 Zsuzsa Szondy
Zsuzsa Szondy Éva Garabuczi
Éva Garabuczi Gergely Joós
Gergely Joós Gregory J. Tsay
Gregory J. Tsay Zsolt Sarang
Zsolt Sarang- 1Department of Dental Biochemistry, Faculty of Dentistry, University of Debrecen, Debrecen, Hungary
- 2Department of Internal Medicine, Faculty of Medicine, Chung Shan Medical University Hospital, Taichung, Taiwan
- 3Department of Biochemistry and Molecular Biology, Faculty of Medicine, University of Debrecen, Debrecen, Hungary
In healthy individuals, billions of cells die by apoptosis every day. Removal of the dead cells by phagocytosis (a process called efferocytosis) must be efficient to prevent secondary necrosis and the consequent release of pro-inflammatory cell contents that damages the tissue environment and provokes autoimmunity. In addition, detection and removal of apoptotic cells generally induces an anti-inflammatory response. As a consequence improper clearance of apoptotic cells, being the result of either genetic anomalies and/or a persistent disease state, contributes to the establishment and progression of a number of human chronic inflammatory diseases such as autoimmune and neurological disorders, inflammatory lung diseases, obesity, type 2 diabetes, or atherosclerosis. During the past decade, our knowledge about the mechanism of efferocytosis has significantly increased, providing therapeutic targets through which impaired phagocytosis of apoptotic cells and the consequent inflammation could be influenced in these diseases.
Introduction
Efficient execution of apoptotic cell death followed by efficient clearance mediated by professional and by non-professional neighboring phagocytes, is a key mechanism in maintaining tissue homeostasis. Every day, billions of our cells die and get cleared without initiating inflammation and an immune response (1). Proper clearance of dead cells also contributes to the initiation of tissue repair processes following injury (2–4). In addition, efficient removal of apoptotic neutrophils is also a key event in the resolution of inflammation (5).
Increasing evidence suggest that improper clearance of apoptotic cells, being the result of either genetic anomalies and/or a persistent disease state, contributes to the establishment and progression of a number of human diseases via affects on the maintenance of tissue homeostasis, tissue repair, and inflammation (6). Autoimmune disorders, in which both animal models and human research indicate a strong relationship between improper clearance and the development of the disease, represent the best characterized example of such diseases. The regulated nature of apoptotic cell death normally prevents the leakage of the immunogenic intracellular contents. If, however, these cells are not promptly cleared, they undergo secondary necrosis leading to the release of the intracellular antigens and DNA, which in the long-term provoke an auto-inflammatory response (7). Thus, in most of the knock out mice in which efferocytosis is impaired, systemic lupus erythematosus (SLE) like autoimmunity develops (8–13). Human SLE is also accompanied by improper efferocytosis (7), and can develop also as a result of a genetic deficiency of the phagocytosis process (13).
While in SLE improper clearance of apoptotic cells affects all the tissues, in several chronic inflammatory respiratory diseases, such as chronic obstructive pulmonary disease (COPD), cystic fibrosis, and asthma, increased numbers of apoptotic cells are seen only in the sputum and lung tissue (14). Though so far no evidence was provided for a definite linkage between genetic anomalies affecting efferocytosis and lung disease, inefficient apoptotic clearance in the lung was detected in all these respiratory diseases (15).
Macrophages play a key role in the development of atherosclerosis, and impaired clearance of apoptotic macrophages characterizes the late plaques, in which uncleared apoptotic cells undergo secondary necrosis leading to the formation of an unstable necrotic core and the maintenance of inflammation (16). Impaired efferocytosis, however, might also contribute to the development of the disease, as knock out mice deficient in efferocytosis are prone to develop atherosclerosis on LDL or ApoE null genetic backgrounds (17–20). An excess of apoptotic cells was detected in a numerous neurodegenerative diseases as well, such as Parkinson’s, Alzheimer’s, and Huntington’s disease (21). Though the elevated levels of apoptotic cells might also be the result of an increased neuronal cell death, in these diseases loss of signaling by fractalkine (an apoptotic cell “find me” signal) resulted in an increase in the number of dying cells and worsening of the disease (22).
Interestingly, type 2 diabetes and obesity were also shown to be associated with impaired phagocytosis of apoptotic β-cells in the pancreas in autoimmune diabetes-prone rats (23) and in ob/ob and db/db mice (24). The phenomenon seems to be related to an enhanced saturated and/or decreased ω-3 fatty acid composition of the plasma membrane, which leads to a decreased phosphatidylinositol 3-kinase activation during the uptake of apoptotic cells (24).
Mechanisms Contributing to Efficient Phagocytosis of Apoptotic Cells
“Find Me” and “Eat Me” Signals
To ensure effective removal, apoptotic cells recruit phagocytes by releasing various soluble “find me” signals. These signals include lysophosphatidylcholine (25), CX3CL1/fractalkine (26), sphingosine-1-phosphate (27), the nucleotides ATP and UTP (28), thrombospondin-1 (TSP-1) (29), and cleaved human tyrosyl-tRNA synthetase (30). Upon arrival at the target cells, phagocytes must distinguish between apoptotic and viable cells. Apoptotic cells display apoptotic cell-associated molecular patterns (ACAMPs), which includes the appearance of “eat me” signals on their cell surface (5). These can bind either directly or through bridging molecules to receptors on phagocytes (Figure 1). Externalization of phosphatidylserine (PS) on the outer leaflet of the cell membrane is the best characterized “eat me” signal during apoptosis. The T-cell immunoglobulin- and mucin-domain-containing molecule (Tim4), stabilin-2, and brain-specific angiogenesis inhibitor 1 (BAI1) were reported to directly recognize PS on dying cells (31–33), while other receptors such as Mer tyrosine kinase (MerTk), scavenger receptor SCARF1, CD36, and integrin αv/β3/β5 together with CD36 or tissue transglutaminase (TG2) recognize apoptotic cells through bridging molecules. Gas6 and protein S were found to facilitate apoptotic cell clearance by recognizing PS on apoptotic cells and MerTk receptor on phagocytes (34, 35). TSP-1 and milk-fat globulin-E8 (MFG-E8) also bind to PS and are recognized by the integrin αv/β3/CD36 or integrin αv/β3/TG2 receptor complexes, respectively (36–38). The collectin family member serum protein C1q also serves as a bridging molecule by recognizing annexin A2 and A5 on the apoptotic cells (39) and binding either SCARF1 scavenger receptor or the calreticulin associated LRP1/CD91 receptor on phagocytes (39, 40). The LPS coreceptor CD14 can also act as a tethering receptor for apoptotic cells, albeit its exact ligand remains unknown (41). Distinguishing between apoptotic and viable cells is further ensured by the “do not eat me” signals, which inhibit the uptake of living cells. CD47, activating SIRPα receptor, is one of these signals being expressed on living cells but altered or diminished on apoptotic cell surface (42). Additionally, homophilic interaction between CD31 on the target cells and macrophages was shown to mediate cell detachment from phagocytes, thus inhibiting phagocytosis of living cells (43).
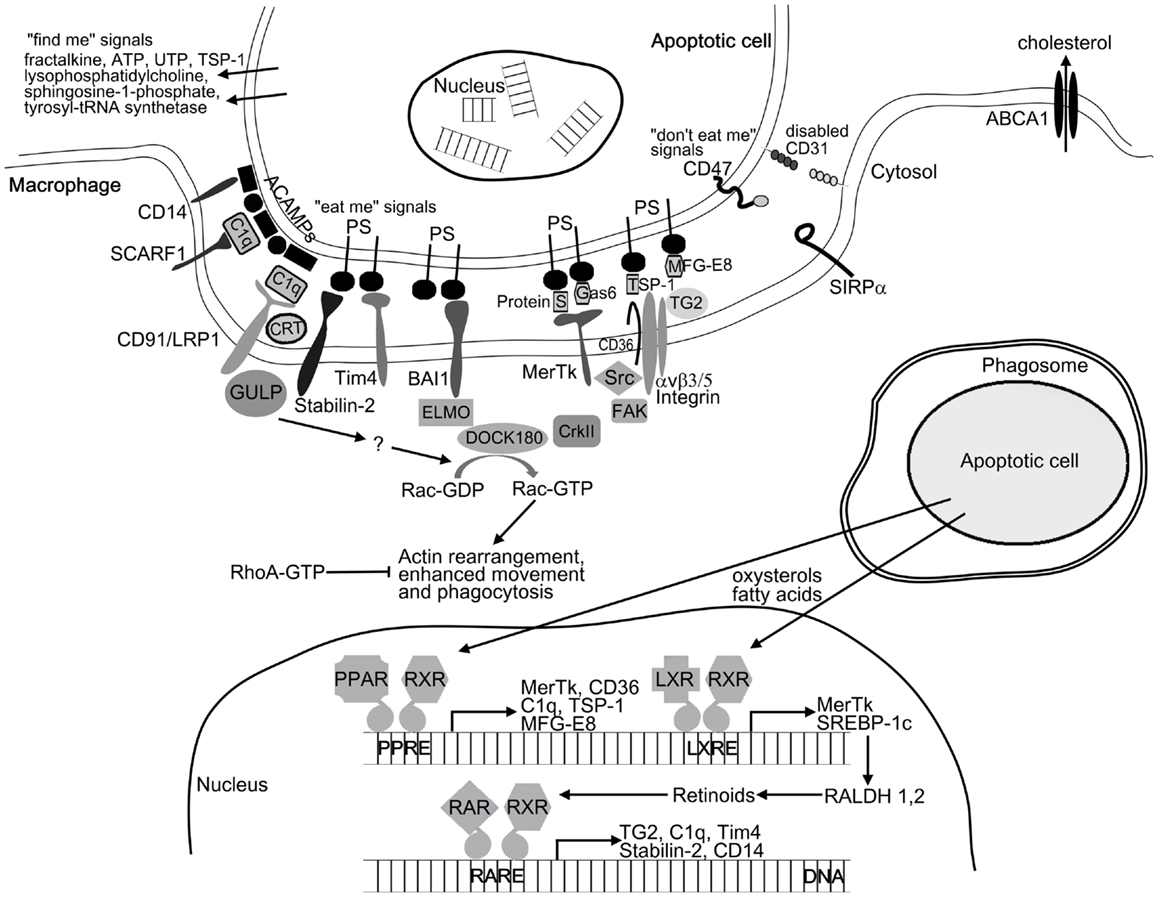
Figure 1. Mechanism of apoptotic cell clearance. For initiating phagocytosis apoptotic cells release “find me” signals for the phagocytes. After finding the recognition of apoptotic cells by phagocyte receptors is mediated by the display of “eat me” signals (e.g., PS and ACAMPs) and the disappearance of the so-called “do not eat me” signals (e.g., CD31 and CD47) on the apoptotic cell surface. Among others, these receptors include the PS receptors (Tim4, stabilin-2, and BAI1) and receptors such as MerTk, SCARF1, CD36, and integrin αvβ5 together with TG2 recognizing apoptotic cells through bridging molecules (e.g., TSP-1, C1q, Gas6, MFG-E8, and protein S). Binding of apoptotic cells to the phagocytic receptors triggers two evolutionary conserved signaling pathways. MerTk, BAI1, and αvβ3/5 receptors will activate the DOCK180/CrkII/ELMO complex, while CD91/LPR1 and stabilin-2 will activate the adaptor protein GULP. Both pathways converge on the small GTPase Rac, which initiates actin rearrangement and phagocytosis. Following engulfment, apoptotic cell derived lipids (oxysterols and fatty acids) trigger the lipid-sensing LXR and PPAR receptors leading to enhanced retinoid production. Retinoid receptors together with LXR and PPARs upregulate a number of phagocytic receptors to further enhance the engulfing capacity of macrophages under conditions when the rate of apoptosis is increased.
Engulfment and Ingestion of the Apoptotic Corpses
Uptake of the apoptotic cells requires the reorganization of the actin filament network, which drives the movement of the cell, formation of the phagocytic cup and the phagosome. This process is regulated by the small GTPases RhoA, Cdc42, and Rac. While RhoA activation was found to inhibit apoptotic cell phagocytosis, Cdc42, and Rac were shown to enhance it (44). Phagocytic receptors activate two evolutionary conserved pathways both converging on the activation of Rac-1, a small GTPase (45) (Figure 1). The first pathway is initiated by MerTk or integrin αv/β5 receptors (46, 47), resulting in association of the adaptor protein ELMO with the Rac GEF DOCK180 forming a bipartite GEF (48). Recruitment of the ELMO/DOCK180 complex to the cell membrane might require the adaptor protein CrkII, but binding of ELMO to the carboxyl terminus of BAI1 also recruits DOCK180 to the phagocytic membranes (33). The second pathway activating the Rac is initiated by LRP1 (CD91) (49) or by stabilin-2 receptors followed by recruitment of the adaptor protein GULP (50). Further steps, resulting in the activation of Rac are still unclear. The newly formed phagosome must fuse with lysosomes to degrade the dead cells. Recently, several autophagic genes were described to participate in phagosome maturation (51, 52). Following phagolysosomal fusion, lysosomal enzymes degrade the content of phagolysosomes. Lysosomal cathepsin protease CPL-1 was found to be indispensable in the digestion of apoptotic cell derived proteins (53), while lysosomal DNase II degrades the DNA content (54).
Reprograming of Phagocytes by Apoptotic Cell Content
Engulfment of apoptotic cells delivers excess materials to the phagocytes. Some of these materials can be completely degraded, while the excess of non-digestible cholesterol is removed via ATP-binding cassette (ABC) transporters (Figure 1). Both PS (55) and lipid-sensing nuclear receptors (56, 57) can upregulate the levels of the ABCA1 transporter. The ingested macromolecules provide the extra energy required for prolonged phagocytosis. However, if too much energy is generated, engulfing cells upregulate the mitochondrial uncoupling protein 2 (UCP2) and dissipate H+ gradient to reduce mitochondrial membrane potential (58). UCP2 also decreases reactive oxygen species formation.
To ensure efficient long-term phagocytosis, apoptotic cells reprogram macrophages not only by altering their metabolism but also by increasing the expression of a number of phagocytic receptors via activating peroxisome proliferator-activated receptor (PPAR)δ/γ and liver X receptor (LXR)α/β receptors by their lipid content (59–61). This process is partially mediated via upregulation of endogenous retinoid synthesis (62, 63).
While the phagocytosis of a variety of pathogenic targets normally triggers a pro-inflammatory response in macrophages, ingestion of apoptotic cells by macrophages induces an anti-inflammatory phenotype. The earliest anti-inflammatory activity of the apoptotic cell is manifest as an immediate-early inhibition of macrophage pro-inflammatory cytokine gene transcription and is exerted directly upon binding to the macrophage (64). Subsequently, both nuclear receptors (65, 66) are activated and soluble mediators are released from macrophages, which act in a paracrine or autocrine fashion to amplify and sustain the anti-inflammatory response (67, 68). During the resolution of inflammation the reprogramed macrophages appear as pro-resolving CD11blow macrophages (69) that express immunoregulatory 12/15-lipoxygenase (70) involved in the formation of pro-resolving lipid mediators, termination of phagocytosis, and emigration to lymphoid organs (69) required for the proper termination of the inflammatory program. This process is regulated by the expression of a typical chemokine receptor D6 on the surface of apoptotic neutrophils (71).
Since improper efferocytosis might contribute to both the initiation and the maintenance of human diseases, enhancing phagocytosis might provide a therapeutic possibility to influence the progression of these diseases.
Therapeutic Possibilities for Enhancing Efferocytosis in Diseases in Which Clearance of Apoptotic Cells is Impaired
Affecting Recognition and Binding of Apoptotic Cells
If lack of sufficient MFG-E8 production leading to improper efferocytosis participates in the pathomechanism of a disease, providing MFG-E8 in recombinant protein form to the site of acute inflammation might enhance the efficiency of efferocytosis. Indeed, a decreased MFG-E8 expression was found in inflamed colons during the acute phase of murine experimental colitis, and intrarectal treatment with recombinant MFG-E8 ameliorated colitis by reducing inflammation and improving disease parameters (72). Alternatively, both prolactin (73) and glucocorticoids (74) can enhance MFG-E8 production providing a theoretical possibility for enhancing its expression in macrophages systematically.
MFG-E8 contains a PS binding domain, as well as an arginine–glycine–aspartic acid (RGD) motif, which enables its binding to integrins. Opsonization of the apoptotic cells and binding to integrins on the surface of phagocytic cells, mediates the engulfment of the dead cell. Based on this observation, an RGD–anxA5 was designed, and it was shown that introduction of RGD transformed the annexin A5, a molecule that binds to PS of apoptotic cells, from an inhibitor into a stimulator of efferocytosis (75). While recombinant MFG-E8 or the RGD–anxA5 could be utilized in acute inflammation, long-term administration of MFG-E8 leads to obesity, because it stimulates the fatty acid uptake of adipocytes (76). It is an open question, whether chronic administration of RGD–anxA5 would have the same side effects.
While MFG-E8 acts as a bridging molecule for integrins, Gas6, and protein S are bridging molecules for MerTk. Thus in cases, where MerTk plays a driving role in efferocytosis, such as cardiac repair after myocardial infarction (4), provision of Gas6 or protein S could similarly accelerate phagocytosis of apoptotic cells and tissue repair. Glucocorticoids enhance phagocytosis by making efferocytosis MerTK dependent (77), thus combining glucocorticoids and Gas6 or protein S might have a synergistic effect.
Other bridging molecules, such as collectins, were also reported to promote efferocytosis. Macrolide antibiotics, which have wide-ranging anti-inflammatory effects, were found to enhance efferocytosis by enhancing the expression of collectins (78). The therapeutic potential of these drugs has already been recognized, as they are successfully used in the treatment of COPD, cystic fibrosis, or asthma (79).
Targeting Lipid-Sensing Nuclear Receptors with the Aim of Increasing the Expression of Phagocytic Receptors or Their Bridging Molecules
Since nuclear receptor signaling is strongly associated with enhanced efferocytosis and suppression of inflammation, glucocorticoids, PPARγ, PPARδ, and LXR agonists or retinoids are logical therapeutic targets in diseases in which efferocytosis is impaired.
Glucocorticoids, the most widely used anti-inflammatory drugs, were shown to enhance phagocytosis of apoptotic cells by increasing the expression of the phospholipid binding protein annexin A1 and its receptor ALXR (6, 80), as well as that of MerTK (73, 81). Long-term effects of glucocorticoids were reported to be mediated by PPARγ (82).
LXR agonists were shown to be effective in the treatment of mouse models of atherosclerosis and inflammation. Thus, LXR agonists [hypocholamide, T0901317, GW3965, or N,N-dimethyl-3β-hydroxy-cholenamide (DMHCA)] lower the serum cholesterol, and inhibit the development of atherosclerosis in murine models of atherosclerosis (83), while GW3965 inhibits the expression of inflammatory mediators in cultured macrophages as well as during in vivo inflammation (84). In addition, ligation of LXR was shown to prevent the development of SLE like autoimmunity in lpr mice (61) and decrease the disease severity in Alzheimer disease (85).
While all LXR ligands are effective in enhancing efferocytosis, T0901317, and GW3965 have been reported to increase plasma and liver triglycerides in some mouse models (86). DMHCA, however, reduced atherosclerosis in apolipoprotein E-deficient mice without inducing hypertriglyceridemia and liver steatosis (87). Thus, developing new potent and effective LXR agonists without the undesirable side effects may be beneficial for clinical usage (88). In this aspect, it is worth noting that we found daidzein, which is a plant-derived diphenolic isoflavone present in a number of plants and herbs (89) and has LXR and PPARγ activating activity (90), to enhance efferocytosis efficiently. Daidzein, similar to LXR agonists (91) induced the expression of TG2, as well as decreased the mitochondrial membrane potential (92).
In addition to LXR agonists, PPARγ agonists were also shown to reduce the neutrophil numbers in rodent models of acute inflammation, such as asthma and COPD (93) and to increase efferocytosis and therapeutic efficacy in a mouse model of chronic granulomatosis (94). PPARγ and PPARδ agonists were also shown to attenuate disease severity in experimental autoimmune encephalomyelitis, a murine model of multiple sclerosis (95, 96).
Both RAR and RXR ligands promote efferocytosis, but their effect is more pronounced if both receptors are activated (63). The effect of in vivo all-trans retinoic acid (ATRA) treatment on the development of lupus nephritis has already been tested in both mouse models (97, 98) and humans (99). Lupus nephritis is a major cause of morbidity and mortality in patients with SLE (100). Long-term ATRA treatment in SLE-prone mice resulted in longer survival, significant reduction of proteinuria, renal pathological findings, and glomerular IgG deposits. In humans, it also reduced proteinuria.
Affecting the Rac-1/RhoA Balance
Since previous studies have shown that Rac activation is required, while RhoA activation is inhibitory for effective clearance of apoptotic cells (44), compounds that alter the Rac-1/RhoA balance, by either increasing the level of active Rac-1 or decreasing the levels and/or activity of RhoA/Rho kinase, would be potential candidates for use in therapy. Among the anti-inflammatory drugs glucocorticoids were shown to alter the Rac-1/RhoA balance in macrophages (101). Another molecule that was shown to affect the Rac-1/RhoA balance is lipoxin A4, which enhances phagocytosis via a protein kinase A-dependent manner (102). Though lipoxin A4 activates both Rac-2 and RhoA, its positive effect on efferocytosis suggests that the ultimate balance favors Rac activation. Lipoxins have already been shown to reduce inflammation and tissue damage in a variety of rodent models (103), and their levels are low in cystic fibrosis patients (104). In addition, exposure to daidzein also enhances Rac activity (92).
Statins are 3-hydroxy-3-methylglutaryl coenzyme A-reductase inhibitors with potent anti-inflammatory effects, largely due to their ability to inhibit the prenylation of Rho GTPases, including Rac-1 and RhoA. Since proper membrane localization of these proteins determines their function, statins inhibit the effectiveness of G protein signaling. Lovastatin was shown to enhance efferocytosis in vitro both in naïve murine lung and in alveolar macrophages taken from COPD patients (105). It was demonstrated that its effect is related to a disproportional deactivation of the RhoGTPases favoring the activity of Rac-1, as well as to the activation of PPARγ (106).
During inflammation oxidant-mediated activation of RhoA and inhibition of efferocytosis might be reversed by antioxidant treatment. Thus, in an LPS-induced lung injury model, antioxidants enhanced efferocytosis and reduced inflammation by inhibiting RhoA activation (107).
Affecting Phagosome Maturation
Increasing evidence suggests that autophagy and phagocytosis processes are interactive and co-regulated. Thus, activation of autophagy during salivary gland cell death in the Drosophila requires the engulfment receptor Draper (108). In addition, association of LC3 with intracellular membranes described originally during autophagy was observed during phagocytosis as well (109). In line with these observations, oridonin, an active diterpenoid isolated from Rabdosia rubesens, was able to induce both autophagy and enhance efferocytosis in the human macrophage-like U937 cells. Moreover, enhancing autophagy by rapamycin also enhanced phagocytosis of apoptotic cells by U937 cells (110). Thus, autophagy inducers might also promote efferocytosis. Though rapamycin and the so-called rapalogs are the most effective clinically used inducers of autophagy, they have severe immunosuppressive effects (111). That is why alternative, non-toxic autophagy inducers (such as rilmenidine or carbamazepine) are being characterized for their pharmacological profile in suitable preclinical models (112, 113). In addition, other non-toxic compounds, such as resveratrol and spermidine, are also being evaluated for their potential to induce autophagy in vivo (114, 115). These two latter compounds were shown to induce autophagy by distinct pathways converging on the acetylproteome (116). Resveratrol was suggested to mediate the cardioprotective effect of red wine (117), while spermidine was shown to prolong the life span of various organisms in an autophagy-dependent manner (114). Though the effect of the latter compounds on efferocytosis has not been tested yet, it is interesting to speculate whether enhanced efferocytosis contributes to their observed beneficial in vivo effects.
Altering the Membrane Lipid Composition of Macrophages
Finally, studies on ob/ob and db/db mice indicate that in type 2 diabetes, obesity, or atherosclerosis impaired efferocytosis might be related to altered membrane lipid compositions of macrophages. In these cases, fish oil diet had a reversal effect (24). ω-3 fatty acids provided by fish oil are known substrates for the biosynthesis of pro-resolving mediators, such as resolvins, protectins, and maresin which, similar to glucocorticoids or opsonization of apoptotic cells by iC3b (69, 118), act as enhancers of efferocytosis as well as promote the formation of CD11blow macrophages (119).
Concluding Remarks
Apoptotic cell death is an integral part of the cell turnover in many tissues. If, however, dead cells are not properly cleared, their content is released and induces tissue damage, as well as long-term inflammation. It is increasingly recognized that improper phagocytosis of apoptotic cells contributes to the establishment and progression of a number of human chronic inflammatory diseases. During the past decade, our knowledge about the mechanisms involved in efferocytosis increased significantly providing potential pharmacological targets through which the efficiency of apoptotic clearance could be increased. Since enhanced phagocytosis is coupled to an enhanced anti-inflammatory response, targeting efferocytosis might provide an additional possibility in the treatment of a numerous human chronic inflammatory diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by Hungarian grants from the National Research Fund OTKA (K104228, K83865, and NK105046) and the TÁMOP 4.2.2.A-11/1/KONV-2012-0023 “VÉD-ELEM” project. The project is implemented through the New Hungary Development Plan co-financed by the European Social Fund and the European Regional Development Fund.
References
1. Hart SP, Dransfield I, Rossi AG. Phagocytosis of apoptotic cells. Methods (2008) 44:280–5. doi: 10.1016/j.ymeth.2007.11.009
2. Rigamonti E, Zordan P, Sciorati C, Rovere-Querini P, Brunelli S. Macrophage plasticity in skeletal muscle repair. Biomed Res Int (2014) 2014:560629. doi:10.1155/2014/560629
3. Thorp EB. Contrasting inflammation resolution during atherosclerosis and post myocardial infarction at the level of monocyte/macrophage phagocytic clearance. Front Immunol (2012) 3:39. doi:10.3389/fimmu.2012.00039
4. Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res (2013) 113:1004–12. doi:10.1161/CIRCRESAHA.113.301198
5. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol (2002) 2:965–75. doi:10.1038/nri957
6. Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol (2010) 189:1059–70. doi:10.1083/jcb.201004096
7. Muñoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol (2010) 6:280–9. doi:10.1038/nrrheum.2010.46
8. Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (2004) 304:1147–50. doi:10.1126/science.1094359
9. Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature (2001) 411:207–11. doi:10.1038/35079659
10. Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet (1998) 19:56–9. doi:10.1038/ng0598-56
11. Ramirez-Ortiz ZG, Pendergraft WF III, Prasad A, Byrne MH, Iram T, Blanchette CJ, et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol (2013) 14:917–26. doi:10.1038/ni.2670
12. Szondy Z, Sarang Z, Molnar P, Nemeth T, Piacentini M, Mastroberardino PG, et al. Transglutaminase 2-/- mice reveal a phagocytosis-associated crosstalk between macrophages and apoptotic cells. Proc Natl Acad Sci U S A (2003) 100:7812–7. doi:10.1073/pnas.0832466100
13. Lewis MJ, Botto M. Complement deficiencies in humans and animals: links to autoimmunity. Autoimmunity (2006) 39:367–78. doi:10.1080/08916930600739233
14. Henson PM, Tuder RM. Apoptosis in the lung: induction, clearance and detection. Am J Physiol Lung Cell Mol Physiol (2008) 294:L601–11. doi:10.1152/ajplung.00320.2007
15. McCubbrey AL, Curtis JL. Efferocytosis and lung disease. Chest (2013) 143:1750–7. doi:10.1378/chest.12-2413
16. Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res (2009) 50(Suppl):S382–7. doi:10.1194/jlr.R800032-JLR200
17. Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe-/- mice. Arterioscler Thromb Vasc Biol (2008) 28:1421–8. doi:10.1161/ATVBAHA.108.167197
18. Bhatia VK, Yun S, Leung V, Grimsditch DC, Benson GM, Botto MB, et al. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol (2007) 170:416–26. doi:10.2353/ajpath.2007.060406
19. Boisvert WA, Rose DM, Boullier A, Quehenberger O, Sydlaske A, Johnson KA, et al. Leukocyte transglutaminase 2 expression limits atherosclerotic lesion size. Arterioscler Thromb Vasc Biol (2006) 26:563–9. doi:10.1161/01.ATV.0000203503.82693.c1
20. Moura R, Tjwa M, Vandervoort P, Van Kerckhoven S, Holvoet P, Hoylaerts MF. Thrombospondin-1 deficiency accelerates atherosclerotic plaque maturation in ApoE-/- mice. Circ Res (2008) 103:1181–9. doi:10.1161/CIRCRESAHA.108.185645
21. Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol (2000) 1:120–9. doi:10.1038/35040009
22. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci (2006) 9:917–24. doi:10.1038/nn1715
23. O’Brien BA, Fieldus WE, Field CJ, Finegood DT. Clearance of apoptotic beta-cells is reduced in neonatal autoimmune diabetes-prone rats. Cell Death Differ (2002) 9:457–64. doi:10.1038/sj.cdd.4400973
24. Li S, Sun Y, Liang CP, Thorp EB, Han S, Jehle AW, et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ Res (2009) 105:1072–82. doi:10.1161/CIRCRESAHA.109.199570
25. Lauber K, Bohn E, Kröber SM, Xiao YJ, Blumenthal SG, Lindemann RK, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell (2003) 113:717–30. doi:10.1016/S0092-8674(03)00422-7
26. Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood (2008) 112:5026–36. doi:10.1182/blood-2008-06-162404
27. Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J (2008) 22:2629–38. doi:10.1096/fj.08-107169
28. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature (2009) 461:282–6. doi:10.1038/nature08296
29. Moodley Y, Rigby P, Bundell C, Bunt S, Hayashi H, Misso N, et al. Macrophage recognition and phagocytosis of apoptotic fibroblasts is critically dependent on fibroblast-derived thrombospondin 1 and CD36. Am J Pathol (2003) 162:771–9. doi:10.1016/S0002-9440(10)63874-6
30. Wakasugi K, Schimmel P. Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science (1999) 284:147–51. doi:10.1126/science.284.5411.147
31. Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature (2007) 450:435–9. doi:10.1038/nature06307
32. Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ (2008) 15:192–201. doi:10.1038/sj.cdd.4402242
33. Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature (2007) 450:430–4. doi:10.1038/nature06329
34. Chen J, Carey K, Godowski PJ. Identification of Gas6 as a ligand for Mer, a neural cell adhesion molecule related receptor tyrosine kinase implicated in cellular transformation. Oncogene (1997) 14:2033–9. doi:10.1038/sj.onc.1201039
35. Hall MO, Obin MS, Heeb MJ, Burgess BL, Abrams TA. Both protein S and Gas6 stimulate outer segment phagocytosis by cultured rat retinal pigment epithelial cells. Exp Eye Res (2005) 81:581–91. doi:10.1016/j.exer.2005.03.017
36. Savill J, Hogg N, Ren Y, Haslett C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J Clin Invest (1992) 90:1513–22. doi:10.1172/JCI116019
37. Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature (2002) 417:182–7. doi:10.1038/417182a
38. Tóth B, Garabuczi E, Sarang Z, Vereb G, Vámosi G, Aeschlimann D, et al. Transglutaminase 2 is needed for the formation of an efficient phagocyte portal in macrophages engulfing apoptotic cells. J Immunol (2009) 182:2084–92. doi:10.4049/jimmunol.0803444
39. Martin M, Leffler J, Blom AM. Annexin A2 and A5 serve as new ligands for C1q on apoptotic cells. J Biol Chem (2012) 287:33733–44. doi:10.1074/jbc.M112.341339
40. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med (2001) 194:781–95. doi:10.1084/jem.194.6.781
41. Thomas L, Bielemeier A, Lambert PA, Darveau RP, Marshall LJ, Devitt A. The N-terminus of CD14 acts to bind apoptotic cells and confers rapid-tethering capabilities on non-myeloid cells. PLoS One (2013) 8:e70691. doi:10.1371/journal.pone.0070691
42. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell (2005) 123:321–34. doi:10.1016/j.cell.2005.08.032
43. Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, Savill J. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature (2002) 418:200–3. doi:10.1038/nature00811
44. Nakaya M, Tanaka M, Okabe Y, Hanayama R, Nagata S. Opposite effects of rho family GTPases on engulfment of apoptotic cells by macrophages. J Biol Chem (2006) 281:8836–42. doi:10.1074/jbc.M510972200
45. Kinchen JM, Cabello J, Klingele D, Wong K, Feichtinger R, Schnabel H, et al. Two pathways converge at CED-10 to mediate actin rearrangement and corpse removal in C. elegans. Nature (2005) 434:93–9. doi:10.1038/nature03263
46. Wu Y, Singh S, Georgescu MM, Birge RB. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci (2005) 118:539–53. doi:10.1242/jcs.01632
47. Albert ML, Kim JI, Birge RB. Alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat Cell Biol (2000) 2:899–905. doi:10.1038/35046549
48. Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, Tosello-Trampont AC, et al. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol (2002) 4:574–82. doi:10.1038/ncb824
49. Su HP, Nakada-Tsukui K, Tosello-Trampont AC, Li Y, Bu G, Henson PM, et al. Interaction of CED-6/GULP, an adapter protein involved in engulfment of apoptotic cells with CED-1 and CD91/low density lipoprotein receptor-related protein (LRP). J Biol Chem (2002) 277:11772–9. doi:10.1074/jbc.M109336200
50. Park SY, Kang KB, Thapa N, Kim SY, Lee SJ, Kim IS. Requirement of adapter protein GULP-6 for stabilin-2 mediated cell corpse engulfment. J Biol Chem (2008) 283:10593–600. doi:10.1074/jbc.M709105200
51. Zou W, Wang X, Vale RD, Ou G. Autophagy genes promote apoptotic cell corpse clearance. Autophagy (2012) 8:1267–8. doi:10.4161/auto.20786
52. Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A (2011) 108:17396–401. doi:10.1073/pnas.1113421108
53. Xu M, Liu Y, Zhao L, Gan Q, Wang X, Yang C. The lysosomal cathepsin protease CPL-1 plays a leading role in phagosomal degradation of apoptotic cells in Caenorhabditis elegans. Mol Biol Cell (2014) 25(13):2071–83. doi:10.1091/mbc.E14-01-0015
54. Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature (2006) 443:998–1002. doi:10.1038/nature05245
55. Kiss RS, Elliott MR, Ma Z, Marcel YL, Ravichandran KS. Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr Biol (2006) 16:2252–8. doi:10.1016/j.cub.2006.09.043
56. Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, et al. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc Natl Acad Sci U S A (2000) 97:12097–102. doi:10.1073/pnas.200367697
57. Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell (2001) 7:161–71. doi:10.1016/S1097-2765(01)00164-2
58. Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature (2011) 477:220–4. doi:10.1038/nature10340
59. Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med (2009) 15:1266–72. doi:10.1038/nm.2048
60. Rőszer T, Menéndez-Gutiérrez MP, Lefterova MI, Alameda D, Núñez V, Lazar MA, et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol (2011) 186:621–31. doi:10.4049/jimmunol.1002230
61. A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity (2009) 31:245–58. doi:10.1016/j.immuni.2009.06.018
62. Garabuczi É, Kiss B, Felszeghy S, Tsay GJ, Fésüs L, Szondy Z. Retinoids produced by macrophages engulfing apoptotic cells contribute to the appearance of transglutaminase 2 in apoptotic thymocytes. Amino Acids (2013) 44:235–44. doi:10.1007/s00726-011-1119-4
63. Sarang Z, Joós G, Rühl R, Gregory CD, Garabuczi E, Szondy Z. Macrophages engulfing apoptotic cells produce non-classical retinoids to enhance their phagocytic capacity. J Immunol (2014) 192(12):5730–8. doi:10.4049/jimmunol.1400284
64. Cvetanovic M, Ucker DS. Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled to specific recognition. J Immunol (2004) 172:880–9. doi:10.4049/jimmunol.172.2.880
65. Kidani Y, Bensinger SJ. Liver X receptor and peroxisome proliferator-activated receptor as integrators of lipid homeostasis and immunity. Immunol Rev (2012) 249:72–83. doi:10.1111/j.1600-065X.2012.01153.x
66. Ipseiz N, Uderhardt S, Scholtysek C, Steffen M, Schabbauer G, Bozec A, et al. The nuclear receptor Nr4a1 mediates anti-inflammatory effects of apoptotic cells. J Immunol (2014) 192:4852–8. doi:10.4049/jimmunol.1303377
67. McDonald PP, Fadok VA, Bratton D, Hanson PM. Transcriptional and translational regulation of inflammatory mediator production by endogenous TGF-β in macrophages that have ingested apoptotic cells. J Immunol (1999) 163:6164–72.
68. Köröskényi K, Duró E, Pallai A, Sarang Z, Kloor D, Ucker DS, et al. Involvement of adenosine A2A receptors in engulfment-dependent apoptotic cell suppression of inflammation. J Immunol (2011) 186:7144–55. doi:10.4049/jimmunol.1002284
69. Schif-Zuck S, Gross N, Assi S, Rostoker R, Serhan CN, Ariel A. Saturated-efferocytosis generates pro-resolving CD11b low macrophages: modulation by resolvins and glucocorticoids. Eur J Immunol (2010) 41:366–79. doi:10.1002/eji.201040801
70. Uderhardt S, Krönke G. 12/15-Lipoxygenase during the regulation of inflammation, immunity, and self-tolerance. J Mol Med (2012) 90:1247–56. doi:10.1007/s00109-012-0954-4
71. Pashover-Schallinger E, Aswad M, Schif-Zuck S, Shapiro H, Singer P, Ariel A. The atypical chemokine receptor D6 controls macrophage efferocytosis and cytokine secretion during the resolution of inflammation. FASEB J (2012) 26:3891–900. doi:10.1096/fj.11-194894
72. Otani A, Ishihara S, Aziz MM, Oshima N, Mishima Y, Moriyama I, et al. Intrarectal administration of milk fat globule epidermal growth factor-8 protein ameliorates murine experimental colitis. Int J Mol Med (2012) 29:349–56. doi:10.3892/ijmm.2011.866
73. Aziz MM, Ishihara S, Rumi MA, Mishima Y, Oshima N, Kadota C, et al. Prolactin induces MFG-E8 production in macrophages via transcription factor C/EBPbeta-dependent pathway. Apoptosis (2008) 13:609–20. doi:10.1007/s10495-008-0201-1
74. Lauber K, Keppeler H, Munoz LE, Koppe U, Schröder K, Yamaguchi H, et al. Milk fat globule-EGF factor 8 mediates the enhancement of apoptotic cell clearance by glucocorticoids. Cell Death Differ (2013) 20:1230–40. doi:10.1038/cdd.2013.82
75. Schutters K, Kusters DH, Chatrou ML, Montero-Melendez T, Donners M, Deckers NM, et al. Cell surface-expressed phosphatidylserine as therapeutic target to enhance phagocytosis of apoptotic cells. Cell Death Differ (2013) 20:49–56. doi:10.1038/cdd.2012.107
76. Khalifeh-Soltani A, McKleroy W, Sakuma S, Cheung YY, Tharp K, Qiu Y, et al. Mfge8 promotes obesity by mediating the uptake of dietary fats and serum fatty acids. Nat Med (2014) 20:175–83. doi:10.1038/nm.3450
77. McColl A, Bournazos S, Franz S, Perretti M, Morgan BP, Haslett C, et al. Glucocorticoids induce protein S-dependent phagocytosis of apoptotic neutrophils by human macrophages. J Immunol (2009) 183:2167–75. doi:10.4049/jimmunol.0803503
78. Hodge S, Hodge G, Jersmann H, Matthews G, Ahern J, Holmes M, et al. Azithromycin improves macrophage phagocytic function and expression of mannose receptor in chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2008) 178:139–48. doi:10.1164/rccm.200711-1666OC
79. Yamaryo T, Oishi K, Yoshimine H, Tsuchihashi Y, Matsushima K, Nagatake T. Fourteen-member macrolides promote the phosphatidylserine receptor-dependent phagocytosis of apoptotic neutrophils by alveolar macrophages. Antimicrob Agents Chemother (2003) 47:48–53. doi:10.1128/AAC.47.1.48-53.2003
80. Maderna P, Yona S, Perretti M, Godson C. Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac(2-26). J Immunol (2005) 174:3727–33. doi:10.4049/jimmunol.174.6.3727
81. Zahuczky G, Kristóf E, Majai G, Fésüs L. Differentiation and glucocorticoid regulated apopto-phagocytic gene expression patterns in human macrophages. Role of Mertk in enhanced phagocytosis. PLoS One (2011) 6:e21349. doi:10.1371/journal.pone.0021349
82. Majai G, Sarang Z, Csomós K, Zahuczky G, Fésüs L. PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur J Immunol (2007) 37:1343–54. doi:10.1002/eji.200636398
83. Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A (2002) 99:7604–9. doi:10.1073/pnas.112059299
84. Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med (2003) 9:213–9. doi:10.1038/nm820
85. Sodhi RK, Singh N. Liver X receptors: emerging therapeutic targets for Alzheimer’s disease. Pharmacol Res (2013) 72:45–51. doi:10.1016/j.phrs.2013.03.008
86. Im SS, Osborne TF. Liver X receptors in atherosclerosis and inflammation. Circ Res (2011) 108:996–1001. doi:10.1161/CIRCRESAHA.110.226878
87. Kratzer A, Buchebner M, Pfeifer T, Becker TM, Uray G, Miyazaki M, et al. Synthetic LXR agonist attenuates plaque formation in apoE-/- mice without inducing liver steatosis and hypertriglyceridemia. J Lipid Res (2009) 50:312–26. doi:10.1194/jlr.M800376-JLR200
88. Loren J, Huang Z, Laffitte BA, Molteni V. Liver X receptor modulators: a review of recently patented compounds (2009-2012). Expert Opin Ther Pat (2013) 23:1317–35. doi:10.1517/13543776.2013.814640
89. Franke AA, Custer LJ, Wang W, Shi CY. HPLC analysis of isoflavonoids and other phenolic agents from foods and from human fluids. Proc Soc Exp Biol Med (1998) 217:263–73. doi:10.3181/00379727-217-44231
90. Ronis MJ, Chen Y, Badeaux J, Badger TM. Dietary soy protein isolate attenuates metabolic syndrome in rats via effects on PPAR, LXR, and SREBP signaling. J Nutr (2009) 139:1431–8. doi:10.3945/jn.109.107029
91. Rébé C, Raveneau M, Chevriaux A, Lakomy D, Sberna AL, Costa A, et al. Induction of transglutaminase 2 by a liver X receptor/retinoic acid receptor alpha pathway increases the clearance of apoptotic cells by human macrophages. Circ Res (2009) 105:393–401. doi:10.1161/CIRCRESAHA.109.201855
92. Yen JH, Yang DJ, Chen MC, Yi-Ying W, Hsieh YF, Cheng YM, et al. Daidzein enhances efferocytosis via transglutaminase 2 and augmentation of Rac1 activity. Mol Immunol (2014) 60:135–42. doi:10.1016/j.molimm.2014.04.006
93. Belvisi MG, Hele DJ. Peroxisome proliferator-activated receptors as novel targets in lung disease. Chest (2008) 134:152–7. doi:10.1378/chest.08-0019
94. Fernandez-Boyanapalli R, Frasch SC, Riches DW, Vandivier RW, Henson PM, Bratton DL. PPARγ activation normalizes resolution of acute sterile inflammation in murine chronic granulomatous disease. Blood (2010) 116:4512–22. doi:10.1182/blood-2010-02-272005
95. Feinstein DL, Galea E, Gavrilyuk V, Brosnan CF, Whitacre CC, Dumitrescu-Ozimek L, et al. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol (2002) 51:694–702. doi:10.1002/ana.10206
96. Polak PE, Kalinin S, Dello Russo C, Gavrilyuk V, Sharp A, Peters JM, et al. Protective effects of a peroxisome proliferator-activated receptor-beta/delta agonist in experimental autoimmune encephalomyelitis. J Neuroimmunol (2005) 168:65–75. doi:10.1016/j.jneuroim.2005.07.006
97. Kinoshita K, Yoo BS, Nozaki Y, Sugiyama M, Ikoma S, Ohno M, et al. Retinoic acid reduces autoimmune renal injury and increases survival in NZB/W F1 mice. J Immunol (2003) 170:5793–8. doi:10.4049/jimmunol.170.11.5793
98. Pérez de Lema G, Lucio-Cazaña FJ, Molina A, Luckow B, Schmid H, de Wit C, et al. Retinoic acid treatment protects MRL/lpr lupus mice from the development of glomerular disease. Kidney Int (2004) 66:1018–28. doi:10.1111/j.1523-1755.2004.00850.x
99. Kinoshita K, Kishimoto K, Shimazu H, Nozaki Y, Sugiyama M, Ikoma S, et al. Successful treatment with retinoids in patients with lupus nephritis. Am J Kidney Dis (2010) 55:344–7. doi:10.1053/j.ajkd.2009.06.012
100. Walsh SJ, Algert C, Gregorio DI, Reisine ST, Rothfield NF. Divergent racial trends in mortality from systemic lupus erythematosus. J Rheumatol (1995) 22:1663–8.
101. Giles KM, Ross K, Rossi AG, Hotchin NA, Haslett C, Dransfield I. Glucocorticoid augmentation of macrophage capacity for phagocytosis of apoptotic cells is associated with reduced p130Cas expression, loss of paxillin/pyk2 phosphorylation, and high levels of active Rac. J Immunol (2001) 167:976–86. doi:10.4049/jimmunol.167.2.976
102. Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol (2000) 164:1663–7. doi:10.4049/jimmunol.164.4.1663
103. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol (2008) 8:349–61. doi:10.1038/nri2294
104. Karp CL, Flick LM, Park KW, Softic S, Greer TM, Keledjian R, et al. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nat Immunol (2004) 5:388–92. doi:10.1038/ni1056
105. Morimoto K, Janssen WJ, Fessler MB, McPhillips KA, Borges VM, Bowler RP, et al. Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J Immunol (2006) 176:7657–65. doi:10.4049/jimmunol.176.12.7657
106. Yano M, Matsumura T, Senokuchi T, Ishii N, Murata Y, Taketa K, et al. Statins activate peroxisome proliferator-activated receptor gamma through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Circ Res (2007) 100:1442–51. doi:10.1161/01.RES.0000268411.49545.9c
107. Moon C, Lee YJ, Park HJ, Chong YH, Kang JL. N-acetylcysteine inhibits RhoA and promotes apoptotic cell clearance during intense lung inflammation. Am J Respir Crit Care Med (2010) 181:374–87. doi:10.1164/rccm.200907-1061OC
108. McPhee CK, Logan MA, Freeman MR, Baehrecke EH. Activation of autophagy during cell death requires the engulfment receptor Draper. Nature (2010) 465:1093–6. doi:10.1038/nature09127
109. Romao S, Münz C. LC3-associated phagocytosis. Autophagy (2014) 10:526–8. doi:10.4161/auto.27606
110. Zang L, Xu Q, Ye Y, Li X, Liu Y, Tashiro S, et al. Autophagy enhanced phagocytosis of apoptotic cells by oridonin-treated human histocytic lymphoma U937 cells. Arch Biochem Biophys (2012) 518:31–41. doi:10.1016/j.abb.2011.11.019
111. Hartford CM, Ratain MJ. Rapamycin: something old, something new, sometimes borrowed and now renewed. Clin Pharmacol Ther (2007) 82:381–8. doi:10.1038/sj.clpt.6100317
112. Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science (2010) 329:229–32. doi:10.1126/science.1190354
113. Rose C, Menzies FM, Renna M, Acevedo-Arozena A, Corrochano S, Sadiq O, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum Mol Genet (2010) 19:2144–53. doi:10.1093/hmg/ddq093
114. Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol (2009) 11:1305–14. doi:10.1038/ncb1975
115. Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, et al. The life span-prolonging effect of sirtuin-1 is mediated by autophagy. Autophagy (2010) 6:186–8. doi:10.4161/auto.6.1.10817
116. Morselli E, Mariño G, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol (2011) 192:615–29. doi:10.1083/jcb.201008167
117. Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov (2006) 5:493–506. doi:10.1038/nrd2060
118. Verbovetski I, Bychkov H, Trahtemberg U, Shapira I, Hareuveni M, Ben-Tal O, et al. Opsonization of apoptotic cells by autologous iC3b facilitates clearance by immature dendritic cells, down-regulates DR and CD86, and up-regulates CC chemokine receptor 7. J Exp Med (2002) 196:1553–61. doi:10.1084/jem.20020263
Keywords: apoptotic cell, phagocytosis, inflammation, autoimmunity, atherosclerosis, obesity, type 2 diabetes, therapy
Citation: Szondy Z, Garabuczi É, Joós G, Tsay GJ and Sarang Z (2014) Impaired clearance of apoptotic cells in chronic inflammatory diseases: therapeutic implications. Front. Immunol. 5:354. doi: 10.3389/fimmu.2014.00354
Received: 18 June 2014; Accepted: 09 July 2014;
Published online: 01 August 2014.
Edited by:
Martin Herrmann, Universitätsklinikum Erlangen, GermanyCopyright: © 2014 Szondy, Garabuczi, Joós, Tsay and Sarang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zsuzsa Szondy, Department of Biochemistry and Molecular Biology, University of Debrecen, Nagyerdei krt. 98, Debrecen H-4012, Hungary e-mail:c3pvbmR5QG1lZC51bmlkZWIuaHU=