools (98).
Concluding Remarks
The study of TRM cells is in its infancy. As we continue to analyze this unique lineage of memory cells, we will certainly deepen our understanding of TRM biology in unique sites such as the respiratory tract and perhaps better understand how to selectively manipulate this pool for development of vaccines. While the defining characteristic of what makes a cell a TRM cell is quite clear (i.e., long-term residence at a site), some of the markers currently used to distinguish TRM cells, most notably CD103, only recognize a subset of TRM cells localized to the (respiratory) epithelium. This leaves a large population (anywhere from 50 to 90% of TRM cells in the lung) excluded from studies. Thus, overall TRM frequency can only be confirmed using complicated transfer and cell tracking experiments, warranting the need for more definitive phenotypic markers to readily identify TRM. Moreover, understanding the environment in which TRM cells at specific sites reside will be key to developing phenotypic definitions of these cells, as markers vary between anatomical locations. In the case of the lung, this particular environment has many mechanisms in place to suppress inflammation and any inadvertent immunopathology. Thus, while higher numbers of TRM cells at the site of infection may be ideal for protection against disease, tight regulation of the number, and longevity of TRM cells at this site may be essential for tissue function. This may be especially relevant in the context of human disease, where respiratory infections are commonplace and populations of TRM are not only numerically enhanced but very likely dynamically regulated.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by the National Institutes of Health (AI081800 to Kimberly D. Klonowski)
References
1. Woodland DL, Hogan RJ, Zhong W. Cellular immunity and memory to respiratory virus infections. Immunol Res (2001) 24(1):53–67. doi: 10.1385/IR:24:1:53
2. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401(6754):708–12. doi:10.1038/44385
3. Pham TH, Okada T, Matloubian M, Lo CG, Cyster JG. S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity (2008) 28(1):122–33. doi:10.1016/j.immuni.2007.11.017
4. Klonowski KD, Williams KJ, Marzo AL, Blair DA, Lingenheld EG, Lefrancois L. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity (2004) 20(5):551–62. doi:10.1016/S1074-7613(04)00103-7
5. Masopust D, Vezys V, Usherwood EJ, Cauley LS, Olson S, Marzo AL, et al. Activated primary and memory CD8 T cells migrate to nonlymphoid tissues regardless of site of activation or tissue of origin. J Immunol (2004) 172(8):4875–82. doi:10.4049/jimmunol.172.8.4875
6. Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med (2010) 207(3):553–64. doi:10.1084/jem.20090858
7. Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature (2012) 483(7388):227–31. doi:10.1038/nature10851
8. Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A (2010) 107(42):17872–9. doi:10.1073/pnas.1010201107
9. Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature (2012) 491(7424):463–7. doi:10.1038/nature11522
10. Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A (2012) 109(18):7037–42. doi:10.1073/pnas.1202288109
11. Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, et al. Cutting edge: intravascular staining redefines lung CD8 T cell responses. J Immunol (2012) 189(6):2702–6. doi:10.4049/jimmunol.1201682
12. Ziegler SF, Ramsdell F, Alderson MR. The activation antigen CD69. Stem Cells (1994) 12(5):456–65. doi:10.1002/stem.5530120502
13. Schon MP, Arya A, Murphy EA, Adams CM, Strauch UG, Agace WW, et al. Mucosal T lymphocyte numbers are selectively reduced in integrin alpha E (CD103)-deficient mice. J Immunol (1999) 162(11):6641–9.
14. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol (2013) 14(12):1294–301. doi:10.1038/ni.2744
15. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol (2009) 10(5):524–30. doi:10.1038/ni.1718
16. Wakim LM, Woodward-Davis A, Liu R, Hu Y, Villadangos J, Smyth G, et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol (2012) 189(7):3462–71. doi:10.4049/jimmunol.1201305
17. Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, Lefrancois L. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity (2014) 40(5):747–57. doi:10.1016/j.immuni.2014.03.007
18. Liang S, Mozdzanowska K, Palladino G, Gerhard W. Heterosubtypic immunity to influenza type A virus in mice. Effector mechanisms and their longevity. J Immunol (1994) 152(4):1653–61.
19. Hogan RJ, Usherwood EJ, Zhong W, Roberts AA, Dutton RW, Harmsen AG, et al. Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J Immunol (2001) 166(3):1813–22. doi:10.4049/jimmunol.166.3.1813
20. Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol (2014) 95(2):215–24. doi:10.1189/jlb.0313180
21. Obar JJ, Lefrancois L. Early events governing memory CD8+ T-cell differentiation. Int Immunol (2010) 22(8):619–25. doi:10.1093/intimm/dxq053
22. Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, et al. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev (2006) 211:81–92. doi:10.1111/j.0105-2896.2006.00382.x
23. Youngblood B, Hale JS, Ahmed R. T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology (2013) 139(3):277–84. doi:10.1111/imm.12074
24. Kim TS, Braciale TJ. Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS One (2009) 4(1):e4204. doi:10.1371/journal.pone.0004204
25. Kim TS, Gorski SA, Hahn S, Murphy KM, Braciale TJ. Distinct dendritic cell subsets dictate the fate decision between effector and memory CD8(+) T cell differentiation by a CD24-dependent mechanism. Immunity (2014) 40(3):400–13. doi:10.1016/j.immuni.2014.02.004
26. del Rio ML, Bernhardt G, Rodriguez-Barbosa JI, Forster R. Development and functional specialization of CD103+ dendritic cells. Immunol Rev (2010) 234(1):268–81. doi:10.1111/j.0105-2896.2009.00874.x
27. Gallichan WS, Rosenthal KL. Long-lived cytotoxic T lymphocyte memory in mucosal tissues after mucosal but not systemic immunization. J Exp Med (1996) 184(5):1879–90. doi:10.1084/jem.184.5.1879
28. Ferko B, Stasakova J, Sereinig S, Romanova J, Katinger D, Niebler B, et al. Hyperattenuated recombinant influenza A virus nonstructural-protein-encoding vectors induce human immunodeficiency virus type 1 Nef-specific systemic and mucosal immune responses in mice. J Virol (2001) 75(19):8899–908. doi:10.1128/JVI.75.19.8899-8908.2001
29. Gherardi MM, Perez-Jimenez E, Najera JL, Esteban M. Induction of HIV immunity in the genital tract after intranasal delivery of a MVA vector: enhanced immunogenicity after DNA prime-modified vaccinia virus Ankara boost immunization schedule. J Immunol (2004) 172(10):6209–20. doi:10.4049/jimmunol.172.10.6209
30. Ruane D, Brane L, Reis BS, Cheong C, Poles J, Do Y, et al. Lung dendritic cells induce migration of protective T cells to the gastrointestinal tract. J Exp Med (2013) 210(9):1871–88. doi:10.1084/jem.20122762
31. Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol (2012) 188(10):4866–75. doi:10.4049/jimmunol.1200402
32. Venturi V, Davenport MP, Swan NG, Doherty PC, Kedzierska K. Consequences of suboptimal priming are apparent for low-avidity T-cell responses. Immunol Cell Biol (2012) 90(2):216–23. doi:10.1038/icb.2011.36
33. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity (2007) 27(2):281–95. doi:10.1016/j.immuni.2007.07.010
34. Cui W, Kaech SM. Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol Rev (2010) 236:151–66. doi:10.1111/j.1600-065X.2010.00926.x
35. Schluns KS, Lefrancois L. Cytokine control of memory T-cell development and survival. Nat Rev Immunol (2003) 3(4):269–79. doi:10.1038/nri1052
36. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol (2003) 4(12):1191–8. doi:10.1038/ni1009
37. Obar JJ, Jellison ER, Sheridan BS, Blair DA, Pham QM, Zickovich JM, et al. Pathogen-induced inflammatory environment controls effector and memory CD8+ T cell differentiation. J Immunol (2011) 187(10):4967–78. doi:10.4049/jimmunol.1102335
38. Monteiro JM, Harvey C, Trinchieri G. Role of interleukin-12 in primary influenza virus infection. J Virol (1998) 72(6):4825–31.
39. Fujimoto K, Karuppuchamy T, Takemura N, Shimohigoshi M, Machida T, Haseda Y, et al. A new subset of CD103+CD8alpha+ dendritic cells in the small intestine expresses TLR3, TLR7, and TLR9 and induces Th1 response and CTL activity. J Immunol (2011) 186(11):6287–95. doi:10.4049/jimmunol.1004036
40. Slutter B, Pewe LL, Kaech SM, Harty JT. Lung airway-surveilling CXCR3(hi) memory CD8(+) T cells are critical for protection against influenza A virus. Immunity (2013) 39(5):939–48. doi:10.1016/j.immuni.2013.09.013
41. Obar JJ, Lefrancois L. Early signals during CD8 T cell priming regulate the generation of central memory cells. J Immunol (2010) 185(1):263–72. doi:10.4049/jimmunol.1000492
42. Verbist KC, Field MB, Klonowski KD. Cutting edge: IL-15-independent maintenance of mucosally generated memory CD8 T cells. J Immunol (2011) 186(12):6667–71. doi:10.4049/jimmunol.1004022
43. Schluns KS, Williams K, Ma A, Zheng XX, Lefrancois L. Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J Immunol (2002) 168(10):4827–31. doi:10.4049/jimmunol.168.10.4827
44. Bedoui S, Gebhardt T. Interaction between dendritic cells and T cells during peripheral virus infections: a role for antigen presentation beyond lymphoid organs? Curr Opin Immunol (2011) 23(1):124–30. doi:10.1016/j.coi.2010.11.001
45. McGill J, Legge KL. Cutting edge: contribution of lung-resident T cell proliferation to the overall magnitude of the antigen-specific CD8 T cell response in the lungs following murine influenza virus infection. J Immunol (2009) 183(7):4177–81. doi:10.4049/jimmunol.0901109
46. Fraser KA, Schenkel JM, Jameson SC, Vezys V, Masopust D. Preexisting high frequencies of memory CD8+ T cells favor rapid memory differentiation and preservation of proliferative potential upon boosting. Immunity (2013) 39(1):171–83. doi:10.1016/j.immuni.2013.07.003
47. Cui W, Joshi NS, Jiang A, Kaech SM. Effects of Signal 3 during CD8 T cell priming: bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine (2009) 27(15):2177–87. doi:10.1016/j.vaccine.2009.01.088
48. Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity (2010) 32(1):91–103. doi:10.1016/j.immuni.2009.11.010
49. Kohlmeier JE, Woodland DL. Immunity to respiratory viruses. Annu Rev Immunol (2009) 27:61–82. doi:10.1146/annurev.immunol.021908.132625
50. Sun J, Madan R, Karp CL, Braciale TJ. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med (2009) 15(3):277–84. doi:10.1038/nm.1929
51. Palmer EM, Holbrook BC, Arimilli S, Parks GD, Alexander-Miller MA. IFNgamma-producing, virus-specific CD8+ effector cells acquire the ability to produce IL-10 as a result of entry into the infected lung environment. Virology (2010) 404(2):225–30. doi:10.1016/j.virol.2010.05.004
52. Sun J, Dodd H, Moser EK, Sharma R, Braciale TJ. CD4+ T cell help and innate-derived IL-27 induce Blimp-1-dependent IL-10 production by antiviral CTLs. Nat Immunol (2011) 12(4):327–34. doi:10.1038/ni.1996
53. Bedoya F, Cheng GS, Leibow A, Zakhary N, Weissler K, Garcia V, et al. Viral antigen induces differentiation of Foxp3+ natural regulatory T cells in influenza virus-infected mice. J Immunol (2013) 190(12):6115–25. doi:10.4049/jimmunol.1203302
54. Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity (2011) 35(5):792–805. doi:10.1016/j.immuni.2011.09.017
55. Plumb AW, Patton DT, Seo JH, Loveday EK, Jean F, Ziegler SF, et al. Interleukin-7, but not thymic stromal lymphopoietin, plays a key role in the T cell response to influenza A virus. PLoS One (2012) 7(11):e50199. doi:10.1371/journal.pone.0050199
56. Shane HL, Klonowski KD. A direct and nonredundant role for thymic stromal lymphopoietin on antiviral CD8 T cell responses in the respiratory mucosa. J Immunol (2014) 192(5):2261–70. doi:10.4049/jimmunol.1302085
57. Yadava K, Sichelstiel A, Luescher IF, Nicod LP, Harris NL, Marsland BJ. TSLP promotes influenza-specific CD8+ T-cell responses by augmenting local inflammatory dendritic cell function. Mucosal Immunol (2013) 6(1):83–92. doi:10.1038/mi.2012.50
58. McGill J, Van Rooijen N, Legge KL. IL-15 trans-presentation by pulmonary dendritic cells promotes effector CD8 T cell survival during influenza virus infection. J Exp Med (2010) 207(3):521–34. doi:10.1084/jem.20091711
59. Gray H, Standring S, Ellis H, Berkovitz BKB. Gray’s Anatomy: the Anatomical Basis of Clinical Practice. Edinburgh (2005).
60. Miyasaka M, Tanaka T. Lymphocyte trafficking across high endothelial venules: dogmas and enigmas. Nat Rev Immunol (2004) 4(5):360–70. doi:10.1038/nri1354
61. Ray SJ, Franki SN, Pierce RH, Dimitrova S, Koteliansky V, Sprague AG, et al. The collagen binding alpha1beta1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity (2004) 20(2):167–79. doi:10.1016/S1074-7613(04)00021-4
62. Shin H, Iwasaki A. Tissue-resident memory T cells. Immunol Rev (2013) 255(1):165–81. doi:10.1111/imr.12087
63. Masopust D, Jiang J, Shen H, Lefrancois L. Direct analysis of the dynamics of the intestinal mucosa CD8 T cell response to systemic virus infection. J Immunol (2001) 166(4):2348–56. doi:10.4049/jimmunol.166.4.2348
64. Lefrancois L, Parker CM, Olson S, Muller W, Wagner N, Schon MP, et al. The role of beta7 integrins in CD8 T cell trafficking during an antiviral immune response. J Exp Med (1999) 189(10):1631–8. doi:10.1084/jem.189.10.1631
65. Svensson M, Marsal J, Ericsson A, Carramolino L, Broden T, Marquez G, et al. CCL25 mediates the localization of recently activated CD8alphabeta(+) lymphocytes to the small-intestinal mucosa. J Clin Invest (2002) 110(8):1113–21. doi:10.1172/JCI0215988
66. Fadel SA, Bromley SK, Medoff BD, Luster AD. CXCR3-deficiency protects influenza-infected CCR5-deficient mice from mortality. Eur J Immunol (2008) 38(12):3376–87. doi:10.1002/eji.200838628
67. Lee LN, Ronan EO, de Lara C, Franken KL, Ottenhoff TH, Tchilian EZ, et al. CXCR6 is a marker for protective antigen-specific cells in the lungs after intranasal immunization against Mycobacterium tuberculosis. Infect Immun (2011) 79(8):3328–37. doi:10.1128/IAI.01133-10
68. Kohlmeier JE, Reiley WW, Perona-Wright G, Freeman ML, Yager EJ, Connor LM, et al. Inflammatory chemokine receptors regulate CD8(+) T cell contraction and memory generation following infection. J Exp Med (2011) 208(8):1621–34. doi:10.1084/jem.20102110
69. Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity (2009) 31(1):131–44. doi:10.1016/j.immuni.2009.04.020
70. Hu Y, Cauley L. Antigen and transforming growth factor Beta receptors contribute to long term functional and phenotypic heterogeneity of memory CD8 T cells. Front Immunol (2013) 4:227. doi:10.3389/fimmu.2013.00227
71. Sheridan BS, Lefrancois L. Regional and mucosal memory T cells. Nat Immunol (2011) 12(6):485–91. doi:10.1038/ni.2029
72. Carlson CM, Turpin EA, Moser LA, O’Brien KB, Cline TD, Jones JC, et al. Transforming growth factor-beta: activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog (2010) 6(10):e1001136. doi:10.1371/journal.ppat.1001136
73. Schultz-Cherry S, Hinshaw VS. Influenza virus neuraminidase activates latent transforming growth factor beta. J Virol (1996) 70(12):8624–9.
74. Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol (2009) 2(2):103–21. doi:10.1038/mi.2008.85
75. Ely KH, Cookenham T, Roberts AD, Woodland DL. Memory T cell populations in the lung airways are maintained by continual recruitment. J Immunol (2006) 176(1):537–43. doi:10.4049/jimmunol.176.1.537
76. Wakim LM, Gupta N, Mintern JD, Villadangos JA. Enhanced survival of lung tissue-resident memory CD8(+) T cells during infection with influenza virus due to selective expression of IFITM3. Nat Immunol (2013) 14(3):238–45. doi:10.1038/ni.2525
77. Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL. Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol (2011) 187(11):5510–4. doi:10.4049/jimmunol.1102243
78. Turner DL, Bickham KL, Thome JJ, Kim CY, D’Ovidio F, Wherry EJ, et al. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol (2014) 7(3):501–10. doi:10.1038/mi.2013.67
79. Belkin VM, Belkin AM, Koteliansky VE. Human smooth muscle VLA-1 integrin: purification, substrate specificity, localization in aorta, and expression during development. J Cell Biol (1990) 111(5 Pt 1):2159–70. doi:10.1083/jcb.111.5.2159
80. Hemler ME. VLA proteins in the integrin family: structures, functions, and their role on leukocytes. Annu Rev Immunol (1990) 8:365–400. doi:10.1146/annurev.iy.08.040190.002053
81. Miner JH, Sanes JR. Collagen IV alpha 3, alpha 4, and alpha 5 chains in rodent basal laminae: sequence, distribution, association with laminins, and developmental switches. J Cell Biol (1994) 127(3):879–91. doi:10.1083/jcb.127.3.879
82. Sado Y, Kagawa M, Naito I, Ueki Y, Seki T, Momota R, et al. Organization and expression of basement membrane collagen IV genes and their roles in human disorders. J Biochem (1998) 123(5):767–76. doi:10.1093/oxfordjournals.jbchem.a022003
83. Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol (2005) 5(1):58–68. doi:10.1038/nri1528
84. Qaseem AS, Sonar S, Mahajan L, Madan T, Sorensen GL, Shamji MH, et al. Linking surfactant protein SP-D and IL-13: implications in asthma and allergy. Mol Immunol (2013) 54(1):98–107. doi:10.1016/j.molimm.2012.10.039
85. Zaid A, Mackay LK, Rahimpour A, Braun A, Veldhoen M, Carbone FR, et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci U S A (2014) 111(14):5307–12. doi:10.1073/pnas.1322292111
86. Verbist KC, Klonowski KD. Functions of IL-15 in anti-viral immunity: multiplicity and variety. Cytokine (2012) 59(3):467–78. doi:10.1016/j.cyto.2012.05.020
87. Shen CH, Ge Q, Talay O, Eisen HN, Garcia-Sastre A, Chen J. Loss of IL-7R and IL-15R expression is associated with disappearance of memory T cells in respiratory tract following influenza infection. J Immunol (2008) 180(1):171–8. doi:10.4049/jimmunol.180.1.171
88. Kohlmeier JE, Miller SC, Woodland DL. Cutting edge: antigen is not required for the activation and maintenance of virus-specific memory CD8+ T cells in the lung airways. J Immunol (2007) 178(8):4721–5. doi:10.4049/jimmunol.178.8.4721
89. Taylor BC, Zaph C, Troy AE, Du Y, Guild KJ, Comeau MR, et al. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J Exp Med (2009) 206(3):655–67. doi:10.1084/jem.20081499
90. Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol (2006) 176(4):2079–83. doi:10.4049/jimmunol.176.4.2079
91. Schenkel JM, Fraser KA, Masopust D. Cutting edge: resident memory CD8 T cells occupy frontline niches in secondary lymphoid organs. J Immunol (2014) 192(7):2961–4. doi:10.4049/jimmunol.1400003
92. Zammit DJ, Turner DL, Klonowski KD, Lefrancois L, Cauley LS. Residual antigen presentation after influenza virus infection affects CD8 T cell activation and migration. Immunity (2006) 24(4):439–49. doi:10.1016/j.immuni.2006.01.015
93. Kim TS, Hufford MM, Sun J, Fu YX, Braciale TJ. Antigen persistence and the control of local T cell memory by migrant respiratory dendritic cells after acute virus infection. J Exp Med (2010) 207(6):1161–72. doi:10.1084/jem.20092017
94. Moyron-Quiroz JE, Rangel-Moreno J, Kusser K, Hartson L, Sprague F, Goodrich S, et al. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat Med (2004) 10(9):927–34. doi:10.1038/nm1091
95. de Bree GJ, van Leeuwen EM, Out TA, Jansen HM, Jonkers RE, van Lier RA. Selective accumulation of differentiated CD8+ T cells specific for respiratory viruses in the human lung. J Exp Med (2005) 202(10):1433–42. doi:10.1084/jem.20051365
96. Piet B, de Bree GJ, Smids-Dierdorp BS, van der Loos CM, Remmerswaal EB, von der Thusen JH, et al. CD8(+) T cells with an intraepithelial phenotype upregulate cytotoxic function upon influenza infection in human lung. J Clin Invest (2011) 121(6):2254–63. doi:10.1172/JCI44675
97. Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS. Resident memory T cells (T(RM)) are abundant in human lung: diversity, function, and antigen specificity. PLoS One (2011) 6(1):e16245. doi:10.1371/journal.pone.0016245
Keywords: CD8+ T cells, memory T cells, tissue-resident memory cells, influenza A virus, lung
Citation: Shane HL and Klonowski KD (2014) Every breath you take: the impact of environment on resident memory CD8 T cells in the lung. Front. Immunol. 5:320. doi: 10.3389/fimmu.2014.00320
Received: 03 June 2014; Accepted: 24 June 2014;
Published online: 08 July 2014.
Edited by:
Kimberly Sue Schluns, University of Texas MD Anderson Cancer Center, USAReviewed by:
Karl Kai McKinstry, University of Massachusetts Medical School, USAJacob Kohlmeier, Emory University, USA
Copyright: © 2014 Shane and Klonowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kimberly D. Klonowski, Department of Cellular Biology, University of Georgia, 724 Biological Sciences Building, Athens, GA 30602-2607, USA e-mail:a2xvbm93c2tAdWdhLmVkdQ==