 Michael L. Walker1
Michael L. Walker1 Kathryn E. Holt2,3
Kathryn E. Holt2,3 Gary P. Anderson4
Gary P. Anderson4 Shu Mei Teo1,2
Shu Mei Teo1,2 Peter D. Sly5
Peter D. Sly5 Patrick G. Holt3,5*
Patrick G. Holt3,5* Michael Inouye1,3,6
Michael Inouye1,3,6
- 1Medical Systems Biology, Department of Pathology, The University of Melbourne, Parkville, VIC, Australia
- 2Department of Biochemistry and Molecular Biology, Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Melbourne, VIC, Australia
- 3Telethon Kids Institute, The University of Western Australia, West Perth, WA, Australia
- 4Department of Pharmacology and Therapeutics, Lung Health Research Centre, The University of Melbourne, Melbourne, VIC, Australia
- 5Queensland Children’s Medical Research Institute, The University of Queensland, Brisbane, QLD, Australia
- 6Medical Systems Biology, Department of Microbiology and Immunology, The University of Melbourne, Parkville, VIC, Australia
Asthma is a genetically complex, chronic lung disease defined clinically as episodic airflow limitation and breathlessness that is at least partially reversible, either spontaneously or in response to therapy. Whereas asthma was rare in the late 1800s and early 1900s, the marked increase in its incidence and prevalence since the 1960s points to substantial gene × environment interactions occurring over a period of years, but these interactions are very poorly understood (1–6). It is widely believed that the majority of asthma begins during childhood and manifests first as intermittent wheeze. However, wheeze is also very common in infancy and only a subset of wheezy children progress to persistent asthma for reasons that are largely obscure. Here, we review the current literature regarding causal pathways leading to early asthma development and chronicity. Given the complex interactions of many risk factors over time eventually leading to apparently multiple asthma phenotypes, we suggest that deeply phenotyped cohort studies combined with sophisticated network models will be required to derive the next generation of biological and clinical insights in asthma pathogenesis.
Wheezing Diseases in Early Childhood
In common with many chronic diseases, the early stages of asthma development can frequently be recognized during childhood. In particular, the wheezing symptoms that are the hallmark of this disease typically manifest initially during the first 1–3 years of life, and at the population level their frequency is highest in this age group. A range of distinct childhood wheezing phenotypes are now recognized (7), the most important of which are
(i) Transient early wheeze, involving infants with repeated symptoms, which resolve by age ~3 years and recur only infrequently (if at all) thereafter.
(ii) Atopic (allergic) wheeze, in which children express intermittent lower respiratory symptoms associated with sensitization and subsequent exposure to aeroallergens.
(iii) Virus-associated wheeze, in which children experience episodic symptoms in association with respiratory infections but remain wheeze-free at other times.
Sex Differences
There is a clear sex difference in transient early wheeze prevalence during the first years of life, with higher rates observed in boys (8). It is speculated that this is due to smaller airway diameter relative to overall lung size in boys (9). Given equal exposure, this alone would render boys more susceptible than girls to the airway-narrowing effects of the inflammation and associated edema that accompanies activation of host anti-viral defense in the respiratory tract. A similar situation likely applies to allergic airways inflammation, given that aeroallergen sensitization rates are also usually initially higher in boys than in girls (10). With increasing postnatal age these anatomical differences are thought to become much less prominent. This is reflected in the increasingly more uniform rates of wheeze across the genders, with progression through the school years. In general terms, wheezing prevalence at the population level declines after infancy. As discussed below, it is the subset of boys and girls in whom symptoms are maintained that are at highest risk of development of persistent and clinically severe asthma.
Clinical and Pathological Features of Asthma as it Develops
The general consensus among pediatric respiratory physicians is that, because of the high frequency of transient early wheeze among preschoolers, “asthma” cannot be reliably diagnosed until at least age 5 years in the majority of patients. Affected subjects are characterized by the persistence of their early wheezing symptoms to this age. Similar to the picture gleaned from experimental animal models, the most consistent marker of persistent wheeze is sensitization to aeroallergens, but beyond this comparison the human situation becomes increasingly complex. In particular, while >90% of young asthmatics are atopic, <25% of the overall pediatric atopic population develops asthma despite in many cases virtually continuous aeroallergen exposure [reviewed in Ref. (11)]. By contrast, in the majority of animal models virtually all pre-sensitized animals respond to aerosol challenge via high-level inflammatory responses in the airways, probably reflecting the genetic uniformity of the mouse strains used in these studies, which are pre-selected for responsiveness (12). This suggests that additional mechanisms downstream of allergic sensitization operate in humans that determine (a) the strength of airways inflammation following individual exposure events, and/or (b) the degree to which airways inflammation can be tolerated before it provokes significant clinical symptoms.
A number of innate and adaptive immune mechanisms have been identified as major controllers of the intensity/duration of inflammatory reactions in the airways and a range of evidence suggests that these may malfunction in asthmatics (13–15). Some hints as to the types of additional factors that might be involved can be gleaned from the symptom profiles of children with early stage asthma. A clinical hallmark in these subjects is variable degrees of hypersensitivity to excessively cold, dry, or dusty (particularly irritant laden) air, which provokes cough and/or lower respiratory symptoms, a classic example being “exercise induced wheeze” triggered by the increased ventilatory requirements during energy intensive sports inducing a switch from nasal to oral breathing (16). This is coupled, again to varying degrees among young asthmatics, with exaggerated responsiveness to lower respiratory infections, which frequently triggers wheezing attacks, whereas the same infections elicit milder non-wheezy symptoms in the rest of the population (17).
Lung Function
An important risk factor for persistent asthma is low-lung function, with most longitudinal cohort studies showing a deficit in lung function in asthmatic children when it is first measured (18, 19). In some cases, this may reflect the effects of maternal exposures to environmental toxicants during pregnancy and this is discussed in a subsequent section of this review. However, lung function is also partially under genetic influence and this may contribute to low-lung function in asthmatics (20, 21) and to the familial risk seen in offspring of asthmatic parents. The extent to which genetically determined lung function is altered by postnatal environmental exposures is uncertain. However, as detailed below, longitudinal birth cohort studies have shown that wheezing in early life, especially when associated with respiratory infection, is both a risk factor for subsequent asthma and is associated with lower lung function in childhood (22). The question of the degree to which airway inflammation induced by viruses and other environmental agents is able to damage lungs and further compromise postnatal lung growth has not been fully answered (23), but understanding of the underlying mechanisms is growing, and relevant recent findings are discussed below.
Airway Damage
While histological evidence of airway inflammation and remodeling in infants and young children with recurrent wheeze/asthma is limited, what little evidence there is suggests that recurrent wheeze is associated with thickening of the epithelial reticular basement membrane and airway inflammation (24). These changes seem to relate to the severity of symptoms (24), the need for anti-asthma medication (25) and long-term evidence of airway inflammation (26). However, studies in older children with asthma, cystic fibrosis, and non-cystic fibrosis bronchiectasis show similar histological changes; an increase in smooth muscle mass is observed in all three groups when compared to controls (27), suggesting that these changes may be related to chronic inflammation rather than being disease specific. In addition, recent data from challenge tests in atopic asthmatic adults suggest that bronchoconstriction per se, regardless of the mechanism inducing it, increases the secretion of pro-fibrotic cytokines and the deposition of sub-epithelial collagen (28). These factors include TGFbeta, connective tissue growth factor (CTGF), and “TH2” cytokines, most notably IL-13, which is profibrotic, directly and via induced periostin (29–32). The deposition of collagen, a major component of airway remodeling, has been shown to increase airway stiffness and reduce dynamic changes in airway caliber during breathing in adults (33).
Chronic Pathology
Current understanding of how this pathological state is maintained in adult asthmatics is incomplete, but likely involves recurrent cycles of airway tissue injury and repair/remodeling associated with asthma exacerbations, which are triggered by environmental stimuli similar to those discussed below, which are believed to be responsible for initiation of the disease at younger ages. It is increasingly recognized that in adulthood several different asthma phenotypes exist (34–36), some of which are defined via biomarkers indicative of hyper-expression of Th1-, Th2-, and/or Th-17-polarized immunity in airway tissues. How these chronic phenotypes evolve from those observed in children is unknown, but the limited biopsy data available on pediatric asthmatics are consistent with the premise that underlying pathological processes that lead eventually to chronicity are initiated many years before the disease finally consolidates (37), and emerging data from the birth cohort studies are beginning to unravel some of the underlying mechanisms.
Tracking Wheezing Phenotypes in Early Life: Prospective Birth Cohort Studies
The research strategy that has proven most fruitful in progressively elucidating the initiation phase of asthma involves utilization of large cohorts of children, either randomly selected populations or specific “high-risk” groups recruited on the basis of family history of disease susceptibility. These cohorts are tracked throughout childhood and, where feasible, into early adulthood (38–47). In some instances, exemplified by the “farmer-mother” studies in northern Europe whose offspring express very low susceptibility to asthma/allergy (48), this has additionally involved collecting data on maternal environmental exposures during pregnancy and assessing, the impact of these on subsequent disease risk in their offspring. These cohort studies have involved prospective multidisciplinary/multiparameter phenotypic assessments of subjects across a range of socioeconomic and ethnic groups, in some cases for periods in excess of 20 years, and they are beginning to build a cogent picture of the determinants of asthma susceptibility and resistance.
Interaction between Viral Infection and Aeroallergen Sensitization
Collectively, the two most prominent asthma risk factors identified from the collective birth cohort studies are lower respiratory viral infections and sensitization to perennial aeroallergens [reviewed in Ref. (11, 49)]. In relation to viral pathogens, the principal focus of current research attention on asthma susceptibility is upon rhinovirus infections occurring in the first 2 years of life (23, 40, 50), in particular rhinovirus type C (51, 52). However, respiratory syncytial virus (RSV) has also been identified as a risk factor in this context (40, 53–55), and to a lesser extent parainfluenza, adenovirus, metapneumovirus, and influenza (11), all of which have shown association with symptomatic early infections in cohort studies (40). Viral infections, which occur within the ensuing preschool years also contribute to persistence of wheeze, but the available data are currently insufficient to reliably compute the relative risk for asthma associated with these later infections.
As noted above, symptoms in the majority of children exhibiting virus-associated wheeze during infancy disappear by the early school years (39, 56) by which time they display normal lung function, and these transient wheezers are also usually non-atopic (39). In contrast, children who become sensitized to aeroallergens and who exhibit early infection-associated wheezing symptoms are much more likely to remain symptomatic and have lower lung function by early school age (7, 39, 40, 43). A key issue in this regard is the nature of the relationship between the atopic phenotype and susceptibility to virus-induced wheeze – is it associative or causal?
It has been argued that the atopic state involves intrinsically “unbalanced” immune responses against all forms of environmental antigens, in particular, responses that are skewed toward selective production of cytokines (such as IL-4) that are antagonistic toward development of Th1-dependent sterilizing immunity. There is some support for this in the form of evidence showing diminished and Th2-biased immune responses to rhinovirus by peripheral blood mononuclear cells from adult atopics (57–59), and moreover, innate anti-viral immune functions expressed by airway epithelial cells in established atopic asthmatics may also be defective (60). Thus, it is plausible that immune functions unrelated to allergic sensitization may contribute to increased susceptibility to severe viral infections in subjects with established atopic disease including atopic asthma, but to what extent is unknown.
Emerging evidence from the cohort studies suggests strongly that with respect to disease initiation during early childhood, some of the links between atopy and asthma risk are direct and causal:
1. While early lower respiratory viral infections and development of aeroallergen sensitization are associated with asthma diagnosis by school age, the highest risk is observed in children experiencing both exposures (17, 40, 41, 61) suggesting synergistic interactions between underlying viral- and allergen-triggered inflammatory pathways.
2. For this interaction to occur, it is necessary that aeroallergen sensitization either precedes or is contemporaneous with the relevant viral infections (40, 61, 62).
The level of resultant risk for asthma development is related quantitatively to (a) the level of sensitization achieved during infancy as measured by aeroallergen-specific IgE titres (61), and (b) the intensity of inflammation-associated lower respiratory symptoms expressed during these infections (63). With regard to (b), the relevant asthma-risk-associated infections are those that spread to the lower respiratory tract and trigger symptoms of wheeze and particularly fever (40, 61, 63). Thus, the mechanism by which expression of the atopic phenotype amplifies the asthmatogenic potential of early lower respiratory infections is likely to involve pro-inflammatory effector pathway that is IgE-dependent. The nature of this mechanism is discussed further in a subsequent section of this review.
Host Factors Influencing Susceptibility to Early Sensitization and Early Infections in Infants
The interim conclusions from the discussion above are that risk for eventual development of asthma by school age is positively associated with concomitant high susceptibility to both aeroallergen sensitization and severe lower respiratory tract infections during infancy. It is well established that the early postnatal period represents the life phase during which resistance to respiratory infections is lowest (64). Similarly, it is now widely recognized that the initiation of allergic sensitization occurs frequently during the first 1–2 years of life, and further that priming of aeroallergen-specific Th2-memory responses during this period can result in allergies that persist into adulthood (11). One of the most detailed pictures of the dynamics of this process has been provided via the Childhood Asthma Study (CAS) cohort in Perth, tracking aeroallergen-specific IgE and associated Th2-memory responses in ~180 subjects at high-genetic risk of allergy/asthma via annual blood samples. This demonstrated initiation of aeroallergen-specific IgE production before age 2 years in virtually all children who attained persistent sensitization status by age 5 years (61).
Postnatal Maturation of the Immune System
Heightened susceptibility to infections and allergy in early postnatal life has been linked to the functionally immature state of both the innate and adaptive arms of the immune system at birth (65–67). In the protected in utero environment, the fetal immune system is maintained in a relatively quiescent state, and T-helper cell function is subtly skewed to favor production of Th2 cytokines while limiting Th1 response capacity, principally because of the necessity to protect the placenta against the toxic effects of inflammatory cytokines exemplified by IFNγ (68). This form of developmental-associated immune homeostasis is maintained via a multi-layered set of Th2-trophic control mechanisms [reviewed in Ref. (69)], the most direct of which involves transient hypermethylation of the IFNγ promoter in CD4+ Th-cells that are released from the thymus over this period and during early postnatal life (70). It is of interest to note that in contrast, production of IL-17 by Th-cells displays an inverse relationship with developmental age, and is maximal in the neonatal period (71). Th-17 cells are recognized to play a significant protective role in immunity to infections via assumption of Th-1-like effector functions to promote pathogen clearance by enhancement of neutrophil recruitment to infection sites and ensuing activation of macrophages (72, 73); their hyperactivity during early life may represent an evolutionary adaptation to the necessity for transient attenuation of Th1 functions during this period. Innate immune responses in early life are also distinct from those in adulthood. In particular, triggering of most toll-like receptors (TLR) ligands in neonatal leukocyte populations yields less IL-12, type 1 IFN, and TNFα, but enhanced levels of IL-1, IL-6, IL-23, and IL-10 relative to corresponding adult cells; overall, the neonatal innate response profile supports robust Th-2 and Th-17 immunity while Th-1 immunity is attenuated (74).
Survival in the microbially hostile extra-uterine environment necessitates upregulation of both innate and adaptive immune response capacity, in particular Th1-associated functions, as the effectiveness of short-term protective humoral mechanisms operative during the neonatal period – such as transplacentally transferred IgG and allied immunoactive factors provided in colostrum and milk, and hyperactivity of endogenous Th-17 cells – progressively wane with age. It is clear that the kinetics of the overall postnatal immune maturation process is highly variable across the human population, and within individuals the rates of maturation in different effector and regulatory pathways are rarely synchronous. Of particular interest are observations linking delayed postnatal maturation of certain key immune functions with increased susceptibility to subsequent development of atopic and/or asthma-associated phenotypes. This has been demonstrated both in prospective studies tracking cohorts of children through to disease outcomes, and in cross sectional studies comparing groups of children at low versus high-genetic risk of atopy/asthma, prior to disease onset. Immunological parameters, which have been implicated in this context include IFNγ response capacity (75–77), IL-12 production (78, 79), HLA-DR expression (80), and the numbers/functions of T-regulatory cells (81) and dendritic cells (DC) (82–84). Increased susceptibility to severe lower respiratory tract infections during infancy has likewise been linked with developmental deficiency in the circulating DC compartment (84), with decreased capacity for production of IFNγ (85) and/or IL-12 (86, 87), and also with an imbalance between production of pro-inflammatory and regulatory cytokines by circulating Th-cells (88). Moreover, young children at high-risk of atopic diseases additionally display attenuated responses to both DTaP (89) and pneumococcal polysaccharide vaccine (90).
Variability in Early Immune Development
Collectively, these findings suggest that susceptibility to early infections, allergic sensitization, and subsequent asthma are linked to a common set of subtle developmental deficiencies related to postnatal maturation of innate and adaptive immune functions. It is important to note that the available data indicate that these deficiencies, in particular, those related to postnatal expression of Th1-associated functions, are transient and typically resolve after infancy (89). Of additional interest, in many cases the children exhibiting this at-risk immunophenotype display a prominent “rebound” in immunocompetence by the early preschool years and become hyper producers of both Th1 and Th2 cytokines in Th-memory responses (69, 89, 91), and it appears possible that this heightened immunoactivity state may contribute to subsequent symptom expression once asthma becomes consolidated (92, 93). However, the short-term consequences of these transient developmental deficiencies may be even more important in relation to subsequent asthma pathogenesis, and this will be discussed further below.
Impact of Environmental Factors: The “Hygiene Hypothesis,” Postnatal Maturation of Immune Function, and the Microbiota
The attention of the allergy/asthma research community to the potential role of environmental microbial factors as determinants of disease susceptibility was first raised via publication of the “hygiene hypothesis” by Strachan in 1989 (94). In the original iteration of this theory, high rates of infections in children from large families were proposed to protect against allergy development by some form of “bystander” immunostimulation. However, this concept has evolved considerably over time. As noted above, the transition from the relatively microbial-free maternal environment to the outside world necessitates radical changes in both the “polarity” and overall activity state of the immune system. This includes but is not restricted to reversal of the Th2/Th1 balance that characterizes the neonatal immune system toward a more Th1-dominant state to facilitate programing of efficient Th1-dependent sterilizing immunity, at the same time increasing resistance to development of the excessively Th2-polarized memory responses to non-pathogenic antigens that are associated with allergy.
Human Microbiome
A broad range of evidence suggests that postnatal maturation of immune function in mammals is driven by environmental microbial derived stimuli, and the strongest signals appear to come from the commensal microbiota of the gastrointestinal tract [reviewed in Ref. (95)], the seeding of which is normally initiated during the birth process (96). Exemplary of this relationship, immune function in mice maintained in a gnotobiotic (germ-free) state postnatally remains highly Th2 polarized, and they are unable to develop protective immunological tolerance to exogenous non-pathogenic antigens and instead default to allergy-associated Th2-dependent IgE responses following exposure (97). It is pertinent to note in this context the epidemiological evidence indicating that cesarean delivery in humans is associated with increased risk both for subsequent allergy development (98–100) and for severe lower respiratory tract infections during infancy (101), and it is widely speculated that this may be the result of differing patterns of gut bacterial colonization in the immediate postnatal period relative to newborns delivered vaginally.
Airborne Microbes
It is additionally of interest to note a series of recent findings relating to northern European “farmer mothers” and their offspring, who are heavily exposed to airborne microbial stimuli (exemplified by bacterial lipopolysaccharide) in dust from animal-holding barns in which the mothers work (including during pregnancy) for several hours per day (48). Earlier studies have shown that postnatal exposure to lipopolysaccharide in household dust is associated with a moderate but significant reduction in risk for development of allergy in early childhood (102). However, the tracking of postnatal asthma and allergy development in the offspring of the heavily exposed farmer mothers has provided strong evidence [reviewed in Ref. (48)] that the offspring of these mothers develop strikingly higher levels of resistance to both asthma and allergy relative to the household dust-exposed populations. Furthermore, the development of resistance was independently associated with exposure of the children themselves to barn dust postnatally, and with exposure of their mothers during pregnancy, the latter, resulting in more robust immunoregulatory function in their newborns (103). Follow-up animal studies pinpointed the mechanism underlying this maternal effect on immune development in their offspring as low-intensity innate immune responses to microbial products in inhaled barn dust, which result in indirect attenuation of potentially pro-inflammatory TLR functions in the maternal decidua (103), likely operating via a bone marrow-placental axis (104). It is speculated that this quiescent state at the feto-maternal interface is protective toward placental function, thus, promoting optimization of the growth and development of the fetal immune system, resulting in higher levels of immunocompetence (and hence disease resistance) in the offspring at birth (104, 105).
Development of Persistent Asthma: The “Critical Window” Concept
In common with the immune system, postnatal survival dictates that the respiratory system of newborns must rapidly adapt to the demands placed on it by the outside world, in particular, it needs to establish response thresholds to inhaled stimuli that will enable it to “tolerate” normal levels of exposure to ubiquitous environmental irritants. This adaptation includes the gross structural changes associated with physical growth, which inter alia progressively increases airway diameter, thus, reducing susceptibility to the physical obstruction that frequently accompanies episodic airways inflammation in infants. A range of more subtle processes are involved in parallel including alveolarization and accompanying changes in the airway epithelium, which continue beyond infancy. Local growth and differentiation of non-adrenergic non-cholinergic nerves also establishes neural control of irritant receptor systems and airway smooth muscle during this period (106). These pathways are particularly prominent in rodent animal model where both inhibitory and excitatory NANC pathways have strong effects on airway caliber and antidromic activation of neuropeptideric excitatory NANC pathways induces neurogenic inflammation (107).
Parenchymal lung tissues undergo comparably profound changes in this early postnatal period, and any aberrations in normal growth patterns in this compartment, which result in abnormalities in parenchymal lung mechanics in infants, are postulated to contribute to the lung function changes that are part of the persistent asthma phenotype in later life (108). More direct support for the general concept that structure–function changes initiated during this early postnatal phase of rapid growth can exert long-term effects on the respiratory system comes from studies in infants that demonstrate stable “tracking” of lung function over the course of early childhood, i.e., while lung function progressively increases in all subjects in absolute terms as they mature, the relative position of individuals within the overall population distribution tends to remain close to where they were at birth (109).
Long-Term Impacts of Events in the Early Postnatal Period
Moreover, the available evidence suggests that this tracking phenomenon also applies to the sequelae of events associated with respiratory stress or trauma to the respiratory system, particularly severe lower respiratory tract viral infections, which can interrupt normal lung growth and result in a drop to a lower centile within the respiratory function population distribution, which is then maintained. An archetypal example is RSV infection in infancy, which is a major risk factor for wheeze by the end of the preschool years, and this is associated with persistence of low-baseline lung function that is correctable by bronchodilator (56), implying that the early infection can result in increased airway smooth muscle tone, which tracks for years thereafter. This process is illustrated in Figure 1. Thus, respiratory function can be expressed as population centiles (dark lines in Figure 1), and during the preschool years individual children typically track on the same level defined by their personal centile at birth. Experiencing recurrent inflammatory events of sufficient intensity and duration to perturb ongoing growth/differentiation of lung tissues can result in non-reversible tissue remodeling, accompanied by stepwise dropping to progressively lower levels of lung function. The example illustrated (red line in Figure 1) exemplifies an infant who is in the 49th centile at birth, who after three such severe events has declined to the 44th centile. In principle, this down-stepping process can continue over a period of years, but the impact of individual inflammatory episodes is likely inversely related to the postnatal age at which they occur.
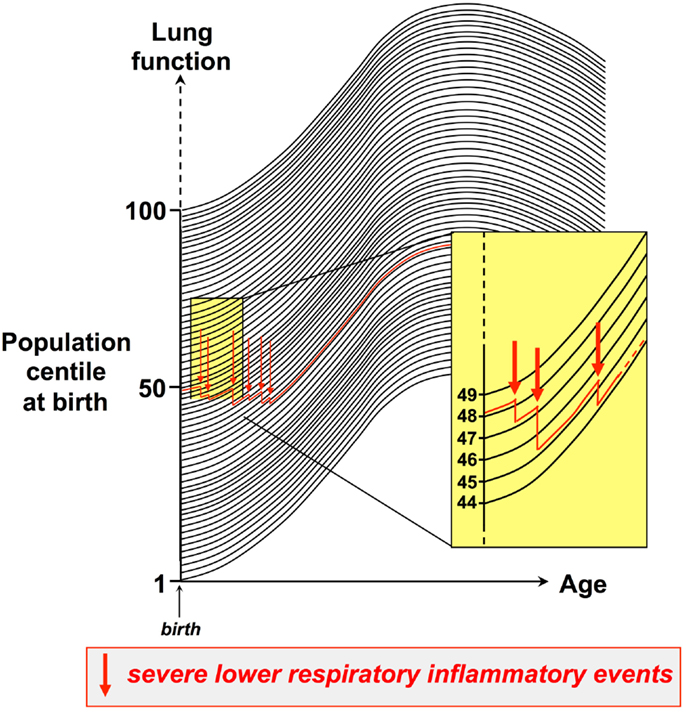
Figure 1. Establishing trajectories for maturation of respiratory function during childhood. Respiratory function can be expressed as population centiles, and during the preschool years individual children typically track on the same respiratory function centile (dark lines) defined by their personal centile at birth. Experiencing recurrent inflammatory events of sufficient intensity and duration to perturb ongoing growth/differentiation of lung tissues can result in non-reversible tissue remodeling, accompanied by stepwise dropping to progressively lower levels of lung function. The figure exemplifies an infant who is in the 49th centile at birth, who after three such severe events has declined to the 44th centile.
Additional studies have shown that respiratory function also tracks from childhood into adulthood (110, 111). This finding is very consistent with long-term asthma cohort studies, which indicated that for the vast majority of patients, the degree of severity they acquire as children will remain a constant in their lives (111). Very few patients show progressive worsening of disease when assessed on a population basis.
Antenatal Versus Postnatal Factors
There is additionally growing evidence that similar processes can occur at earlier stages of development, as illustrated in Figure 2. In particular, dietary factors, which are likely to include microbial products in unpasteurized milk, have been identified as an important component of the “asthma/allergy-protective” signal present in the environment of the northern European farmer mothers (48, 112). Maternal environmental exposures can also impact negatively on these developmental processes. A well documented example relates to the offspring of mothers who smoke during pregnancy: these infants typically have reduced lung function at birth relative to the overall population; this reduced function tracks into adolescence (113–116) and is associated with persistent wheeze and asthma (117, 118). Maternal smoking during pregnancy is also a risk factor for asthma persisting into adolescence, independent of effects on lung function and immune-phenotype (110). It is important to note that the same general principles apply to the immune system, which also displays dynamic maturational changes during fetal development, which are further refined postnatally (Figure 2). In this regard, the offspring of the smoking mothers also show altered patterns of cytokine production at birth and associated increased risk for postnatal allergic sensitization (118), and reduced numbers of circulating T-regulatory cells (119), indicating effects on early immune development. Of particular note, the protective “farmer-mother” environment described above promotes reciprocal effects on T-regulatory cells in their asthma-resistant offspring (100).
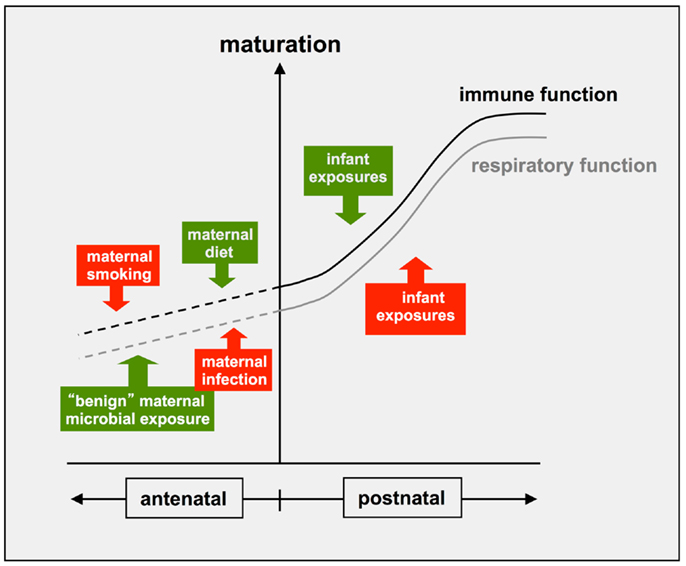
Figure 2. Antenatal and postnatal factors influencing development if immune and respiratory functions. Recent evidence indicates that the postnatal maturation of both respiratory and immune functions can be influenced by positive (green) and negative (red) environmental influences, which can exert their effects after birth and/or antenatally.
Maternal exposure to indoor air pollution especially that is associated with biomass fuel use for cooking and heating, in developing countries is a particular problem and associated with both acute and chronic respiratory diseases in the offspring (120, 121). Maternal infections during pregnancy, particularly those associated with antibiotic use, have also been associated with increased propensity for later wheeze in offspring (122, 123). Thus, the temporal window period during which functional programing of the key physiological processes underpinning subsequent risk for asthma spans both the antenatal and early postnatal periods.
Progression toward Persistent Asthma
A diagnosis of asthma at the end of the preschool years implies increased risk for progression to persistent asthma in the teens and beyond, but at the population level only 50–60% of early-onset asthmatics follow this track, and at least through to the end of the teenage years the majority of the latter are atopics (93, 124). In these subjects, cumulative pathological changes resulting from episodic cycles of airway tissue damage and ensuing repair/remodeling continue to drive pathogenesis, progressively eroding levels of respiratory function relative to their non-asthmatic counterparts (11, 49). The most overt manifestations of this process are asthma exacerbation events, particularly those occasioning physician visits, and the decrement in ensuing lung function after each episode reflects their intensity and duration (125). It is now well established that as per the initiation phase of asthma in preschoolers, the principal environmental triggers of asthma exacerbations in older children and adults with established disease are aeroallergens and respiratory viral infections, acting independently and/or in synergy (11, 49) (Box 1).
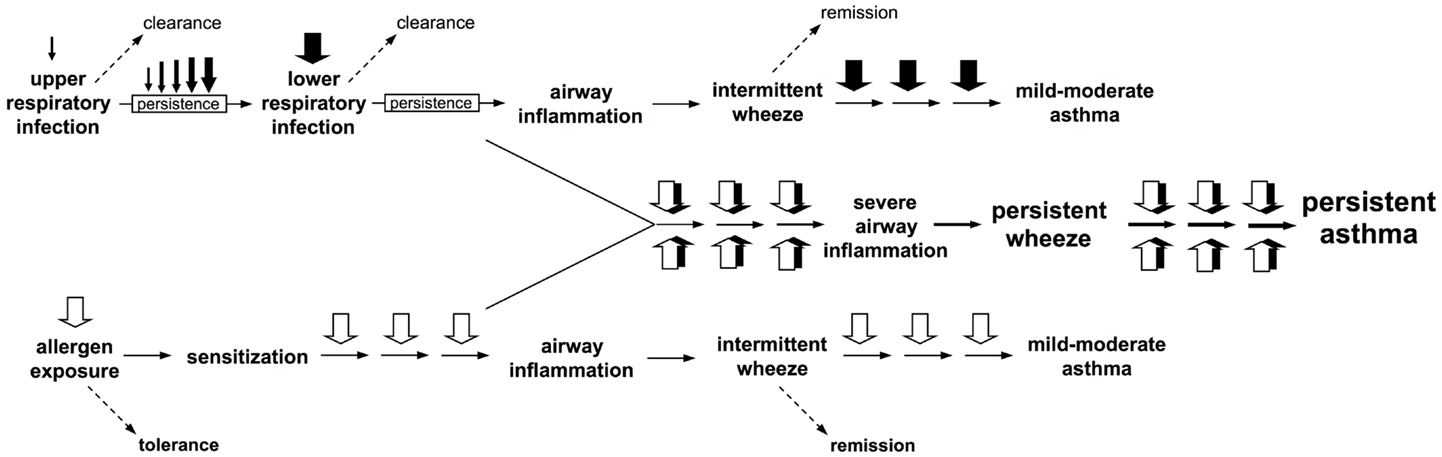
Box 1. Interactions between atopic and anti-viral inflammatory pathways during exacerbations drive progression to persistent asthma. Two clear pathways to wheeze development are now recognized. Firstly, upper respiratory viral infections which evade clearance, and instead persist and intensify and spread to the small airways of the lower respiratory tract where they trigger intermittent inflammatory responses and associated wheeze. These symptoms frequently remit, but recurrence of such episodes can result in their persistence and this is associated with heightened risk for asthma development. Secondly, sensitization to perennial (especially indoor) allergens which are present continuously in the domestic environment can also lead to airway inflammation of generally lower intensity than those associated with infections, but such events are likely to occur considerably more frequently, and can also be associated with asthma risk. However, the highest risk is associated with concomitant exposure to virus and aeroallergen against a background of aeroallergen sensitization, resulting in interaction(s) between the pro-inflammatory pathways triggered by these agents that appear synergistic.
Recent studies from our group and others have elucidated a series of interrelated mechanisms that underlie interactions between viral and allergen-triggered inflammatory pathways [reviewed in Ref. (49)]. The key mechanism involves viral triggered upregulation of FcER1-expression on local airway mucosal (AM) DC that results in recruitment of “bystander” aeroallergen-specific Th2-memory responses into the inflammatory milieu at the infection site, which in turn attenuates Th1-dependent anti-viral defense mechanisms and delays viral clearance. Ensuing type 1 IFN- and IL-4/IL-13 signaling to the bone marrow results in upregulation of FcER1-expression on DC precursors prior to their release into the circulation for migration to the infected airway mucosa to replenish the rapidly turning over AMDC population. This further enhances local aeroallergen-specific Th2-memory cell activation, amplifying inflammation, and further retarding viral clearance. Consistent with this sequence, gene co-expression network analysis of sputum cells collected from exacerbating children indicated expression of Th2-associated effector genes in conjunction with attenuated expression of Th-1/cytotoxic pathway genes, and linked the level of this attenuation with the degree of chronic airflow obstruction, which followed the event (126).
It is important to note that this interaction between atopy/viral infection appears operative across the full spectrum of virus-associated wheezing events including those in mild asthmatics (17). Moreover, IL-4/IL-13 signaling from sites of allergy-induced inflammation to the bone marrow clearly occurs in the absence of viral infection, resulting in upregulation of FcER1-expression in the circulating DC precursor pool that is likely related to the intensity/duration of the inducing event (49). The same precursor pool continuously replenishes mucosal DC populations throughout the body, and this process hence provides a putative mechanism through which allergic manifestations in one tissue can promote the spread of allergic disease to other (previously unaffected) tissues by enhancing the baseline Th2-stimulatory potential of DCs in the latter (49). Such cross-tissue relationships have long been recognized in the allergy field, exemplified in this context by the epidemiological data linking allergic rhinitis in young adults with risk for subsequent development of asthma (127, 128). The “Allergic March” concept in pediatric allergy in which food allergy during infancy precedes rhinitis, which in turn precedes asthma [reviewed in Ref. (49)] would appear to represent another example of this process in action.
Additional Complexities in Asthma Development
The pathways illustrated in Figures 1 and 2 and Box 1 provide plausible mechanistic explanations for some of the major epidemiological associations observed in relation to risk for asthma development, but cannot accommodate important emerging data from other crucial areas.
Genetic Susceptibility to Disease
Susceptibility to asthma is recognized to be in part genetically determined and, while asthma heritability is less than many other complex diseases, to date variants in excess of 100 genes have been identified as potential contributors to this risk (1, 3, 4, 6). Moreover, specific gene × environment interactions exemplified by those involving microbial exposures and asthma/allergy-related outcomes (2, 5, 129–131) are increasingly being identified in the context.
Childhood Obesity
Obesity is increasingly prevalent in children and there is increasing evidence that obesity affects childhood asthma and its severity (132). The nature of the association between obesity and asthma remains obscure in children and appears to be stronger for non-atopic asthma. Longitudinal studies suggest that high body weight precedes asthma symptoms. However, it is not clear how body mass may influence primary acquisition of persistent wheeze. It is also of interest that body mass can exert cross generational epigenetic effects so it is possible that parental obesity may affect their offspring.
Airway Microbiota
Until very recently, it was assumed that at baseline the lower airways of normal immunocompetent individuals is maintained in a sterile condition, punctuated by only occasional incursions in the form of viral infections accompanied in some instances by opportunistic bacterial pathogens derived mainly from the nasopharyngeal flora. However, a recent study using bacterial metagenomics and applied in conjunction with sterile fiber-optic bronchoscopic sampling of the lower airways has demonstrated the presence of complex bacterial flora extending from the nasopharynx to the alveolar spaces in healthy individuals (133). There is emerging evidence that in diseases such as asthma and COPD the density and complexity of this airway flora may be increased relative to the overall population, but the currently available data must be considered principally qualitative rather than quantitative and reflects the very early stage of research in this area.
We have recently reviewed the theoretical implications of these new findings (134). Notably, data exemplified by the CAS birth cohort (61) demonstrate that it is common for infants within the high-risk “window period” for asthma development to experience three to six moderate–severe lower respiratory viral infections per year, and it must now be accepted that virtually every one of these episodes will involve some level of breaching of airway epithelial barriers by “bystander” bacterial flora, which are present in fluids overlying the airway mucosa at the time of viral infection. As noted (134), this carries the attendant risk of bacterial-induced amplification of local inflammatory tissue damage at the viral infection site, and/or potential secondary bacterial infection as pneumonia or sepsis, unless clearance is rapid. In this regard, it is of interest that early colonization of the nasopharynx with bacterial pathogens has been linked with increased risk for subsequent development of asthma in preschoolers (135). It is therefore relevant to question whether variations in antibacterial host defense mechanisms may also contribute to asthma susceptibility in young children. Preliminary evidence from the CAS birth cohort (134, 136) suggests that the efficiency of immune surveillance for common nasopharyngeal bacterial species during infancy as measured via specific IgG1 antibody titers is inversely related to risk for subsequent development of atopic asthma in children, and this may reflect the role of this antibody in bacterial clearance. Moreover, we have recently reported that Th2-polarized immunity in school children against these bacterial species is protective against expression of asthma symptoms (137). The surrogate marker for this latter effect is IL-4/IL-13-dependent IgE antibody against bacterial-specific particulate antigens, but we theorize that protection is mediated by bacterial-specific IL-4/IL-13-producing Th2-memory cells that employ these same cytokines to inhibit bacterial-induced activation of airway tissue macrophages at sites of low-level bacterial incursions (134, 137).
These new findings have collectively opened, yet another chapter in the already highly complex story of the immunopathogenesis of human asthma. It is clear that high-precision experimental models, which focus on individual inflammatory effector pathways can provide invaluable understanding relating to their targeted mechanisms, but applying this information to the development of treatment strategies that must account for variations in patient age, disease stage, and in particular, disease sub-phenotype has proven to be a major and still outstanding challenge (49).
Need for Complex Models
Table 1 summarizes the multiple known and presumptive risk factors for the development of asthma discussed above. Given the breadth and diversity of these factors and the relative long-time frame over which their effects may be exerted, the relevance of cohort studies where objective longitudinal data collected over decades becomes immediately clear. One of the most important recent innovations has been the application of mathematical modeling methods to cohort data in an effort to parse out the important interactions and driving mechanism leading to persistent asthma.
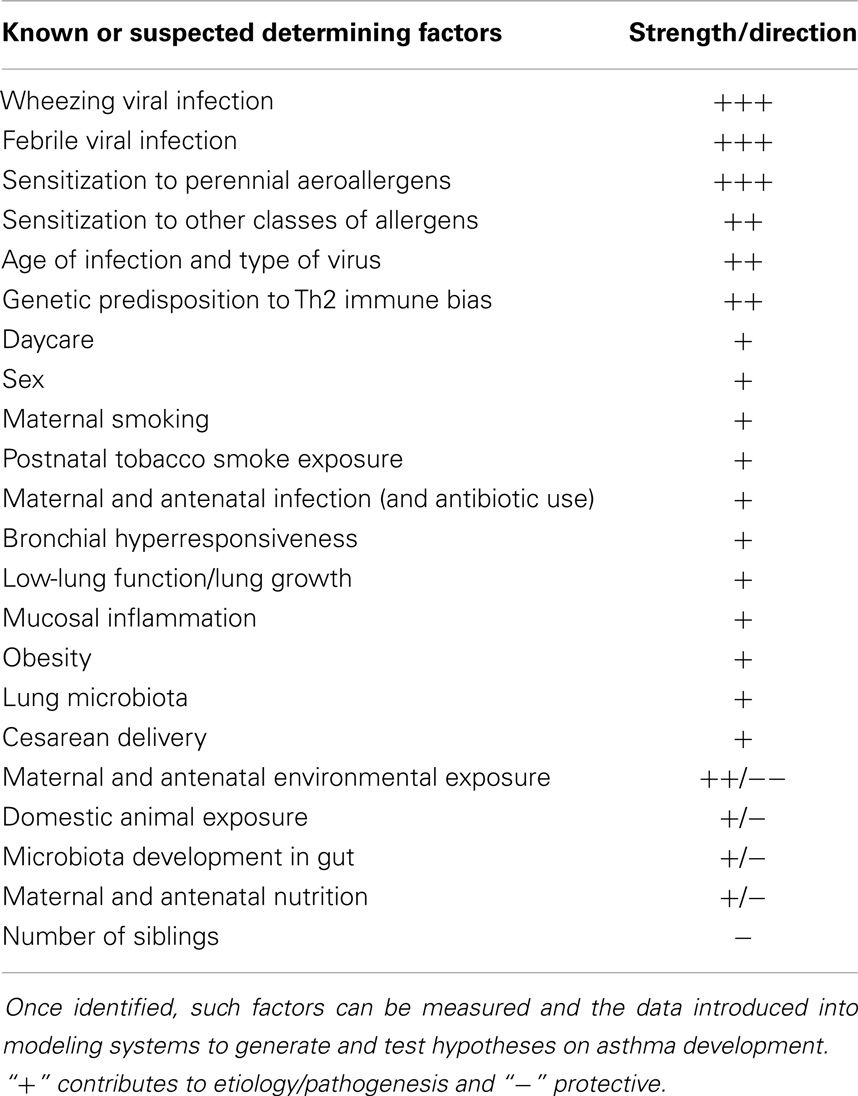
Table 1. Known and presumptive factors though to influence the development and persistence of asthma during early childhood.
Modeling Birth Cohorts Using Networks
A useful way of thinking about human development in infancy and childhood is that of a (spatio)temporally dynamic network of interacting systems. The multi-factorial nature of asthma pathogenesis implies that many such systems need to be considered, including the respiratory, immune, and microbial systems. These systems are constrained by the genomes of their respective organisms and are impacted by environmental factors, including viral infections and inhaled exposures.
An accurate and robust network model of the factors comprising asthma pathogenesis may be effective in identifying and prioritizing prevention and intervention strategies at the population and individual level. Indeed, such an approach has already been advocated for modeling the immune system (138). Deeply phenotyped cohort studies, such as CAS (40, 139), MAS (39, 140), and URECA (141), will be critical in the building and validation of such network models. These studies present a broad range of detailed data with the corresponding time order, allowing researchers to map the paths of disease pathogenesis and gain insight into causation, prevention, and possible interventions. Further, network modeling of such cohorts would take advantage of the wealth of expert knowledge in asthma pathogenesis, such as that in prior sections of this review.
Network Models
The behaviors of biological networks can be complex. For this reason, as well as a human predilection for pairwise relationships, network models are still rarely the first approach for many researchers. Yet, tools for studying networks have so far found a diverse range of applications including the imputation of missing data from an asthma intervention/education program (142), the relationship between ozone point data and regional asthma (143), the role of cytokine networks in asthma (144) and the identification and characterization of transcriptional networks for IgE signaling (145, 146). Further, with the increasing utilization and integration of genomic and other omics profiling, a network modeling framework offers a powerful approach for extracting maximum biological information for massive datasets (147).
A network graph is a versatile means of representing the multiplicity of relationships that can exist, whether they are simple correlations or of a directed, often causal, nature. Graphs of networks, comprising nodes (representing entities such as phenotypes), and connecting lines called “edges” (e.g., representing relationships between phenotypes), can be an intuitive way of capturing complex interactions; for example, most would find the graph rather than the table in Figure 3A easier to comprehend, and graphs can also visually indicate the strength and sign of relationships between nodes (Figure 3B).
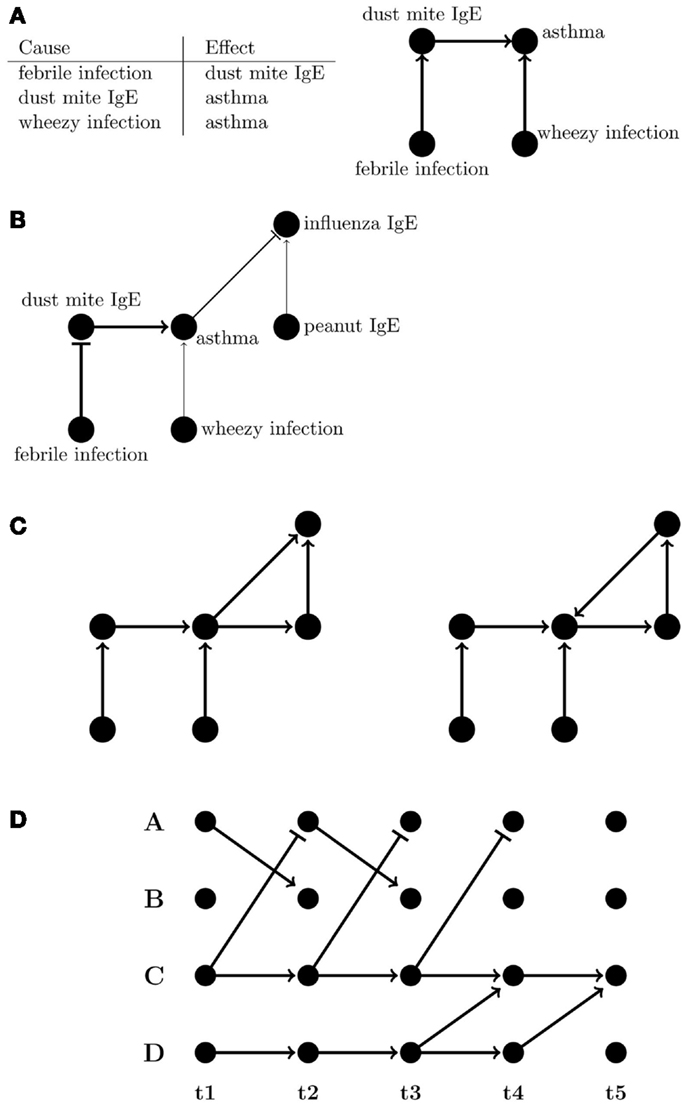
Figure 3. Representations of directed networks. (A) Tabular and graphical (left and right, respectively) displays of directional flow between variables. (B) Graph showing variations in strength and sign of effects between variables. The flattened edges indicate negative relationships. (C) Directed acyclic (DAG) and directed cyclic graphs (left and right, respectively). (D) A dynamic Bayesian network where edges are only between subsequent time points.
Bayesian Network Models
A popular and mathematically rigorous example of a network, which seeks to model causal relationships, is a Bayesian network (BN), a type of directed acyclic graph (DAG) (Figure 3C). BNs are particularly useful for biological and clinical data because they are inherently probabilistic, tolerant of incomplete or missing data, and there are wide array of robust tools available to researchers wishing to use them. Importantly, they explicitly allow for integration of prior expert knowledge into the model. BNs have also been applied to biological and clinical problems such as diagnostics (148), cell signaling pathways (149, 150), asthma exacerbations in a pediatric emergency department (151), and transcriptional networks associated with asthma susceptibility (152). For cohort studies, a useful feature of BNs is that mutual interactions and self-interactions can still be represented if the factors involved have corresponding nodes at multiple time points. Longitudinal data allow for better inference of causal relationships, allowing one to infer the relationship between node A at time ti and node B at subsequent time ti + 1 even if A and B are the same phenotype. So-called “un-rolled” BNs, i.e., those that allow their topology to change over time, are referred to as dynamic Bayesian networks (DBNs) (153–157) (Figure 3D). While homeostatic self-regulating systems like immune networks can have multiple cyclical feedbacks, DBNs, and other models for how multiple susceptibilities and risks affect disease over time can account for cyclic feedback by making the assumption that time itself always moves forward. Events in the future do not influence events in the present or past.
Model Inference
While an expert can manually construct networks from known relationships, efficient algorithms can infer them from raw data (158–163). Many are of a “hill-climbing” variety (164), in which a graph/network is varied in some way, usually (semi-) randomly, evaluated for its fit to the data and accepted if the resulting network is found to be an improvement. Repeating this process, the algorithm finds a succession of networks of increasingly better fit to the data. To avoid getting “stuck” on a suboptimal network, which happens to be better than those most similar to it, it is possible to run the algorithm multiple times from different starting points (156) or to allow (165) limited excursions “downhill” so that higher hills (which better fit the data) might be found.
Another generic approach is to infer appropriate networks using Bayesian inference (166–174). The operating principle for Bayesian inference may be reasonably characterized as saying that a network is more likely to be correct if it is more likely to generate the data. These so-called marginal likelihoods are weighted by any information already available, which might indicate the true values. This ability to incorporate prior information can be a major advantage of the Bayesian approach (166, 170). Of particular relevance to clinical application, Bayesian inference has long been utilized for classification and prediction (175–177). Bayesian classification has been applied to such quality of care assessment for hospitals (178) prediction of outcomes of medical procedures (158, 178–180), and the association of genetic data with phenotypes (181).
Naïve Bayesian networks
The simplest BN, successfully used for many classification and prediction applications (175–177, 182–184), assumes that the probability distributions of the variables of interest are independent. While this assumption is clearly strong for a complex disease such as asthma and the deeply phenotyped cohorts needed for its study, the so-called naïve Bayes model is still instructive as a basic BN, is relatively accessible as a model for clinical and biological researchers, and can be illustrative of the effects, or lack thereof, of modeling assumptions. Indeed, the naïve Bayes model can be more accurate as a classifier than one might reasonably expect. While violations of the independence assumption can be detrimental to the algorithm (185), it has often been observed that naïve Bayes performs comparably to more sophisticated approaches with fewer assumptions (183). Yet, more sophisticated approaches that, for example, estimate joint, conditional and marginal probability distributions from the data are usually not solved exactly due to computational limitations so approximation methods must be used to infer the network (186–191).
Comparison of a Simple (Naïve) Bayesian Network to Logistic Regression Using the Perth-Based Childhood Asthma Study
To demonstrate the utility of a simple BN on cohort data, we performed a comparison of the performance of the naïve Bayes and logistic regression models. Performance in this case has been defined as prediction of a child’s wheezing phenotype at 5 years of age without regard to network topology (as the true network is not known) and we caution that the performance in this case is not a fully realized prediction model, and as such is suitable for method comparison only. In this case, naïve Bayes simplifies the problem of BN inference and illustrates the advantage of the prior. To perform the analyses, we utilized the open-source R statistical package klaR (192) and the glm function in stats. Since asthma and its characteristic wheezing phenotype are complex multi-factorial diseases whose pathogenesis often begins in childhood, we simultaneously model a wide cross-section of 45 CAS phenotypes collected in the first 2 years of life (Table S1 in Supplementary Material). These include measures of respiratory infection, including virus-specific confirmation, circulating antibody profiles, and environmental variables, as well as previously published risk factors (40, 61). For simplicity, we assume that all variables are Gaussian and each prior is set to the mean across all the data.
Model Construction
Construction of the models proceeded via cross-validation. We used two-thirds of the CAS data to train both logistic regression and naïve Bayes models to predict wheeze status at 5 years given data. The resulting models were then externally tested on the remaining third of the data (the “test” set) to determine their performance in predicting subsequent wheeze. We minimized the effect of stochasticity from sampling variation by repeating this process 100 times for each model, taking different training and testing datasets each time, then averaging the result. We also ensured that the proportion of wheezing individuals in both training and testing datasets reflected that of the overall study and missing data was replaced with corresponding mean values.
Performance
The relative performance of the two approaches was assessed using the area under the ROC curve (AUC), a measure, which considers both sensitivity and specificity and can be interpreted as the probability of a given model correctly ranking a disease case higher than a healthy individual. The ROC curves for logistic regression and naïve Bayes models are given in Figure 4. From these analyses, the naïve Bayes was the better predictor (i.e., explained more variance in the wheeze phenotype) with an AUC of 0.64 compared to an AUC of 0.57 for logistic regression. These results have implications for modern day cohort studies where the number of phenotypes, especially those generated from omics technologies, will usually be greater than the study’s sample size and, further, inevitably there will be a large tradeoff between the number of individuals and the extent of phenotyping (for large phenotype networks there are likely a relatively small number of individuals).
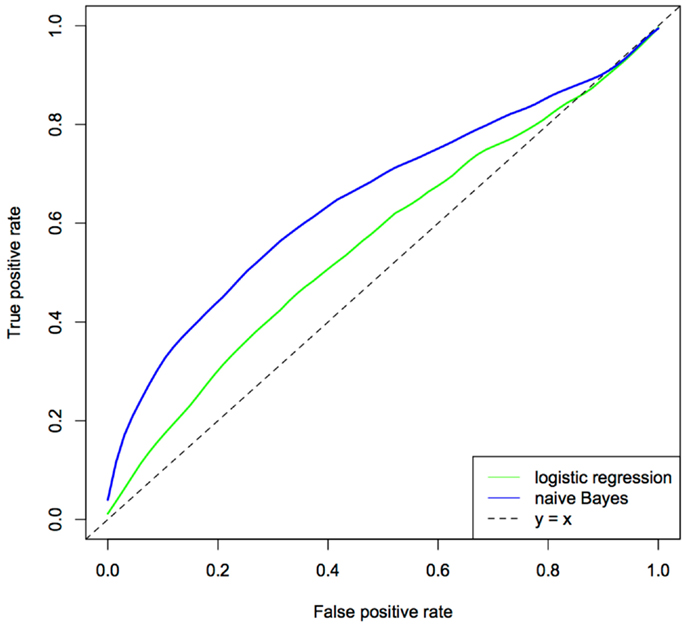
Figure 4. Comparison of logistical regression and naïve Bayes ROC curves. The predictor set contains 45 different predictors, including measures of respiratory infections, immune function, lung function, and maternal and environmental factors. The ROC curve was generated using 100 × 3-fold cross-validation.
The naïve Bayes model clearly outperformed logistic regression in this setting, showing that the integration of a large number of phenotypes with prior information has had a decidedly positive effect on the performance of the model. Yet, it is likely that the assumptions of naïve Bayes are detrimental to its overall performance. More sophisticated approaches, which are able to incorporate prior information as well as remove the assumption of independence among phenotypes, such as a DBN, are likely to yield superior models and improve stratification of those at risk of asthma and those who are not. Further, models that allow topological changes in the network, are likely to better reflect the pathogenesis of the disease.
Areas of Development
Harnessing the full power of a network-based approach raises several difficult issues. Perhaps the most basic is the comparison of networks describing corresponding data from different studies. For the large, hybrid networks envisaged here, the issue of replicating networks and quantifying their differences is a complex problem and one requiring further research. A second issue is the harmonization of clinical and biological measures between studies as well as the incorporation of disparate variable types, including continuous, discrete/binary data into the network. This complication alone requires advanced statistical methods to “augment” non-continuous variables (193). Asthma pathogenesis is further complicated by the existence of multiple asthma phenotypes, as described above. Since these different phenotypes are likely to follow differing paths of development, their corresponding networks are likely different and the interventions required to prevent them may also be different. An obvious approach to this problem is to construct different networks for each phenotype, but this approach would neglect the insight that might be gleaned from modeling all asthma phenotypes simultaneously within the same network. Finally, the inference of causal relationships from statistical data is a difficult and subtle task. There are some tools for causal inference, which can be adopted from other fields, such as Granger causality (194, 195); however, the limitations of causal inference from observational data alone should be emphasized and, ultimately, definitive demonstration of causality will require experimental interventions.
Conclusion
Asthma is a complex disease with multiple factors acting in concert over long-time periods. Here, we have reviewed known asthma pathways, characterized the clear complexity of the asthma phenotype, and proposed that prospective birth cohorts offer an attractive way forward, via tracking of disease development and a broad range of patient-associated clinical, physiological, biochemical, immunological, and microbiological markers in large human populations over time. On the resultant datasets, statistical modeling that integrates the deep phenotyping/genetic profiling with expert human knowledge is likely to provide valuable insights for the next generation of studies into asthma’s pathogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the NHMRC of Australia [Project Grant #1049539; Fellowships #1061435 (Michael Inouye, co-funded with the Australian Heart Foundation) and #1061409 (Kathryn E. Holt)]. Michael L. Walker is supported by the 2013 and 2014 Beaney Fellowship from the University of Melbourne.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fimmu.2014.00447/abstract
References
1. Ober C, Hoffjan S. Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun (2006) 7:95–100. doi: 10.1038/sj.gene.6364284
2. Ege MJ, Strachan DP, Cookson WO, Moffatt MF, Gut I, Lathrop M, et al. Gene-environment interaction for childhood asthma and exposure to farming in Central Europe. J Allergy Clin Immunol (2011) 127:e1–4. doi:10.1016/j.jaci.2010.09.041
3. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med (2010) 363:1211–21. doi:10.1056/NEJMoa0906312
4. Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature (2007) 448:470–3. doi:10.1038/nature06014
5. Renz H, von Mutius E, Brandtzaeg P, Cookson WO, Autenrieth IB, Haller D. Gene-environment interactions in chronic inflammatory disease. Nat Immunol (2011) 12:273–7. doi:10.1038/ni0411-273
6. Spycher BD, Henderson J, Granell R, Evans DM, Smith GD, Timpson NJ, et al. Genome-wide prediction of childhood asthma and related phenotypes in a longitudinal birth cohort. J Allergy Clin Immunol (2012) 130(503–9):e7. doi:10.1016/j.jaci.2012.06.002
7. Martinez FD, Wright AL, Taussig LM, Holberg CJ, Halonen M, Morgan WJ. Asthma and wheezing in the first six years of life. The group health medical associates. N Engl J Med (1995) 332:133–8. doi:10.1056/NEJM199501193320301
8. Anderson HR, Pottier AC, Strachan DP. Asthma from birth to age 23: incidence and relation to prior and concurrent atopic disease. Thorax (1992) 47:537–42. doi:10.1136/thx.47.7.537
9. Tepper RS, Morgan WJ, Cota K, Wright A, Taussig LM. Physiologic growth and development of the lung during the first year of life [erratum appears in Am Rev Respir Dis 1987;136(3):800]. Am Rev Respir Dis (1986) 134:513–9.
10. Collins RA, Parsons F, Deverell M, Hollams EM, Holt PG, Sly PD. Risk factors for bronchial hyperresponsiveness in teenagers differ with sex and atopic status. J Allergy Clin Immunol (2011) 128(301–7):e1. doi:10.1016/j.jaci.2011.03.016
11. Sly P, Boner A, Bjorksten B, Bush A, Custovic A, Eigenmann PA, et al. Early identification of atopy in the prediction of persistent asthma in children. Lancet (2008) 372:1100–6. doi:10.1016/S0140-6736(08)61451-8
12. Himes BE, Sheppard K, Berndt A, Leme AS, Myers RA, Gignoux CR, et al. Integration of mouse and human genome-wide association data identifies KCNIP4 as an asthma gene. PLoS One (2013) 8:e56179. doi:10.1371/journal.pone.0056179
13. Akdis M, Akdis CA. Therapeutic manipulation of immune tolerance in allergic disease. Nat Rev Drug Discov (2009) 8:645–60. doi:10.1038/nrd2653
14. Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol (2008) 8:142–52. doi:10.1038/nri2236
15. Strickland D, Holt P. T-regulatory cells in childhood asthma. Trends Immunol (2011) 32:420–7. doi:10.1016/j.it.2011.06.010
17. Olenec JP, Kim WK, Lee WM, Vang F, Pappas TE, Salazar LE, et al. Weekly monitoring of children with asthma for infections and illness during common cold seasons. J Allergy Clin Immunol (2010) 125(1001–6):e1. doi:10.1016/j.jaci.2010.01.059
18. Morgan WJ, Stern DA, Sherrill DL, Guerra S, Holberg CJ, Guilbert TW, et al. Outcome of asthma and wheezing in the first 6 years of life: follow-up through adolescence. Am J Respir Crit Care Med (2005) 172:1253–8. doi:10.1164/rccm.200504-525OC
20. Soler Artigas M, Loth DW, Wain LV, Gharib SA, Obeidat M, Tang W, et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat Genet (2011) 43:1082–90. doi:10.1038/ng.941
21. van Putte-Katier N, Koopmans M, Uiterwaal CS, de Jong BM, Kimpen JL, Verheij TJ, et al. Relationship between parental lung function and their children’s lung function early in life. Eur Respir J (2011) 38:664–71. doi:10.1183/09031936.00034210
22. Guilbert TW, Singh AM, Danov Z, Evans MD, Jackson DJ, Burton R, et al. Decreased lung function after preschool wheezing rhinovirus illnesses in children at risk to develop asthma. J Allergy Clin Immunol (2011) 128:532.e–8.e. doi:10.1016/j.jaci.2011.06.037
23. Sly PD, Kusel M, Holt PG. Do early-life viral infections cause asthma? J Allergy Clin Immunol (2010) 125:1202–5. doi:10.1016/j.jaci.2010.01.024
24. Saglani S, Payne DN, Zhu J, Wang Z, Nicholson AG, Bush A, et al. Early detection of airway wall remodelling and eosinophilic inflammation in preschool wheezers. Am J Respir Crit Care Med (2007) 176:858–64. doi:10.1164/rccm.200702-212OC
25. Malmström K, Pelkonen AS, Malmberg LP, Sarna S, Lindahl H, Kajosaari M, et al. Lung function, airway remodelling and inflammation in symptomatic infants: outcome at 3 years. Thorax (2011) 66:157–62. doi:10.1136/thx.2010.139246
26. Sonnappa S, Bastardo C, Saglani S, Bush A, Aurora P. Relationship between past airway pathology and current lung function in preschool wheezers. Eur Respir J (2011) 38:1431–6. doi:10.1183/09031936.00164910
27. Regamey N, Ochs M, Hilliard TN, Mühlfeld C, Cornish N, Fleming L, et al. Increased airway smooth muscle mass in children with astham, cystic fibrosis and non-cytsic fibrosis bronchiectasis. Am J Respir Crit Care Med (2008) 177:837–43. doi:10.1164/rccm.200707-977OC
28. Grainge C, Lau L, Ward J, Dulay V, Lahiff G, Wilson S, et al. Effect of bronchoconstriction on airway remodelling in asthma. N Engl J Med (2011) 364:2006–15. doi:10.1056/NEJMoa1014350
29. Burgess JK, Johnson PR, Ge Q, Au WW, Poniris MH, McParland BE, et al. Expression of connective tissue growth factor in asthmatic airway smooth muscle cells. Am J Respir Crit Care Med (2003) 167:71–7. doi:10.1164/rccm.200205-416OC
30. Balzar S, Chu HW, Silkoff P, Cundall M, Trudeau JB, Strand M, et al. Increased TGF-β2 in severe asthma with eosinophilia. J Allergy ClinImmunol (2005) 115:110–7. doi:10.1016/j.jaci.2004.09.034
31. Takayama G, Arima K, Kanaji T, Toda S, Tanaka H, Shoji S, et al. Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J Allergy ClinImmunol (2006) 118:98–104. doi:10.1016/j.jaci.2006.02.046
32. Doucet C, Brouty-Boyé D, Pottin-Clémenceau C, Canonica GW, Jasmin C, Azzarone B. Interleukin (IL) 4 and IL-13 act on human lung fibroblasts. Implication in asthma. J Clin Invest (1998) 101:2129. doi:10.1172/JCI741
33. Ward C, Johns DP, Bish R, Pais M, Reid DW, Ingram C, et al. Reduced airway distensibility, fixed airflow limitation, and airway wall remodelling in asthma. Am J Respir Crit Care Med (2001) 164:1718–21. doi:10.1164/ajrccm.164.9.2102039
34. Choy DF, Modrek B, Abbas AR, Kummerfeld S, Clark HF, Wu LC, et al. Gene expression patterns of Th2 inflammation and intercellular communication in asthmatic airways. J Immunol (2011) 186:1861–9. doi:10.4049/jimmunol.1002568
35. Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med (2010) 181:315–23. doi:10.1164/rccm.200906-0896OC
36. Newcomb DC, Peebles RS Jr. Th17-mediated inflammation in asthma. Curr Opin Immunol (2013) 25:755–60. doi:10.1016/j.coi.2013.08.002
37. Pohunek P, Roche WR, Turzikova J, Kudrmann J, Warner JO. Eosinophilic inflammation in the bronchial mucosa of children with bronchial asthma. Eur Respir J (1997) 10:160.
38. Gern JE, Brooks GD, Meyer P, Chang A, Shen K, Evans MD, et al. Bidirectional interactions between viral respiratory illnesses and cytokine responses in the first year of life. J Allergy Clin Immunol (2006) 117:72–8. doi:10.1016/j.jaci.2005.10.002
39. Illi S, von Mutius E, Lau S, Niggemann B, Gruber C, Wahn U. Perennial allergen sensitisation early in life and chronic asthma in children: a birth cohort study. Lancet (2006) 368:763–70. doi:10.1016/S0140-6736(06)69286-6
40. Kusel MM, de Klerk NH, Kebadze T, Vohma V, Holt PG, Johnston SL, et al. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol (2007) 119:1105–10. doi:10.1016/j.jaci.2006.12.669
41. Oddy WH, de Klerk NH, Sly PD, Holt PG. The effects of respiratory infections, atopy, and breastfeeding on childhood asthma. Eur Respir J (2002) 19:899–905. doi:10.1183/09031936.02.00103602
42. Peat JK, Salome CM, Woolcock AJ. Longitudinal changes in atopy during a 4-year period: relation to broncial hyperresponsiveness and respiratory symptoms in a population sample of Australian schoolchildren. J Allergy Clin Immunol (1990) 85:65–74. doi:10.1016/0091-6749(90)90223-Q
43. Rhodes HL, Thomas P, Sporik R, Holgate ST, Cogswell JJ. A birth cohort study of subjects at risk of atopy: twenty-two-year follow-up of wheeze and atopic status. Am J Respir Crit Care Med (2002) 165:176–80. doi:10.1164/ajrccm.165.2.2104032
44. Sherrill D, Stein R, Kurzius-Spencer M, Martinez F. Early senstization to allergens and development of respiratory symptoms. Clin Exp Allergy (1999) 29:905–11. doi:10.1046/j.1365-2222.1999.00631.x
45. Croner S, Kjellman NIM. Development of atopic disease in relation to family history and cord blood IgE levels. Eleven year follow-up in 1654 children. Pediatr Allergy Immunol (1990) 1:14–20. doi:10.1111/j.1399-3038.1990.tb00003.x
46. Stern DA, Morgan WJ, Halonen M, Wright AL, Martinez FD. Wheezing and bronchial hyper-responsiveness in early childhood as predictors of newly diagnosed asthma in early adulthood: a longitudinal birth-cohort study. Lancet (2008) 372:1058–64. doi:10.1016/S0140-6736(08)61447-6
47. Simpson A, Tan VY, Winn J, Svensén M, Bishop CM, Heckerman DE, et al. Beyond atopy: multiple patterns of sensitization in relation to asthma in a birth cohort study. Am J Respir Crit Care Med (2010) 181:1200–6. doi:10.1164/rccm.200907-1101OC
48. von Mutius E, Radon K. Living on a farm: impact on asthma induction and clinical course. Immunol Allergy Clin North Am (2008) 28:631–47. doi:10.1016/j.iac.2008.03.010
49. Holt PG, Sly PD. Viral infections and atopy in asthma pathogenesis: new rationales for asthma prevention and treatment. Nat Med (2012) 18:726–35. doi:10.1038/nm.2768
50. Rakes GP, Arruda E, Ingram JM, Hoover GE, Zambrano JC, Hayden FG, et al. Rhinovirus and respiratory syncytial virus in wheezing children requiring emergency care. IgE and eosinophil analyses. Am J Respir Crit Care Med (1999) 159:785–90. doi:10.1164/ajrccm.159.3.9801052
51. Bizzintino J, Lee WM, Laing IA, Vang F, Pappas T, Zhang G, et al. Association between human rhinovirus C and severity of acute asthma in children. Eur Respir J (2011) 37:1037–42. doi:10.1183/09031936.00092410
52. Bochkov YA, Palmenberg AC, Lee WM, Rathe JA, Amineva SP, Sun X, et al. Molecular modeling, organ culture and reverse genetics for a newly identified human rhinovirus C. Nat Med (2011) 17:627–32. doi:10.1038/nm.2358
53. James KM, Gebretsadik T, Escobar GJ, Wu P, Carroll KN, Li SX, et al. Risk of childhood asthma following infant bronchiolitis during the respiratory syncytial virus season. J Allergy Clin Immunol (2013) 132:227–9. doi:10.1016/j.jaci.2013.01.009
54. Sigurs N, Aljassim F, Kjellman B, Robinson PD, Sigurbergsson F, Bjarnason R, et al. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax (2010) 65:1045–52. doi:10.1136/thx.2009.121582
55. Wu P, Dupont WD, Griffin MR, Carroll KN, Mitchel EF, Gebretsadik T, et al. Evidence of a causal role of winter virus infection during infancy in early childhood asthma. Am J Respir Crit Care Med (2008) 178:1123–9. doi:10.1164/rccm.200804-579OC
56. Stein RT, Sherrill D, Morgan WJ, Holberg CJ, Halonen M, Taussig LM, et al. Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years [see comment]. Lancet (1999) 354:541–5. doi:10.1016/S0140-6736(98)10321-5
57. Xatzipsalti M, Psarros F, Konstantinou G, Gaga M, Gourgiotis D, Saxoni-Papageorgiou P, et al. Modulation of the epithelial inflammatory response to rhinovirus in an atopic environment. Clin Exp Allergy (2008) 38:466–72. doi:10.1111/j.1365-2222.2007.02906.x
58. Papadopoulos NG, Stanciu LA, Papi A, Holgate ST, Johnston SL. A defective type 1 response to rhinovirus in atopic asthma. Thorax (2002) 57:328–32. doi:10.1136/thorax.57.4.328
59. Papadopoulos NG, Stanciu LA, Papi A, Holgate ST, Johnston SL. Rhinovirus-induced alterations on peripheral blood mononuclear cell phenotype and costimulatory molecule expression in normal and atopic asthmatic subjects. Clin Exp Allergy (2002) 32:537–42. doi:10.1046/j.0954-7894.2002.01313.x
60. Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med (2005) 201:937–47. doi:10.1084/jem.20041901
61. Holt PG, Rowe J, Kusel M, Parsons F, Hollams EM, Bosco A, et al. Toward improved prediction of risk for atopy and asthma amongst preschoolers: a prospective cohort study. J Allergy Clin Immunol (2010) 125:643–51. doi:10.1016/j.jaci.2009.12.018
62. Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Pappas TE, Lee WM, et al. Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in early life. Am J Respir Crit Care Med (2012) 185:281–5. doi:10.1164/rccm.201104-0660OC
63. Kusel MMH, Kebadze T, Johnston SL, Holt PG, Sly PD. Febrile respiratory illnesses in infancy and atopy are risk factors for persistent asthma and wheeze. Eur Respir J (2012) 39:876–82. doi:10.1183/09031936.00193310
64. Schelonka RL, Infante AJ. Neonatal immunology. Semin Perinatol (1998) 22:2–14. doi:10.1016/S0146-0005(98)80003-7
65. Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol (2004) 4:553–64. doi:10.1038/nri1394
66. Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat Rev Immunol (2007) 7:379–90. doi:10.1038/nri2075
67. Siegrist CA. Vaccination in the neonatal period and early infancy. Int Rev Immunol (2000) 19:195–219. doi:10.3109/08830180009088505
68. Wegmann TG, Lin H, Guilbert L, Mosmann TR. Bidirectional cytokine interactions in the maternal-fetal relationship: is successful pregnancy a Th2 phenomenon? Immunol Today (1993) 14:353–6. doi:10.1016/0167-5699(93)90235-D
69. Holt PG, Jones CA. The immunology of fetuses and infants: the development of the immune system during pregnancy and early life [review]. Allergy (2000) 55:688–97. doi:10.1034/j.1398-9995.2000.00118.x
70. White GP, Watt PM, Holt BJ, Holt PG. Differential patterns of methylation of the IFNg promoter at CpG and non-CpG sites underlie differences in IFNg gene expression between human neonatal and adult CD45RO- T-cells. J Immunol (2002) 168:2820–7. doi:10.4049/jimmunol.168.6.2820
71. Black A, Bhaumik S, Kirkman RL, Weaver CT, Randolph DA. Developmental regulation of Th17-cell capacity in human neonates. Eur J Immunol (2012) 42:311–9. doi:10.1002/eji.201141847
72. Mills KH. Induction, function and regulation of IL-17-producing T cells. Eur J Immunol (2008) 38:2636–49. doi:10.1002/eji.200838535
73. Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol (2007) 25:821–52. doi:10.1146/annurev.immunol.25.022106.141557
74. Kollmann TR, Crabtree J, Rein-Weston A, Blimkie D, Thommai F, Wang XY, et al. Neonatal innate TLR-mediated responses are distinct from those of adults. J Immunol (2009) 183:7150–60. doi:10.4049/jimmunol.0901481
75. Holt PG, Clough JB, Holt BJ, Baron-Hay MJ, Rose AH, Robinson BW, et al. Genetic ‘risk’ for atopy is associated with delayed postnatal maturation of T-cell competence. Clin Exp Allergy (1992) 22:1093–9. doi:10.1111/j.1365-2222.1992.tb00135.x
76. Tang MLK, Kemp AS, Thorburn J, Hill DJ. Reduced interferon-gamma secretion in neonates and subsequent atopy. Lancet (1994) 344:983–6. doi:10.1016/S0140-6736(94)91641-1
77. Rinas U, Horneff G, Wahn V. Interferon-gamma production by cord-blood mononuclear cells is reduced in newborns with a family history of atopic disease and is independent from cord blood IgE-levels. Pediatr Allergy Immunol (1993) 4:60–4. doi:10.1111/j.1399-3038.1993.tb00068.x
78. Nilsson C, Larsson AK, Hoglind A, Gabrielsson S, Troye Blomberg M, Lilja G. Low numbers of interleukin-12-producing cord blood mononuclear cells and immunoglobulin E sensitization in early childhood. Clin Exp Allergy (2004) 34:373–80. doi:10.1111/j.1365-2222.2004.01896.x
79. Prescott SL, Taylor A, King B, Dunstan J, Upham JW, Thornton CA, et al. Neonatal interleukin-12 capacity is associated with variations in allergen-specific immune responses in the neonatal and postnatal periods. Clin Exp Allergy (2003) 33:566–72. doi:10.1046/j.1365-2222.2003.01659.x
80. Upham JW, Holt PG, Taylor A, Thornton CA, Prescott SL. HLA-DR expression on neonatal monocytes is associated with allergen-specific immune responses. J Allergy ClinImmunol (2004) 114:1202–8. doi:10.1016/j.jaci.2004.06.051
81. Schaub B, Liu J, Höppler S, Haug S, Sattler C, Lluis A, et al. Impairment of T-regulatory cells in cord blood of atopic mothers. J Allergy ClinImmunol (2008) 121:e1–13. doi:10.1016/j.jaci.2008.04.010
82. Hagendorens MM, Ebo DG, Schuerwegh AJ, Huybrechs A, Van Bever HP, Bridts CH, et al. Differences in circulating dendritic cell subtypes in cord blood and peripheral blood of healthy and allergic children. Clin Exp Allergy (2003) 33:633–9. doi:10.1046/j.1365-2222.2003.01649.x
83. Silver E, Yin-Declue H, Schechtman KB, Grayson MH, Bacharier LB, Castro M. Lower levels of plasmacytoid dendritic cells in peripheral blood are associated with a diagnosis of asthma 6 yr after severe respiratory syncytial virus bronchiolitis. Pediatr Allergy Immunol (2009) 20(5):471–6. doi:10.1111/j.1399-3038.2008.00818.x
84. Upham JW, Zhang G, Rate A, Yerkovich ST, Kusel M, Sly PD, et al. Plasmacytoid dendritic cells during infancy are inversely associated with childhood respiratory tract infections and wheezing. J Allergy Clin Immunol (2009) 124(707–13):e2. doi:10.1016/j.jaci.2009.07.009
85. Copenhaver CC, Gern JE, Li Z, Shult PA, Rosenthal LA, Mikus LD, et al. Cytokine response patterns, exposure to viruses, and respiratory infections in the first year of life. Am J Respir Crit Care Med (2004) 170:175–80. doi:10.1164/rccm.200312-1647OC
86. Blanco-Quiros A, Gonzalez H, Arranz E, Lapena S. Decreased interleukin-12 levels in umbilical cord blood in children who developed acute bronchiolitis. Pediatr Pulmonol (1999) 28:175–80. doi:10.1002/(SICI)1099-0496(199909)28:3<175::AID-PPUL3>3.0.CO;2-U
87. Bont L, Kavelaars A, Heijnen CJ, van Vught AJ, Kimpen JL. Monocyte interleukin-12 production is inversely related to duration of respiratory failure in respiratory syncytial virus bronchiolitis. J Infect Dis (2000) 181:1772–5. doi:10.1086/315433
88. Zhang G, Rowe J, Kusel M, Bosco A, McKenna K, de Klerk N, et al. Interleukin-10/Interleukin-5 responses at birth predict risk for respiratory infections in children with atopic family history. Am J Respir Crit Care Med (2009) 179:205–11. doi:10.1164/rccm.200803-438OC
89. Rowe J, Macaubas C, Monger T, Holt BJ, Harvey J, Poolman JT, et al. Heterogeneity in diphtheria-tetanus-acellular pertussis vaccine-specific cellular immunity during infancy: relationship to variations in the kinetics of postnatal maturation of systemic Th1 function. J Infect Dis (2001) 184:80–8. doi:10.1086/320996
90. Arkwright PD, Patel L, Moran A, Haeney MR, Ewing CI, David TJ. Atopic eczema is associated with delayed maturation of the antibody response to pneumococcal vaccine. Clin Exp Immunol (2000) 122:16–9. doi:10.1046/j.1365-2249.2000.01338.x
91. Smart JM, Kemp AS. Increased Th1 and Th2 allergen-induced cytokine responses in children with atopic disease. Clin Exp Allergy (2002) 32:796–802. doi:10.1046/j.1365-2222.2002.01391.x
92. Heaton T, Rowe J, Turner S, Aalberse RC, de Klerk N, Suriyaarachchi D, et al. An immunoepidemiological approach to asthma: identification of in-vitro T-cell response patterns associated with different wheezing phenotypes in children. Lancet (2005) 365:142–9. doi:10.1016/S0140-6736(05)17704-6
93. Hollams EM, Deverell M, Serralha M, Suriyaarachchi D, Parsons F, Zhang G, et al. Elucidation of asthma phenotypes in atopic teenagers through parallel immunophenotypic and clinical profiling. J Allergy Clin Immunol (2009) 124:e1–16. doi:10.1016/j.jaci.2009.06.019
94. Strachan DP. Hay fever, hygiene, and household size. Br Med J (1989) 299:1259–60. doi:10.1136/bmj.299.6710.1259
95. Holt PG. Environmental factors and primary T-cell sensitization to inhalant allergens in infancy: reappraisal of the role of infections and air pollution [review]. Pediatr Allergy Immunol (1995) 6:1–10. doi:10.1111/j.1399-3038.1995.tb00250.x
96. Adlerberth I, Wold AE. Establishment of the gut microbiota in western infants. Acta Paediatr (2009) 98:229–38. doi:10.1111/j.1651-2227.2008.01060.x
97. Sudo N, Sawamura S-A, Tanaka K, Aiba Y, Kubo C, Koga Y. The requirement of intestinal bacterial flora for the development of an IgE production system fully susceptible to oral tolerance induction. J Immunol (1997) 159:1739–45.
98. Koplin J, Allen K, Gurrin L, Osborne N, Tang ML, Dharmage S. Is caesarean delivery associated with sensitization to food allergens and IgE-mediated food allergy: a systematic review. Pediatr Allergy Immunol (2008) 19:682–7. doi:10.1111/j.1399-3038.2008.00731.x
99. Thavagnanam S, Fleming J, Bromley A, Shields MD, Cardwell CR. A meta-analysis of the association between Caesarean section and childhood asthma. Clin Exp Allergy (2008) 38:629–33. doi:10.1111/j.1365-2222.2007.02780.x
100. Xu B, Pekkanen J, Hartikainen AL, Jarvelin MR. Caesarean section and risk of asthma and allergy in adulthood. J Allergy Clin Immunol (2001) 107:732–3. doi:10.1067/mai.2001.113048
101. Moore HC, de Klerk NH, Holt PG, Richmond PC, Lehmann D. Hospitalisation for bronchiolitis in infants is more common after elective caesarean delivery. Arch Dis Child (2012) 97:410–4. doi:10.1136/archdischild-2011-300607
102. Gereda JE, Leung DY, Thatayatikom A, Streib JE, Price MR, Klinnert MD, et al. Relation between house-dust endotoxin exposure, type 1 T-cell development, and allergen sensitisation in infants at high risk of asthma. Lancet (2000) 355:1680–3. doi:10.1016/S0140-6736(00)02239-X
103. Schaub B, Liu J, Höppler S, Schleich I, Huehn J, Olek S, et al. Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. J Allergy ClinImmunol (2009) 123(774–82):e5. doi:10.1016/j.jaci.2009.01.056
104. Conrad ML, Ferstl R, Teich R, Brand S, Blümer N, Yildirim AO, et al. Maternal TLR signaling is required for prenatal asthma protection by the nonpathogenic microbe Acinetobacter lwoffii F78. J Exp Med (2009) 206:2869–77. doi:10.1084/jem.20090845
105. Holt PG, Strickland DH. Soothing signals: transplacental transmission of resistance to asthma and allergy. J Exp Med (2009) 206:2861–4. doi:10.1084/jem.20092469
106. Larsen GL, Colasurdo GN. Neural control mechanisms within airways: disruption by respiratory syncytial virus. J Pediatr (1999) 136:s21–7.
107. Barnes P. Neurogenic inflammation in airways. Int Arch Allergy Immunol (1991) 94:303–9. doi:10.1159/000235392
108. Hall GL, Hantos Z, Sly PD. Altered respiratory tissue mechanics in asymptomatic wheezy infants. Am J Respir Crit Care Med (2001) 164:1387–91. doi:10.1164/ajrccm.164.8.2012148
109. Hoo AF, Dezateux C, Henschen M, Costeloe K, Stocks J. The development of airway function in infancy following preterm delivery. J Pediatr (2002) 141:652–8. doi:10.1111/1440-1681.12068
110. Hibbert ME, Lannigan A, Landau LI, Phelan PD. Lung function values from a longitudinal study of healthy children and adolescents [erratum appears in Pediatr Pulmonol 1990;8(1):68]. Pediatr Pulmonol (1989) 7:101–9. doi:10.1002/ppul.1950070209
111. Phelan PD, Robertson CF, Olinsky A. The Melbourne asthma study: 1964-1999. J Allergy ClinImmunol (2002) 109:189–94. doi:10.1067/mai.2002.120951
112. Loss G, Apprich S, Waser M, Kneifel W, Genuneit J, Büchele G, et al. The protective effect of farm milk consumption on childhood asthma and atopy: the GABRIELA study. J Allergy Clin Immunol (2011) 128(766–73):e4. doi:10.1016/j.jaci.2011.07.048
113. Hanrahan JP, Tager IB, Segal MR, Tosteson TD, Castile RG, Van Vunakis H, et al. The effect of maternal smoking during pregnancy on early infant lung function. Am Rev Respir Dis (1992) 145:1129–35. doi:10.1164/ajrccm/145.5.1129
114. Wang X, Wypij D, Gold DR, Speizer FE, Ware JH, Ferris BG Jr, et al. A longitudinal study of the effects of parental smoking on pulmonary function in children 6-18 years. Am J Respir Crit Care Med (1994) 149:1420–5. doi:10.1164/ajrccm.149.6.8004293
115. Hollams E, de Klerk N, Holt P, Sly P. Persistent effects of maternal smoking during pregnancy on lung function and asthma in adolescents. Am J Respir Crit Care Med (2014) 189:401–7. doi:10.1164/rccm.201302-0323OC
116. Turner S, Fielding S, Mullane D, Cox DW, Goldblatt J, Landau L, et al. A longitudinal study of lung function from 1 month to 18 years of age. Thorax (2014). doi:10.1136/thoraxjnl-2013-204931
117. Gilliland FD, Li YF, Peters JM. Effects of maternal smoking during pregnancy and environmental tobacco smoke on asthma and wheezing in children. Am J Respir Crit Care Med (2001) 163:429–36. doi:10.1164/ajrccm.163.2.2006009
118. Macaubas C, de Klerk NH, Holt BJ, Wee C, Kendall G, Firth M, et al. Association between antenatal cytokine production and the development of atopy and asthma at age 6 years. Lancet (2003) 362:1192–7. doi:10.1016/S0140-6736(03)14542-4
119. Hinz D, Bauer M, Röder S, Olek S, Huehn J, Sack U, et al. Cord blood Tregs with stable FOXP3 expression are influenced by prenatal environment and associated with atopic dermatitis at the age of one year. Allergy (2012) 67:380–9. doi:10.1111/j.1398-9995.2011.02767.x
120. Bruce N, Perez-Padilla R, Albalak R. Indoor air pollution in developing countries: a major environmental and public health challenge. Bull WHO (2000) 78:1078–92.
121. Smith K, Samet J, Romieu I, Bruce N. Indoor air pollution and acute lower respiratory infections in children. Thorax (2000) 55:518–32. doi:10.1136/thorax.55.6.518
122. McKeever TM, Lewis SA, Smith C, Hubbard R. The importance of prenatal exposures on the development of allergic disease: a birth cohort study using the west midlands general practice database. Am J Respir Crit Care Med (2002) 166:827–32. doi:10.1164/rccm.200202-158OC
123. Xu B, Pekkanen J, Jarvelin MR, Olsen P, Hartikainen AL. Maternal infections in pregnancy and the development of asthma among offspring. Int J Epidemiol (1999) 28:723–7. doi:10.1093/ije/28.4.723
124. Burrows B, Martinez F, Halonen M, Barbee R, Cline M. Association of asthma with serum IgE levels and skin-test reactivity to allergens. N Engl J Med (1989) 320:271–7. doi:10.1056/NEJM198902023200502
125. O’Byrne PM, Pedersen S, Lamm CJ, Tan WC, Busse WW. Severe exacerbations and decline in lung function in asthma. Am J Respir Crit Care Med (2009) 179:19–24. doi:10.1164/rccm.200807-1126OC
126. Bosco A, Ehteshami S, Stern DA, Martinez FD. Decreased activation of inflammatory networks during acute asthma exacerbations is associated with chronic airflow obstruction. Mucosal Immunol (2010) 3:399–409. doi:10.1038/mi.2010.13
127. Guerra S, Sherrill DL, Martinez FD, Barbee RA. Rhinitis as an independent risk factor for adult-onset asthma. J Allergy Clin Immunol (2002) 109:419–25. doi:10.1067/mai.2002.121701
128. Togias A. Rhinitis and asthma: evidence for respiratory system integration. J Allergy Clin Immunol (2003) 111:1171–83. doi:10.1067/mai.2003.1592
129. Custovic A, Rothers J, Stern D, Simpson A, Woodcock A, Wright AL, et al. Effect of day care attendance on sensitization and atopic wheezing differs by toll-like receptor 2 genotype in 2 population-based birth cohort studies. J Allergy Clin Immunol (2011) 127(390–7):e1–9. doi:10.1016/j.jaci.2010.10.050
130. Lauener RP, Birchler T, Adamski J, Braun-Fahrländer C, Bufe A, Herz U, et al. Expression of CD14 and toll-like receptor 2 in farmers’ and non-farmers’ children. Lancet (2002) 360:465–6. doi:10.1016/S0140-6736(02)09641-1
131. Simpson A, John SL, Jury F, Niven R, Woodcock A, Ollier WE, et al. . Endotoxin exposure, CD14, and allergic disease: an interaction between genes and the environment [see comment]. Am J Respir Crit Care Med (2006) 174:386–92. doi:10.1164/rccm.200509-1380OC
132. Papoutsakis C, Priftis KN, Drakouli M, Prifti S, Konstantaki E, Chondronikola M, et al. Childhood overweight/obesity and asthma: is there a link? A systematic review of recent epidemiologic evidence. J Acad Nutr Diet (2013) 113:77–105. doi:10.1016/j.jand.2012.08.025
133. Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS One (2010) 5:e8578. doi:10.1371/journal.pone.0008578
134. Holt PG, Strickland DH, Hales BJ, Sly PD. Defective respiratory tract immume surveillance in asthma. Chest (2014) 145:370–8. doi:10.1378/chest.13-1341
135. Bisgaard H, Hermansen MN, Buchvald F, Loland L, Halkjaer LB, Bønnelykke K, et al. Childhood asthma after bacterial colonization of the airway in neonates. N Engl J Med (2007) 357:1487–95. doi:10.1056/NEJMoa052632
136. Hales BJ, Chai LY, Elliot CE, Pearce LJ, Zhang G, Heinrich TK, et al. Antibacterial antibody responses associated with the development of asthma in house dust mite-sensitised and non-sensitised children. Thorax (2012) 67:321–7. doi:10.1136/thoraxjnl-2011-200650
137. Hollams EM, Hales BJ, Bachert C, Huvenne W, Parsons F, de Klerk NH, et al. Th2-associated immunity to bacteria in teenagers and susceptibility to asthma. Eur Respir J (2010) 36:509–16. doi:10.1183/09031936.00184109
138. Kidd BA, Peters LA, Schadt EE, Dudley JT. Unifying immunology with informatics and multiscale biology. Nat Immunol (2014) 15:118–27. doi:10.1038/ni0914-894c
139. Kusel MMH, de Klerk NH, Holt PG, Kebadze T, Johnston SL, Sly PD. Role of respiratory viruses in acute upper and lower respiratory tract illness in the first year of life: a birth cohort study. Pediatr Infect Dis J (2006) 25:680–6. doi:10.1097/01.inf.0000226912.88900.a3
140. Illi S, von Mutius E, Lau S, Bergmann R, Niggemann B, Sommerfeld C, et al. Early childhood infectious diseases and the development of asthma up to school age: a birth cohort study. BMJ (2001) 322:390–5. doi:10.1136/bmj.322.7283.390
141. Gern JE, Visness CM, Gergen PJ, Wood RA, Bloomberg GR, O’Connor GT, et al. The urban environment and childhood asthma (URECA) birth cohort study: design, methods, and study population. BMC Pulm Med (2009) 9:17. doi:10.1186/1471-2466-9-17
142. Kaciroti NA, Raghunathan TE, Schork MA, Clark NM, Gong MA. Bayesian approach for clustered longitudinal ordinal outcome with nonignorable missing data: evaluation of an asthma education program. J Am Stat Assoc (2006) 101:435–46. doi:10.1198/016214505000001221
143. Zhu L, Carlin BP, Gelfand AE. Hierarchical regression with misaligned spatial data: relating ambient ozone and pediatric asthma ER visits in Atlanta. Environmetrics (2003) 14:537–57. doi:10.1002/env.614
144. Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest (2008) 118:3546. doi:10.1172/JCI36130
145. Inouye M, Kettunen J, Soininen P, Silander K, Ripatti S, Kumpula LS, et al. Metabonomic, transcriptomic, and genomic variation of a population cohort. Mol Syst Biol (2010) 6:441. doi:10.1038/msb.2010.93
146. Inouye M, Silander K, Hamalainen E, Salomaa V, Harald K, Jousilahti P, et al. An immune response network associated with blood lipid levels. PLoS Genet (2010) 6:e1001113. doi:10.1371/journal.pgen.1001113
147. Ala-Korpela M, Kangas AJ, Inouye M. Genome-wide association studies and systems biology: together at last. Trends Genet (2011) 27:493–8. doi:10.1016/j.tig.2011.09.002
148. Nikovski D. Constructing Bayesian networks for medical diagnosis from incomplete and partially correct statistics. IEEE Trans (2000) 12:509–16. doi:10.1109/69.868904
149. Needham CJ, Bradford JR, Bulpitt AJ, Westhead DR. Inference in Bayesian networks. Nat Biotechnol (2006) 24:51–3. doi:10.1038/nbt0106-51
150. Friedman N. Inferring cellular networks using probabilistic graphical models. Science (2004) 303:799–805. doi:10.1126/science.1094068
151. Sanders DL, Aronsky D. Prospective evaluation of a Bayesian network for detecting asthma exacerbations in a pediatric emergency department. AMIA Annu Symp Proc (2006) 2006:1085.
152. Hao K, Bossé Y, Nickle DC, Paré PD, Postma DS, Laviolette M, et al. Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS Genet (2012) 8:e1003029. doi:10.1371/journal.pgen.1003029
153. Fujita A, Sato JR, Garay-Malpartida HM, Morettin PA, Sogayar MC, Ferreira CE. Time-varying modeling of gene expression regulatory networks using the wavelet dynamic vector autoregressive method. Bioinformatics (2007) 23:1623–30. doi:10.1093/bioinformatics/btm151
154. Lebre S, Becq J, Devaux F, Stumpf M, Lelandais G. Statistical inference of the time-varying structure of gene-regulation networks. BMC Syst Biol (2010) 4:130. doi:10.1186/1752-0509-4-130
155. Sachs K, Perez O, Pe’er D, Lauffenburger DA, Nolan GP. Causal protein-signaling networks derived from multiparameter single-cell data. Science (2005) 308:523–9. doi:10.1126/science.1105809
156. Yu J, Smith VA, Wang PP, Hartemink AJ, Jarvis ED. Advances to Bayesian network inference for generating causal networks from observational biological data. Bioinformatics (2004) 20:3594–603. doi:10.1093/bioinformatics/bth448
157. Rao A, Hero AO III, States DJ, Engel JD. Using directed information to build biologically relevant influence networks. Comput Syst Bioinformatics Conf (2007) 6:145–56. doi:10.1142/S0219720008003515
158. Sesen MB, Nicholson AE, Banares-Alcantara R, Kadir T, Brady M. Bayesian networks for clinical decision support in lung cancer care. PLoS One (2013) 8:e82349. doi:10.1371/journal.pone.0082349
159. Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. J Roy Stat Soc (1977) 39:1–38.
160. Pearl J, Verma TS. A theory of inferred causation. In: Dag Prawitz BS, Westerståhl D, editors. Logic, Methodology and Philosophy of Science IX Proceedings of the Ninth International Congress of Logic, Methodology and Philosophy of Science. Amsterdam: Elsevier (1995). p. 789–811.
162. Wallace CS, Freeman PR. Estimation and inference by compact coding. J Roy Stat Soc Ser B (Methodological) (1987) 1987:240–65.
163. Cooper GF, Herskovits E. A Bayesian method for the induction of probabilistic networks from data. Mach Learn (1992) 9:309–47. doi:10.1023/A:1022649401552
164. Tsamardinos I, Brown LE, Aliferis CF. The max-min hill-climbing Bayesian network structure learning algorithm. Mach Learn (2006) 65:31–78. doi:10.1007/s10994-006-6889-7
165. Ram DJ, Sreenivas TH, Subramaniam KG. Parallel simulated annealing algorithms. J Parallel Distr Comput (1996) 37:207–12. doi:10.1006/jpdc.1996.0121
166. Heckerman D. A Tutorial on Learning with Bayesian Networks. Technical Report. One Microsoft Way. Redmond, WA: Microsoft Research, Advanced Technology Division, Microsoft Corporation (1995).
167. Heckerman D, Geiger D. Likelihoods and parameter priors for Bayesian networks. Tech MSRTR-95-54 Microsoft Research. (1995).
168. Andrieu C, Doucet A. Joint Bayesian model selection and estimation of noisy sinusoids via reversible jump MCMC. IEEE Trans (1999) 47:2667–76.
169. Talih M, Hengartner N. Structural learning with time-varying components: tracking the cross-section of financial time series. J R Stat Soc Series B Stat Methodol (2005) 67:321–41. doi:10.1111/j.1467-9868.2005.00504.x
170. Peterson C, Stingo F, Vannucci M. Bayesian inference of multiple Gaussian graphical models. J Am Stat Assoc (2014). doi:10.1080/01621459.2014.896806
171. Guimerà R, Sales-Pardo MA. Network inference method for large-scale unsupervised identification of novel drug-drug interactions. PLoS Comput Biol (2013) 9:e1003374. doi:10.1371/journal.pcbi.1003374
172. Demissie K, White N, Joseph L, Ernst P. Bayesian estimation of asthma prevalence, and comparison of exercise and questionnaire diagnostics in the absence of a gold standard. Ann Epidemiol (1998) 8:201–8. doi:10.1016/S1047-2797(97)00168-3
173. Gilbert R, Auchincloss JH Jr. Post-test probability of asthma following methacholine challenge. CHEST J (1990) 97:562–5. doi:10.1378/chest.97.3.562
174. Perpiñá M, Pellicer C, de Diego A, Compte L, Macián V. Diagnostic value of the bronchial provocation test with methacholine in asthma. A Bayesian analysis approach. CHEST (1993) 104:149–54.
175. Clark P, Niblett T. The CN2 induction algorithm. Mach Learn (1989) 3:261–83. doi:10.1023/A:1022641700528
176. Friedman N, Geiger D, Goldszmidt M. Bayesian network classifiers. Mach Learn (1997) 29:131–63. doi:10.1023/A:1007465528199
177. Hanson R, Stutz J, Cheeseman P. Bayesian Classification Theory: NASA Ames Research Center, Artificial Intelligence Research Branch. (1991).
178. Draper D, Fouskakis DA. Case study of stochastic optimization in health policy: problem formulation and preliminary results. J Global Optim (2000) 18:399–416. doi:10.1023/A:1026504402220
179. Blanco R, Inza I, Merino M, Quiroga J, Larrañaga P. Feature selection in Bayesian classifiers for the prognosis of survival of cirrhotic patients treated with ∖TIPS∖. J Biomed Inform (2005) 38:376–88. doi:10.1016/j.jbi.2005.05.004
181. Yan A, Laird NM, Li C. Identifying rare variants using a Bayesian regression approach. BMC Proc (2011) 5(Suppl 9):S99. doi:10.1186/1753-6561-5-S9-S99
182. Domingos P, Pazzani M. On the optimality of the simple Bayesian classifier under zero-one loss. Mach Learn (1997) 29:103–30. doi:10.1023/A:1007413511361
183. Dougherty J, Kohavi R, Sahami M. Supervised and unsupervised discretization of continuous features. ICML (1995) 1995:194–202.
184. Rish I. An empirical study of the naive Bayes classifier. IJCAI 2001 Workshop on Empirical Methods in Artificial Intelligence. (2001). p. 41–6.
185. Langley P, Iba W, Thompson K. An analysis of Bayesian classifiers. Proceedings of the Tenth National Conference on Artificial Intelligence. (Vol. 90), San Jose, CA: AAAI Press (1992). p. 223–8.
186. Andrieu C, de Freitas N, Doucet A, Jordan MI. An introduction to MCMC for machine learning. Mach Learn (2003) 50:5–43. doi:10.1023/A:1020281327116
187. Geman S, Geman D. Stochastic relaxation, Gibbs distributions, and the Bayesian restoration of images. IEEE Trans (1984) PAMI-6:721–41.
188. Hastings WK. Monte Carlo sampling methods using Markov chains and their applications. Biometrika (1970) 57:97–109. doi:10.1093/biomet/57.1.97
189. Madigan D, York J, Allard D. Bayesian graphical models for discrete data. Int Stat Rev (1995) 63:215–32.
190. Metropolis N, Ulam S. The Monte Carlo method. J Am Stat Assoc (1949) 44:335–41. doi:10.1080/01621459.1949.10483310
191. Tierney L. Markov chains for exploring posterior distributions. Ann Stat (1994) 22:1701–28. doi:10.1214/aos/1176325750
192. Weihs C, Ligges U, Luebke K, Raabe N. klaR analyzing German business cycles. In: Baier D, Decker R, Schmidt-Thieme L, editors. Data Analysis and Decision Support. Berlin: Springer-Verlag (2005). p. 335–43.
193. Albert JH, Chib S. Bayesian analysis of binary and polychotomous response data. J Am Stat Assoc (1993) 88:669–79. doi:10.1080/01621459.1993.10476321
194. Granger CWJ. Causality, cointegration, and control. J Econ Dyn Control (1988) 12:551–9. doi:10.1016/0165-1889(88)90055-3
Keywords: allergy, asthma, systems biology, virus infection, birth cohort, childhood, immune function, epidemiology
Citation: Walker ML, Holt KE, Anderson GP, Teo SM, Sly PD, Holt PG and Inouye M (2014) Elucidation of pathways driving asthma pathogenesis: development of a systems-level analytic strategy. Front. Immunol. 5:447. doi: 10.3389/fimmu.2014.00447
Received: 18 July 2014; Accepted: 01 September 2014;
Published online: 23 September 2014.
Edited by:
Tobias R. Kollmann, University of British Columbia, CanadaReviewed by:
Mario Barreto, University of Rome La Sapienza, ItalyBeatrice Jahn-Schmid, Medical University of Vienna, Austria
Copyright: © 2014 Walker, Holt, Anderson, Teo, Sly, Holt and Inouye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick G. Holt, Telethon Kids Institute, 100 Roberts Road, Subiaco, WA 6008, Australia e-mail:cGF0cmlja0BpY2hyLnV3YS5lZHUuYXU=