 Andrea Kinga Marias Furuya
Andrea Kinga Marias Furuya Hamayun J. Sharifi
Hamayun J. Sharifi Carlos M. C. de Noronha
Carlos M. C. de NoronhaHuman immunodeficiency virus (HIV) remains a significant public health challenge. According to the World Health Organization, there were approximately 35.3 million people living with HIV/AIDS worldwide in 2012, with Sub-Saharan Africa being the most affected region (1). HIV has claimed 27 million lives. It is estimated that two million people die from HIV/AIDS each year.
Highly active antiretroviral therapies (HAART) now allow HIV-infected individuals to live longer and healthier lives. HAART is, however, burdened with side effects and high costs, and threatened by evolving viral resistance. A hallmark of both HIV and simian immunodeficiency virus (SIV) infection is chronic immune activation together with uncontrollable viremia and lymphocyte apoptosis that progresses to acquired immunodeficiency syndrome (AIDS) (2, 3). The deleterious role for immune activation in HIV infection is supported by animal studies. Although the exact mechanism underlying AIDS resistance of natural hosts for SIV, like sooty mangabey (SM) monkeys, is unknown, reports support the idea that disease progression in non-natural hosts such as rhesus macaques (RM) is mainly due to immune system dysfunction triggered by HIV/SIV infections (4, 5). In non-natural hosts, SIV causes progressive impairment of the immune system characterized by high viremia, CD4+ T cell depletion, and loss of T cell function. Continuous CD4+ T cell loss eventually leads to AIDS as the regenerative capacity of the immune system gradually decreases despite the excess of homeostatic cytokines (6). This exhaustion and increased T cell apoptosis, a phenomenon seen in pathogenic infections, may be due to direct and indirect killing by the virus. Indirect- or bystander-killing, is the loss of uninfected and abortively infected T cells possibly due to generalized immune activation (7, 8). It is widely accepted that type I interferon-producing (IFN I) plasmacytoid dendritic cells (pDC) play a central role in this generalized immune activation (9–11). On the other hand, natural simian hosts preserve T lymphocyte populations despite high viremia and show attenuated immune activation that favors the maintenance of CD4+ T cells; hence, SMs do not develop AIDS (7). Although HIV preferentially infects CD4+ T cells and macrophages, efficient binding and infection of pDCs by HIV have been demonstrated and may contribute to AIDS pathogenesis (12, 13).
Plasmacytoid dendritic cells are specialized cells found in blood and lymphoid tissues. The main function of pDCs is to produce IFN in response to bacterial and viral DNA. Following activation, these produce 1000-fold more IFN I than other IFN producing cells (11). Upon HIV infection, pCDs become activated and express CCR7 and CXCR3, migration markers that induce redistribution to the lymph nodes (LN) (14–16). Further, recent reports suggest that HIV-induced pathogenesis occurs mainly in LNs where the IFN I that pDCs produce elevates serum levels during both acute and chronic infections (13, 17–19).
Type I IFNs are powerful cytokines and adaptive immune system modulators. They are produced upon viral infection, replication, and/or the introduction of double-stranded RNA (20). IFNs trigger antiviral activity and induce the maturation of effector T cells. Therefore, an interesting immunoregulatory role for type I IFN is in lymphocyte activation during viral infection (21). There is, however, evidence that this otherwise beneficial interferon can become detrimental to the host during chronic HIV infection. Persistent levels of IFN I induce apoptosis in both HIV-infected CD4+ T cells and in those that do not become productively infected (bystander-killing) leading to accelerated depletion of CD4+ T cells during pathogenic infection (22). This effect is due to type I IFN triggered apoptosis of uninfected CD4+ T cells via TNF-related apoptosis inducing ligand (TRAIL) (17). Originally, SM, which are natural SIV hosts, were thought to have reduced immune system activation during both acute and chronic SIV infections. Moreover, SM pDCs produce less IFN I ex vivo in response to SIV, which leads to less immune activation during chronic infection (23). Other works, however, have shown that natural hosts exhibit an initially strong, but rapidly controlled, IFN I response (24–26). Therefore, both natural and non-natural hosts mount strong type I IFN responses during the acute stage of infection but only natural hosts suppress the response by the chronic stage, 4–16 months after infection. This downregulation occurs despite sustained high levels of viremia. Unlike natural hosts, non-natural hosts maintain high levels of IFN I production at all times. It is important to note that cells from SIV-infected natural hosts can be repeatedly stimulated in vitro to produce type I IFN and to upregulate interferon-stimulated genes (ISG) during the chronic stage of SIV infections suggesting that these cells are neither more refractory nor resistant to re-stimulation (26). Therefore, the downmodulation of IFN I in natural hosts is likely due to negative control mechanisms. The exact mechanism underlying the downregulation of IFN I is, however, not fully understood. The regulatory complexity of the IFN pathway and its overall effects on target cells make pinpointing a resolution mechanism challenging. A role for immunomodulatory proteins, negative regulation of IFN responses, and other mechanisms have been suggested [reviewed in Ref. (2)]. Efforts have been made to discover the IFN I downregulation pathway by examining the regulation of ISGs as well as immunosuppressive genes (24, 26). One common denominator is the myxovirus resistance protein (MxA or Mx1) gene. This finding was supported by a recent clinical study in which Chang et al. found that persistent higher expression of type I IFN and ISGs, including MxA, may explain, at least in part, the increased immune activation and more rapid disease progression in females with chronic HIV infections when compared to males with similar viral loads (27). Harris et al. convincingly showed that natural hosts African green monkeys (AGM) and SM begin to downregulate MxA responses by 28 dpi whereas non-natural hosts (RM) maintained high levels of MxA in LN. This work, however, did not reveal a mechanism of IFN I resolution (24). Further, it is unclear whether IFN is directly downregulated or whether downstream signaling negatively regulates IFN. Understanding the mechanism of Mx regulation in the transition phase, from acute to chronic infection of natural hosts, should reveal new targets for therapies to block the chronic immune system activation associated with disease progression.
MxA is an IFN induced protein expressed in cells like macrophages and hepatocytes. It is best known for its antiviral activity against orthomyxoviruses (28). MxA was first described in 1962 when Lindenmann showed that A2G inbred mice were resistant to doses of mouse-adapted influenza virus that were lethal to other inbred mice. This resistance was dependent on a single dominant locus named Mx1 and was exquisitely specific for orthomyxoviruses (29). Later, it was discovered that Mx1 was the first member of a small gene family and that the spectrum of antiviral activity is in fact much larger than originally thought. Most species have one to three Mx protein isoforms with different antiviral activity depending on their intracellular localization (28). Mouse Mx1 protein is found primarily in the nucleus whereas human MxA is cytoplasmic. Hence, each protein blocks influenza virus at a different stage of the viral replication cycle (30). Of note, Mx2, an interferon-induced protein, has recently been shown to inhibit HIV infection after entry (31, 32). Mx2, however, differs from Mx1 in that Mx2 localizes to the nucleus and its antiviral activity relies on a nuclear localization signal (32).
What are Mx proteins and how do they exert their antiviral activity? Most importantly, how are Mx proteins involved in SIV/HIV infections? Mx proteins belong to the superfamily of GTPases, which includes dynamins, dynamin-like proteins, and mitofusins [described in more detail in Ref. (33)]. These proteins are involved in endocytosis, intracellular vesicle transport, and mitochondria distribution. Mx proteins are mainly characterized by three conserved domains: an N-terminal GTPase domain (GTP binding), a middle domain responsible for interaction with the GTPase effector domain (GED), and the C-terminus GED domain, which recognizes the virus. Two amphipatic α-helices form leucine zippers in the C-terminus. Furthermore, Mx proteins can self-assemble into higher order ring-like structures to form a helical stack (28). The higher order structures may represent a storage form whereas the monomers are likely the active form of MxA (34). Human MxA can be induced by IFN or directly by the virus through different pathways. Activation of Mx by IFN involves the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway and the formation of an IFN-stimulated gene factor 3 (ISGF3) multimeric complex, which in turn migrates into the nucleus and binds an IFN-stimulated response element (ISRE) upstream of the Mx gene (35, 36). It is not yet clear how viral infection activates Mx but it seems to be independent of ISGF3, involving STAT1 instead and possibly the IFN-regulatory factors 1 and 3 (IRF1 and IRF3) (37–39). Mx gene activation is fast. Its protein product is detectable within 4 h (40). It is thus likely that Mx gene activation in response to both virus and IFN through two different pathways is evolutionarily advantageous to the host. Indeed HIV can induce MxA transcription and activate ISGs independently of IFN (41). As mentioned previously, in contrast to non-natural hosts, natural SIV hosts downregulate MxA responses during the transition to chronic infection despite high viremia (24). Moreover with every upregulation/activation, there must be a downregulation of the response in order to maintain homeostasis. Indeed overexpression of MxA is a common pathogenic link in Fanconi Anemia (FA), which consists of a group of at least five autosomal recessive disorders. Overexpression of MxA can lead to cancer susceptibility, apoptosis, bone marrow failure, and abnormal instability in cells (42). One potential mechanism for regulation of Mx gene expression relies on the IRF-1 and IRF-2 proteins. The most probable mediator of Mx induction is IRF-1, which is increased in the presence of virus and after IFN treatment (43, 44). This suggests that the functional synergy between the two independent pathways converges in upregulation of Mx. Additionally, the virus may synergize with IFN to increase Mx levels. IRF-1 induces IRF-2 production to repress ISG transcription. This in turn inhibits IRF-1 function. IRF-1 and IRF-2 share homology in their DNA binding domain; however, IRF-2 has higher affinity for binding and a longer half-life (45). Interestingly, IRF-2 also protects quiescent hematopoietic stem cells (HSC) from type I IFN exhaustion (46). In order to avoid cancer susceptibility, increased apoptosis, bone marrow failure, and HSC depletion, natural SIV hosts apparently evolved to control the IFN responses (Figure 1); however, the exact mechanism is not yet clear. Studying variations in the Mx genes, simultaneously with standardized screening of their antiviral properties in natural and non-natural SIV hosts, could explain why natural hosts can downregulate MxA responses with the onset of the chronic stage of infection. Additionally, mining for polymorphisms in regulatory genes, such as IRF-2, which correlate with enhanced DNA binding or half-life of the activated protein may offer an avenue for designing new therapeutics for controlling the immune system hyperactivation that is associated with AIDS progression.
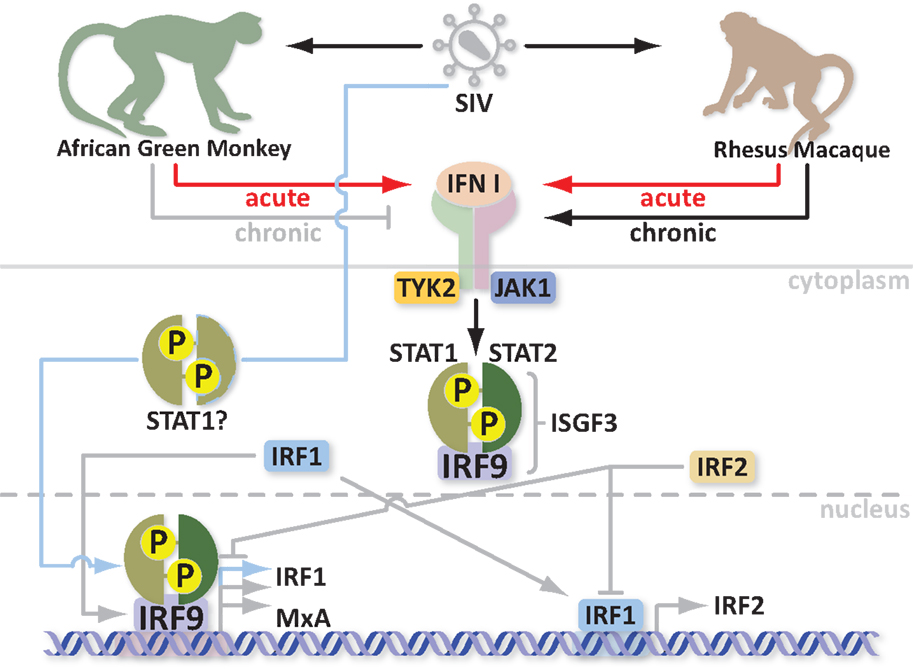
Figure 1. Schematic of IFN and Mx protein regulation in acute and chronic SIV infection models.
Conclusion
Studies in non-human primates have revealed the importance of immune activation in HIV/SIV pathogenesis. Chronic immune activation, associated with CD4+ T cell depletion and loss of T cell function, is likely due to increased serum levels of type I IFN. Although type I IFN is necessary to control viral infections, its detrimental effects during the chronic stage of HIV/SIV infection has been well documented. Natural SIV hosts, in sharp contrast to non-natural SIV hosts, control IFN responses with active regulatory mechanisms. Currently, it is not clear how this regulation is achieved on the molecular level. Understanding the basis of active IFN response repression is crucial as these may play an important role in protection against disease progression. Regulation of the type I IFN pathway is very complex making it hard to narrow down the exact factor or factors responsible for repression of this pathway. Evidence supporting a role for MxA genes, warrants studying polymorphisms in MxA genes, which may shed light on the mechanism of active IFN downregulation. Additionally, better understanding of how Mx genes are regulated will further broaden our perspective for understanding the evolution of host–virus interactions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. WHO. HIV/AIDS Fact Sheet. (2013). Available from: http://www.who.int/mediacentre/factsheets/fs360/en/. English
2. Bosinger SE, Sodora DL, Silvestri G. Generalized immune activation and innate immune responses in simian immunodeficiency virus infection. Curr Opin HIV AIDS (2011) 6(5):411–8. doi: 10.1097/COH.0b013e3283499cf6
3. Sodora DL, Silvestri G. Immune activation and AIDS pathogenesis. AIDS (2008) 22(4):439–46. doi:10.1097/QAD.0b013e3282f2dbe7
4. Picker LJ. Immunopathogenesis of acute AIDS virus infection. Curr Opin Immunol (2006) 18(4):399–405. doi:10.1016/j.coi.2006.05.001
5. Letvin NL, Walker BD. Immunopathogenesis and immunotherapy in AIDS virus infections. Nat Med (2003) 9(7):861–6. doi:10.1038/nm0703-861
6. Biancotto A, Grivel JC, Iglehart SJ, Vanpouille C, Lisco A, Sieg SF, et al. Abnormal activation and cytokine spectra in lymph nodes of people chronically infected with HIV-1. Blood (2007) 109(10):4272–9. doi:10.1182/blood-2006-11-055764
7. Silvestri G, Sodora DL, Koup RA, Paiardini M, O’Neil SP, McClure HM, et al. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity (2003) 18(3):441–52. doi:10.1016/S1074-7613(03)00060-8
8. Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature (2014) 505(7484):509–14. doi:10.1038/nature12940
9. Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol (2004) 5(12):1219–26. doi:10.1038/ni1141
10. Benlahrech A, Patterson S. HIV-1 infection and induction of interferon alpha in plasmacytoid dendritic cells. Curr Opin HIV AIDS (2011) 6(5):373–8. doi:10.1097/COH.0b013e328349592a
11. Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science (1999) 284(5421):1835–7. doi:10.1126/science.284.5421.1835
12. Fong L, Mengozzi M, Abbey NW, Herndier BG, Engleman EG. Productive infection of plasmacytoid dendritic cells with human immunodeficiency virus type 1 is triggered by CD40 ligation. J Virol (2002) 76(21):11033–41. doi:10.1128/JVI.76.21.11033-11041.2002
13. Moris A, Pajot A, Blanchet F, Guivel-Benhassine F, Salcedo M, Schwartz O. Dendritic cells and HIV-specific CD4+ T cells: HIV antigen presentation, T-cell activation, and viral transfer. Blood (2006) 108(5):1643–51. doi:10.1182/blood-2006-02-006361
14. Yonezawa A, Morita R, Takaori-Kondo A, Kadowaki N, Kitawaki T, Hori T, et al. Natural alpha interferon-producing cells respond to human immunodeficiency virus type 1 with alpha interferon production and maturation into dendritic cells. J Virol (2003) 77(6):3777–84. doi:10.1128/JVI.77.6.3777-3784.2003
15. Fonteneau JF, Larsson M, Beignon AS, McKenna K, Dasilva I, Amara A, et al. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J Virol (2004) 78(10):5223–32. doi:10.1128/JVI.78.10.5223-5232.2004
16. Schmidt B, Ashlock BM, Foster H, Fujimura SH, Levy JA. HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology (2005) 343(2):256–66. doi:10.1016/j.virol.2005.09.059
17. Herbeuval JP, Nilsson J, Boasso A, Hardy AW, Kruhlak MJ, Anderson SA, et al. Differential expression of IFN-alpha and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc Natl Acad Sci U S A (2006) 103(18):7000–5. doi:10.1073/pnas.0600363103
18. Lehmann C, Lafferty M, Garzino-Demo A, Jung N, Hartmann P, Fatkenheuer G, et al. Plasmacytoid dendritic cells accumulate and secrete interferon alpha in lymph nodes of HIV-1 patients. PLoS One (2010) 5(6):e11110. doi:10.1371/journal.pone.0011110
19. Dave B, Kaplan J, Gautam S, Bhargava P. Plasmacytoid dendritic cells in lymph nodes of patients with human immunodeficiency virus. Appl Immunohistochem Mol Morphol (2012) 20(6):566–72. doi:10.1097/PAI.0b013e318251d8a4
20. Haller O, Kochs G, Weber F. The interferon response circuit: induction and suppression by pathogenic viruses. Virology (2006) 344(1):119–30. doi:10.1016/j.virol.2005.09.024
21. Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity (2006) 25(3):373–81. doi:10.1016/j.immuni.2006.08.007
22. Herbeuval JP, Grivel JC, Boasso A, Hardy AW, Chougnet C, Dolan MJ, et al. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood (2005) 106(10):3524–31. doi:10.1182/blood-2005-03-1243
23. Mandl JN, Barry AP, Vanderford TH, Kozyr N, Chavan R, Klucking S, et al. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med (2008) 14(10):1077–87. doi:10.1038/nm.1871
24. Harris LD, Tabb B, Sodora DL, Paiardini M, Klatt NR, Douek DC, et al. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J Virol (2010) 84(15):7886–91. doi:10.1128/JVI.02612-09
25. Diop OM, Ploquin MJ, Mortara L, Faye A, Jacquelin B, Kunkel D, et al. Plasmacytoid dendritic cell dynamics and alpha interferon production during Simian immunodeficiency virus infection with a nonpathogenic outcome. J Virol (2008) 82(11):5145–52. doi:10.1128/JVI.02433-07
26. Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G, et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest (2009) 119(12):3544–55. doi:10.1172/JCI40093
27. Chang JJ, Woods M, Lindsay RJ, Doyle EH, Griesbeck M, Chan ES, et al. Higher expression of several interferon-stimulated genes in HIV-1-infected females after adjusting for the level of viral replication. J Infect Dis (2013) 208(5):830–8. doi:10.1093/infdis/jit262
28. Lee SH, Vidal SM. Functional diversity of Mx proteins: variations on a theme of host resistance to infection. Genome Res (2002) 12(4):527–30. doi:10.1101/gr.20102
29. Lindenmann J. Resistance of mice to mouse-adapted influenza A virus. Virology (1962) 16:203–4. doi:10.1016/0042-6822(62)90297-0
30. Pavlovic J, Haller O, Staeheli P. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J Virol (1992) 66(4):2564–9.
31. Goujon C, Moncorge O, Bauby H, Doyle T, Ward CC, Schaller T, et al. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature (2013) 502(7472):559–62. doi:10.1038/nature12542
32. Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature (2013) 502(7472):563–6. doi:10.1038/nature12653
33. Urrutia R, Henley JR, Cook T, McNiven MA. The dynamins: redundant or distinct functions for an expanding family of related GTPases? Proc Natl Acad Sci U S A (1997) 94(2):377–84. doi:10.1073/pnas.94.2.377
34. Janzen C, Kochs G, Haller O. A monomeric GTPase-negative MxA mutant with antiviral activity. J Virol (2000) 74(17):8202–6. doi:10.1128/JVI.74.17.8202-8206.2000
35. Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (1994) 264(5164):1415–21. doi:10.1126/science.8197455
36. von Wussow P, Jakschies D, Hochkeppel HK, Fibich C, Penner L, Deicher H. The human intracellular Mx-homologous protein is specifically induced by type I interferons. Eur J Immunol (1990) 20(9):2015–9. doi:10.1002/eji.1830200920
37. Bandyopadhyay SK, Leonard GT Jr, Bandyopadhyay T, Stark GR, Sen GC. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J Biol Chem (1995) 270(33):19624–9. doi:10.1074/jbc.270.33.19624
38. Pine R. Constitutive expression of an ISGF2/IRF1 transgene leads to interferon-independent activation of interferon-inducible genes and resistance to virus infection. J Virol (1992) 66(7):4470–8.
39. Grant CE, Vasa MZ, Deeley RG. cIRF-3, a new member of the interferon regulatory factor (IRF) family that is rapidly and transiently induced by dsRNA. Nucleic Acids Res (1995) 23(12):2137–46. doi:10.1093/nar/23.12.2137
40. Arnheiter H, Meier E. Mx proteins: antiviral proteins by chance or by necessity? New Biol (1990) 2(10):851–7.
41. Baca LM, Genis P, Kalvakolanu D, Sen G, Meltzer MS, Zhou A, et al. Regulation of interferon-alpha-inducible cellular genes in human immunodeficiency virus-infected monocytes. J Leukoc Biol (1994) 55(3):299–309.
42. Li Y, Youssoufian H. MxA overexpression reveals a common genetic link in four Fanconi anemia complementation groups. J Clin Invest (1997) 100(11):2873–80. doi:10.1172/JCI119836
43. Ronni T, Sareneva T, Pirhonen J, Julkunen I. Activation of IFN-alpha, IFN-gamma, MxA, and IFN regulatory factor 1 genes in influenza A virus-infected human peripheral blood mononuclear cells. J Immunol (1995) 154(6):2764–74.
44. Pine R, Canova A, Schindler C. Tyrosine phosphorylated p91 binds to a single element in the ISGF2/IRF-1 promoter to mediate induction by IFN alpha and IFN gamma, and is likely to autoregulate the p91 gene. EMBO J (1994) 13(1):158–67.
45. Harada H, Takahashi E, Itoh S, Harada K, Hori TA, Taniguchi T. Structure and regulation of the human interferon regulatory factor 1 (IRF-1) and IRF-2 genes: implications for a gene network in the interferon system. Mol Cell Biol (1994) 14(2):1500–9.
Keywords: HIV, SIV, IFN, MxA
Citation: Furuya AKM, Sharifi HJ and de Noronha CMC (2014) The curious case of type I IFN and MxA: tipping the immune balance in AIDS. Front. Immunol. 5:419. doi: 10.3389/fimmu.2014.00419
Received: 30 May 2014; Accepted: 18 August 2014;
Published online: 02 September 2014.
Edited by:
Blossom Damania, University of North Carolina at Chapel Hill, USAReviewed by:
Dietmar Fuchs, Innsbruck Medical University, AustriaLishan Su, The University of North Carolina at Chapel Hill, USA
Copyright: © 2014 Furuya, Sharifi and de Noronha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:ZGVub3JvY0BtYWlsLmFtYy5lZHU=