 Cheri L. Lubahn1†
Cheri L. Lubahn1† Dianne Lorton
Dianne Lorton Denise L. Bellinger
Denise L. Bellinger- 1College of Arts and Sciences, Kent State University, Kent, OH, USA
- 2Department of Pathology and Human Anatomy, Loma Linda University School of Medicine, Loma Linda, CA, USA
The sympathetic nervous system (SNS) regulates host defense responses and restores homeostasis. SNS-immune regulation is altered in rheumatoid arthritis (RA) and rodent models of RA, characterized by nerve remodeling in immune organs and defective adrenergic receptor (AR) signaling to immune cell targets. The SNS typically promotes or suppresses inflammation via α- and β2-AR activation, respectively, and indirectly drives humoral immunity by blocking Th1 cytokine secretion. Here, we investigate how β2-AR stimulation and/or α-AR blockade at disease onset affects disease pathology and cytokine profiles in relevant immune organs from male Lewis rats with adjuvant-induced arthritis (AA). Rats challenged to induce AA were treated with terbutaline (TERB), a β2-AR agonist (600 μg/kg/day) and/or phentolamine (PHEN), an α-AR antagonist (5.0 mg/kg/day) or vehicle from disease onset through severe disease. We report that in spleen, mesenteric (MLN) and draining lymph node (DLN) cells, TERB reduces proliferation, an effect independent of IL-2. TERB also fails to shift T helper (Th) cytokines from a Th1 to Th2 profile in spleen and MLN (no effect on IFN-γ) and DLN (greater IFN-γ) cells. In splenocytes, TERB, PHEN, and co-treatment (PT) promotes an anti-inflammatory profile (greater IL-10) and lowers TNF-α (PT only). In DLN cells, drug treatments do not affect inflammatory profiles, except PT, which raised IL-10. In MLN cells, TERB or PHEN lowers MLN cell secretion of TNF-α or IL-10, respectively. Collectively, our findings indicate disrupted β2-AR, but not α-AR signaling in AA. Aberrant β2-AR signaling consequently derails the sympathetic regulation of lymphocyte expansion, Th cell differentiation, and inflammation in the spleen, DLNs and MLs that is required for immune system homeostasis. Importantly, this study provides potential mechanisms through which reestablished balance between α- and β2-AR function in the immune system ameliorates inflammation and joint destruction in AA.
Introduction
Autonomic and immune dysfunction, and dysregulation of their cross-communication in autoimmune diseases, are hallmarks of many autoimmune diseases, including rheumatoid arthritis (RA) (1–4). In RA, chronic inflammation, “self” targeted arthritogenic T cells and autoantibodies drive the damage in the affected joints and visceral organs (5). Thus, altered innate and adaptive immunity are key mediators of the disease process (5, 6). Dysregulation of the immune system is coupled with hyperactivity of the sympathetic nervous system (SNS). Dysregulation of the SNS in the immune system contributes significantly to disease onset, and greater severity in RA and experimentally-induced arthritis (7, 8). The contribution of a dysregulated SNS and the altered cross-communication between SNS and the immune system in autoimmune diseases is complex. In adjuvant- and collagen-induced arthritis models [adjuvant-induced arthritis (AA) and CIA, respectively], the SNS exerts a pro-inflammatory influence during the inductive, asymptomatic phase, but suppresses the inflammatory and cell-mediated response after disease onset (9, 10). Thus, we still do not understand the complex and diverse roles of the SNS in regulating innate and adaptive immunity that mediate rheumatic disease mechanisms prior to and after disease onset. Growing evidence indicates that the SNS is an important target for developing disease amelioration, or even preventing disease onset, supporting the need to understand sympathetic-immune system cross-talk in RA (2).
We have shown disease promoting effects of sympathetic innervation of secondary lymphoid organs during the induction phase, a time of antigen processing, and disease enhancing effects in the effector phase of AA (11, 12). This finding was subsequently confirmed using the collagen-induced arthritis model of RA (13). In these studies, sympathetic nerves were destroyed by selective toxins (11, 13) or β2-adrenergic receptor (AR) agonist and/or α-AR antagonist treatment (12) prior to the autoimmune inducing challenge or after disease onset. Treatment with a β2-AR agonist and/or α-AR antagonist shifts the balance of pro- and anti-inflammatory cytokine production toward increased disease severity if administered prior to or reduced disease activity after disease onset, respectively. These findings indicate that changes in the nerves and receptors contribute to the opposite disease outcome effects of the SNS observed after challenge and disease onset. Consistent with this notion, we (14–16) and others (17) previously reported dramatic changes in the density and distribution of sympathetic nerves to discrete compartments in secondary immune organs in rodent models of RA, and in the affected joints of RA patients. These anatomical changes suggest that sympathetic nerves interact with various immune cell populations in different activational states and functional compartments. They also implicate a link between consequent changes in sympathetic-immune cell signaling with disease development and progression.
The SNS regulates the functions of immune cells that mediate rheumatic disease through the activation of α- and β2-ARs expressed on their cell surfaces (10, 17–19). Sympathetic nerves target immune cells in secondary lymphoid organs (15, 16), and the immune cell infiltrates and tertiary lymph nodules that occur in the affected joints (20). In secondary lymphoid organs, neurotransmitters, predominantly norepinephrine released from sympathetic nerves, activate β2-ARs expressed in T cells to inhibit IL-2 production, which subsequently suppresses lymphocyte proliferation required for clonal expansion [reviewed in Ref. (21)]. Activation of β2-ARs also inhibit cellular and promote humoral immunity by regulating the phenotypic differentiation of CD4+ T helper (Th) cells in response to challenge with T-cell-dependent antigens (21). This occurs by norepinephrine activating Th0/Th1 cell β2-ARs, which increase intracellular cAMP production. Cyclic AMP then inhibits IFN-γ production. The decrease in IFN-γ reduces the inhibitory cross-regulation of IFN-γ on IL-4 production, thus promoting IL-4 synthesis by Th2 cells. In this manner, the SNS provides a negative feedback mechanism to restore immune system homeostasis after antigen challenges that activate cellular immunity (21).
Recently, we found that activation of β2-ARs no longer induces cAMP in immune cells in the spleen and lymph nodes that drain the arthritic hind-limbs in AA during severe disease (18). Altered β2-AR responses occurred concomitantly with altered receptor-ligand affinity and lymphoid tissue changes in receptor phosphorylation. These findings predict that a negative feedback mechanism required to restore immune system homeostasis after adjuvant challenge is lost after disease onset. However, the impact of sympathetic regulation of IL-2, IFN-γ, and IL-4 production in disease-relevant secondary lymphoid organs has not been determined.
The SNS also time-dependently regulates pro- and anti-inflammatory cytokine production relative to the antigen challenge [reviewed in Ref. (21)]. After antigen challenge, norepinephrine released from sympathetic nerves at the site of challenge promotes TNF-α and suppresses IL-10 production by activating α-ARs expressed in macrophages to amplify the ensuing inflammatory response. Similarly, norepinephrine promotes TNF-α and suppresses IL-10 production in dendritic cells from secondary lymphoid tissue via α-ARs. In contrast, activation of β2-AR in macrophages and dendritic cells suppresses TNF-α and promotes IL-10 production. The balance between the anti- and pro-inflammatory effects of the SNS is dependent upon the balance between α- and β2-AR expression, intracellular signaling pathways they activate and the dynamic local environment that follows the arthritis inducing challenge (9, 12). In this manner, the SNS regulates the initiating and shutting off of inflammatory and innate immune responses. In animal models of RA, α-AR antagonists and β2-AR agonists promote or inhibit joint inflammation if administered prior to or after disease onset, respectively. The effects of altered sympathetic innervation and changes in β2-AR function on SNS regulation of pro- and anti-inflammatory cytokines seen previously in secondary lymphoid organs of arthritic rats have not been examined.
The purpose of this study was to determine how the SNS affects ex vivo cytokine production in secondary lymphoid organs during the effector phase of disease. Specifically, we examined the effect of AR selective drug treatments in (1) modulating IL-2 and proliferation, and (2) the balance between IFN-γ and IL-4 and between TNF-α and IL-10 after AA development. A specific β2-AR agonist and an α-AR antagonist, alone and in combination [previously designated SH1293 (12)] were administered in vivo after disease onset. These treatments were used to determine the contribution of each receptor subtype in altering cytokine production and disease outcome. T cell and macrophage cytokines were measured ex vivo for each tissue collected from each treatment group. Cytokines with crucial roles in Th cell differentiation and clonal expansion or inflammation were selected for assessment: (1) immune cell production of IL-2, an important cytokine for development and differentiation of Th cells and proliferative responses required for clonal expansion (22); (2) IL-4 and IL-10, cytokines, which promote Th2 cell development and have anti-inflammatory functions; (3) IFN-γ and TNF-α, which promote Th1 cell development and which drive inflammation (23, 24).
Materials and Methods
Supplies, Drugs, and Adjuvant Preparation
All tissue culture media and supplements were obtained from Gibco BRL (Rockville, MD, USA) unless otherwise stated. OPTIA sandwich ELISA kits for IL-2, IL-4, IL-10, IFN-γ, and TNF-α were purchased from BD Pharmingen (San Diego, CA, USA). The non-specific α-AR antagonist, phentolamine, and the β2-AR agonist, terbutaline, were obtained from Sigma Chemical Company (St. Louis, MO, USA). All adrenergic drugs were dissolved in 0.01 mM ascorbic acid in 0.9% sterile, endotoxin-free saline. Complete Freund’s adjuvant (CFA; 0.03 g dried and heat-killed Mycobacterium butyricum; Difco, Detroit, MI, USA) was emulsified in 10 ml sterile mineral oil, as previously described (18). The suspension was treated with a sonic dismembraner for 5 min to keep the bacterial cell wall in suspension for animal injections. A single preparation of CFA was used to minimize variability, and 100% of the animals developed arthritis.
Animals
Male Lewis rats (200–250 g) were obtained from Charles River Laboratories (Raleigh, NC, USA) and housed two per cage for 3 weeks prior to the start of each experiment. The animals were maintained on a 12-h off/on light schedule, and food (Purina Lab Diet 5001) and water were available ad libitum. For AA rats, the food was placed in the bottom of the cage, and water was supplied using long-stemmed sipper tubes. All rats were observed to eat and drink throughout the study period. Animals were weighed biweekly to monitor adequate weight gain. All animals were treated in the same manner. Other than the development of arthritis, rats were healthy throughout the experiment. All protocols for the use and care of the animals in the study were approved by our Animal Use and Care Committee, and complied with NIH guidelines for the humane use and care of research animals.
Thirty-two male Lewis rats were given 100 μl of CFA by intradermal injection into the base of the tail. Twelve days later, rats were randomly assigned to one of four groups (N = 8 per group): (1) phentolamine [PHEN; 125 μg/day intraperitoneal (i.p.)]; (2) terbutaline [TERB; 1200 μg/day (i.p.)]; (3) phentolamine and terbutaline (PT, same dose as PHEN and TERB); or (4) vehicle (VEH). Drug treatment was divided into two injections in a total volume of 250 μl per injection given at 7 a.m. and 6 p.m., as previously described (12). This treatment regimen was based on previous reports demonstrating predictable pharmacological effects on disease severity in arthritic rats (9, 12). Adrenergic therapies began 12 days after immunization, the time of disease onset, and continued until sacrifice. All animals were weighed prior to sacrifice on day 28 using an overdose of 8% chloral hydrate 10.0 ml/kg body weight.
At sacrifice, the inguinal and popliteal lymph nodes (DLN, lymph nodes that drain the site of antigen challenge), mesenteric lymph nodes (MLN), and spleen were dissected and weighed, and peripheral blood was collected. Cell suspensions from all immune tissues were prepared for cell culture. All methods used in this study have been previously described in detail (12, 18). Non-arthritic animals were not included in this study, but reference ranges from non-arthritic rats from our database are shown as light gray horizontal bars to indicate normal values or non-specific activity, as appropriate.
Assessment of Disease Outcome
The inflammatory response in the arthritic rats was assessed by routine methods previously described (12, 18). Dorsoplantar widths of the hind feet were measured using a Mitutoyo Corporation dial thickness gage on the day of sacrifice. Prior to sacrifice, radiographs of the hind-limbs were taken to assess disease severity. Radiographs were taken using the following settings: 400 nN, 50 kVp, 0.4 s exposure time at 40 cm, using an X-OMAT processor. Using a grading scale modified from Ackerman and coworkers (25), the radiographs were coded to obscure the treatment groups, then two independent observers subjectively rated each of the radiographs on the scale: 0 (normal), 1 (slight), 2 (mild), 3 (moderate), and 4 (severe) abnormalities in the tissue. The radiographs were scored for each of the following characteristics: (1) soft tissue swelling as indicated by the width of soft tissue shadows and alterations in the normal configuration of the soft tissue planes; (2) osteoporosis (recognized by increases in radiolucency relative to uninvolved adjacent bone); (3) cartilage loss shown by narrowing of the joint spaces; (4) heterotopic ossification defined as proliferation of new bone tissue (fine ossified line paralleling normal bone, but not contiguous with calcified area of the bone itself); and (5) bone erosions. The radiographic scores for each category were added for both hind-limbs giving a maximum score of 40.
Immune Cell Cultures
Spleens were placed in a stomacher bag and homogenized in Hank’s balanced salt solution (HBSS) for 30 s. Spleen cells were washed with HBSS, and passed through a nylon mesh (Fisher Scientific, St Louis, MO, USA) to remove the connective tissue. Spleen cells were centrifuged and the pellet was resuspended in 5 ml NH4Cl hypotonic buffer for 3 min to lyse red blood cells. The cells were washed 2× with 10 ml HBSS, centrifuged, and resuspended in complete RPMI 1640 media (Gibco BRL, Rockville, MD, USA) supplemented with 5% fetal calf serum and 1% antibiotic/antimycotic (complete media). Using forceps, MLNs and DLNs were teased apart in HBSS. Each homogenate was passed through a nylon mesh, washed in 5 ml HBSS, centrifuged, and resuspended into complete media, as described above. Lymph node and spleen cells were counted using a hemocytometer and adjusted to 2 × 106 cells/ml in complete media.
Blood was collected using cardiac puncture into 7-ml lithium heparin vacutainer tubes. After tube inversion (7×), blood cells were pelleted at 1,000 rpm for 15 min at 10°C. The buffy coat was removed and placed into 10 ml NH4Cl hypotonic buffer for 3 min to lyse red blood cells. Peripheral blood mononuclear cells (PBMCs) were centrifuged, and the cell pellet was resuspended and washed 3× in 10 ml HBSS. Following the last centrifugation, the cells were resuspended in complete media. PBMCs were counted and suspended at 2 × 106 cells/ml in complete media. In this study, additional immune challenge was omitted to better mimic in vivo disease conditions.
Ex vivo Cellular Proliferation
Immune cells in supplemented complete RPMI 1640 were plated in 96-well, flat-bottom plates (Falcon, Oxnard, CA, USA) at 2 × 105 cells/well in triplicate without further stimulation. In this study, additional immune challenge was omitted to better mimic in vivo disease conditions. Cultures were maintained for 72 h in a humidified, 7% CO2 incubator at 37°C. [3H]Thymidine (0.5 μCi/10 μl; Amersham, Arlington Heights, IL, USA) was added the last 24 h of culture. Cells were harvested onto glass fiber filters (Brandel, Gaithersburg, MD, USA) using a cell harvester (Brandel, Gaithersburg, MD, USA). The filters were placed in 5 ml of scintillation fluid (Fisher Scientific, Tustin, CA, USA), and [3H]Thymidine incorporation was determined using a liquid scintillation counter (Beckman, Brea, CA, USA).
Cytokine Production
Two milliliters of each cell suspension were plated into 24-well plates (Falcon, Oxnard, CA, USA), and incubated in 7% CO2 at 37°C for 24 h. After 24 h, the supernatants were harvested and stored at −80°C. Cytokine ELISAs were run according to the manufacturer’s instructions. Cytokine levels in unknown samples were determined through comparison with a standard curve obtained from known concentrations of the cytokines run in duplicate on each plate using a Ceres 900 HDI plate reader at 450 nm (Bio Tek Instruments Incorporated, Winooski, VT, USA).
Statistical Analysis
All data are expressed as a mean ± standard error of the mean (SEM), unless otherwise stated. For disease outcome measurements the right and left footpads from each animal were averaged, and the individual means per group determined (mean of a mean in millimeters). Group differences were determined by two-way ANOVA with repeated measures. Means found to be significantly different (p < 0.05) were subjected to Bonferroni post hoc analyses. Mean radiograph scores from two scorers completed without knowledge of the treatment group were averaged for each group, and then subjected to Kruskal–Wallis statistical analysis with Dunn post hoc testing. Body and spleen weights were averaged for each treatment group (mean expressed in grams).
For cell proliferation, counts from triplicate wells were averaged after subtraction of background radioactivity. Group means were determined, and data were expressed as [3H]thymidine incorporation in counts per minute (cpm). Cytokine concentrations from duplicate wells were averaged, group means were calculated, and the data expressed as a mean in picograms per milliliter. The individual cytokine and the cytokine ratios were determined for each animal by comparing the samples to the standard curve on each plate. Group differences in body and spleen weights, cytokine production, cytokine ratios, and cellular proliferation were determined by one-way ANOVA. Means found to be significantly different (p < 0.05) were subjected to Bonferroni post hoc analyses.
Results
Adrenergic Drugs Attenuated Inflammation and Bone Loss
Footpad widths and X-ray scores from drug- and VEH-treated rats are presented in Figures 1A,B, respectively. All adrenergic interventions significantly reduced the dorsoplantar footpad width compared with VEH-treated arthritic rats (p < 0.001) (Figure 1A). All arthritic rats had greater footpad widths compared with non-AA rats (normal range indicated by the gray horizontal bar shown in Figure 1A). There was also a dramatic reduction in the radiographic scores from the arthritic rats treated with TERB compared with VEH-treated arthritic rats (Figure 1B; pTERB < 0.05, pPHEN < 0.05, or pPT < 0.01). X-rays of ankle joints from VEH-treated arthritic rats showed greater soft tissue swelling and joint destruction compared with arthritic rats treated with TERB, PHEN, or PT (Figures 1D–H). These observations were confirmed with X-ray analysis (Figure 1C). Figure 1C demonstrates the adrenergic drug-induced reduction in the components that comprise the radiographic scores. Adrenergic drug treatments reduced periosteal bone formation (POBF) 260–490%, bone erosions 170–270%, osteoporosis 190–320%, cartilage loss (JT NR) 140–220%, and soft tissue swelling 140–290% compared with VEH-treated arthritic animals. The rank order of potency for reducing soft tissue swelling and lower X-ray scores for the treatments was PT > TERB = PHEN > VEH.
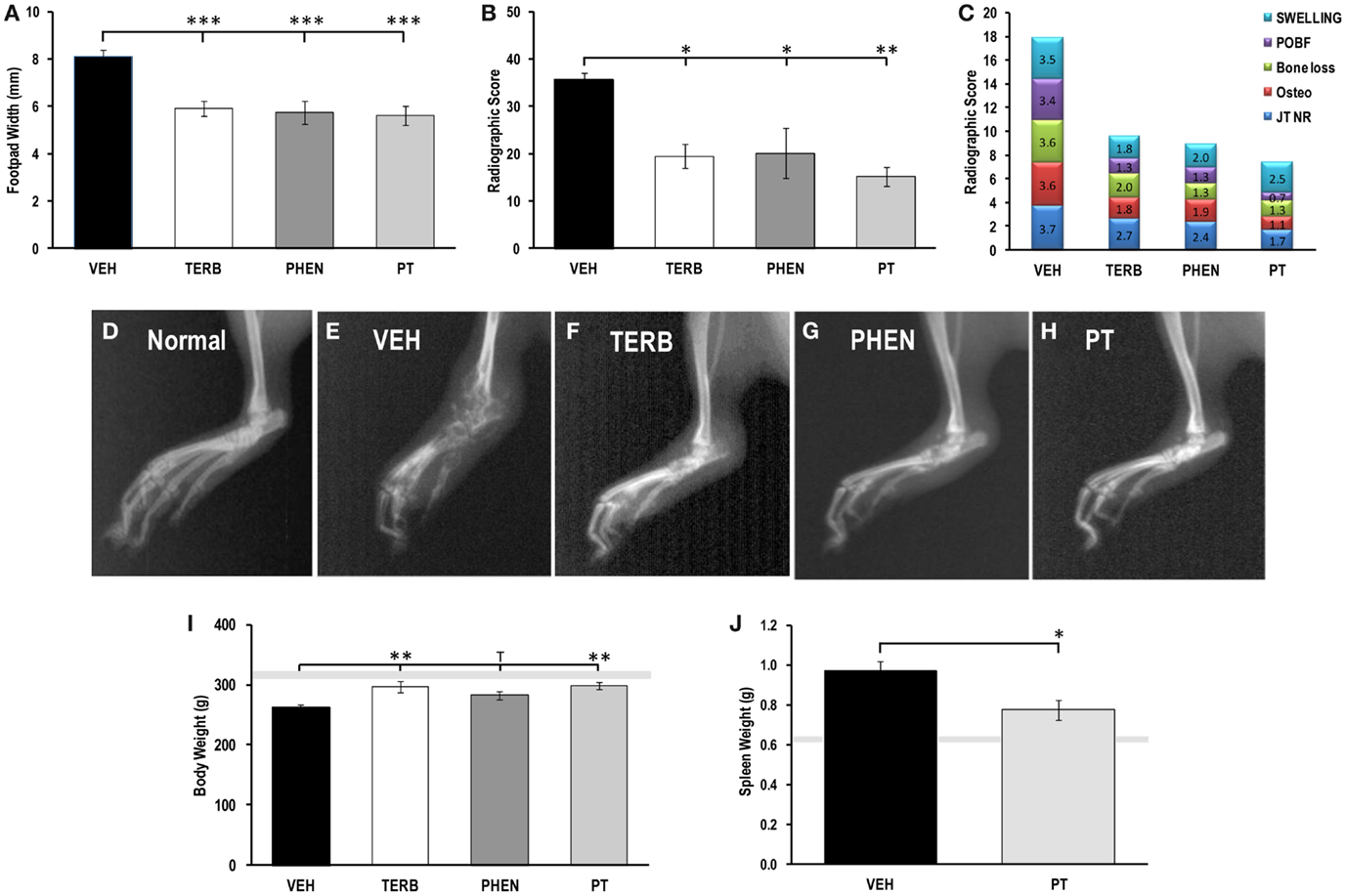
Figure 1. Indices of disease severity. Animals were treated with twice-daily i.p. injections of vehicle (VEH, black bars), terbutaline (TERB), phentolamine (PHEN), or phentolamine and terbutaline (PT) initiated 12 days after adjuvant challenge. (A) Mean Footpad widths ± SEM were significantly reduced in TERB (white bar), PHEN (dark gray bar), and PT (light gray bar) treated compared with VEH (black bar) treated rats. Light gray horizontal bar represents the mean footpad width ± SEM of naïve rat. N = 8. ***p < 0.001. (B) Mean radiographic scores ± SEM for, AA rats treated with TERB (white bar), PHEN (dark gray bar), and PT (light gray bar) were decreased compared with VEH (black bar). N = 8. *p < 0.05; **p < 0.01. (C) Stack-plots showing the contribution of each radiographic score component to the mean total radiographic score for rats treated with VEH, TERB, PHEN, or PT. Swelling, blue; joint narrowing (JT NR), red; osteoporosis, green; bone loss, purple; periosteal bone formation (POBF), turquoise. (D) Radiograph of the hind limb representative of normal untreated rats. (E) Radiograph of the hind limb representative of vehicle (VEH)-treated rats. (F) Radiograph of the hind limb representative TERB-treated rats. (G) Radiograph of the hind limb representative of PHEN-treated rats. (H) Radiograph of the hind limb representative of PT-treated rats. (I) Mean body weights in grams (g) ±SEM for rats treated with vehicle (VEH), terbutaline (TERB), phentolamine (PHEN), or phentolamine and terbutaline (PT). The light gray bar represents the range of body weights for untreated non-arthritic rats. N = 8. **p < 0.01. (J) Mean spleen weights in grams (g) ±SEM for rats challenged with adjuvant and treated with vehicle (VEH) or phentolamine and terbutaline (PT). The light gray bar represents the range for normal spleen weights of untreated non-arthritic rats. N = 8. *p < 0.05.
Adrenergic Drugs Prevented Loss of Body Weight and Increased Spleen Weight
Between day 8 and 12 post-CFA challenge, there was a sharp decline in body weight (~10–15%) that leveled off between days 12 and 16, and then was maintained through the effector phase of the disease (data not shown). In rats treated with TERB and PT, weight loss was prevented compared with VEH-treated control animals at day 28 post-CFA challenge (Figure 1I, p < 0.01). There was a trend (p < 0.1) toward weight loss prevention in rats treated with PHEN. Additionally, disease development resulted in greater spleen weights compared with the normal range (light gray horizontal bar; p < 0.001; Figure 1J). Combined drug treatment, PT, partially reversed the disease-induced increase in spleen mass compared with the VEH-treated AA rats (p < 0.05); however, the PT-treated animals still had spleens with significantly greater mass than the non-arthritic controls (light gray horizontal bar; p < 0.05).
No Effects of Adrenergic Drugs on PBMC Proliferation Despite Lower IL-2 Production by α-AR Blockade
On day 28 post-CFA challenge, the proliferative activity of PBMCs from arthritic rats (Figure 2A) was low in all the treatment groups (~130–200 cpm), but was slightly higher than the level seen in untreated PBMCs from non-AA rats (light gray horizontal bar). There were no differences in PBMC proliferation among the drug treatment groups or when compared with VEH treatment. Despite the absence of drug-induced effects on PBMC proliferative responses, ex vivo IL-2 production was affected by drug treatment on day 28 post-CFA challenge (Figure 2B). PBMCs from VEH- and TERB-treated arthritic rats released relatively similar levels of IL-2 (Figure 2B). Unlike TERB, IL-2 concentration was reduced in both PHEN- (p < 0.01) and PT- (p < 0.01) treated PBMC cultures compared with VEH-treated AA controls.
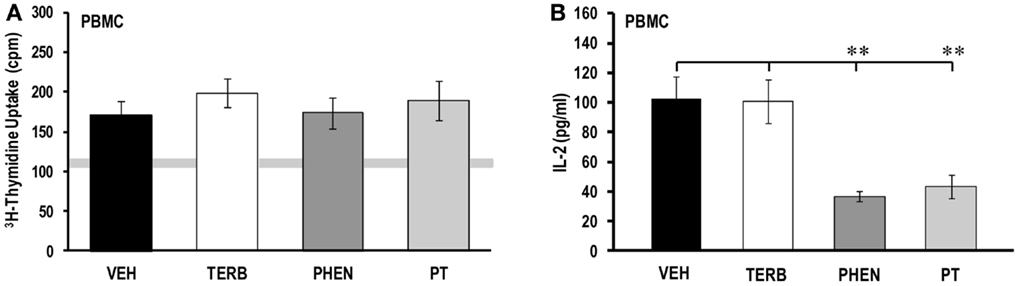
Figure 2. Proliferative response and IL-2 production in peripheral blood mononuclear cells (PBMC). Animals were treated with twice-daily i.p. injections of vehicle (VEH, black bars), terbutaline (TERB, white bars), phentolamine (PHEN, dark gray bars), or phentolamine and terbutaline (PT, light gray bars) initiated 12 days after adjuvant challenge. (A) Proliferative responses 72 h post-PBMC cell culture did not differ between treatment groups. N = 8. Gray horizontal bar represents the range for 3H-thymidine incorporation in unstimulated PBMCs. (B) IL-2 production 24 h-post PBMC culture were reduced in PHEN- and PT-treated compared with VEH-treated AA rats. N = 8. All data represent means ± SEM. **p < 0.01.
α-AR Adrenergic Therapies Reduced PBMC Th1 and Pro-Inflammatory Cytokine Production
In ex vivo cultures of PBMCs from all treatment groups, IL-4 concentrations were similar (Figure 3A). PBMCs from VEH-treated arthritic rats secreted more IFN-γ (319 ± 76 pg/ml) (Figure 3B) compared with IL-2 (103 ± 15 pg/ml, Figure 2B) and IL-4 (49 ± 11 pg/ml, Figure 3A). Treatment with TERB alone did not affect IFN-γ secretion. However, PHEN and PT significantly reduced the IFN-γ concentration compared with VEH- or TERB-treated AA rats (PHEN vs. VEH or TERB, p < 0.01; PT vs. VEH or TERB, p < 0.05).
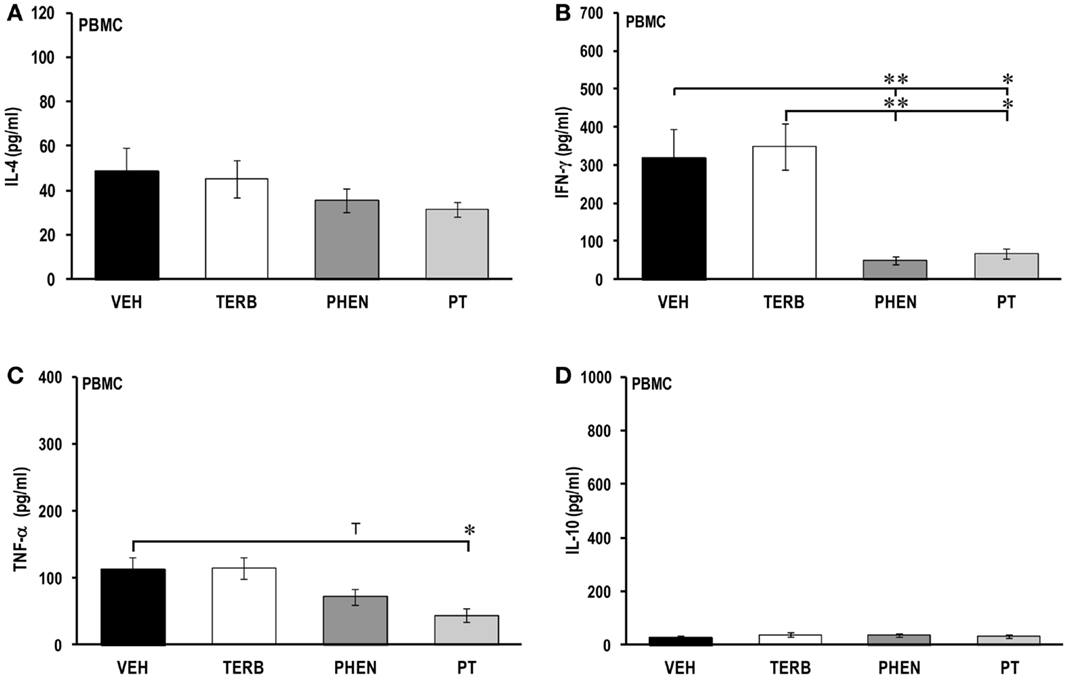
Figure 3. Cytokine production in peripheral blood mononuclear cells (PBMC). Animals were treated with twice-daily i.p. injections of vehicle (VEH, black bars), terbutaline (TERB, white bars), phentolamine (PHEN, dark gray bars), or phentolamine and terbutaline (PT, light gray bars) initiated 12 days after adjuvant challenge. (A) IL-4 concentrations 24 h after PBMC culture did not differ among the treatment groups. N = 8. (B) IFN-γ production 24 h post-PBMC culture was reduced by either PHEN or PT compared with VEH treatment. N = 8. *p < 0.05; **p < 0.01. (C) TNF-α secretion from PBMCs was reduced by PT compared with VEH treatment 24 h post-culture. N = 8. *p < 0.05. (D) IL-10 production in PBMCs was similar in all treatment groups after 24 h of cell culture. N = 8.
PBMCs from VEH-treated arthritic rats secreted low levels of TNF-α (123 ± 21 pg/ml) (Figure 3C) and IL-10 (28 ± 5 pg/ml) (Figure 3D). There was no difference in TNF-α production in TERB- or VEH-treated arthritic rats (Figure 3C). However, PHEN treatment showed a trend (p < 0.1) toward reduced TNF-α production compared with VEH-treated arthritic rats (Figure 3C) and PT treatment significantly inhibited TNF-α release compared with VEH- or TERB-treated arthritic rats (p < 0.05) (Figure 3C). Treatment with TERB, PHEN, or PT had no effects on IL-10 concentration compared with VEH-treated arthritic rats (Figure 3D).
Adrenergic Therapies Reduced ex vivo Spleen Cell Proliferative Response and Differentially Affected IL-2 Production
Thymidine incorporation was greater in the spleen cells from VEH-treated arthritic than untreated non-arthritic rats (light gray horizontal bar; Figure 4A). Splenocyte proliferation was 28-fold greater than PBMC proliferation (Figure 2A). In vivo treatment with TERB, PHEN, or PT markedly reduced spleen cell proliferation compared with VEH treatment (Figure 4A; p < 0.001). IL-2 levels were low (28 ± 5 pg/ml), but detectable in the VEH-treated arthritic animals (Figure 4B). Despite the drug-induced suppression in proliferation (Figure 4A), IL-2 concentrations (Figure 4B) were no different in cultures from TERB- or PT-treated rats, and elevated in cultures from PHEN-treated rats compared with VEH treatment (p < 0.05).
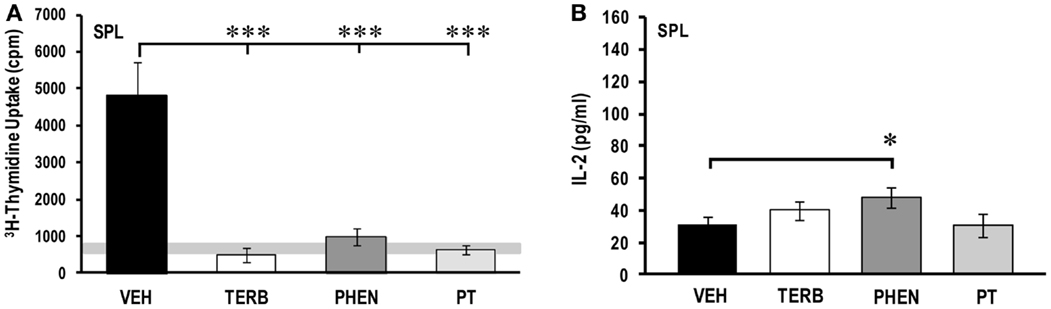
Figure 4. Ex vivo proliferation and IL-2 production by spleen (SPL) cells 72 and 24 h post-culture, respectively. Animals were treated with twice-daily i.p. injections of vehicle (VEH, black bars), terbutaline (TERB, white bars), phentolamine (PHEN, dark gray bars), or phentolamine and terbutaline (PT, light gray bars) initiated 12 days after adjuvant challenge. (A)3H-Thymidine incorporation into spleen cells was significantly lower in all drug-treated groups compared with VEH controls. The horizontal light gray bar represents the range of non-specific background in untreated non-arthritic rats. N = 8. ***p < 0.001. (B) IL-2 secreted by spleen cells was greater with PHEN compared with VEH treatment, but IL-2 concentrations in the other treatments did not differ from VEH-treated rats. N = 8. *p < 0.05.
Adrenergic Therapy Reduced ex vivo Pro-Inflammatory and Increased Th2 Cytokine Production in Spleen Cells
Figures 5A–D show the ex vivo secretion of IL-4 (Figure 5A), IFN-γ (Figure 5B), TNF-α (Figure 5C), and IL-10 (Figure 5D). There was no effect of any of the drug treatments on IL-4 or IFN-γ production compared with VEH controls (Figures 5A–B). Treatment with PT significantly reduced TNF-α production by spleen cells compared with VEH-treated arthritic rats (Figure 5C; p < 0.01). Spleen cells from rats receiving PHEN tended to produce less TNF-α than rats treated with VEH (p < 0.1), while TERB had no effect compared to VEH treatment. Spleen cells from VEH-treated arthritic rats secreted ~4-fold more IL-10 (Figure 5D) compared with PBMCs (Figure 3D). Interestingly, TERB, PHEN, or PT treatment markedly enhanced splenocyte IL-10 production compared with VEH-treated arthritic rats (Figure 5D; pTERB < 0.05; pPHEN < 0.05; pPT < 0.01). There were no significant differences in splenocyte IL-10 production between any of the adrenergic drug-treated animals.
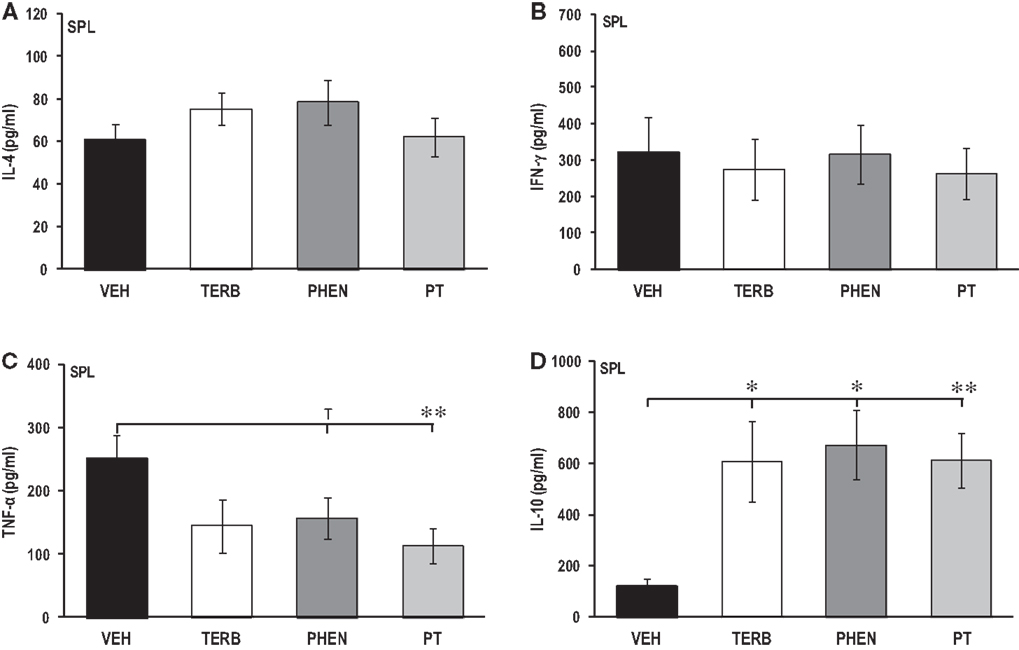
Figure 5. Cytokine production by spleen (SPL) cells after 24 h of culture. Animals were treated with twice-daily i.p. injections of vehicle (VEH, black bars), terbutaline (TERB, white bars), phentolamine (PHEN, dark gray bars), or phentolamine and terbutaline (PT, light gray bars) initiated 12 days after adjuvant challenge. (A) IL-4 production. N = 8. (B) IFN-γ levels. N = 8. (C) TNF-α secretion. N = 8. **p < 0.01. (D) IL-10 release. N = 8. *p < 0.05; **p < 0.001.
Adrenergic Therapies Suppressed ex vivo Cell Proliferation in DLN, but Differentially Affected IL-2 Production
Proliferative responses of DLN cells from VEH-treated arthritic animals were elevated compared with DLN cells from non-arthritic rats (light gray horizontal bars; Figure 6A). Additionally, DLN cell proliferation in VEH-treated rats was reduced 1.5-fold compared with the spleen (Figure 4A), but 19- or 6.7-fold higher than for PBMCs (Figure 2A) or MLN cells (Figure 8A), respectively. In vivo treatment with TERB, PHEN, or PT dramatically (>600%) reduced DLN cell proliferation compared with the VEH-treated arthritic animals (Figure 6A; p < 0.001). In VEH-treated arthritic rats, IL-2 concentrations from DLN cell cultures were similar to spleen cell cultures (Figure 6B), and much lower than PBMC cell cultures (Figure 4B). Treatment with TERB or PHEN tended to increase IL-2 concentrations (p < 0.1; Figure 6B), but PT treatment significantly elevated IL-2 production compared with DLN cells from VEH-treated arthritic rats (p < 0.05) (Figure 6B).
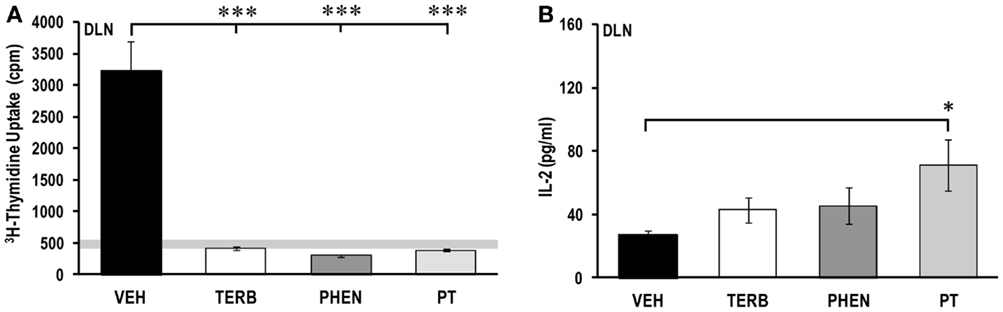
Figure 6. Proliferation of draining lymph node (DLN) cells and IL-2 production. Animals were treated with twice-daily i.p. injections of vehicle (VEH, black bars), terbutaline (TERB, white bars), phentolamine (PHEN, dark gray bars), or phentolamine and terbutaline (PT, light gray bars) initiated 12 days after adjuvant challenge. (A) All drug treatments suppressed proliferation of DLN cells compared with VEH treatment. Horizontal light gray bar demonstrates non-specific background. (B) IL-2 production by DLN cells is elevated in PT compared with VEH treatment. N = 8. *p < 0.5; **p < 0.01.
Adrenergic Interventions Differentially Affected ex vivo Cytokine Secretion in DLN Cells
In vivo administration of PT significantly increased IL-4 levels over levels in VEH-treated arthritic rats (Figure 7A; p < 0.05), but there was no effect of TERB or PHEN treatment on IL-4 concentrations. Interestingly, TERB and PT, but not PHEN, increased IFN-γ production in DLN cells compared with VEH treatment (pTERB < 0.05; pPT < 0.01) (Figure 7B). In contrast, none of the adrenergic treatments altered TNF-α production in DLN cells compared with VEH-treated arthritic rats (Figure 7C). PT treatment significantly increased the IL-10 concentration compared with VEH-treated arthritic rats (pPT < 0.05) (Figure 7D), but treatment with TERB or PHEN had no effect compared with VEH-treated arthritic rats.
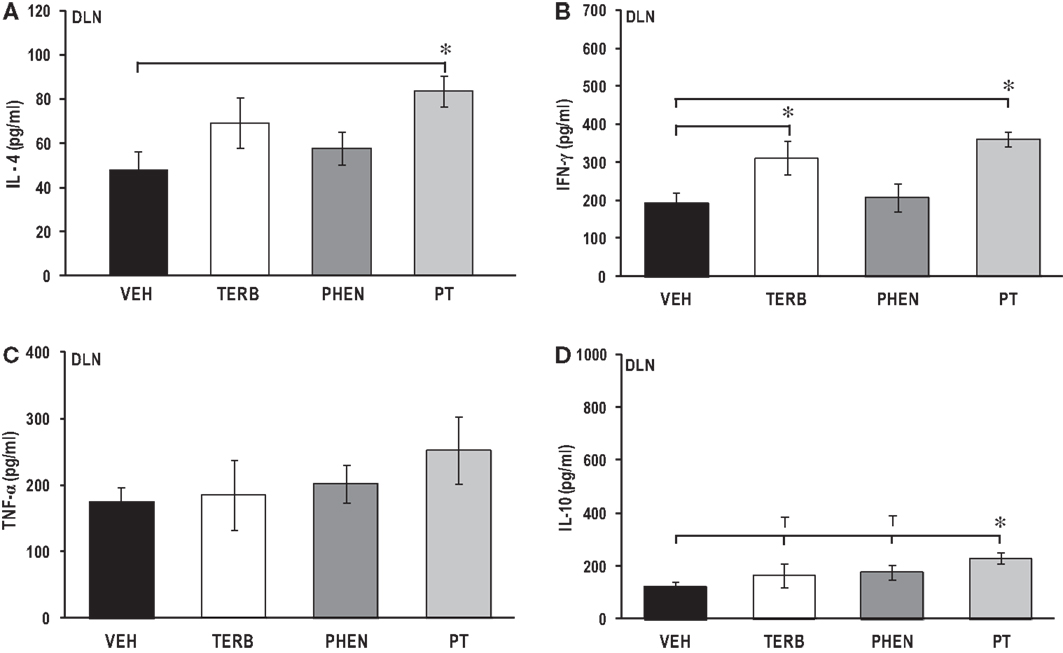
Figure 7. Cytokine production by draining lymph node (DLN) cells after 24 h of culture. Animals were treated with twice-daily i.p. injections of vehicle (VEH, black bars), terbutaline (TERB, white bars), phentolamine (PHEN, dark gray bars), or phentolamine and terbutaline (PT, light gray bars) initiated 12 days after adjuvant challenge. (A) IL-4 production was augmented by treatment with PT over that found in VEH controls. N = 8. *p < 0.05. (B) IFN-γ concentrations were higher in TERB- and PT- than with VEH-treated rats. N = 8. *p < 0.05; **p < 0.01. (C) TNF-α levels were similar in all groups. N = 8. p < 0.05. (D) IL-10 secretion was elevated in rats treated with PT over that seen with VEH treatment. N = 8. *p < 0.05.
Adrenergic Therapies Suppressed Proliferation in MLN Cells without Affecting IL-2 Production
Thymidine incorporation by MLN cells was greater for all treatment groups than unstimulated non-arthritic rats (indicated by gray horizontal bars; Figure 8A). Drug-treated MLN cultures had lower cell proliferation (>600%) than VEH-treated rats (p < 0.001). Proliferative responses in MLN (Figure 8A) were 10-fold lower than observed for spleen (Figure 4A), but 2.8-fold greater than observed for the PBMCs (Figure 2A). IL-2 production was low, with no differences between treatment groups (Figure 8B).
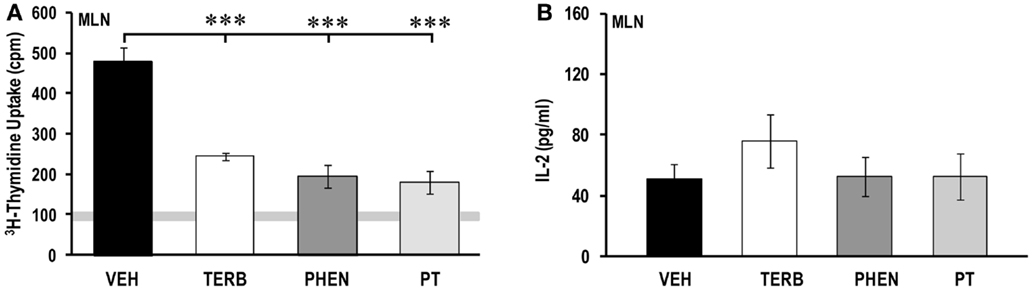
Figure 8. Proliferation of mesenteric lymph node (MLN) cells and IL-2 production. Animals were treated with twice-daily i.p. injections of vehicle (VEH, black bars), terbutaline (TERB, white bars), phentolamine (PHEN, dark gray bars), or phentolamine and terbutaline (PT, light gray bars) initiated 12 days after adjuvant challenge. (A) All drug treatments suppressed proliferation of MLN cells compared with VEH treatment. Horizontal light gray bar demonstrates non-specific background. (B) IL-2 production did not differ between treatment groups. N = 8. ***p < 0.001.
Adrenergic Treatment-Induced Suppression of Pro-Inflammatory and Th Cytokine Production in Cultured MLN Cells
There were no significant effects of any of the drug treatments on IL-4 concentrations in MLN cell cultures compared with VEH treatment (Figure 9A). IL-4 concentrations from MLN cell cultures were similar to levels observed in spleen (Figure 5A), but almost double values observed in PBMC or DLN cell cultures (Figures 3A and 7A, respectively). In vivo treatment with TERB or PHEN alone had no effect on the IFN-γ production in MLN cells compared with the VEH-treated arthritic rats (Figure 9B). However, PT treatment significantly reduced IFN-γ production compared with VEH-treated arthritic rats (p < 0.05). TERB and PT treatment reduced TNF-α production compared with VEH treatment (Figure 9C; p < 0.05); however, PHEN treatment alone had no effect on TNF-α secretion. In vivo treatment with TERB did not significantly alter IL-10 production compared with VEH-treated controls; however, PHEN treatment inhibited IL-10 production by MLN cells compared with VEH-treated arthritic rats (Figure 9D; p < 0.05). There was a trend (p < 0.1) for lower IL-10 levels in the arthritic rats receiving PT treatment compared with the VEH treatment. IL-10 production in MLN cells (Figure 9D) from VEH-treated arthritic rats was 17-, 4-, and 4-fold greater compared with PBMC, spleen, and DLN levels, respectively (Figures 3D, 5D, and 7D, respectively).
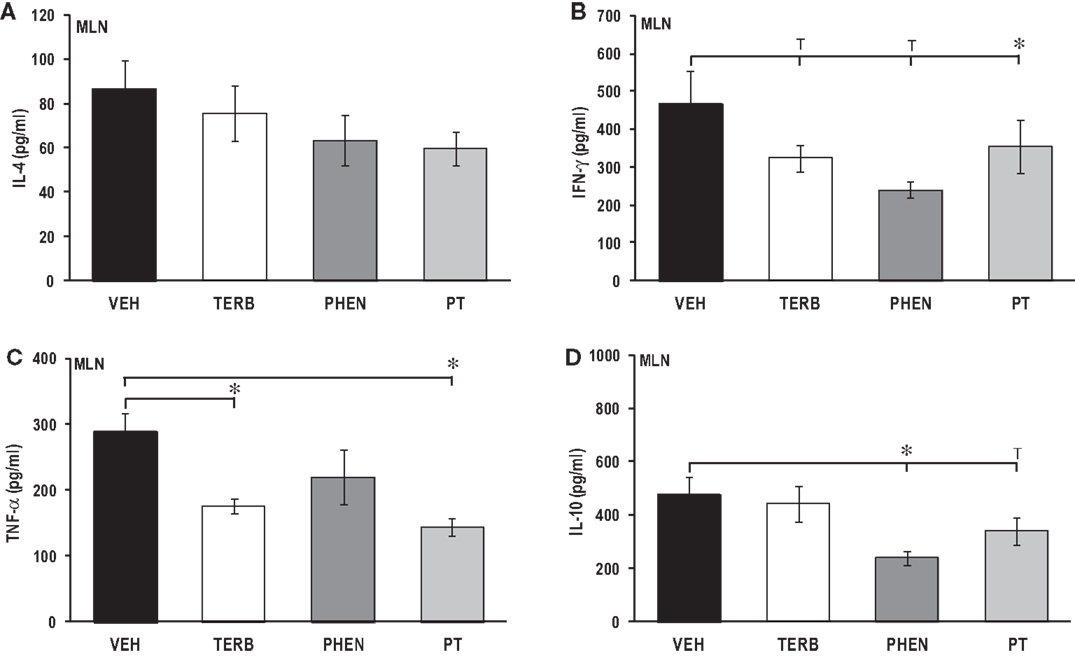
Figure 9. Cytokine production by mesenteric lymph node (MLN) cells after 24 h of culture. Animals were treated with twice-daily i.p. injections of vehicle (VEH, black bars), terbutaline (TERB, white bars), phentolamine (PHEN, dark gray bars), or phentolamine and terbutaline (PT, light gray bars) initiated 12 days after adjuvant challenge. (A) IL-4 production was comparable in all groups. N = 8. (B) IFN-γ concentrations were lower in PT- than with VEH-treated rats. N = 8. *p < 0.05. (C) TNF-α levels were reduced with TERB and PT treatment compared with VEH controls. N = 8. *p < 0.05. (D) IL-10 secretion was suppressed in rats treated with PHEN over that seen with VEH treatment. N = 8. *p < 0.05.
Lymphoid Organ-Dependent Effects of Adrenergic Therapies on Th1/Th2 Cytokine Ratios
Th1/Th2 cytokine ratio (IFN-γ/IL-4) from the PMBCs did not differ between arthritic rats treated with TERB or VEH (Figure 10A); however, treatment with PHEN or PT reduced the Th1/Th2 cytokine ratio compared with VEH-treated rats (p < 0.001). In contrast, adrenergic drug treatments failed to affect the Th1/Th2 ratio in spleen, DLN or MLN cell cultures compared with arthritic rats treated with the VEH (Figures 10B–D, respectively).
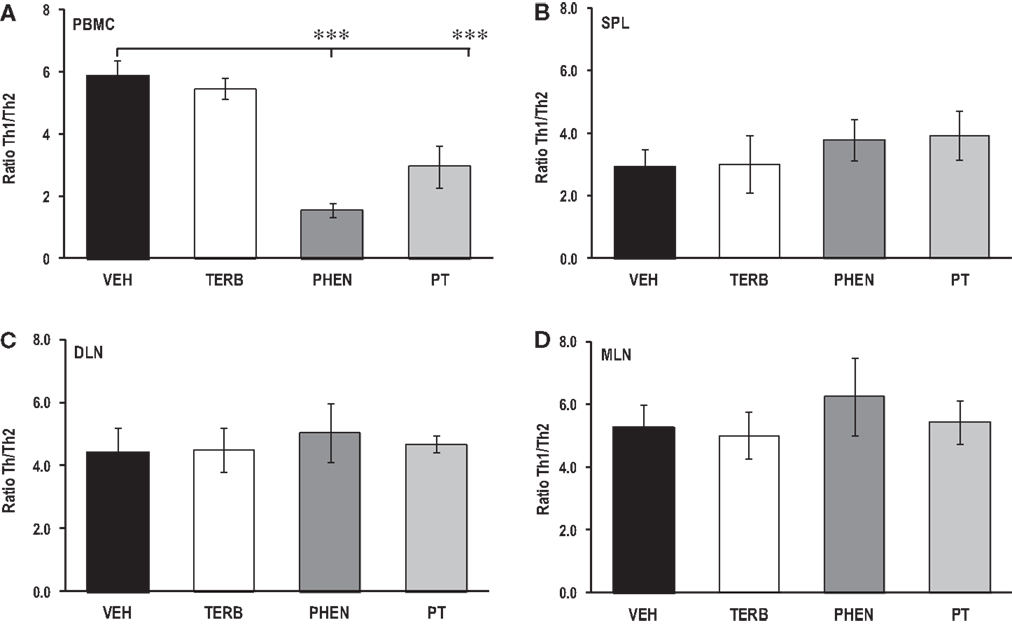
Figure 10. Ratios of Th1 to Th2 cytokines in secondary immune compartments. Animals were treated with twice-daily i.p. injections of vehicle (VEH, black bars), terbutaline (TERB, white bars), phentolamine (PHEN, dark gray bars), or phentolamine and terbutaline (PT, light gray bars) initiated 12 days after adjuvant challenge. (A) PBMC: PHEN and PT treatments lowered the Th1/Th2 cytokine ratio in PBMC cultures compared with VEH treatment. PT Th1/Th2 cytokine ratios were higher than with PHEN treatment. N = 8. ***p < 0.001. (B) Spleen:Th1/Th2 cytokine ratios did not differ between AA rats treated with any of the drug treatments compared with VEH treatment. (C) DLN, similar ratios were found in all groups. (D) MLN, no differences in Th1/Th2 cytokine ratios were found between treatment groups.
Lymphoid Organ-Dependent Effects of Adrenergic Therapies on Pro-Inflammatory/Anti-Inflammatory Cytokine Ratios
The ratio of pro- to anti-inflammatory cytokine production (TNF-α/IL-10) in PBMCs and DLN cells revealed that while TERB or PHEN had minimal effects on the TNF-α/IL-10 ratio, PT treatment significantly reduced the ratio of TNF-α/IL-10 in both cell populations (p < 0.05, Figures 11A,C). In contrast, spleen cells from all three adrenergic therapies had reduced TNF-α/IL-10 ratios compared with VEH-treated arthritic rats (Figure 11B; p < 0.01). Interestingly, treatment with PHEN increased the ratio of TNF-α/IL-10 over VEH control levels in MLN cell cultures (Figure 11D; p < 0.05). Finally, there was a trend (p < 0.1) for reduced TNF-α/IL-10 ratios in the MLN cell cultures following in vivo TERB or PT treatment.
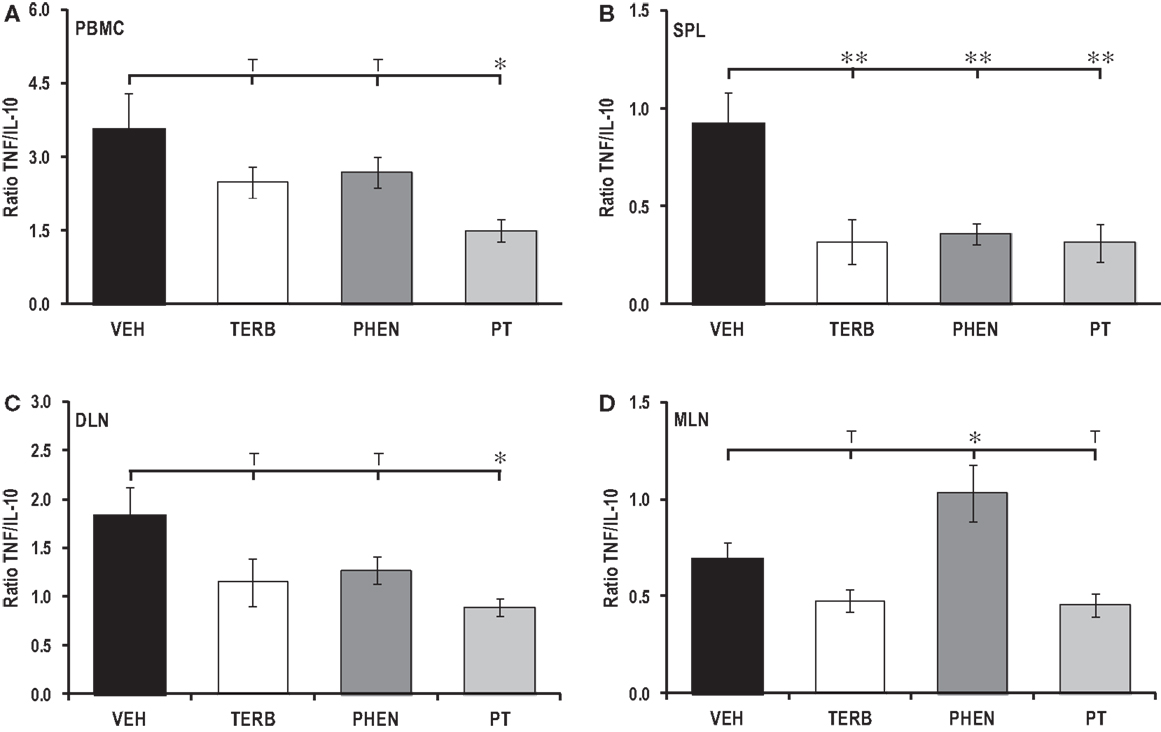
Figure 11. TNF/IL-10 ratios in secondary immune compartments. Animals were treated with twice-daily i.p. injections of vehicle (VEH, black bars), terbutaline (TERB, white bars), phentolamine (PHEN, dark gray bars), or phentolamine and terbutaline (PT, light gray bars) initiated 12 days after adjuvant challenge. (A) PBMC: PT treatments lowered the TNF/IL-10 ratio in PBMC cultures compared with VEH treatment. N = 8. *p < 0.05. (B) Spleen: all drug treatments lowered the TNF/IL-10 ratio compared with VEH treatment. N = 8. **p < 0.01. (C) DLN, PT suppressed the TNF/IL-10 ratio compared with VEH controls. N = 8. *p < 0.05. (D) MLN, the TNF/IL-10 ratio was greater in rats treated with PHEN than in VEH controls. N = 8. *p < 0.08.
Discussion
In this study, we confirm and extend our previous findings of the disease-modifying effects of a β2-AR agonist and/or α-AR antagonist treatment in AA after disease onset. Consistent with previous findings, TERB, PHEN, or PT dramatically ameliorates AA in male Lewis rats when treatment begins at disease onset (12). Our data extend these findings to show that all drug treatments suppressed proliferative responses in cell cultures from all lymphoid tissues examined. However, IL-2 production is not coupled with proliferation regardless of the adrenergic therapy. Immune cells in secondary lymphoid organs and peripheral blood are largely non-responsive to treatment with the β2-AR agonist. These findings support that the SNS loses its ability to regulate immune cell functions via normal β-AR signal transduction in secondary lymphoid organs in AA and by extension RA. Importantly, the exceptions to these findings are the dramatic β2-AR-mediated increase in spleen cell IL-10 and DLN cell IFN-γ production, respectively. Surprisingly, treatment with the α-AR antagonist produces greater changes in cytokine profiles than targeting β2-ARs, suggesting a shift in the class of AR that predominantly regulates immune function during chronic disease. In sites not expected to elicit robust immune responses to CFA challenge (i.e., PBMCs and MLN cells), PHEN and PT suppresses IFN-γ and TNF-α cytokine production. In spleen cells, the major effect of PHEN or PT was to drive IL-10 and suppress TNF-α production. This cytokine profile is expected to suppress inflammation and drive Th2 responses, which are consistent with improved disease outcome of PHEN or PT treatment. In contrast to single drug treatments, PT increases IL-4 and IL-10 production in DLN cells, which is expected to suppress inflammation and promote Th2 immunity. This difference in cytokine profiles may account for PT having the greatest ameliorating effects on disease, particularly its bone sparing effects.
All adrenergic therapies reduce disease severity. Radiographs and scoring provide support for a direct and/or indirect effect of adrenergic drugs on all scoring components used to assess soft tissue swelling (i.e., inflammation) and bone destruction/remodeling in the arthritic hind-limbs. Direct effects of adrenergic agents have been reported for both cartilage and bone. Both chondrocytes and bone cells (osteoclasts and osteoblasts) express functional ARs (26, 27) and can alter their growth and resorption activity (28–31). Additionally, immune cells (i.e., macrophages, T, B, and NK cells) and their products (i.e., TNF-α, IL-1, IFN-γ, and IL-2) coordinate changes in bone cell function and are regulated by ARs (32–34). Our findings support direct and/or indirect effects of TERB, PHEN, and PT treatments in ameliorating inflammation and joint destruction, and indicate these treatments are disease-modifying, anti-rheumatic drugs (DMARDs). X-ray scores suggest that TERB was more effective at reducing swelling than either PHEN or PT based on soft tissue assessment, in agreement with other reports demonstrating β2-AR-mediated anti-inflammatory effects (35–37). The β2-AR-mediated decrease in inflammation may be mediated by inhibiting macrophage TNF-α production and/or reducing reactive oxygen species (35–37). Surprisingly, PT was much less effective in reducing soft tissue swelling than specifically targeting β2-, or α-ARs. This finding suggests opposing actions of α- and β-ARs in the inflamed hind limbs. Whether this interaction involves multiple cell types and/or direct or indirect interplay between α- and β-ARs in the same cell types is unclear. Provocative new data (38, 39) indicate that β- and α-AR subtypes can form heterodimers that affect downstream signaling to regulate the inflammatory response. Similar research is warranted in our model to better understand β- and α-AR-mediated mechanisms regulating soft tissue swelling.
Immune cells cultured from lymphoid organs of CFA-challenged rats proliferate ex vivo without any additional immune challenge, an approach that better mimics the in vivo setting. Immune cell proliferation was most pronounced in DLNs and spleen. This finding is consistent with reports of persistent high antigen load in these tissues (40) and the significant involvement of both the DLNs and spleen in disease induction and immune dysregulation during the effector phase of the immune response. In response to CFA challenge, arthritogenic T cells that can transfer disease to naïve rats are generated in the spleen and DLNs (41). In agreement with this, we observed the greatest proliferative responses in DLNs and spleen cells, whereas proliferation was barely above background in PBMCs and MLN cells. TERB, PHEN, or PT treatment significantly reduced cellular proliferation in the spleen, DLN, and MLN, but had no effect on PBMC proliferation. This is consistent with the normalization of spleen weights after adrenergic treatments and with secondary lymphoid organs being sites of lymphocyte clonal expansion.
The reduced proliferative responses with TERB treatment are also consistent with other studies that demonstrate β2-AR agonists and norepinephrine treatment reduce proliferation of antigen- and mitogen-challenged lymphocytes from secondary immune organs [reviewed in Ref. (21)]. Upon antigen challenge, T cell proliferation is promoted by the binding of IL-2 to the IL-2R expressed on T cells [reviewed in Ref. (21)]. Numerous studies have shown that norepinephrine/epinephrine decrease the proliferative responses in T cells by a β2-AR-mediated reduction in IL-2 production following mitogen, anti-CD3 antibody, or antigen challenge (21). However, TERB treatment had no significant effects on IL-2 in our study, despite the β2-AR-mediated reduction in proliferation. These findings indicate that IL-2 does not drive the lymphocyte proliferation observed in secondary lymphoid compartments during the effector phase of AA, and that TERB-induced decreases in lymphocyte proliferation are not mediated by IL-2. Reports that chronic secretion of TNF-α can cause IL-2 deficiency at sites of inflammation (42–44) is consistent with our high TNF levels, low IL-2 production and uncoupling of IL-2-induced proliferation. Moreover, our findings are consistent with T cell hyporesponsiveness to T cell receptor (TCR) engagement (42), with a switch from an IL-2-to-inflammatory-driven immune response that suppresses IL-2 gene transcription (44). Finally, the inability of TERB to reduce IL-2 production in spleen and DLN cells from AA rats is consistent with recent findings from our lab that β2-ARs are uncoupled from cAMP (18). Others have reported that IL-2 decreases β-AR density, removing a negative control over cellular proliferation (45). Similarly, in certain cell populations, IL-1β can impair β2-AR coupling with adenylate cyclase (46, 47). Future studies are needed to determine the mechanisms for the β2-AR-mediated suppression of proliferative responses observed in secondary lymphoid organs in AA rats.
Surprisingly, treatment with the α-AR antagonist also reduced lymphocyte proliferation, an effect also observed when combined with TERB. These findings indicate that norepinephrine interaction with immune cell α- and β-ARs have opposing influences on lymphocyte proliferation, with α-ARs promoting and β-ARs opposing lymphocyte expansion during the effector phase of AA. Studies examining the effects of α-AR agonists on lymphocyte proliferation have reported increased, decreased, or no effect [reviewed in Ref. (48)]. Given the current evidence to date, lymphocytes express primarily β2-ARs, while innate immune cells appear to express β2-, α1-, and α2-ARs. Thus, effects of the α-AR antagonist, PHEN, on lymphocyte proliferation are likely mediated indirectly via α-AR-induced changes in functions of innate immune cells, such as macrophages, and/or by altering norepinephrine availability through presynaptic α2-ARs. Given the multiple potential targets of α-AR ligand, the mixed results regarding α-AR-mediated effects on proliferation is not surprising. Discrepancies regarding the direction of the impact of α-ARs-targeting drugs on lymphocyte proliferation are likely due to experimental differences, such as the type of antigen challenge, in vivo or in vitro drug administration, strain differences, and timing of administration relative to immune challenge. Collectively, our findings support that in contrast to β2-ARs, α-ARs suppress lymphocyte cell proliferation during the effector phase of AA, an effect that is blocked by α-AR antagonists during the effector phase. The extent to which the effects of TERB, PHEN or PT on proliferative responses contributes to disease reduction is unclear. However, inhibition of clonal expansion of arthritogenic T cells would be expected to reduce disease severity. Future studies will explore this possibility, as well as, the cell types targeted by these adrenergic treatments.
In contrast to TERB, PHEN treatment increases the production of IL-2 in spleen cells. Interestingly, PT also elevated IL-2 in DLN cells, but had no effect in other lymphoid compartments. The importance of these findings for reduced pathology is not clear. IL-2 is a pleiotropic cytokine. Besides its potent T-cell growth factor activity, IL-2 is essential for promoting the differentiation of Th1 (49) and Th2 cells (50). While IL-2 inhibits Th17 (51) and T follicular helper (Tfh) cell development (52), it does promote Th17 cell expansion once these cells develop (53). IL-2 is also involved in activation-induced cell death (AICD), which is important for homeostasis and the elimination of potentially harmful auto-reactive cells, at least in part by a Fas and FasL-dependent mechanism (54). IL-2 promotes antibody production and proliferation by B cells (55) and drives the development of CD4+FOXP3+ regulatory T cells (Treg cells), which have suppressor functions and mediate tolerance (56–58). Thus, IL-2 has broad essential biological actions, not only driving T cell proliferation and modulating effector cell differentiation, but also limiting potentially dangerous autoimmune reactions. Further research is required to understand the impact of α- and β-AR-mediated changes in IL-2 production on disease outcome in our disease model.
Regarding IL-2 and leukocyte proliferation responses, our findings are consistent with reports of increased SNS activity, decreased IL-2 production, and reduced proliferative responses in RA patients. Serum levels of IL-2 are reduced in patients with active disease, particularly in those with extra-articular manifestations (59, 60). Further, in RA patients mitogen-stimulated PBMCs produce less IL-2 and proliferate less robustly than PBMCs from controls (59). Low IL-2 and proliferative responses inversely correlate with disease activity and positively correlate with the proportion of CD4+ T cells (59), suggesting their importance in the pathophysiology of RA. Moreover, monocytes may play a role in regulating IL-2, as their depletion and partial reconstitution increases IL-2 production and proliferation in RA patients. Circulating cytokines that are indicators of general immune activation (including IL-2) increase prior to disease onset in RA patients (61), consistent with their importance in disease onset. Our data showing the ability of an α-AR antagonist or combined β2-AR agonist and α-AR antagonist treatment to increase splenic and DLN IL-2, respectively, indicate the complexity of regulatory mechanisms for this cytokine by the SNS. IL-2 is at the “cross-roads” of both effector T-cell responses and tolerance [reviewed in Ref. (62)], and the development of lethal autoimmune disease in IL-2 knock-out mice (63). We have speculated that this particular cytokine may be the key to explaining opposite effects of adrenergic agonists on disease severity when administered prior to vs. after disease onset (12). If this hypothesis is true, then understanding of the mechanisms for these contradictory effects of adrenergic treatments relative to disease onset has implications for prevention and therapeutic treatment of RA.
In the present study, TERB treatment had no effect on IFN-γ production in spleen, MLN and PBMC, despite the well accepted role of norepinephrine via β2-ARs to inhibit and enhance the production of the Th1 and Th2 cytokines, IFN-γ and IL-4, respectively (64, 65). The inability of TERB treatment to impact Th cell IFN-γ and IL-4 production in spleen, MLN, and PBMCs are consistent with findings by Heijink and coworkers (66) demonstrating that polarized Th1 and Th2 cells are less responsive to negative feedback by receptors coupled to the adenylate cyclase/cAMP pathway compared with freshly isolated T cells. Sanders and coworkers (67) also showed that in freshly isolated CD4+ T cells, a β2-AR agonist could activate the cAMP pathway to increase IL-2 and IFN-γ, however, this response is attenuated in differentiated Th1 and Th2 cells. In these studies, specialized Th subset cells were stimulated by anti-CD3/anti-CD28 or in vitro differentiation of Th0 cells under Th1 or Th2 polarizing conditions. Similar to the differentiated Th cells, we also observed that treatment with β2-AR agonists is no longer able to induce an increase in intracellular cAMP in splenocytes from arthritic rats 28 days post-adjuvant challenge (18). Further, lymphocyte β2-ARs in spleen cells from arthritic rats were phosphorylated at a site known to induce receptor desensitization. Our findings are consistent with the loss of control over cytokine production by the β2-AR-coupled cAMP pathway in Th effector cells after CFA challenge. These findings are also consistent with the differential effects on disease outcome in AA rats when a β-agonist is administered at different times across the time course of AA. Thus, differential effects of the agonist after CFA challenge and after disease onset may be due to targeting of differentiating Th0 cells versus fully differentiated Th cells.
Despite the known inhibitory effects of β2-AR agonists on Th1 cell production of IFN-γ (64, 65), TERB treatment in arthritic rats elevated IFN-γ production in the DLNs. This finding suggests that β2-AR signaling is not only uncoupled from its normal cAMP-PKA pathway, but also switches receptor coupling to an alternate second messenger. Recent findings from our lab indicate that in DLNs, β2-ARs are phosphorylated at a site known to switch signaling from cAMP to mitogen-activated protein kinase (MAPK) pathways (18). In AA rats, DLNs drain a site of chronic inflammation, and thus, are exposed to high concentrations of inflammatory cytokines. This may explain, in part, the different β2-AR responses in spleen and DLN cells. Consistent with this hypothesis, IL-1β is reported to cause a concentration- and time-dependent decrease in responses of airway smooth muscle cell and cardiac myocyte to a β-AR agonist that is mediated by uncoupling β-AR from Gs-induced activation of adenylyl cyclase (47, 68, 69). This response was accompanied by an increase in membrane Gi expression (69), a G-protein coupled with MAPK activation. The DLNs also receive a much higher antigen load than other secondary lymphoid organs after base of the tail CFA challenge. Additionally, the slow release of the M. butyricum cell wall components from the adjuvant may result in the persistent sub-optimal challenge in the DLNs. Interestingly, low persistent challenge with environmental adjuvant exposure has been linked to failed tolerance and the development of autoimmunity (70).
Neither TERB nor PHEN treatment altered IL-4 production in any of the tissues compared with VEH-treated arthritic rats. Our findings are consistent with studies showing that treatment with either norepinephrine or TERB induces IL-4 production from Th2 clones or Th2 effector cells derived from β2-AR-stimulated naïve CD4+ T cells (67, 71, 72). In contrast, other studies have shown that elevations in cAMP were able to increase IL-4 production in Th2 cells (66). It is unlikely that β2-ARs directly mediate increased IL-4 production by Th2 cells as the differentiation of Th0 to Th2 cells induces the loss of β2-AR expression (67, 71, 72). Notably, PT treatment increased IL-4 production in the DLNs. Whether this is a direct or indirect result of TERB or PHEN, the result of an interaction between the two drugs on Th2 cell IL-4 production, or due to TERB- or PT-induced effects on other immune cell populations will require additional studies.
The most dramatic effects of PHEN treatment during the effector phase were the reductions of IFN-γ and IL-2 in the PBMCs. Since T cells do not normally express α-ARs, the effects of PHEN or PT on IL-2 or IFN-γ cytokine production could be indirect. PHEN binding to α2-ARs present in presynaptic terminals is expected to increase availability of norepinephrine for interaction with T cell β2-ARs. A change in circulating norepinephrine due to spillover from organs that are innervated and altered epinephrine release from the adrenal medulla could alter the circulating concentrations of norepinephrine and epinephrine. If this occurs, then circulating T cell β2-AR stimulation could induce changes in T cell cytokines in this compartment. However, since TERB had no effect on PBMC IFN-γ release, the decreased IFN-γ production observed following PHEN or PT treatment is unlikely to be mediated indirectly through presynaptic α2-ARs. Alternatively, these treatments could be targeting α-ARs expressed in monocytes to alter their production of cytokines that then induce changes in T cell cytokine production. This notion is consistent with a report by Heijnen and coworkers (73) showing functional α1-ARs in peripheral blood leukocytes (presumably monocytes) of patients with polyarticular juvenile RA. Finally, the altered PBMC T cell cytokine production observed after PHEN or PT treatment could alter the T cell subtypes that enters the circulation, increasing subtypes that produce higher amounts of IFN-γ and IL-2. Mechanisms responsible for the effects of PHEN treatment on IFN-γ and IL-2 production in PBMCs during the effector phase of AA requires further investigation.
In this study, TERB or PHEN treatment did not alter production of the pro-inflammatory cytokine, TNF-α in spleen, lymph nodes, or PBMCs in arthritic rats after disease onset. This is in contrast to reports that β2-AR agonists and α-AR antagonists inhibit macrophage TNF-α production, respectively [reviewed in Ref. (74–77)]. These findings suggest an impairment of signaling via both β2- and α-ARs that occurs during the effector phase of AA. Interestingly, treatment with PT significantly reduced production of TNF-α in the spleen, MLN, and PBMCs. It is not clear why the combination drug treatment reduces TNF-α, in the absence of effects by the individual treatments, but the findings suggest interactions between β2- and α-AR signaling. Collectively, these findings indicated that SNS signaling to immune cells via β2- and α-ARs is impaired, or induces alternative second messengers in monocytes/macrophages in these organs in rats with established AA.
The greatest effect of TERB and/or PHEN on cytokine production was observed for splenic IL-10 production, where all three adrenergic treatments induced a significant increase in IL-10 production. This finding is consistent with the known effects of β2-AR agonists and α-AR antagonists to increase IL-10 production (78, 79). The combination treatment increased IL-10 production to the same extent as treatment with each drug alone, suggesting that the effects of each of the components were neither additive nor synergistic. Thus, at least in the spleen, these receptors are capable of inducing production of second messengers, despite the lack of AR-induced responses for TNF-α production. In DLNs, IL-10 production increased significantly only after treatment with combined TERB and PHEN, although there was a trend for increased IL-10 with each individual adrenergic drug. We observed no effect for any of the adrenergic treatments in IL-10 production by PBMCs. In contrast, PHEN significantly reduced IL-10 levels in the MLNs, an effect that is inconsistent with reports that α-AR agonists suppress IL-10 production (78). Thus, the pattern of IL-10 production after adrenergic treatments was dependent upon the secondary lymphoid tissue examined.
Data supports that sympathetic dysfunctions significantly alter the balance between Th1-Th2 cell differentiation in RA (18, 80). RA is dominated by a Th1 response with selective accumulation of Th1 cells within the synovial compartment (81). RA and juvenile chronic arthritis co-exist with chronically elevated sympathetic activity [reviewed in Ref. (8)]. We and others have demonstrated that sympathetic innervation of the lymphoid organs in arthritic rodents (14, 15) and in arthritic joints of RA patients (17, 82) are lost and/or reorganized. Indeed, in this study, treatment with a β2-AR agonist fails to shift T lymphocyte cytokine responses toward a Th2 profile in any of the secondary lymphoid organs of AA rats. However, in vivo treatment with the α-AR antagonist shifts the ex vivo cytokine profile from Th1 to a Th2 response, indicating that α-AR stimulation inhibits this shift. These findings are consistent with a recent report showing that catecholamines fail to shift T cell cytokines from a Th1 to a Th2 profile in PBMCs from RA patients, as seen in healthy donors (83). Thus, impaired sympathetic functioning is intimately linked with disease pathology.
Stimulating β2-ARs and/or blocking α-ARs reduces the ratio of pro-to-anti-inflammatory cytokine production by PBMCs, spleen, and DLN cells. The reduced pro-to-anti-inflammatory cytokine ratios are consistent with the well-known effects of β2-AR stimulation or α-AR blockade to inhibit TNF-α and promote IL-10 production in macrophages and dendritic cells (74–79). Spleen cells displayed the greatest drug-induced changes in anti-inflammatory profiles, largely due to greater IL-10 production. In contrast, TERB or PHEN treatment, tend to induce an anti-inflammatory secretory profile in PBMCs and DLN cells. However, dual drug treatment that augment β2- and dampen α-AR activation-induced anti-inflammatory profiles. These changes were due to reduced TNF-α secretion in PBMCs and increased IL-10 in DLN cells. Therefore, TERB, PHEN, or PT-induced anti-inflammatory profiles may be responsible, at least in part, for improved disease outcomes in our study.
Interestingly, PHEN treatment induced a pro-inflammatory cytokine profile in MLN cells, an effect due to lower IL-10 in the absence of an effect on TNF-α production. Beta2-AR stimulation inhibited TNF-α secretion, but was unable to significantly reduce the pro-to-anti-inflammatory ratio. Thus, blocking α-AR activation in the MLNs had an opposing effect on IL-10 production to that seen in the spleen. The reason for this difference may lie in the cellular source of IL-10 in these immune organs, but requires further investigation. Moreover, our data suggest that activation of either α- or β-ARs have anti-inflammatory effects in the MLNs. Why our drug treatments differentially affected inflammatory drive depending on the specific lymphoid tissue is unclear and warrants further study. Perhaps these differences reflect the unique functions of each lymphoid organ/tissue in response to the CFA challenge and the site of injection. The primary function of DLNs is to respond to the intradermal challenge, whereas the spleen participates in the systemic response. As part of the systemic response in AA, the spleen filters the blood, and is a major source of arthritogenic T cells that infiltrate affected joints. In contrast, the blood serves as a conduit for trafficking cells of the immune system. Finally, the MLNs are most responsive to antigens that travel in lymphatic vessels from the gut. Collectively, our findings indicate that the SNS maintains the ability to inhibit (via β2-ARs) or promote (via α-ARs) inflammation in PBMCs, splenocytes, and DLN cells, but not in MLN cells.
Body weights are reduced 28 days after disease induction. This finding concurs with the well-documented AA-, CIA- and RA-induced cachexia (loss of body mass without altered diet or increased activity) due to inflammation that causes hypermetabolic protein and fat breakdown, skeletal muscle wasting, and to a lesser extent, lost white adipose tissue mass (12, 84–86). Moreover, treatment with PHEN, TERB, or PT significantly reduced disease-associated cachexia. The significance of these findings is underscored by cachexia occurring in ~2/3rds of all RA patients, and is a major contributor in greater morbidity and mortality in RA patients (84, 87, 88). Here, we did not monitor food intake, but other studies have shown that this factor is not contributory to disease-induced weight loss (89). However, inflammation indirectly reduces cellular mass by protein breakdown due to activation of the ubiquitin–proteasome pathway (90) and lipolysis (86). These effects are mediated at least in part by TNF-α (91). Adrenergic therapies in our study suppresses ex vivo TNF-α production and the pro- to anti-inflammatory ratios in a treatment- and immune organ-specific manner, consistent with a role for inflammation in rheumatoid cachexia.
In summary, it is clear that the critical functions of the SNS to regulate lymphocyte proliferation, Th1/Th2 cell differentiation, and inflammation are impaired/altered in AA rats, due to altered β2- and α-AR functions. Selective α-AR blockade and/or β-AR activation in AA suppresses ex vivo proliferation, but is independent of sympathetic regulation of IL-2 production in AA. This is true in all immune organs examined, except PBMCs, and is consistent with the function of blood as a conduit for immune cell trafficking. Adrenergic treatments affect Th1/Th2 cytokines differently in every tissue examined in AA, perhaps reflecting the different immune functions of each organ. Most striking was the β2-AR-mediated increase in IFN-γ in the DLNs and lack of a β2-AR-mediated effect on IFN-γ in the spleen, particularly since arthritogenic T cells are generated at both sites (92, 93). Regardless of these differences, the overall outcome is a failure to drive Th2-type cytokine expression via β2-AR stimulation, and thus, loss of a negative feedback mechanism for regulating cellular immunity. These findings are consistent with reports of disease-induced elevation in sympathetic tone (8), and high norepinephrine concentration-dependent β2-AR down-regulation, desensitization, and/or shifts in G-protein coupling (18, 82, 94, 95). Indeed, we have previously discovered differences in phosphorylation of β2-ARs in DLN and spleen cells collected from AA rats that may explain the IFN-γ findings in this study (18). Further, essential SNS regulation of innate immunity, via β2- and α-ARs, is also impaired as demonstrated, with inability of β2-AR stimulation or α-AR blockade to reduce TNF-α and increase IL-10 production in DLN cells. Stimulation of β2-ARs similarly fails to reduce TNF-α production in the spleen cells.
Collectively, our findings indicate disrupted β2-ARs, but not α-ARs signaling in AA, which consequently derails sympathetic regulation of lymphocyte expansion, Th cell differentiation, and inflammation required for immune system homeostasis. The clinical relevance of our findings is several-fold. First, they underscore the complexity of the disease’s relevant immune functions, their regulation by the SNS and the limitations of peripheral blood to decipher complex pathology. Secondly, distinct differences in SNS-immune interactions in spleen and lymph nodes likely reflect functional and microenvironmental differences of these immune organs, which currently are under appreciated and sources of discrepancies, inconsistencies, and confusion in the literature. Thus, relevant animal models for RA and novel approaches are required to elucidate both immune and neural-immune mechanisms in disease pathology. Finally, disease-modifying and suppressive inflammatory effects of targeting β2- and α-ARs strongly support that combined AR-targeted therapies have great therapeutic potential to reduce joint destruction and inflammation and complications of RA, like cachexia. While promising, a greater understanding of the mechanisms by which these beneficial effects are realized is essential to evaluate therapeutic potential and guide clinical applications.
Author Contributions
All authors made a substantial contribution to conceiving and/or designing of the research project, the acquiring, analyses, or interpretation of the data presented in this paper. All authors have contributed to writing of this paper and its intellectual content and critical evaluation of content. All authors have approved the final version of this paper, and are accountable for the accuracy and integrity of the research presented in this paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Christine Molinaro for comments and careful editing of this manuscript.
References
1. Ramos-Remus C, Duran-Barragan S, Castillo-Ortiz JD. Beyond the joints: neurological involvement in rheumatoid arthritis. Clin Rheumatol (2012) 31:1–12. doi: 10.1007/s10067-011-1841-z
2. Koopman FA, Stoof SP, Straub RH, Van Maanen MA, Vervoordeldonk MJ, Tak PP. Restoring the balance of the autonomic nervous system as an innovative approach to the treatment of rheumatoid arthritis. Mol Med (2011) 17:937–48. doi:10.2119/molmed.2011.00065
3. del Rey A, Wolff C, Wildmann J, Randolf A, Hahnel A, Besedovsky HO, et al. Disrupted brain-immune system-joint communication during experimental arthritis. Arthritis Rheum (2008) 58:3090–9. doi:10.1002/art.23869
4. Straub RH, Härle P. Sympathetic neurotransmitters in joint inflammation. Rheum Dis Clin North Am (2005) 31:43–59,viii. doi:10.1016/j.rdc.2004.09.003
5. Pablos JL, Cañete JD. Immunopathology of rheumatoid arthritis. Curr Top Med Chem (2013) 13:705–11. doi:10.2174/1568026611313060003
6. Rodríguez-Fernández JL. Antigen presentation by dendritic cells in rheumatoid arthritis. Curr Top Med Chem (2013) 13:712–9. doi:10.2174/1568026611313060004
7. Yadav RK, Gupta R, Deepak KK. A pilot study on short term heart rate variability & its correlation with disease activity in Indian patients with rheumatoid arthritis. Indian J Med Res (2012) 136:593–8.
8. Härle P, Straub RH, Wiest R, Mayer A, Schölmerich J, Atzeni F, et al. Increase of sympathetic outflow measured by neuropeptide Y and decrease of the hypothalamic-pituitary-adrenal axis tone in patients with systemic lupus erythematosus and rheumatoid arthritis: another example of uncoupling of response systems. Ann Rheum Dis (2006) 65:51–6. doi:10.1136/ard.2005.038059
9. Levine JD, Coderre TJ, Helms C, Basbaum AI. Beta 2-adrenergic mechanisms in experimental arthritis. Proc Natl Acad Sci U S A (1988) 85:4553–6. doi:10.1073/pnas.85.12.4553
10. Malfait AM, Malik AS, Marinova-Mutafchieva L, Butler DM, Maini RN, Feldmann M. The beta2-adrenergic agonist salbutamol is a potent suppressor of established collagen-induced arthritis: mechanisms of action. J Immunol (1999) 162:6278–83.
11. Lorton D, Lubahn C, Bellinger DL. Potential use of drugs that target neural-immune pathways in the treatment of rheumatoid arthritis and other autoimmune diseases. Curr Drug Targets Inflamm Allergy (2003) 2:1–30. doi:10.2174/1568010033344499
12. Lubahn CL, Schaller JA, Bellinger DL, Sweeney S, Lorton D. The importance of timing of adrenergic drug delivery in relation to the induction and onset of adjuvant-induced arthritis. Brain Behav Immun (2004) 18:563–71. doi:10.1016/j.bbi.2004.02.004
13. Härle P, Möbius D, Carr DJ, Schölmerich J, Straub RH. An opposing time-dependent immune-modulating effect of the sympathetic nervous system conferred by altering the cytokine profile in the local lymph nodes and spleen of mice with type II collagen-induced arthritis. Arthritis Rheum (2005) 52:1305–13. doi:10.1002/art.20987
14. Lorton D, Lubahn C, Lindquist CA, Schaller J, Washington C, Bellinger DL. Changes in the density and distribution of sympathetic nerves in spleens from Lewis rats with adjuvant-induced arthritis suggest that an injury and sprouting response occurs. J Comp Neurol (2005) 489:260–73. doi:10.1002/cne.20640
15. Lorton D, Lubahn C, Sweeney S, Major A, Lindquist CA, Schaller J, et al. Differences in the injury/sprouting response of splenic noradrenergic nerves in Lewis rats with adjuvant-induced arthritis compared with rats treated with 6-hydroxydopamine. Brain Behav Immun (2009) 23:276–85. doi:10.1016/j.bbi.2008.10.004
16. Lorton D, Lubahn C, Klein N, Schaller J, Bellinger DL. Dual role for noradrenergic innervation of lymphoid tissue and arthritic joints in adjuvant-induced arthritis. Brain Behav Immun (1999) 13:315–34. doi:10.1006/brbi.1999.0564
17. Miller LE, Jüsten HP, Schölmerich J, Straub RH. The loss of sympathetic nerve fibers in the synovial tissue of patients with rheumatoid arthritis is accompanied by increased norepinephrine release from synovial macrophages. FASEB J (2000) 14:2097–107. Erratum in: FASEB J (2003) 17(8):4A. FASEB J (2001) 15(13):2554. doi:10.1096/fj.99-1082com
18. Lorton D, Bellinger DL, Schaller JA, Shewmaker E, Osredkar T, Lubahn C. Altered sympathetic-to-immune cell signaling via β-adrenergic receptors in adjuvant arthritis. Clin Dev Immunol (2013) 2013:764395. doi:10.1155/2013/764395
19. Mishima K, Otani H, Tanabe T, Kawasaki H, Oshiro A, Saito N, et al. Molecular mechanisms for alpha2-adrenoceptor-mediated regulation of synoviocyte populations. Jpn J Pharmacol (2001) 85:214–26. doi:10.1254/jjp.85.214
20. Schaible HG, Straub RH. Function of the sympathetic supply in acute and chronic experimental joint inflammation. Auton Neurosci (2013) 182:55–64. doi:10.1016/j.autneu.2013
21. Bellinger DL, Lorton D. Autonomic regulation of cellular immune function. Auton Neurosci (2014) 182C:15–41. doi:10.1016/j.autneu.2014.01.006
22. Smol’nikova VV, Voznyuk AV, Potapnev MP. Cytokine-induced differentiation and proliferation of human T lymphocytes in vitro: effects of interleukin 2 and interleukin 6. Bull Exp Biol Med (2000) 129:567–70. doi:10.1007/BF02434879
23. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clones. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol (1986) 136:2348–57.
24. Paul WE, Seder RA. Lymphocyte responses and cytokines. Cell (1994) 76:241–51. doi:10.1016/0092-8674(94)90332-8
25. Ackerman NR, Rooks WH II, Shott L, Genant H, Maloney P, West E. Effects of naproxen on connective tissue changes in the adjuvant arthritic rat. Arthritis Rheum (1979) 22:1365–74. doi:10.1002/art.1780221208
26. Aitken SJ, Landao-Bassonga E, Ralston SH, Idris AI. Beta2-adrenoreceptor ligands regulate osteoclast differentiation in vitro by direct and indirect mechanisms. Arch Biochem Biophys (2009) 482:96–103. doi:10.1016/j.abb.2008.11.012
27. Huang HH, Brennan TC, Muir MM, Mason RS. Functional alpha1- and beta2-adrenergic receptors in human osteoblasts. J Cell Physiol (2009) 220:267–75. doi:10.1002/jcp.21761
28. Elefteriou F, Ahn JD, Takeda S, Starbuck M, Yang X, Liu X, et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature (2005) 434:514–20. doi:10.1038/nature03398
29. Suzuki A, Palmer G, Bonjour JP, Caverzasio J. Catecholamines stimulate the proliferation and alkaline phosphatase activity of MC3T3-E1 osteoblast-like cells. Bone (1998) 23:197–203. doi:10.1016/S8756-3282(98)00099-4
30. Lai LP, Mitchell J. Beta2-adrenergic receptors expressed on murine chondrocytes stimulate cellular growth and inhibit the expression of Indian hedgehog and collagen type X. J Cell Biochem (2008) 104:545–53. doi:10.1002/jcb.21646
31. Opolka A, Straub RH, Pasoldt A, Grifka J, Grässel S. Substance P and norepinephrine modulate murine chondrocyte proliferation and apoptosis. Arthritis Rheum (2012) 64:729–39. doi:10.1002/art.33449
32. Wythe SE, Nicolaidou V, Horwood NJ. Cells of the immune system orchestrate changes in bone cell function. Calcif Tissue Int (2014) 94:98–111. doi:10.1007/s00223-013-9764-0
33. Pérez E, García-Martínez O, Arroyo-Morales M, Reyes-Botella C, Ruiz C. Modulation of antigenic phenotype in cultured human osteoblast-like cells by FGFb, TGFbeta1, PDGF-BB, IL-2, IL-1beta, LPS and IFNgamma. Biosci Rep (2006) 26:281–9. doi:10.1007/s10540-006-9022-z
34. Tanaka Y, Nakayamada S, Okada Y. Osteoblasts and osteoclasts in bone remodeling and inflammation. Curr Drug Targets Inflamm Allergy (2005) 4:325–8. doi:10.2174/1568010054022015
35. Suleyman H, Halici Z, Cadirci E, Hacimuftuoglu A, Bilen H. Indirect role of beta2-adrenergic receptors the mechanism of anti-inflammatory action of NSAIDS. J Physiol Pharmacol (2008) 59:661–72.
36. Uzkeser H, Cadirci E, Halici Z, Odabasoglu F, Polat B, Yuksel TN, et al. Anti-inflammatory and antinociceptive effects of salbutamol on acute and chronic models of inflammation in rats: involvement of an antioxidant mechanism. Mediators Inflamm (2012) 2012:438912. doi:10.1155/2012/438912
37. Bosmann M, Grailer JJ, Zhu K, Matthay MA, Sarma JV, Zetoune FS, et al. Anti-inflammatory effects of β2 adrenergic receptor agonists in experimental acute lung injury. FASEB J (2012) 26:2137–44. doi:10.1096/fj.11-201640
38. Copik AJ, Ma C, Kosaka A, Sahdeo S, Trane A, Ho H, et al. Facilitatory interplay in alpha 1a and beta 2 adrenoceptor function reveals a non-Gq signaling mode: implications for diversification of intracellular signal transduction. Mol Pharmacol (2009) 75:713–28. doi:10.1124/mol.108.050765
39. Uberti MA, Hague C, Oller H, Minneman KP, Hall RA. Heterodimerization with beta2-adrenergic receptors promotes surface expression and functional activity of alpha1D-adrenergic receptors. J Pharmacol Exp Ther (2005) 313:16–23. doi:10.1124/jpet.104.079541
40. Vernon-Roberts B, Liyanage SP, Currey HL. Adjuvant arthritis in the rat. Distribution of fluorescent material after footpad injection of rhodamine-labelled tubercle bacilli. Ann Rheum Dis (1975) 35:389–97. doi:10.1136/ard.35.5.389
41. Cannon GW, Harper DS, Clayton F, Griffiths MM. Passive transfer of adjuvant-induced arthritis into irradiated DA recipient rats. Autoimmunity (1993) 15:267–74. doi:10.3109/08916939309115748
42. Cope AP, Liblau RS, Yang XD, Congia M, Laudanna C, Schreiber RD, et al. Chronic tumor necrosis factor alters T cell responses by attenuating T cell receptor signaling. J Exp Med (1997) 185:1573–84. doi:10.1084/jem.185.9.1573
43. Kassiotis G, Kollias G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. J Exp Med (2001) 193:427–34. doi:10.1084/jem.193.4.427
44. Clark J, Vagenas P, Panesar M, Cope AP. What does tumour necrosis factor excess do to the immune system long term? Ann Rheum Dis (2005) 64(Suppl 4):iv70–6. doi:10.1136/ard.2005.042523
45. Cazaux CA, Sterin-Borda L, Gorelik G, Cremaschi GA. Down-regulation of beta-adrenergic receptors induced by mitogen activation of intracellular signaling events in lymphocytes. FEBS Lett (1995) 364:120–4. doi:10.1016/0014-5793(95)00366-H
46. Anakwe O, Zhou S, Benovic J, Aksoy M, Kelsen SG. Interleukins impair beta-adrenergic receptor adenylate cyclase (beta AR-AC) system function in human airway epithelial cells. Chest (1995) 107(3 Suppl):138S–9S. doi:10.1378/chest.107.3_Supplement.138S
47. Mak JC, Hisada T, Salmon M, Barnes PJ, Chung KF. Glucocorticoids reverse IL-1beta-induced impairment of beta-adrenoceptor-mediated relaxation and up-regulation of G-protein-coupled receptor kinases. Br J Pharmacol (2002) 135:987–96. doi:10.1038/sj.bjp.0704545
48. Grisanti LA, Perez DM, Porter JE. Modulation of immune cell function by α(1)-adrenergic receptor activation. Curr Top Membr (2011) 67:113–38. doi:10.1016/B978-0-12-384921-2.00006-9
49. Liao W, Lin JX, Leonard WJ. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol (2011) 23:598–604. doi:10.1016/j.coi.2011.08.003
50. Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh TY, et al. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor alpha-chain expression. Nat Immunol (2008) 9:1288–96. doi:10.1038/ni.1656
51. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity (2007) 26:371–81. doi:10.1016/j.immuni.2007.02.009
52. Ballesteros-Tato A, León B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity (2012) 36:847–56. doi:10.1016/j.immuni.2012.02.012
53. Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, et al. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med (2007) 13:711–8. doi:10.1038/nm1585
54. Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, et al. Mature T lymphocyte apoptosis – immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol (1999) 17:221–53. doi:10.1146/annurev.immunol.17.1.221
55. Mingari MC, Gerosa F, Carra G, Accolla RS, Moretta A, Zubler RH, et al. Human interleukin-2 promotes proliferation of activated B cells via surface receptors similar to those of activated T cells. Nature (1984) 312:641–3. doi:10.1038/312641a0
56. Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell (2010) 140:845–58. doi:10.1016/j.cell.2010.02.021
57. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi:10.1016/j.cell.2008.05.009
58. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity (2009) 30:636–45. doi:10.1016/j.immuni.2009.04.010
59. Kitas GD, Salmon M, Farr M, Gaston JS, Bacon PA. Deficient interleukin 2 production in rheumatoid arthritis: association with active disease and systemic complications. Clin Exp Immunol (1988) 73:242–9.
60. Avrămescu C, Vere CC, Mărgăritescu C, Turculeanu A, Bălăsoiu M, Rogoz S. Cytokinic panel in rheumatoid arthritis and correlation with histological patterns of synovitis – active type of disease. Rom J Morphol Embryol (2005) 46: 87–92.
61. Kokkonen H, Söderström I, Rocklöv J, Hallmans G, Lejon K, Rantapää Dahlqvist S. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum (2010) 62:383–91. doi:10.1002/art.27186
62. Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity (2013) 38:13–25. doi:10.1016/j.immuni.2013.01.004
63. Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity (2002) 17:167–78. doi:10.1016/S1074-7613(02)00367-9
64. Straub RH, Rauch L, Rauh L, Pongratz G. Sympathetic inhibition of IL-6, IFN-γ, and KC/CXCL1 and sympathetic stimulation of TGF-β in spleen of early arthritic mice. Brain Behav Immun (2011) 25:1708–15. doi:10.1016/j.bbi.2011.07.001
65. Loza MJ, Foster S, Peters SP, Penn RB. Beta-agonists modulate T-cell functions via direct actions on type 1 and type 2 cells. Blood (2006) 107:2052–60. doi:10.1182/blood-2005-08-3265
66. Heijink IH, Vellenga E, Borger P, Postma DS, Monchy JG, Kauffman HF. Polarized Th1 and Th2 cells are less responsive to negative feedback by receptors coupled to the AC/cAMP system compared to freshly isolated T cells. Br J Pharmacol (2003) 138:1441–50. doi:10.1038/sj.bjp.0705193
67. Ramer-Quinn DS, Swanson MA, Lee WT, Sanders VM. Cytokine production by naïve and primary effector CD4+ T cells exposed to norepinephrine. Brain Behav Immun (2000) 14:239–55. doi:10.1006/brbi.2000.0603
68. Hakonarson H, Herrick DJ, Serrano PG, Grunstein MM. Mechanism of cytokine-induced modulation of beta-adrenoceptor responsiveness in airway smooth muscle. J Clin Invest (1996) 97:2593–600. doi:10.1172/JCI118708
69. Chung MK, Gulick TS, Rotondo RE, Schreiner GF, Lange LG. Mechanism of cytokine inhibition of beta-adrenergic agonist stimulation of cyclic AMP in rat cardiac myocytes. Impairment of signal transduction. Circ Res (1990) 67:753–63. doi:10.1161/01.RES.67.3.753
70. Whitehouse M. Oily adjuvants and autoimmunity: now time for reconsideration? Lupus (2012) 21:217–22. doi:10.1177/0961203311429818
71. Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE. Differential expression of the beta2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help. J Immunol (1997) 158:4200–10.
72. Ramer-Quinn DS, Baker RA, Sanders VM. Activated T helper 1 and T helper 2 cells differentially express the beta-2-adrenergic receptor: a mechanism for selective modulation of T helper 1 cell cytokine production. J Immunol (1997) 159:4857–67.
73. Heijnen CJ, Rouppe van der Voort C, Wulffraat N, van der Net J, Kuis W, Kavelaars A. Functional alpha 1-adrenergic receptors on leukocytes of patients with polyarticular juvenile rheumatoid arthritis. J Neuroimmunol (1996) 71:223–6. doi:10.1016/S0165-5728(96)00125-7
74. Huang JL, Zhang YL, Wang CC, Zhou JR, Ma Q, Wang X, et al. Enhanced phosphorylation of MAPKs by NE promotes TNF-α production by macrophage through α adrenergic receptor. Inflammation (2012) 35:527–34. doi:10.1007/s10753-011-9342-4
75. Donnelly LE, Tudhope SJ, Fenwick PS, Barnes PJ. Effects of formoterol and salmeterol on cytokine release from monocyte-derived macrophages. Eur Respir J (2010) 36:178–86. doi:10.1183/09031936.00158008
76. Tanaka S, Tsutsui M, Kishida T, Souma S, Kuroda J, Yoshida T. Salbutamol inhibits lipopolysaccharide-induced inflammatory responses in rat peritoneal macrophages. J Toxicol Sci (2010) 35:327–34. doi:10.2131/jts.35.327
77. Szelényi J, Kiss JP, Vizi ES. Differential involvement of sympathetic nervous system and immune system in the modulation of TNF-alpha production by alpha2- and beta-adrenoceptors in mice. J Neuroimmunol (2000) 103:34–40. doi:10.1016/S0165-5728(99)00234-9
78. Szelényi J, Kiss JP, Puskás E, Selmeczy Z, Szelényi M, Vizi ES. Opposite role of alpha2- and beta-adrenoceptors in the modulation of interleukin-10 production in endotoxaemic mice. Neuroreport (2000) 11:3565–8. doi:10.1097/00001756-200011090-00032
79. Suberville S, Bellocq A, Fouqueray B, Philippe C, Lantz O, Perez J, et al. Regulation of interleukin-10 production by beta-adrenergic agonists. Eur J Immunol (1996) 26:2601–5. doi:10.1002/eji.1830261110
80. Sanders VM, Straub RH. Norepinephrine, the beta-adrenergic receptor, and immunity. Brain Behav Immun (2002) 16:290–332. doi:10.1006/brbi.2001.0639
81. Fournier C. Where do T cells stand in rheumatoid arthritis? Joint Bone Spine (2005) 72:527–32. doi:10.1016/j.jbspin.2004.12.012
82. Wahle M, Krause A, Pierer M, Hantzschel H, Baerwald CG. Immunopathogenesis of rheumatic diseases in the context of neuroendocrine interactions. Ann N Y Acad Sci (2002) 966:355–64. doi:10.1111/j.1749-6632.2002.tb04235.x
83. Wahle M, Hanefeld G, Brunn S, Straub RH, Wagner U, Krause A, et al. Failure of catecholamines to shift T-cell cytokine responses toward a Th2 profile in patients with rheumatoid arthritis. Arthritis Res Ther (2006) 8:R138. doi:10.1186/ar2028
84. Roubenoff R, Roubenoff RA, Cannon JG, Kehayias JJ, Zhuang H, Dowson-Hughes B, et al. Rheumatoid cachexia: cytokine-driven hypermetabolism accompanying reduced body cell mass in chronic inflammation. J Clin Invest (1994) 93:2379–86. doi:10.1172/JCI117244
85. Granado M, Martín AI, Villanúa MA, López-Calderón A. Experimental arthritis inhibits the insulin-like growth factor-I axis and induces muscle wasting through cyclooxygenase-2 activation. Am J Physiol Endocrinol Metab (2007) 292:E1656–65. doi:10.1152/ajpendo.00502.2006
86. Martín AI, Castillero E, Granado M, López-Menduiña M, Villanúa MA, López-Calderón A. Adipose tissue loss in adjuvant arthritis is associated with a decrease in lipogenesis, but not with an increase in lipolysis. J Endocrinol (2008) 197:111–9. doi:10.1677/JOE-07-0491
87. Summers GD, Deighton CM, Rennie MJ, Booth AH. Rheumatoid cachexia: a clinical perspective. Rheumatology (Oxford) (2008) 47:1124–31. doi:10.1093/rheumatology/ken146
88. Filippin LI, Teixeira VN, Viacava PR, Lora PS, Xavier LL, Xavier RM. Temporal development of muscle atrophy in murine model of arthritis is related to disease severity. J Cachexia Sarcopenia Muscle (2013) 4:231–8. doi:10.1007/s13539-013-0102-1
89. Roubenoff R, Freeman LM, Smith DE, Abad LW, Dinarello CA, Kehayias JJ. Adjuvant arthritis as a model of inflammatory cachexia. Arthritis Rheum (1997) 40:534–9. doi:10.1002/art.1780400320
90. Granado M, Martín AI, Priego T, López-Calderón A, Villanúa MA. Tumour necrosis factor blockade did not prevent the increase of muscular muscle RING finger-1 and muscle atrophy F-box in arthritic rats. J Endocrinol (2006) 191:319–26. doi:10.1677/joe.1.06931
91. Llovera M, García-Martínez C, Agell N, López-Soriano FJ, Argilés JM. TNF can directly induce the expression of ubiquitin-dependent proteolytic system in rat soleus muscles. Biochem Biophys Res Commun (1997) 230:238–41. doi:10.1006/bbrc.1996.5827
92. Holm BC, Lorentzen JC, Bucht A. Adjuvant oil induces waves of arthritogenic lymph node cells prior to arthritis onset. Clin Exp Immunol (2004) 137:59–64. doi:10.1111/j.1365-2249.2004.02498.x
93. Holmberg J, Tuncel J, Yamada H, Lu S, Olofsson P, Holmdah R. Pristane, a non-antigenic adjuvant, induces MHC class II-restricted, arthritogenic T cells in the rat. J Immunol (2006) 176:1172–9. doi:10.4049/jimmunol.176.2.1172
94. Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, et al. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem (2006) 281:1261–73. doi:10.1074/jbc.M506576200
Keywords: sympathetic nervous system, rheumatoid arthritis, macrophage, T cell, cytokine balance, spleen, lymph nodes, peripheral blood mononuclear cells
Citation: Lubahn CL, Lorton D, Schaller JA, Sweeney SJ and Bellinger DL (2014) Targeting α- and β-adrenergic receptors differentially shifts Th1, Th2, and inflammatory cytokine profiles in immune organs to attenuate adjuvant arthritis. Front. Immunol. 5:346. doi: 10.3389/fimmu.2014.00346
Received: 01 May 2014; Accepted: 07 July 2014;
Published online: 11 August 2014.
Edited by:
Oreste Gualillo, Santiago University Clinical Hospital, SpainReviewed by:
Fulvio D’Acquisto, Queen Mary University of London, UKGiuseppe Matarese, Università di Salerno, Italy
Copyright: © 2014 Lubahn, Lorton, Schaller, Sweeney and Bellinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dianne Lorton, College of Arts and Sciences, Kent State University, 113R Bowman Hall, 850 University Esplanade, Kent, OH 44242, USA e-mail:ZGRpZnJhbjdAa2VudC5lZHU=
†Co-First Authors