Abstract
Pattern recognition receptors (PRRs) expressed on immune cells are crucial for the early detection of invading pathogens, in initiating early innate immune response and in orchestrating the adaptive immune response. PRRs are activated by specific pathogen-associated molecular patterns that are present in pathogenic microbes or nucleic acids of viruses or bacteria. However, inappropriate activation of these PRRs, such as the Toll-like receptors (TLRs), due to genetic lesions or chronic inflammation has been demonstrated to be a major cause of many hematological malignancies. Gain-of-function mutations in the TLR adaptor protein MYD88 found in 39% of the activated B cell type of diffuse large B cell lymphomas and almost 100% of Waldenström’s macroglobulinemia further highlight the involvement of TLRs in these malignancies. MYD88 mutations result in the chronic activation of TLR signaling pathways, thus the constitutive activation of the transcription factor NFκB to promote cell survival and proliferation. These recent insights into TLR pathway driven malignancies warrant the need for a better understanding of TLRs in cancers and the development of novel anti-cancer therapies targeting TLRs. This review focuses on TLR function and signaling in normal or inflammatory conditions, and how mutations can hijack the TLR signaling pathways to give rise to cancer. Finally, we discuss how potential therapeutic agents could be used to restore normal responses to TLRs and have long lasting anti-tumor effects.
Introduction
Pattern recognition receptors (PRRs) are germline-encoded receptors with the ability to relay “danger signals” to the host in order to mediate an early innate immune response. The term “pattern recognition receptors” comes from their ability to recognize specific pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) (1, 2). PRRs can be broadly divided into five distinct subfamilies: Toll-like receptors (TLRs), C-type lectin receptors (CLRs), NOD-like receptors (NLRs), RIG-1-like receptors (RLRs), and AIM2-like receptors (ALRs). These PRR subfamilies differ in their structures, localization patterns, the distinct types of ligands they recognize, and the activation of specific intracellular signaling cascades to mediate a range of responses such as the regulation of gene transcription, cell activation, and proliferation, and the production of pro-inflammatory cytokines, chemokines, and anti-viral molecules (3).
One of the most well characterized PRR is the TLR (4). TLRs are type I transmembrane proteins with an extracellular domain consisting of leucine-rich repeats and a cytoplasmic domain homologous to that of the interleukin (IL)-1 receptor (5, 6). These evolutionarily conserved receptors are absolutely critical for the host innate immune response against many pathogens (7). Activation of TLRs depends on the number of different ligands they may encounter, which is by large, governed by their subcellular localization. Much insight has been gained in recent years on the localization and trafficking of TLRs and the important roles their localization play in the way they recognize their ligands. TLRs can be divided into two groups based on their subcellular localization, either on the cell surface or within intracellular compartments (8). Given the ability of TLRs to recognize a large number of pathogen-associated ligands such as glycoproteins, lipopolysaccharides, flagellin, and viral double-strand or single-strand RNAs or DNAs, TLRs have emerged as an important family of PRRs in shaping both the innate and adaptive immunity (7). However, inappropriate activation of these pathways can often lead to chronic inflammatory diseases or cancer.
This review will focus on TLRs and malignancies associated with the dysregulation of TLR signaling pathways. TLR activation by somatic MYD88 mutations and chronic inflammations has been implicated in a number of hematological malignancies. Targeting the TLR signaling network has gained increasing attention from researchers and clinicians seeking strategies to achieve long lasting anti-tumor outcomes. Here, we discuss the signal transduction and immune regulation by TLRs and the immunological malignancies that manifest from dysregulation of TLR pathways, including how targeting these pathways could be an attractive therapeutic regime.
Toll-Like Receptors
Toll-like receptors are probably the best studied PRRs that participate in the first line of host defense against pathogens. TLRs belong to an evolutionarily conserved family of adaptors sharing homology with the Drosophila protein Toll, which is best known for its essential role in establishing dorsoventral polarity during embryogenesis in insects (9). Amino acid sequencing and hydropathy profiling identified Toll as a type I transmembrane protein with a membrane-spanning segment and multiple tandem leucine-rich repeats directed at the extracellular surface (9). Further biochemical and functional studies conducted on the receptor Toll and its leucine repeats established it as a critical pathogen sensing receptor for recognizing bacteria and fungus in Drosophila (10). This study later became critical for the discovery of Toll-like homologs (TLRs) in mammals as mediators of the innate immunity (4, 10, 11).
A total of 10 TLRs have been identified in humans and 12 in mice (7). Due to the small repertoire of TLRs available to recognize a virtually unlimited combination of pathogen-associated patterns, each individual TLR must be able to detect and respond to a large number of pathogens ranging from bacteria, fungi, protozoa, and viruses (12, 13). For instance, TLRs 1, 2, and 6 recognize lipo-, glycol-, and acyl-peptides expressed on the surfaces of many Gram-positive and Gram-negative bacteria and mycobacteria (7). Additional cooperation between TLRs 1, 2, and 6 enables them to further discriminate different microbial components (14). TLR4 recognizes lipopolysaccharide components of the cell wall of Gram-negative bacteria through its co-receptor MD-2 (15, 16). In addition, TLR4 can also recognize endogenous ligands such as heat-shock proteins, extracellular matrix components including fibronectin, hyaluronic acid, and heparin sulfate in response to tissue injury (7). The nucleic acid sensing subfamily of TLRs consists of TLRs 3, 7, and 9 and exhibit unique endosomal localization in contrast to the surface expression of the other TLRs (17). These TLRs have the ability to detect nuclear material such as ssRNAs, dsRNAs, and dsDNAs and are vital for anti-viral responses (18–21). Importantly, these nucleic acid sensing TLRs must discriminate between foreign and self-nuclear material to prevent autoimmunity. Due to the relative lack of specificity of TLRs compared to the B cell receptors (BCRs), restriction of self-TLR activation must be achieved through other means. TLRs are protected from engaging self-nuclear material by Unc93b mediated restriction to the endosome (22). In such way, self-nucleic acids are prevented from entering the endosome, but foreign material can enter via endocytosis and be processed in the acidified endosomes in order to activate the endosomal TLRs (23, 24).
Together, the 10 human TLRs can recognize a virtually unlimited combination of pathogens, however, the downstream signaling pathways they share are striking. All TLRs except for TLR3 signal through the adaptor protein MYD88 (25). Upon ligand binding, TLRs induce the dimerization of their ectodomains, bringing the cytoplasmic TIR domains together, and initiating a signaling cascade via signal adaptor molecules. The four main TLR adaptor molecules are the myeloid differentiation response protein 88 (MYD88), Toll-interleukin 1 receptor (TIR) domain containing adaptor protein (TIRAP; also known as MAL), TIRAP inducing IFN-β (TRIF), and TRIF-related adaptor molecule (TRAM) (Figure 1). These adaptors are used in various combinations by the different TLRs, but these signaling pathways can be broadly classified into either MYD88 dependent or MYD88 independent.
Figure 1
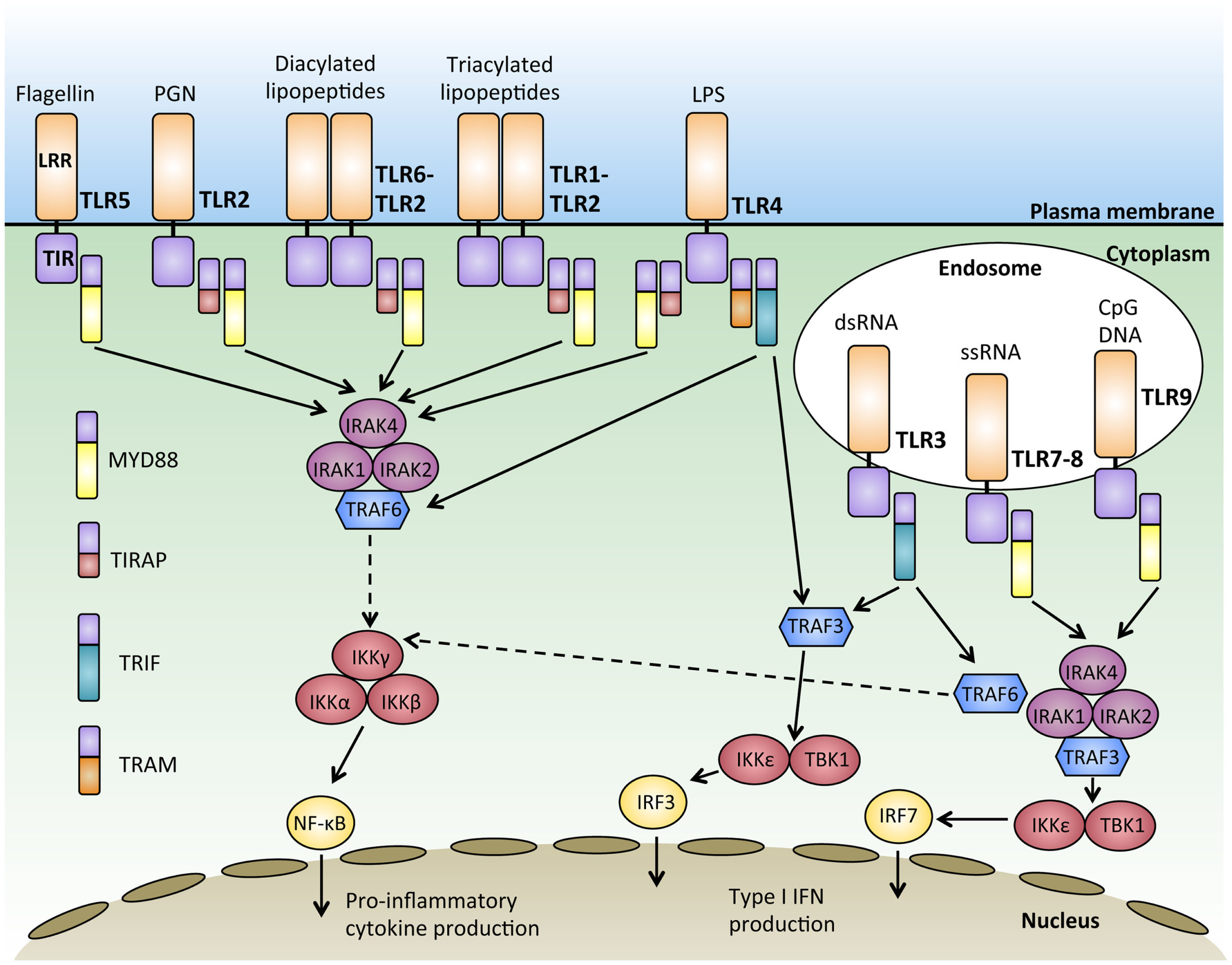
Signal transduction downstream of MYD88-dependent and independent pathways. Activation of Toll-like receptors (TLRs) through binding of their ligand leads to receptor dimerization and the recruitment of adaptor proteins such as MYD88, TIRAP, TRIF, and TRAM. Most of the TLRs form homodimers upon activation while TLR2 can also form heterodimers with either TLR6 or TLR1 to recognize diacylated and triacylated lipopeptides, respectively. Downstream signals are propagated through the activation of IRAKs-TRAF6 and the IKK complex, culminating in the activation of transcription factors such as nuclear factor-κB (NFκB) and interferon-regulatory factors (IRFs), which regulate the production of pro-inflammatory cytokines and type 1 interferon (IFNs).
MYD88 Dependent TLR Signaling
With the exception of TLR3, all TLRs initiate a MYD88-dependent signaling pathway (26). The signal adaptor protein MYD88 contains two main conserved protein domains; a C-terminal TIR and a N-terminal death domain (DD) (27, 28). Upon TLR activation, MYD88 is recruited to the TIR domain of the activated TLR via TIR–TIR interaction (29). The serine–threonine kinase, IL1-receptor associated kinase 4 (IRAK4), is then recruited to MYD88 through the interaction of their DD domains. IRAK4 then recruits and phosphorylates IRAK1 and IRAK2 to form a structure known as the “Myddosome” (30). Phosphorylation of IRAKs 1 and 2 allows them to interact with the E3 ubiquitin ligase, TRAF6, via their TRAF binding domain (31). TRAF6 then ubiquitylates and activates TAK1 (32), which has the dual ability to activate both the NFκB pathway and the mitogen-activated protein kinase (MAPK) pathway (26). In resting cells, NFκB dimers are sequestered in an inactive form in the cytoplasm by the IκB protein (33). During NFκB activation, TAK1 phosphorylates and activates IκB kinase β (IKKβ), which in turn phosphorylates IκB, targeting it for proteosomal degradation (34). The degradation of IκB releases NFκB, enabling it to enter the nucleus and bind to sequences known as κB sites to activate transcription of genes (35). TAK1 also activates the MAPK pathway, leading to the activation of c-Jun N-terminal kinase (JNK), which activates the Jun family of transcription factors (36) (Figure 1).
The MYD88-dependent pathway can be initiated by TLR5 and TLR7-9 using the adaptor MYD88 alone, while the adaptor protein TIRAP is required with MYD88 to initiate signaling downstream of TLR2 and TLR4 (37, 38). In this subset of TLRs, TIRAP acts as a sorting molecule that is necessary for efficient recruitment of MYD88 to the activated TLRs to initiate signal transduction to activate NFκB and produce pro-inflammatory cytokines (39). During TLR 7 and 9 activation, MYD88 also recruits TRAF3 to activate TBK1 and IKKε, which phosphorylates the transcription factor interferon-regulatory factor 7 (IRF7) and leads to IFN-α production (40, 41). IFN-α production, as with production of other IFNs, is particularly important for anti-viral responses (42) (Figure 1).
MYD88 Independent TLR Signaling
MYD88-independent TLR3 signaling requires the adaptor molecule TRIF to activate downstream signaling pathways, including the activation of IRF3 and the production of IFN-β (43). TRIF has also been known to participate in signaling downstream of TLR4 for type 1 interferon responses (44). Upon ligand binding, TRIF recruits TRAF3, which acts as a scaffold for the activation of the IKKs, TBK1, and IKKε, leading to the phosphorylation and activation of the transcription factor IRF3 and IFN-β transcription (45, 46). While TLR3 can activate this pathway using TRIF alone, the adaptor TRAM is required for TLR4, where TRAM facilitates the recruitment of TRIF to TLR4 (47). Upon the activation of TRIF, TRAF6 is recruited, which then activates TAK1 through ubiquitination and leading to the subsequent activation of NFκB (48)(Figure 1). Interestingly, TRAF3 has been shown to play important roles in regulating both the MYD88 dependent and independent response through its differential ubiquitination (49). MYD88-independent signaling triggers the non-degradative self-ubiquitination of TRAF3, promoting IRF3 activation. On the other hand, the MYD88-dependent pathway results in the degradative ubiquitination of TRAF3 and the activation of TAK1 (49). In this manner, TRAF3 acts to balance pro-inflammatory and IFN response by the MYD88 dependent and independent pathways.
Hematological Malignancy and MYD88 Mutation
Inappropriate activation of TLRs due to the somatic acquisition of gain-of-function mutations in the TLR adaptor protein MYD88 has been implicated in many hematological malignancies. Activated B cell type diffuse large B cell lymphoma (ABC-DLBCL), a particularly aggressive subtype of DLBCL whose pathogenesis relies on constitutively active NFκB, frequently accumulates MYD88 mutations. 39% of tumor samples contain mutations in MYD88, and strikingly, 29% of those mutations result in a single nucleotide change from leucine into proline at position 265 (L265P) (50). shRNA knockdown of MYD88 in lymphoma cell lines demonstrated that MYD88 mutations are critical for their survival and high NFκB transcription factor activity (50). A hyperphosphorylated isoform of IRAK1 was strongly associated with the L265P mutant form of MYD88, suggesting that this mutation is a gain-of-function mutation that leads to the constitutive activation of downstream IRAKs (50). The effects of the L265P mutation include increased NFκB activity as well as increased JAK-STAT3 signaling and the production of pro-inflammatory cytokines such as IL6, IL10, and IFN-β (50). The production of these cytokines further activates JAK-STAT3 signaling as part of an autocrine loop that enhances the survival of the lymphoma cells (51, 52) (Figure 2).
Figure 2
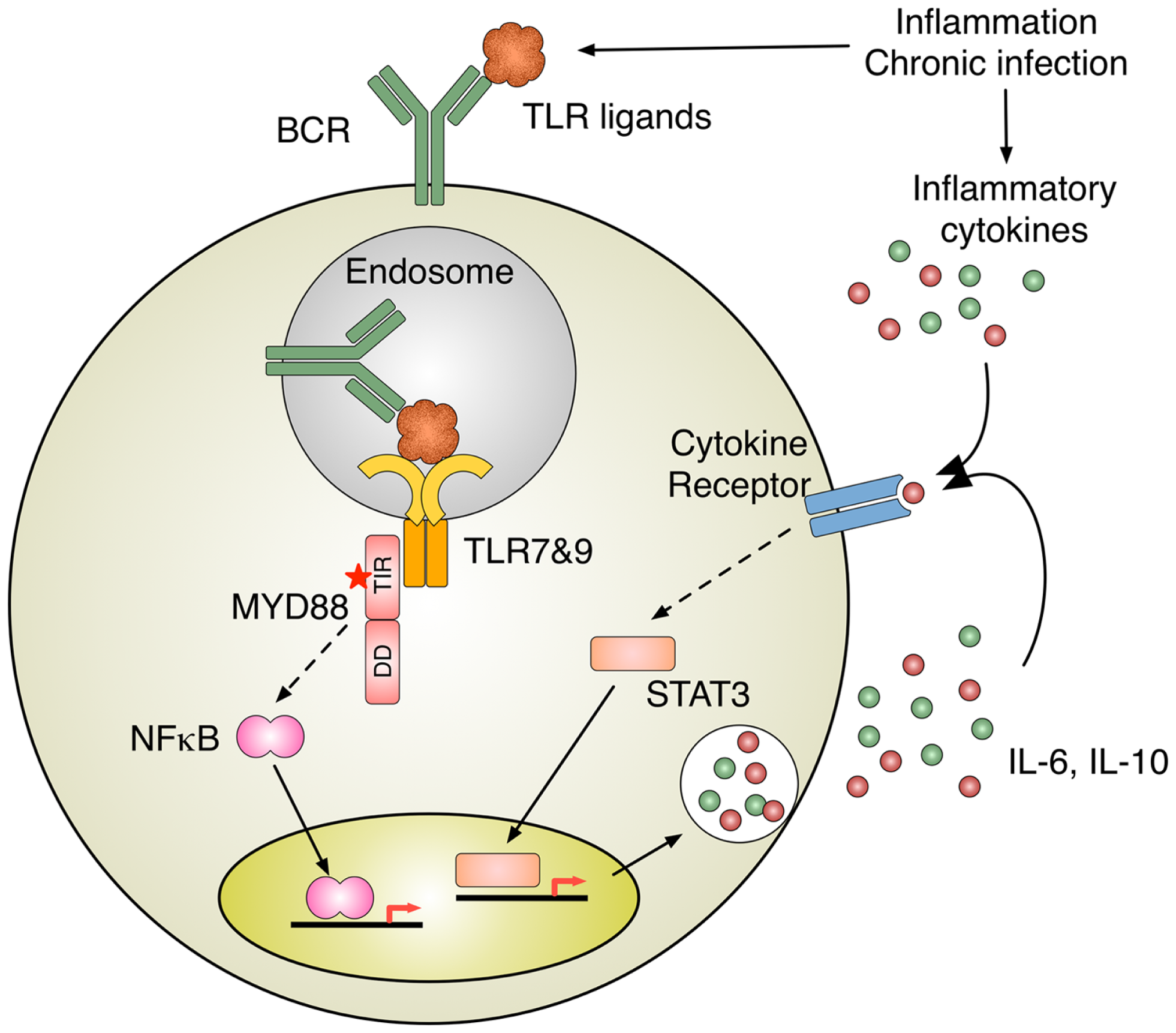
Inflammation induces B cell activation. TLR activation by nucleic acid–protein complexes derived from inflammation and MYD88 mutation B cell antigen receptor (BCR) delivers nucleic acids–protein complexes to TLR-containing endosomes, where MYD88 initiates the activation of intracellular signaling pathways, such as NFκB. Intriguingly, oncogenic MYD88 mutations require intact TLR apparatuses to recognize nucleic acids. One of the potential sources for TLR ligands is nucleic acid–protein complexes derived from inflammation and chronic infection. A subset of ABC-DLBCL shows constitutively activation of JAK-STAT3 pathway, presumably due to autocrine stimulation by IL-6 and IL-10. The activation of cytokine receptor signaling can also be induced by inflammatory cytokines in the milieu of inflammation and chronic infection.
MYD88 mutations have since emerged in a number of other human malignancies, with the L265P mutation found in including almost 100% of Waldenström’s macroglobulinemia (WM), 2–10% of chronic lymphocytic leukemia (CLL), 69% of cutaneous diffuse large B cell lymphoma (CBCL), and 38% of primary central nervous system lymphoma (PCNSL) (previously reviewed in Ref. (53)). However, the effect of single MYD88 L265P mutation on tumor growth is confounded by the accumulation of other potential damaging mutations in the same malignant clones. Recently, a retroviral gene transfer strategy to study the effects of single MYD88 mutation in other wise normal mature B cells found that the MYD88 L265P mutation alone was able to drive limited rounds of mitogen independent B cell proliferation both in vitro and in vivo (54). Nevertheless, the drive for B cell proliferation was dependent on intact nucleic acid sensing TLR activity since Unc93b13d mutation or Tlr9 deficiency inhibited the proliferation of MYD88 L265P B cells in vitro (54). Other studies have also shown that oncogenic MYD88 depends on TLRs by using the depletion of UNC91B1, PRAT4A, and CD14 in ABC-DLBCL lines as well as by using pharmacological inhibitors to TLR7 and TLR9 (55). Given that intact TLR activity is critical for lymphoma cells carrying MYD88 mutations, targeting this pathway appears to be attractive for treating these malignancies. Indeed, blocking endosome acidification using chloroquine selectively inhibits MYD88 L265P mutation driven B cell proliferation in vitro (54). The use of chloroquine to treat hematological malignancies should be further explored, as evidence suggests that there is a strong involvement of the activation of nucleic acid sensing TLRs that depends on normal endosome acidification in promoting proliferative abnormality in these tumors.
Hematological Malignancy and Inflammation
Remarkably, inflammation enables most of the key cellular and molecular capabilities that are required for carcinogenesis, such as genomic instability, proliferative abnormality, and reprograming of the stromal environment (56). Although, the mechanisms by which inflammation promotes neoplastic transformation are not fully understood, it is apparent that, in many cases, tumor development is linked to chronic inflammation (57, 58).
The link between inflammation and tumor formation was first speculated by Virchow in the 1800s as he observed that tissue injury and inflammation induced by irritants could promote cell proliferation (59). Infection has been accepted as a major driver of inflammation-induced tumorigenesis, with up to one-fifth of all cases of cancer associated with infection (60, 61). For instance, persistent Helicobacter pylori infection is associated with gastric cancer and mucosa-associated lymphoid tissue (MALT) lymphoma, infections with hepatitis B and C viruses are associated with hepatocellular carcinoma, and infections with Bacteroides species are linked to colon cancer (62, 63). The inflammatory response triggered by infection is a part of normal host defense to eliminate the pathogen. However, some tumorigenic pathogens subvert host immunity and establish persisting infections, leading to chronic inflammation (64, 65).
Persistent inflammation establishes a microenvironment, which contains macrophages, dendritic cells, natural killer cells, and T and B lymphocytes in addition to the surrounding stroma (66) (Figure 3A). These diverse cells communicate with each other by means of direct contact or cytokine and chemokines, which influence tumor formation and growth (67, 68). This network of inflammatory cells promotes the formation of cancerous cells, which further complicates the initial chronic inflammation induced by infection. The neoplastic cells trigger anti-tumor immunity, which further adds to the established inflammation. Early during tumor formation, whether tumor-promoting inflammation or anti-tumor immunity follows seems to be stochastic and is influenced by a combination of cell-intrinsic and cell-extrinsic processes (69, 70). In established cancers, pro-tumor inflammation seems to be favored, as without therapeutic intervention advanced tumors rarely regress.
Figure 3
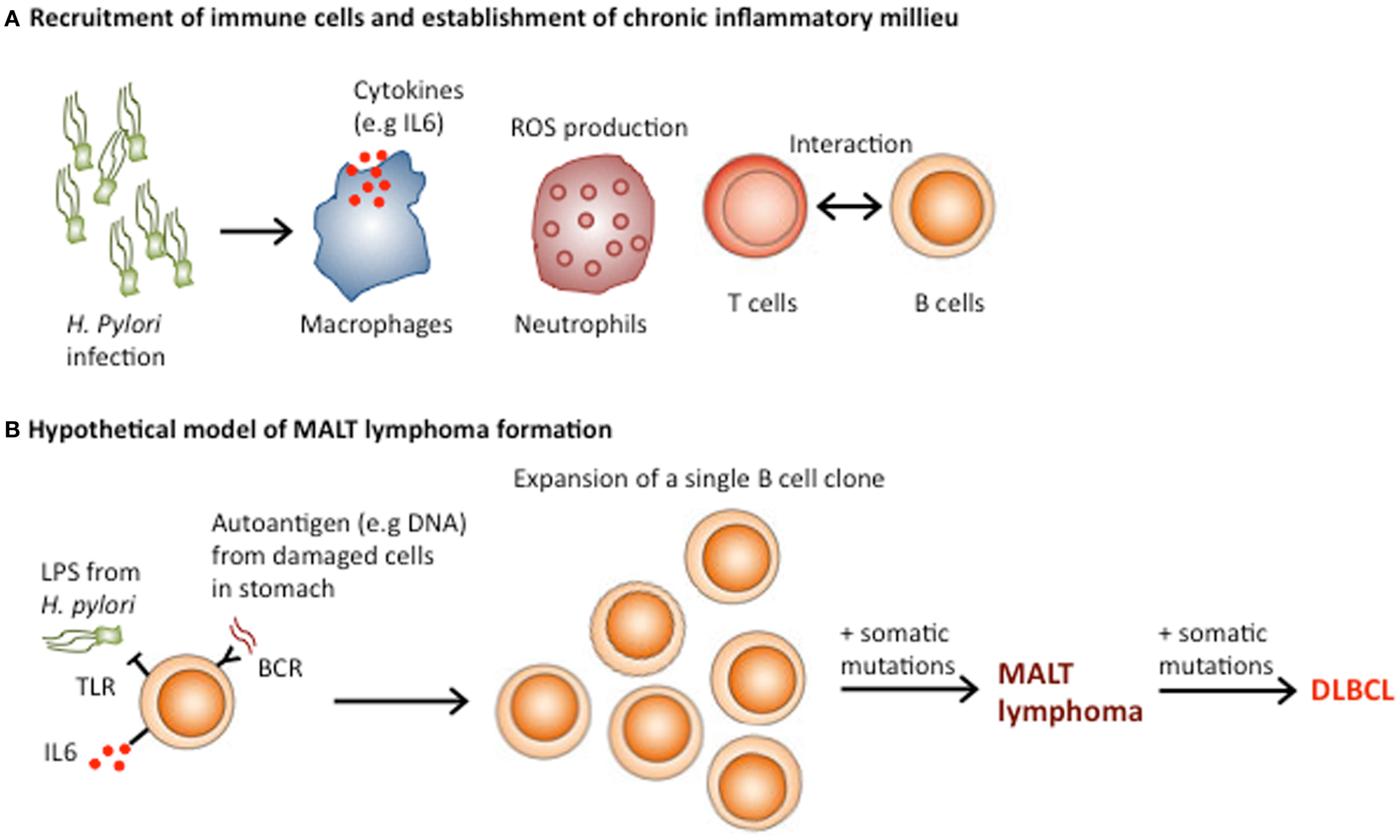
Role of Helicobacter pylori in the pathogenesis of gastric MALT lymphoma. (A)H. pylori infection results in buffering of the gastric pH, which allows immune cell infiltration and the establishment of MALT. The presentation of H. pylori antigens by dendritic cells recruits and activates T cell responses, which enhance B cell activation through CD40–CD40L interactions. (B) MALT lymphoma may result from the transformation of a single B cell clone, which initially formed part of the polyclonal B lymphocyte response against H. Pylori. Both direct activation of TLR signaling by H. pylori and chronic BCR signaling from engagement of autoantigens from damaged stomach cells and the B cell receptor, in addition to T cell-B cell co-stimulation could be involved in the expansion of the single neoplastic B cell clone. Acquisition of additional genomic lesions could transform MALT lymphomas into more aggressive DLBCL.
Continuous stimulation of TLRs by microbial products constitutively engages the activation of the NFκB and STAT3 transcription factors, which exert pro-cancerous activity through multiple effectors (62, 71, 72). Additionally, the production of cytokines by the host inflammatory cells activates these transcription factors (62) (Figure 2). These cytokines facilitate the establishment of feed-forward signal amplification loops, which ultimately promote cell proliferation and resistance to cell death. For instance, the expression of the anti-apoptotic proteins Bcl-2 and Bcl-XL is promoted by both NFκB and STAT3, as is the expression of c-IAP1, c-IAP2, Mcl-1, c-FLIP, and survivin (62, 72). Moreover, both transcription factors interfere with p53 expression and function, representing another potential tumor-promoting mechanism (73).
An additional mechanism linking inflammation to tumor formation is the expression of activation-induced cytidine deaminase (AID), an enzyme that promotes immunoglobulin gene class switching by catalyzing deamination of cytosines in DNA (74). In addition to B lymphocytes, where it was originally discovered, AID is overexpressed in many cancers of diverse origin, and its expression is induced by inflammatory cytokines in a NFκB-dependent manner (74). AID induces genomic instability and increases mutation probability during the error-prone joining of double-stranded DNA breaks. This mutagenic process causes mutations in critical cancer-associated genes such as Tp53 and c-Myc (75, 76).
Helicobacter pylori, Inflammation and MALT Lymphoma
MALT lymphomas, which occur in the context of chronic inflammation caused by infectious agents, such as H. pylori (gastric lymphoma), Chlamydia psittacii (ocular adnexal lymphoma), and Borrelia burgdorferi (cutaneous lymphoma) are a prime example of lymphoid malignancies associated with chronic inflammation (77, 78). Interestingly, in some patients, gastric MALT lymphoma and diffuse large B cell lymphoma (DLBCL) co-occur, indicating that MALT lymphomas can develop into more aggressive DLBCL (79). The pathogenesis of MALT lymphoma involves several steps, which result in the transformation of a single B cell clone, initially part of the polyclonal B lymphocyte response against H. Pylori into a monoclonal tumor (78) (Figure 3B). Under physiological conditions, the stomach lacks MALT because the low pH prevents the survival of lymphocytes in the gastric wall. However, H. pylori infection results in buffering of the gastric pH owing to the secretion of bacterial urease. The decreased acidity of stomach environment, along with the presence of the infection, triggers lymphoid infiltration and the establishment of MALT (78).
Subsequently, the continuous presence of H. pylori induces an upregulation of TLR4 and MD-2 expression in gastric epithelial cells, which contributes to establishing a persistent inflammatory environment (80–82). Although, the role of TLRs in the pathogenesis of MALT lymphoma has been poorly investigated, the immune response to chronic stimulation by H. pylori infection is thought to induce NFκB activation in B cells, which plays a crucial part in the development of MALT lymphoma (83, 84). In addition, the presentation of H. pylori by dendritic cells recruits and activates T cell responses, which enhance B cell activation through CD40–CD40L interactions (85) (Figure 3A). Thus, both direct activation of TLR signaling by H. pylori and T cell-mediated B cell activation could be involved in the pathogenesis of MALT lymphoma (86).
Interestingly, several lines of evidence indicate that chronic antigen stimulation precedes MALT lymphoma pathogenesis. The rearranged IGVH genes from MALT lymphomas have a high frequency of variants, which have been implicated in autoantibody production (87). In addition, approximately half of the MALT lymphoma cases display evidence of intraclonal variation in the IGVH locus, indicating that continued antigenic stimulation is a key driver of clonal B cell expansion (87). As both somatic hypermutation and intraclonal variations are antigen-driven processes, their occurrence in gastric MALT lymphoma strongly indicates a role for antigens during both initiation and progression of this neoplasm.
Remarkably, tumor-derived immunoglobulins from MALT lymphomas bind to various autoantigens as well as H. pylori with varying affinities (87). The autoantigens include DNA and stomach-associated antigens, which could be abundant in the MALT-microenvironment under a situation of continuous inflammation. Given that H. pylori eradication with antibiotics is the preferred therapy for patients with H. pylori-positive gastric MALT lymphoma (88, 89), and the evidence that MALT lymphoma cells proliferate when stimulated with H. pylori in tissue culture, one possible hypothesis is that neoplastic B cells receive proliferative signals from both the B cell receptor and TLRs, which are continuously and simultaneously engaged by self-antigens and LPS from H. pylori respectively. Thus, the eradication of H. pylori by antibiotics disrupts a critical ‘weak’ link in the inflammatory process, which gradually resolves and shuts off the supply of autoantigens available to lymphoma cells.
Role of Inflammation and Cytokines in CLL and Multiple Myeloma
It is apparent that antigenic stimulation, autoimmunity, and inflammation contribute to the development of CLL (90). One mechanism through which these stimuli promote CLL development is induction of B cell activating factor (BAFF), a member of the TNF family, recently shown to accelerate development of CLL-like disease in mice (91). In addition, cytokines such as IL6 and interactions with bone marrow stromal cells support CLL expansion and suppress apoptosis through the expression of Bcl-2, Survivin, and Mcl-1 (92, 93). Increased IL6 production activates the JAK-STAT, MAPK, and PI3K pathways to promote cell survival, proliferation, and resistance to apoptosis (94–96), with the constitutive activation of STAT3 being a hallmark for CLLs (97, 98). Similarly, through the secretion of IL6, TNF-α, and BAFF, bone marrow stromal cells promote the survival of neoplastic plasma cells and also confer drug resistance in multiple myeloma (99). Interestingly, IL6-deficient mice are resistant to induction of multiple myeloma (100, 101). Thus, despite cell-intrinsic constitutive NFκB activation, multiple myeloma cells depend on an extrinsic source of IL6 for their development and survival. High levels of plasma IL6 have been associated with increased disease progression and decreased survival, thus providing the rationale for the evaluation of combination therapies including drugs targeting IL6 for the treatment of this malignancy (102–104).
Targeting Inflammation and TLRs in Cancer
Constitutively, active NFκB signaling due to the aberrant activation of TLRs during chronic inflammation or by MYD88 mutation determines the poor clinical outcome of many hematological malignancies. Desirable outcomes in treating these diseases can be achieved by using a combination of inhibition of signal transducers and transcription factors, sequestration of chemokines and cytokines that sustain inflammatory cells, and the depletion of immune or inflammatory cells that promote tumor development.
Gain-of-function MYD88 mutations have emerged as a potent driver of constitutively active NFκB signaling in many tumors. Targeting this pathway is likely going to be useful as part of a multi-component therapy for many hematological malignancies that are addicted to NFκB activity for their survival. MYD88 signaling is critically dependent on its homo-dimerization through conserved residues within the BB-loop structure of the TIR domain (29, 105). Interfering with this interaction by heptapeptides mimicking the BB-loop has achieved significant reduction in NFκB activity (106). Another novel synthetic compound, ST2825, developed by the same group of researchers is currently under pre-clinical evaluation for the treatment of chronic inflammatory diseases (107). Other peptide-based synthetic small molecule inhibitors such as hydrocinnamoyl-l-valyl pyrrolidine (compound 4a) and Pephinh-MYD88 have also been developed to target MYD88 dimerization in the treatment of lymphoma patients with MYD88 mutations (108). However, these potential MYD88 specific therapeutic options are yet to be trialled in large clinical cohorts.
Constitutive NFκB activity in certain lymphoid tumors suggests that the activation of this pathway is crucial for their survival and thus making them attractive drug targets for anti-cancer therapy (62, 72, 109, 110). However in most cases, such therapy is likely to be effective only in combination with more conventional approaches. Furthermore, as genotoxic therapies often lead to NFκB activation in remaining malignant cells, it makes sense to combine genotoxic dugs with NFκB inhibitors to overcome drug resistance. However, prolonged NFκB inhibition can result in severe immune deficiency and may lead to neutrophilia and greatly enhanced acute inflammation due to enhanced IL1β secretion. Such complications as well as increase propensity for liver damage have hindered the clinical development of NFκB and IKKβ inhibitors (57, 111, 112). An attractive alternative target is the STAT3 transcription factor and the signaling pathway that leads to its activation (113, 114). Several STAT3 and JAK2 inhibitors have been described and shown to inhibit the growth of various cancers that exhibit STAT3 activation (115, 116). So far, none of the complications associated with NFκB inhibitor have been reported for STAT3 or JAK2 inhibitors.
It is unlikely that inhibition of NFκB or STAT signaling alone will be sufficient for tumor regression, yet the combination of an NFκB inhibitor and an apoptosis inducing drug or cytokine could be highly effective. Selective inhibition of NFκB in cancer cells blocks the stimulatory effect of TNF and markedly increases susceptibility to TRAIL-induced cell death, resulting in tumor regression (117, 118). NFκB inhibition and anti-TNF therapy, together with the administration of IFN or TRAIL might offer an attractive combined strategy for immunomodulatory cancer therapy. A recent study has found such synergy between lenalidomide and the BTK inhibitor Ibrutinib in killing ABC-DLBCL by the induction of IRF7 and IFN-β production to cause cell cycle arrest and apoptosis (119, 120). Combinatorial strategies provide a distinct advantage where by certain IFN induced side-effects might be diminished after NFκB inhibitor treatment, shifting the balance of cytokines in the tumor microenvironment to promote tumor regression.
Although it is widely accepted that dampening inflammation and diminishing TLR activity are beneficial for tumor regression, several new lines of evidence have emerged to suggest that TLR agonists could be used as potent anti-tumor agents. When treated with a TLR9 agonist, type B CpG oligodeoxynucleotides (CpG-B ODNs), and CLL B cells that selectively express high levels of TLR9 undergo profound apoptosis by the activation of STAT1, reduction of Bcl-xl pro-survival protein, and elevation of Fas and Fas ligand (121). TLR9 triggered apoptosis seems to be dependent on the altered NFκB status of lymphoma cells compared to normal cells. Moreover, the use of TLR agonists has been known to activate the cognate immune system against cancer cells (122–124). TLRs in lymphoid malignancies appear to be a “double-edged sword” in actively driving disease progression in some but exhibit tumor regressive roles in others. Activation of TLRs by MYD88 mutations has often been associated with poor clinical outcome in lymphoma patients. However, a recent study has reported improved patient survival in a subset of young CLL patients with the identical mutation (125). Interestingly, patients with MYD88 mutations were much younger and had lower expression of CD38 and ZAP-70 than patients with unmutated MYD88. CD38 expression on CLL cells is important for their proliferation and chemotaxis through a signaling pathway involving ZAP-70 (90). Elevated CD38 expression often marks CLL patients with poor clinical outcome and responsiveness to therapy (90). Complex interactions between MYD88 mutation, IGHV mutation status, and CD38 and ZAP-70 levels confound the explanation behind why patients with MYD88 mutations had reduced CD38 expression and show better survival (125).
Conclusion
Pattern recognition receptors protect us from danger and damage associated signals, however, inappropriate activation of these pathways can cause cancer. TLRs can also use ubiquitously available self-ligands such as our own DNA to drive aberrant cell growth when the adaptor protein MYD88 is mutated. This recent finding is one of the many pieces of supportive evidence for Virchow’s hypothesis that chronic inflammation is linked with cancer development. Studies into mutations in the TLR signaling pathways have significantly advanced our understanding on the involvement of TLRs in cancer. However, the potential for targeting TLRs as anti-cancer therapy remains an area that is not yet fully understood. Often TLRs act as a “double-edged sword” in cancer, over active TLR signal provides a microenvironment that is necessary for malignant cell proliferation; on the other hand, TLR agonists can also be used to inhibit cancer cell growth. A better understanding of the involvement of TLRs in cancer would help in tipping the balance between tumor stimulatory and inhibitory effects and the development of novel anti-cancer agents.
Statements
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
JanewayCAJr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol (1989) 54(Pt 1):1–13.10.1101/SQB.1989.054.01.003
2
JanewayCAJrMedzhitovR. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216.10.1146/annurev.immunol.20.083001.084359
3
TakeuchiOAkiraS. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20.10.1016/j.cell.2010.01.022
4
MedzhitovRPreston-HurlburtPJanewayCAJr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature (1997) 388:394–7.10.1038/41131
5
GayNJKeithFJ. Drosophila toll and IL-1 receptor. Nature (1991) 351:355–6.10.1038/351355b0
6
WhithamSDinesh-KumarSPChoiDHehlRCorrCBakerB. The product of the tobacco mosaic virus resistance gene N: similarity to toll and the interleukin-1 receptor. Cell (1994) 78:1101–15.10.1016/0092-8674(94)90283-6
7
AkiraSTakedaK. Toll-like receptor signalling. Nat Rev Immunol (2004) 4:499–511.10.1038/nri1391
8
McGettrickAFO’NeillLA. Localisation and trafficking of Toll-like receptors: an important mode of regulation. Curr Opin Immunol (2010) 22:20–7.10.1016/j.coi.2009.12.002
9
HashimotoCHudsonKLAndersonKV. The Toll gene of Drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein. Cell (1988) 52:269–79.10.1016/0092-8674(88)90516-8
10
LemaitreBNicolasEMichautLReichhartJMHoffmannJA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell (1996) 86:973–83.10.1016/S0092-8674(00)80172-5
11
PoltorakAHeXSmirnovaILiuMYVan HuffelCDuXet alDefective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science (1998) 282:2085–8.10.1126/science.282.5396.2085
12
TakedaKKaishoTAkiraS. Toll-like receptors. Annu Rev Immunol (2003) 21:335–76.10.1146/annurev.immunol.21.120601.141126
13
AkiraS. Mammalian Toll-like receptors. Curr Opin Immunol (2003) 15:5–11.10.1016/S0952-7915(03)00005-0
14
FarhatKRiekenbergSHeineHDebarryJLangRMagesJet alHeterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J Leukoc Biol (2008) 83:692–701.10.1189/jlb.0807586
15
ReFStromingerJL. Monomeric recombinant MD-2 binds toll-like receptor 4 tightly and confers lipopolysaccharide responsiveness. J Biol Chem (2002) 277:23427–32.10.1074/jbc.M202554200
16
NagaiYAkashiSNagafukuMOgataMIwakuraYAkiraSet alEssential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol (2002) 3:667–72.10.1038/ni809
17
BlasiusALBeutlerB. Intracellular toll-like receptors. Immunity (2010) 32:305–15.10.1016/j.immuni.2010.03.012
18
KrugAFrenchARBarchetWFischerJADzionekAPingelJTet alTLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity (2004) 21:107–19.10.1016/j.immuni.2004.06.007
19
KrugALukerGDBarchetWLeibDAAkiraSColonnaM. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood (2004) 103:1433–7.10.1182/blood-2003-08-2674
20
DieboldSSKaishoTHemmiHAkiraSReis e SousaC. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science (2004) 303:1529–31.10.1126/science.1093616
21
TabetaKGeorgelPJanssenEDuXHoebeKCrozatKet alToll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci U S A (2004) 101:3516–21.10.1073/pnas.0400525101
22
TabetaKHoebeKJanssenEMDuXGeorgelPCrozatKet alThe Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol (2006) 7:156–64.10.1038/ni1297
23
GrovesEDartAECovarelliVCaronE. Molecular mechanisms of phagocytic uptake in mammalian cells. Cell Mol Life Sci (2008) 65:1957–76.10.1007/s00018-008-7578-4
24
ConnerSDSchmidSL. Regulated portals of entry into the cell. Nature (2003) 422:37–44.10.1038/nature01451
25
TakedaKAkiraS. TLR signaling pathways. Semin Immunol (2004) 16:3–9.10.1016/j.smim.2003.10.003
26
CompagnoMLimWKGrunnANandulaSVBrahmacharyMShenQet alMutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature (2009) 459:717–21.10.1038/nature07968
27
NishiyaTKajitaEHorinouchiTNishimotoAMiwaS. Distinct roles of TIR and non-TIR regions in the subcellular localization and signaling properties of MyD88. FEBS Lett (2007) 581:3223–9.10.1016/j.febslet.2007.06.008
28
WescheHHenzelWJShillinglawWLiSCaoZ. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity (1997) 7:837–47.10.1016/S1074-7613(00)80402-1
29
LoiarroMVolpeERuggieroVGalloGFurlanRMaiorinoCet alMutational analysis identifies residues crucial for homodimerization of myeloid differentiation factor 88 (MyD88) and for its function in immune cells. J Biol Chem (2013) 288:30210–22.10.1074/jbc.M113.490946
30
LinSCLoYCWuH. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature (2010) 465:885–90.10.1038/nature09121
31
YeHArronJRLamotheBCirilliMKobayashiTShevdeNKet alDistinct molecular mechanism for initiating TRAF6 signalling. Nature (2002) 418:443–7.10.1038/nature00888
32
XiaZPSunLChenXPinedaGJiangXAdhikariAet alDirect activation of protein kinases by unanchored polyubiquitin chains. Nature (2009) 461:114–9.10.1038/nature08247
33
JacobsMDHarrisonSC. Structure of an IkappaBalpha/NF-kappaB complex. Cell (1998) 95:749–58.10.1016/S0092-8674(00)81698-0
34
LiQLuQBotteroVEstepaGMorrisonLMercurioFet alEnhanced NF-kappaB activation and cellular function in macrophages lacking IkappaB kinase 1 (IKK1). Proc Natl Acad Sci U S A (2005) 102:12425–30.10.1073/pnas.0505997102
35
SoltLAMayMJ. The IkappaB kinase complex: master regulator of NF-kappaB signaling. Immunol Res (2008) 42:3–18.10.1007/s12026-008-8025-1
36
LiuWOuyangXYangJLiuJLiQGuYet alAP-1 activated by toll-like receptors regulates expression of IL-23 p19. J Biol Chem (2009) 284:24006–16.10.1074/jbc.M109.025528
37
VerstakBNagpalKBottomleySPGolenbockDTHertzogPJMansellA. MyD88 adapter-like (Mal)/TIRAP interaction with TRAF6 is critical for TLR2- and TLR4-mediated NF-kappaB proinflammatory responses. J Biol Chem (2009) 284:24192–203.10.1074/jbc.M109.023044
38
YamamotoMSatoSHemmiHSanjoHUematsuSKaishoTet alEssential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature (2002) 420:324–9.10.1038/nature01182
39
OhnishiHTochioHKatoZOriiKELiAKimuraTet alStructural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc Natl Acad Sci U S A (2009) 106:10260–5.10.1073/pnas.0812956106
40
NingSPaganoJSBarberGN. IRF7: activation, regulation, modification and function. Genes Immun (2011) 12:399–414.10.1038/gene.2011.21
41
KawaiTAkiraS. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol (2010) 11:373–84.10.1038/ni.1863
42
KawaiTSatoSIshiiKJCobanCHemmiHYamamotoMet alInterferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol (2004) 5:1061–8.10.1038/ni1118
43
DoyleSVaidyaSO’ConnellRDadgostarHDempseyPWuTet alIRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity (2002) 17:251–63.10.1016/S1074-7613(02)00390-4
44
KaganJCSuTHorngTChowAAkiraSMedzhitovR. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol (2008) 9:361–8.10.1038/ni1569
45
HäckerHRedeckeVBlagoevBKratchmarovaIHsuLCWangGGet alSpecificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature (2006) 439:204–7.10.1038/nature04369
46
OganesyanGSahaSKGuoBHeJQShahangianAZarnegarBet alCritical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature (2006) 439:208–11.10.1038/nature04374
47
YamamotoMSatoSHemmiHUematsuSHoshinoKKaishoTet alTRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol (2003) 4:1144–50.10.1038/ni986
48
SatoMSuemoriHHataNAsagiriMOgasawaraKNakaoKet alDistinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity (2000) 13:539–48.10.1016/S1074-7613(00)00053-4
49
TsengPHMatsuzawaAZhangWMinoTVignaliDAKarinM. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol (2010) 11:70–5.10.1038/ni.1819
50
NgoVNYoungRMSchmitzRJhavarSXiaoWLimKHet alOncogenically active MYD88 mutations in human lymphoma. Nature (2011) 470:115–9.10.1038/nature09671
51
LamLTWrightGDavisRELenzGFarinhaPDangLet alCooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood (2008) 111:3701–13.10.1182/blood-2007-09-111948
52
DingBBYuJJYuRYMendezLMShaknovichRZhangYet alConstitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood (2008) 111:1515–23.10.1182/blood-2007-04-087734
53
WangJQJeelallYSHorikawaK. Emerging targets in human lymphoma: targeting the MYD88 mutation. Blood Lymphat Cancer (2013) 2013:53–61.10.2147/BLCTT.S35292
54
WangJQJeelallYSBeutlerBHorikawaKGoodnowCC. Consequences of the recurrent MYD88(L265P) somatic mutation for B cell tolerance. J Exp Med (2014) 211:413–26.10.1084/jem.20131424
55
LimK-HBartonGMStaudtLM. Oncogenic MYD88 mutants require Toll-like receptors [abstract]. In: Proceedings of the 104th Annual Meeting of the American Association for Cancer Research; 2013 Apr 6–10; Washington, DC. Philadelphia: AACR; Cancer Res (2013) 73(8 Suppl):Abst 2332.10.1158/1538-7445.AM2013-2332
56
HanahanDWeinbergRA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74.10.1016/j.cell.2011.02.013
57
GrivennikovSIGretenFRKarinM. Immunity, inflammation, and cancer. Cell (2010) 140:883–99.10.1016/j.cell.2010.01.025
58
MedzhitovR. Origin and physiological roles of inflammation. Nature (2008) 454:428–35.10.1038/nature07201
59
VirchowR. An address on the value of pathological experiments. Br Med J (1881) 2:198–203.10.1136/bmj.2.1075.198
60
KuperHAdamiHOTrichopoulosD. Infections as a major preventable cause of human cancer. J Intern Med (2000) 248:171–83.10.1046/j.1365-2796.2000.00742.x
61
SiegelRMaJZouZJemalA. Cancer statistics, 2014. CA Cancer J Clin (2014) 64:9–29.10.3322/caac.21208
62
KarinM. Nuclear factor-kappaB in cancer development and progression. Nature (2006) 441:431–6.10.1038/nature04870
63
WuSRheeKJAlbesianoERabizadehSWuXYenHRet alA human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med (2009) 15:1016–22.10.1038/nm.2015
64
CoussensLMWerbZ. Inflammation and cancer. Nature (2002) 420:860–7.10.1038/nature01322
65
VirginHWWherryEJAhmedR. Redefining chronic viral infection. Cell (2009) 138:30–50.10.1016/j.cell.2009.06.036
66
de VisserKEEichtenACoussensLM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6:24–37.10.1038/nrc1782
67
BuiJDSchreiberRD. Cancer immunosurveillance, immunoediting and inflammation: independent or interdependent processes?Curr Opin Immunol (2007) 19:203–8.10.1016/j.coi.2007.02.001
68
SwannJBVeselyMDSilvaASharkeyJAkiraSSchreiberRDet alDemonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc Natl Acad Sci U S A (2008) 105:652–6.10.1073/pnas.0708594105
69
LinWWKarinM. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest (2007) 117:1175–83.10.1172/JCI31537
70
SmythMJDunnGPSchreiberRD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol (2006) 90:1–50.10.1016/S0065-2776(06)90001-7
71
KawaiTAkiraS. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34:637–50.10.1016/j.immuni.2011.05.006
72
YuHKortylewskiMPardollD. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol (2007) 7:41–51.10.1038/nri1995
73
ColottaFAllavenaPSicaAGarlandaCMantovaniA. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis (2009) 30:1073–81.10.1093/carcin/bgp127
74
OkazakiIMKotaniAHonjoT. Role of AID in tumorigenesis. Adv Immunol (2007) 94:245–73.10.1016/S0065-2776(06)94008-5
75
LiuMDukeJLRichterDJVinuesaCGGoodnowCCKleinsteinSHet alTwo levels of protection for the B cell genome during somatic hypermutation. Nature (2008) 451:841–5.10.1038/nature06547
76
RamiroARJankovicMCallenEDifilippantonioSChenHTMcBrideKMet alRole of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature (2006) 440:105–9.10.1038/nature04495
77
IsaacsonPWrightDH. Malignant lymphoma of mucosa-associated lymphoid tissue. A distinctive type of B-cell lymphoma. Cancer (1983) 52:1410–6.10.1002/1097-0142(19831015)52:8<1410::AID-CNCR2820520813>3.0.CO;2-3
78
FerreriAJErnbergICopie-BergmanC. Infectious agents and lymphoma development: molecular and clinical aspects. J Intern Med (2009) 265:421–38.10.1111/j.1365-2796.2009.02083.x
79
BarthTFBarthCAKestlerHAMichlPWenigerMABuchholzMet alTranscriptional profiling suggests that secondary and primary large B-cell lymphomas of the gastrointestinal (GI) tract are blastic variants of GI marginal zone lymphoma. J Pathol (2007) 211:305–13.10.1002/path.2096
80
KawaharaTTeshimaSOkaASugiyamaTKishiKRokutanK. Type I Helicobacter pylori lipopolysaccharide stimulates toll-like receptor 4 and activates mitogen oxidase 1 in gastric pit cells. Infect Immun (2001) 69:4382–9.10.1128/IAI.69.7.4382-4389.2001
81
EisenhoferGKopinIJGoldsteinDS. Leaky catecholamine stores: undue waste or a stress response coping mechanism?Ann N Y Acad Sci (2004) 1018:224–30.10.1196/annals.1296.027
82
SchmausserBAndrulisMEndrichSMuller-HermelinkHKEckM. Toll-like receptors TLR4, TLR5 and TLR9 on gastric carcinoma cells: an implication for interaction with Helicobacter pylori. Int J Med Microbiol (2005) 295:179–85.10.1016/j.ijmm.2005.02.009
83
FukataMAbreuMT. Role of Toll-like receptors in gastrointestinal malignancies. Oncogene (2008) 27:234–43.10.1038/sj.onc.1210908
84
FarinhaPGascoyneRD. Molecular pathogenesis of mucosa-associated lymphoid tissue lymphoma. J Clin Oncol (2005) 23:6370–8.10.1200/JCO.2005.05.011
85
GuindiM. Role of activated host T cells in the promotion of MALT lymphoma growth. Semin Cancer Biol (2000) 10:341–4.10.1006/scbi.2000.0351
86
SagaertXVan CutsemEDe HertoghGGeboesKTousseynT. Gastric MALT lymphoma: a model of chronic inflammation-induced tumor development. Nat Rev Gastroenterol Hepatol (2010) 7:336–46.10.1038/nrgastro.2010.58
87
CraigVJArnoldIGerkeCHuynhMQWündischTNeubauerAet alGastric MALT lymphoma B cells express polyreactive, somatically mutated immunoglobulins. Blood (2010) 115:581–91.10.1182/blood-2009-06-228015
88
FerrucciPFZuccaE. Primary gastric lymphoma pathogenesis and treatment: what has changed over the past 10 years?Br J Haematol (2007) 136:521–38.10.1111/j.1365-2141.2006.06444.x
89
ZulloAHassanCAndrianiACristofariFDeFrancescoVIerardiEet alEradication therapy for Helicobacter pylori in patients with gastric MALT lymphoma: a pooled data analysis. Am J Gastroenterol (2009) 104:1932–1937;quiz1938.10.1038/ajg.2009.314
90
ChiorazziNRaiKRFerrariniM. Chronic lymphocytic leukemia. N Engl J Med (2005) 352:804–15.10.1056/NEJMra041720
91
EnzlerTKaterAPZhangWWidhopfGF2ndChuangHYLeeJet alChronic lymphocytic leukemia of Emu-TCL1 transgenic mice undergoes rapid cell turnover that can be offset by extrinsic CD257 to accelerate disease progression. Blood (2009) 114:4469–76.10.1182/blood-2009-06-230169
92
GranzieroLGhiaPCircostaPGottardiDStrolaGGeunaMet alSurvivin is expressed on CD40 stimulation and interfaces proliferation and apoptosis in B-cell chronic lymphocytic leukemia. Blood (2001) 97:2777–83.10.1182/blood.V97.9.2777
93
PedersenIMKitadaSLeoniLMZapataJMKarrasJGTsukadaNet alProtection of CLL B cells by a follicular dendritic cell line is dependent on induction of Mcl-1. Blood (2002) 100:1795–801.
94
HeinrichPCBehrmannIMuller-NewenGSchaperFGraeveL. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J (1998) 334(Pt 2):297–314.
95
MurakamiMHibiMNakagawaNNakagawaTYasukawaKYamanishiKet alIL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science (1993) 260:1808–10.10.1126/science.8511589
96
WegielBBjartellACuligZPerssonJL. Interleukin-6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int J Cancer (2008) 122:1521–9.10.1002/ijc.23261
97
Hazan-HalevyIHarrisDLiuZLiuJLiPChenXet alSTAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood (2010) 115:2852–63.10.1182/blood-2009-10-230060
98
FrankDAMahajanSRitzJ. B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues. J Clin Invest (1997) 100:3140–8.10.1172/JCI119869
99
KastritisEPalumboADimopoulosMA. Treatment of relapsed/refractory multiple myeloma. Semin Hematol (2009) 46:143–57.10.1053/j.seminhematol.2009.01.004
100
HodgeDRHurtEMFarrarWL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer (2005) 41:2502–12.10.1016/j.ejca.2005.08.016
101
GadóKSilvaSPálócziKDomjánGFalusA. Mouse plasmacytoma: an experimental model of human multiple myeloma. Haematologica (2001) 86:227–36.
102
KurzrockRVoorheesPMCasperCFurmanRRFayadLLonialSet alA phase I, open-label study of siltuximab, an anti-IL-6 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma, multiple myeloma, or Castleman disease. Clin Cancer Res (2013) 19:3659–70.10.1158/1078-0432.CCR-12-3349
103
VoorheesPMMangesRFSonneveldPJagannathSSomloGKrishnanAet alA phase 2 multicentre study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with relapsed or refractory multiple myeloma. Br J Haematol (2013) 161:357–66.10.1111/bjh.12266
104
LaiRO’BrienSMaushouriTRogersAKantarjianHKeatingMet alPrognostic value of plasma interleukin-6 levels in patients with chronic lymphocytic leukemia. Cancer (2002) 95:1071–5.10.1002/cncr.10772
105
BurnsKMartinonFEsslingerCPahlHSchneiderPBodmerJLet alMyD88, an adapter protein involved in interleukin-1 signaling. J Biol Chem (1998) 273:12203–9.10.1074/jbc.273.20.12203
106
LoiarroMSetteCGalloGCiacciAFantòNMastroianniDet alPeptide-mediated interference of TIR domain dimerization in MyD88 inhibits interleukin-1-dependent activation of NF-{kappa}B. J Biol Chem (2005) 280:15809–14.10.1074/jbc.C400613200
107
LoiarroMCapolunghiFFantòNGalloGCampoSArseniBet alPivotal advance: inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J Leukoc Biol (2007) 82:801–10.10.1189/jlb.1206746
108
BartfaiTBehrensMMGaidarovaSPembertonJShivanyukARebekJJr. A low molecular weight mimic of the Toll/IL-1 receptor/resistance domain inhibits IL-1 receptor-mediated responses. Proc Natl Acad Sci U S A (2003) 100:7971–6.10.1073/pnas.0932746100
109
MackenzieGGQueisserNWolfsonMLFragaCGAdamoAMOteizaPI. Curcumin induces cell-arrest and apoptosis in association with the inhibition of constitutively active NF-kappaB and STAT3 pathways in Hodgkin’s lymphoma cells. Int J Cancer (2008) 123:56–65.10.1002/ijc.23477
110
CoppoPGouilleux-GruartVHuangYBouhlalHBouamarHBouchetSet alSTAT3 transcription factor is constitutively activated and is oncogenic in nasal-type NK/T-cell lymphoma. Leukemia (2009) 23:1667–78.10.1038/leu.2009.91
111
GretenFRArkanMCBollrathJHsuLCGoodeJMiethingCet alNF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell (2007) 130:918–31.10.1016/j.cell.2007.07.009
112
HeGKarinM. NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res (2011) 21:159–68.10.1038/cr.2010.183
113
KortylewskiMKujawskiMWangTWeiSZhangSPilon-ThomasSet alInhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med (2005) 11:1314–21.10.1038/nm1325
114
YuHPardollDJoveR. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9:798–809.10.1038/nrc2734
115
HedvatMHuszarDHerrmannAGozgitJMSchroederASheehyAet alThe JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell (2009) 16:487–97.10.1016/j.ccr.2009.10.015
116
LinLAminRGallicanoGIGlasgowEJogunooriWJessupJMet alThe STAT3 inhibitor NSC 74859 is effective in hepatocellular cancers with disrupted TGF-beta signaling. Oncogene (2009) 28:961–72.10.1038/onc.2008.448
117
KimYSSchwabeRFQianTLemastersJJBrennerDA. TRAIL-mediated apoptosis requires NF-kappaB inhibition and the mitochondrial permeability transition in human hepatoma cells. Hepatology (2002) 36:1498–508.10.1053/jhep.2002.36942
118
BraeuerSJBunekerCMohrAZwackaRM. Constitutively activated nuclear factor-kappaB, but not induced NF-kappaB, leads to TRAIL resistance by up-regulation of X-linked inhibitor of apoptosis protein in human cancer cells. Mol Cancer Res (2006) 4:715–28.10.1158/1541-7786.MCR-05-0231
119
YangYShafferAL3rdEmreNCCeribelliMZhangMWrightGet alExploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell (2012) 21:723–37.10.1016/j.ccr.2012.05.024
120
HondaKYanaiHNegishiHAsagiriMSatoMMizutaniTet alIRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434:772–7.10.1038/nature03464
121
LiangXMosemanEAFarrarMABachanovaVWeisdorfDJBlazarBRet alToll-like receptor 9 signaling by CpG-B oligodeoxynucleotides induces an apoptotic pathway in human chronic lymphocytic leukemia B cells. Blood (2010) 115:5041–52.10.1182/blood-2009-03-213363
122
VacchelliEGalluzziLEggermontAFridmanWHGalonJSautès-FridmanCet alTrial watch: FDA-approved Toll-like receptor agonists for cancer therapy. Oncoimmunology (2012) 1:894–907.10.4161/onci.20931
123
GalluzziLVacchelliEEggermontAFridmanWHGalonJSautès-FridmanCet alTrial watch: experimental Toll-like receptor agonists for cancer therapy. Oncoimmunology (2012) 1:699–716.10.4161/onci.20696
124
AdamsS. Toll-like receptor agonists in cancer therapy. Immunotherapy (2009) 1:949–64.10.2217/imt.09.70
125
Martínez-TrillosAPinyolMNavarroAAymerichMJaresPJuanMet alMutations in the Toll-like receptor/MYD88 pathway in chronic lymphocytic leukemia identify a subset of young patients with favorable outcome. Blood (2014) 123:3790–6.10.1182/blood-2013-12-543306
Summary
Keywords
cancer, drug targets, inflammation, lymphoma, MYD88 L265P, pattern recognition receptors, self-nucleic acid, Toll-like receptors
Citation
Wang JQ, Jeelall YS, Ferguson LL and Horikawa K (2014) Toll-Like Receptors and Cancer: MYD88 Mutation and Inflammation. Front. Immunol. 5:367. doi: 10.3389/fimmu.2014.00367
Received
28 May 2014
Accepted
16 July 2014
Published
31 July 2014
Volume
5 - 2014
Edited by
Anton G. Kutikhin, Russian Academy of Medical Sciences, Russia
Reviewed by
Ronald B. Corley, Boston University School of Medicine, USA; Muriel Moser, Université Libre de Bruxelles, Belgium
Copyright
© 2014 Wang, Jeelall, Ferguson and Horikawa.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James Q. Wang and Yogesh S. Jeelall, Immunogenomics Laboratory, Department of Immunology, The John Curtin School of Medical Research, Australian National University, Building 131, Garran Road, Canberra, ACT 0200, Australia e-mail: james.wang@anu.edu.au; yogesh.jeelall@anu.edu.au
†James Q. Wang and Yogesh S. Jeelall have contributed equally to this work.
This article was submitted to Tumor Immunity, a section of the journal Frontiers in Immunology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.