 Walker Julliard
Walker Julliard Joshua D. Mezrich
Joshua D. Mezrich- Division of Transplantation, Department of Surgery, University of Wisconsin School of Medicine and Public Health, Madison, WI, USA
The aryl hydrocarbon receptor (AHR) has long been studied by toxicologists as a ligand-activated transcription factor that is activated by dioxin and other environmental pollutants such as polycyclic aromatic hydrocarbons (PAHs). The hallmark of AHR activation is the upregulation of the cytochrome P450 enzymes that metabolize many of these toxic compounds. However, recent findings demonstrate that both exogenous and endogenous AHR ligands can alter innate and adaptive immune responses including effects on T-cell differentiation. Kynurenine, a tryptophan breakdown product, is one such endogenous ligand of the AHR. Expression of indoleamine 2,3-dioxygenase by dendritic cells causes accumulation of kynurenine and results in subsequent tolerogenic effects including increased regulatory T-cell activity. At the same time, PAHs found in pollution enhance Th17 differentiation in the lungs of exposed mice via the AHR. In this perspective, we will discuss the importance of the AHR in the immune system and the role this might play in normal physiology and response to disease.
Introduction
Our laboratory has been actively investigating the role of the aryl hydrocarbon receptor (AHR) in the immune system, and the variable effects seen after binding endogenous and exogenous ligands. Study of this receptor traditionally was in the domain of toxicologists, as it was originally defined as a receptor to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (1). It was further identified that additional toxins, including polycyclic aromatic hydrocarbons (PAHs), also bind to the AHR (2–4). As the mechanism of AHR activation and function was characterized, the importance of this receptor as a transcription factor was realized. The AHR is a cytosolic receptor that after binding migrates to the nucleus where it becomes a transcription factor for cytochrome P450 genes that encode enzymes that metabolize toxins, including those same toxins that bind to the receptor (5). A connection to the immune system had been previously recognized, primarily based on the knowledge that exposure to TCDD led to rapid thymic involution, and animals and human beings exposed to TCDD were known to be immunosuppressed. Effects of TCDD on dendritic cells (DCs) and T-cells have been recognized for years (6), and generation of regulatory T-cells by an AHR-dependent mechanism was identified in 2005 (7). But in general, immunologists and those studying autoimmunity did not become interested in the AHR until 2008, when two high impact papers identified the role of the AHR in T-cell differentiation, with certain ligands enhancing Treg generation, and others enhancing Th17 differentiation, both in vitro and in vivo (8, 9). Since that time, there has been a flood of reports on the role of the AHR in the adaptive immune system and animal models of disease, particularly autoimmune disease (10–12).
The role of the AHR in responding to toxins is thought to have evolved, as invertebrate forms of the AHR do not bind TCDD (13), and the toxins associated with the AHR are man made and were generated long after the origins of the receptor. It has been postulated that the initial importance of the AHR was in embryologic development, and in addition there was (and remains) a requirement for binding of this receptor to endogenous ligands early in development. This is based on the abnormal phenotype of AHR null mice that display a patent ductus venosus (14), as well as the necessity for the hypomorphs to bind AHR ligand early in development to prevent the patent ductus venosus in these transgenic mice (15). In addition to binding to pollution and endogenous ligands, the AHR binds to numerous ligands present in the diet including flavonoids, which are ubiquitous in fruits and vegetables. There has been an ongoing search for relevant endogenous ligands in animals and human beings.
Around the time that the AHR was first recognized to play some role in the acquired immune system by toxicologists, immunologists were investigating indoleamine 2,3-dioxygenase (IDO1), initially recognized as the rate-limiting enzyme in tryptophan metabolism. In 1998, it was reported that tryptophan catabolism by IDO1 and other enzymes was responsible for prevention of allogeneic fetal rejection in mice, and it was further revealed that this enzyme was generated by DCs, and in some way increased the differentiation of Tregs (16). The mechanism was unclear, but the two leading theories were that either tryptophan deprivation from its metabolism decreased the generation and division of effector cells (17), or certain breakdown products of tryptophan, termed kynurenines, were acting through some target on T-cells or other cells to favor Treg differentiation (18, 19). In our own lab, we considered the hypothesis that kynurenine or one of its breakdown products was working through the AHR in T-cells to enhance Treg generation. This was based on the knowledge that indoles and other tryptophan derivatives are often ligands of this receptor, making kynurenine a good candidate as an endogenous ligand. We identified that not only does kynurenine bind to the AHR in the cytosol of T-cells and enhance Treg generation in vitro but additionally kynurenine and other AHR ligands act on BMDCs to upregulate IDO expression in an AHR-dependent manner (20) (Figure 1). The effects of AHR ligands on differentiation and function of natural versus induced Tregs has not yet been delineated.
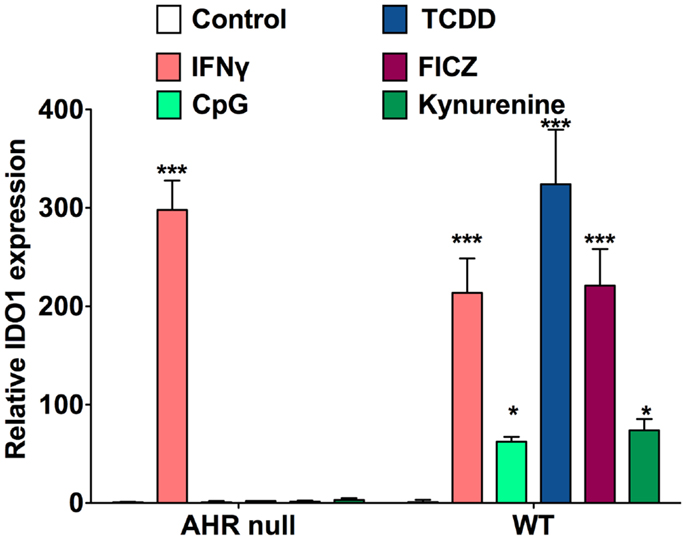
Figure 1. AHR ligands increase IDO1 expression in BMDCs. qRT-PCR analysis of IDO1 mRNA expression in bone marrow-derived DCs treated for 48 h with IFN-γ (10 μg/ml), CpG (2.5 μM), TCDD (10 nM), FICZ (200 nM), or kynurenine (50 μM). Data are from three independent experiments.
One of the controversial and yet unanswered questions about the role of the AHR in the immune system is whether it is probable that different ligands bind to the AHR and lead to different, almost opposite outcomes, perhaps based on some conformational change or transcription of different genes, a finding that would almost be unprecedented in physiology. We theorize that some combination of strength of binding, duration of binding, route of exposure, and surrounding milieu determines whether Treg or Th17 differentiation is enhanced. Although early experience seemed to suggest that ligands were either “regulatory” (meaning they favored differentiation of Tregs) or “effector” (meaning they favored differentiation of effector Th17 cells) (8), more recent studies and our own experience has suggested that this is not likely the case. For instance, the ligand 6-formylindolo[3,2-b]carbazole (FICZ) was first described as enhancing Th17 differentiation in vitro and in vivo, and when administered subcutaneously worsened autoimmunity in an experimental autoimmune encephalomyelitis (EAE) model in mice. However, it has recently been shown that when this same ligand is administered intraperitoneally, Treg differentiation is favored and there is a reduction in EAE disease severity (21, 22). Our own experience has also shown that depending on the culture conditions or inflammatory status of the animals, similar ligands can enhance either Th17 or Treg generation. We have also found that every ligand we have tested stimulates T-cells to generate IL-22, and it has been demonstrated that the AHR is required for IL-22 generation in the majority of scenarios (23–25). IL-22 is an interesting cytokine that is generated by various types of T-cells, but its receptor is found on epithelial cells. In general, IL-22 is thought to be protective against immune damage and helpful in epithelial cell repair (26–28). We have recently shown that particulate matter (PM) in the form of urban dust particles (UDP) from the National Institute of Standards and Technology (NIST; SRM1649b), containing PAHs, increases the Th17 response both in the lung in vivo and in T-cells in vitro (29). The in vitro response was shown to be AHR dependent. Therefore, a connection between the AHR and the immune system seems real, but the effects are complicated and dependent on the model or conditions being examined. This relationship of the AHR with the immune system, where binding of this receptor with different ligands in differing milieus can lead to seemingly opposing responses in T-cell differentiation has allowed us to come up with the following hypothesis:
The AHR as a Sensor
The AHR serves as a sensor that responds to signals, both from the outside environment or internal milieu, to modulate an immune response. In the setting of an endogenous immune response, the AHR helps contain the inflammation and decreases collateral damage, so the response does not cascade in strength and lead to autoimmune destruction. In responding to exogenous ligands, the AHR functions as a sensor to “danger” signals, such as dioxins, PAHs, and other components of pollution. This is of evolutionary benefit for individuals exposed to man-made pollution or other toxins, as illustrated by the following scenario. When someone is exposed to inhaled atmospheric pollution, damage to bacterial clearance mechanisms and the epithelial lining of the airway puts the individual at risk for endogenous bacterial invasion and activation of a reactive airway response. The AHR present in immune cells residing in the lung binds to fractions of PM (PAHs and others), leading to an enhancement of Th17 differentiation that can serve as an immune barrier to the endogenous bacteria found in the lining of the lung. Simultaneously, AHR activation leads to generation of IL-22 that helps with epithelial cell repair and maintenance of tight junctions. Finally, activation of the AHR causes an upregulation of cytochrome P450 enzymes that metabolize the harmful toxicants. Taken together, the AHR aids in tissue protection, repair, and toxin clearance (Figure 2, left).
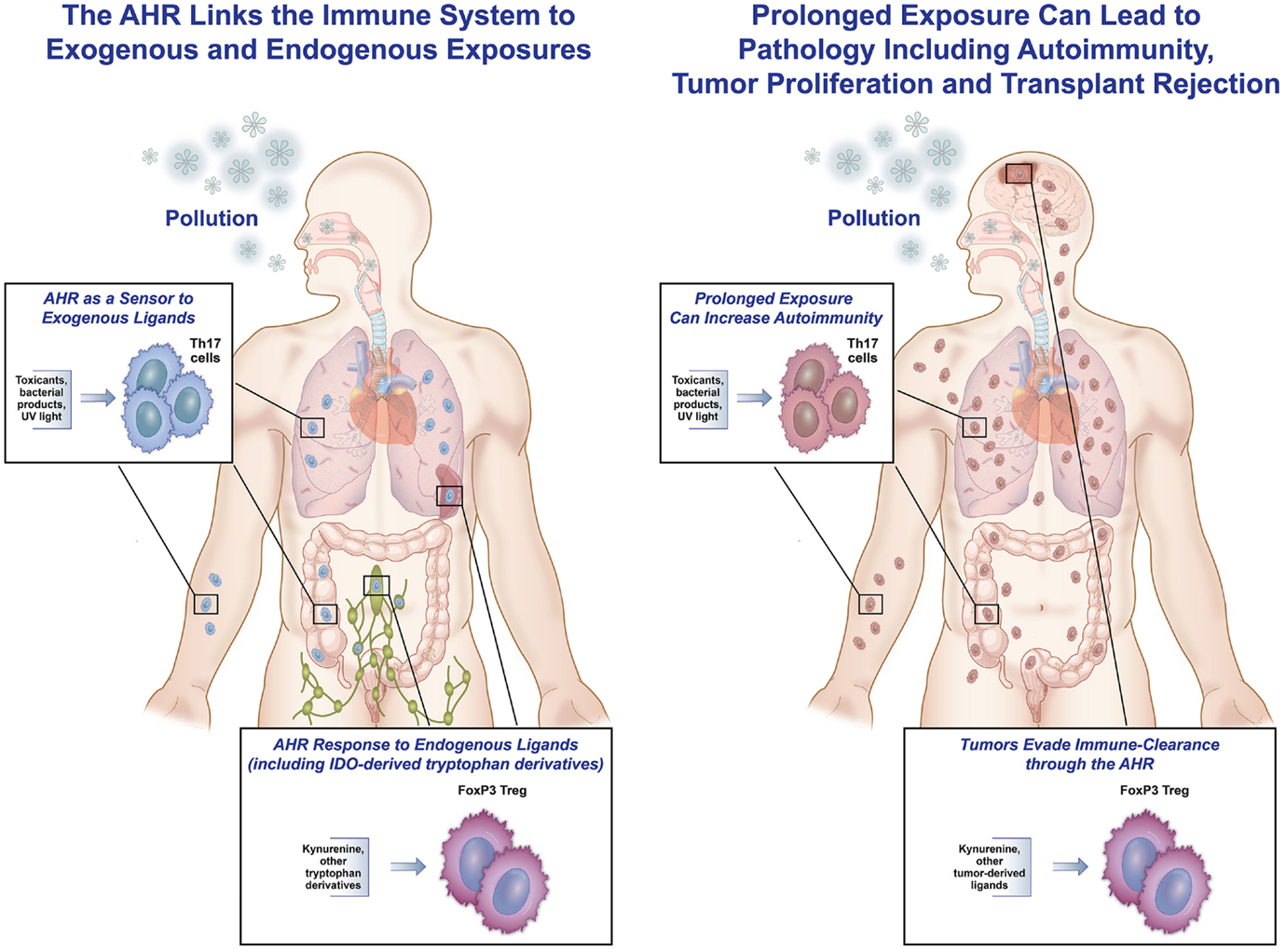
Figure 2. The AHR as friend and foe. This figure represents the hypothesis that the AHR can be both protective and pathologic in responding to endogenous and exogenous ligands. On the left, exposure to inhaled pollution (as well as ingested or topical) can bind to the AHR in immune cells, and in an inflammatory setting, lead to increased Th17 differentiation, IL-22 generation, and cytochrome P450 enzyme production. All of these responses can help protect the host from pathologic inflammation and invasion by endogenous bacteria. Similarly, in response to endogenous ligands during inflammation (in the spleen, lymph node, or throughout the body), AHR ligands can lead to increased IDO production and differentiation of Tregs by binding of kynurenine to the AHR in T-cells, hence controlling an immune response. On the right, examples of binding of the AHR leading to increased pathology are shown. Tumors (in the brain or elsewhere) have been shown to generate TDO, leading to kynurenine production that binds to the AHR in tumor cells, causing increased regulation and decreased tumor destruction by the native immune system. In addition, prolonged exposure to external pollutants can lead to over-differentiation of Th17 cells and aggravation of autoimmunity in the lungs, gut, skin, and elsewhere in susceptible people.
Endogenous Ligands
We hypothesize that as the adaptive immune system developed, it became crucial that there be a way for the body to dampen overly robust responses, thereby preventing reactivity to self-proteins and ensuing autoimmunity. Kynurenine, the first breakdown product of tryptophan in the IDO pathway, became the transmitter that interacts with the AHR in T-cells and enhances the differentiation of Tregs that reduce an immune response. At the same time, kynurenine also interacts with the AHR in DCs to enhance the generation of IDO, leading to further tryptophan break down. This response is self-limited, as once the tryptophan in the inflammatory milieu is dissipated, kynurenine is no longer generated and Treg generation is decreased. This push toward a regulatory response from endogenous ligands makes evolutionary sense, as internal inflammatory responses would need to be controlled to prevent damage to surrounding epithelial and parenchymal cells at the sight of an immune response. Additionally, binding of the AHR to ligand leads to the generation of IL-22 and engenders epithelial cell homeostasis, proliferation, and anti-microbial peptide production (30), further protecting the bystander tissue.
Exogenous Ligands
In contrast to the role that the AHR plays in regulating autoimmunity when presented with endogenous ligands, at interface organs such as the lung, gut, and skin, AHR activation leads to an effector response. At these locations, toxin binding to the AHR in immune cells leads to Th17 differentiation and a heightened inflammatory response. We view this interaction as a “danger” signal that allows activation of the immune system at the same time that P450 enzymes are upregulated to metabolize toxins. When a host is presented with a potentially harmful exposure, early signaling and activation of the immune system occurs concurrently with ligand clearance. We believe that this is the role of exogenous activation of the AHR in immune cells.
It is important to again point out that we do not believe that endogenous ligands such as kynurenine always favor Treg generation, and exogenous ligands including fractions of pollution always favor Th17 differentiation. However, we do believe that in the in vivo inflammatory milieu where exogenous ligands are exposed to interface organs during a toxic environmental exposure, Th17 differentiation is favored, and similarly in the endogenous in vivo milieu of an immune response in the presence of IDO and tryptophan, Treg generation is favored. The mechanistic details of these effects on T-cell differentiation require further delineation and are an exciting area of research.
Pathologic Response
While we hypothesize that the AHR and its role in the immune system evolved to be protective, there are examples where this interaction actually increases pathology. In the setting of an overwhelming exposure to exogenous ligands, generation of ligands by tumors, or changes in the microbiome altering physiologic interactions between products of bacteria and receptors in the gut, the AHR can worsen pathology (Figure 2, right).
Cancer
Certain cancers have developed the ability to evade their own clearance through use of the AHR. In gliomas, constitutive activation of tryptophan 2,3-dioxygenase (TDO-2) leads to high levels of kynurenine production. The TDO-2-derived kynurenine then acts through the AHR in tumor cells in an autocrine manner and promotes tumor survival as well as altering the regional immune response by favoring Treg development. These Tregs hamper the ability of the immune system to clear the tumor. This is particularly effective as the interaction between kynurenine and T-cells in the vicinity of the tumor leads to local regulation in the very location and milieu that the tumor resides in Ref. (31). In addition, there are some examples in human cancer where the tumors show increased copies of the AHR (32), which could increase the autocrine effect of preventing tumor clearance.
Autoimmunity
The immune system is in a constant balance of regulation and inflammation, and loss of this equilibrium can lead to pathology. We hypothesize that exogenous ligands of the AHR found in pollution can aggravate autoimmune conditions in an AHR-dependent mechanism. For many years, there has been speculation that environmental exposures can aggravate or even cause autoimmune disease, and a recent analysis of epidemiologic data has supported this hypothesis. The specific exposures that lead to disease and the mechanisms through which they impart pathology remain unclear. Recent data suggest that modulation of the Treg/Th17 balance may be at the center of environmentally induced autoimmunity. An NIEHS workshop concluded that “dysregulated Th17 activity can lead to pathology” in various autoimmune diseases, and that both smoking and “AHR binding by aromatic hydrocarbons favors differentiation of Th17 cells and can exacerbate autoimmunity” (33). We recently identified that inhaled PM leads to an increase in Th17-driven inflammation (29). In our resultant model, when an individual is exposed to inhaled PM for a prolonged period of time, there is a shift in the Treg/Th17 balance toward Th17 cells, aggravating or initiating autoimmunity in susceptible people. This ubiquitous mechanism will be applicable to multiple environmentally induced autoimmune diseases.
Transplant Rejection
One of the novel theories we have been studying in our lab is the possibility that exposure to pollution is altering the Treg/Th17 balance and leading to chronic rejection after solid organ transplant. This may be most relevant to lung transplantation, where the transplanted organ is at the interface with the outside environment (34). There is growing evidence that inhaled exposure to elevated levels of atmospheric pollution, with increased PM, increases chronic rejection in lung transplants in human beings (35, 36). Bronchiolitic obliterans syndrome (BOS) is the major form of chronic rejection in lung transplantation, and it has been well documented that Th17 cells and the cytokine IL-17 are required for pathology (37–39). Given the link between IL-17 and BOS, the fact that pollution accelerates BOS, and our own work demonstrating that pollutants contain AHR ligands, which favor a Th17 response, it is possible that the AHR is a key factor in pollutant accelerated rejection in lung transplantation.
Gut Immunity
Some of the highest impact papers regarding the AHR in the last couple of years have described the relationship between gut immunity and the AHR. The presence of this receptor is necessary for the establishment of certain populations of immune cells in the gut. Furthermore, both dietary ligands of the AHR as well as ligands produced by gut bacteria interact with and alter the gastrointestinal immune system (40, 41). The healthy microbiome generates ligands that interact with the AHR to maintain gut immune structure and stability. Loss of some of these ligand-producing bacteria may increase the risk of autoimmune colitis (42).
Skin
The AHR is highly expressed in multiple cell types found in skin. As skin is one of the organs at the interface with the outside environment, it is likely that inflammatory disorders are altered by interaction with toxins and other environmental factors. A recent publication did document the protective role of various AHR ligands in reducing inflammation in a model for psoriasis (22). At the same time, exposure to AHR antagonists increased inflammation. This is clinically relevant, as it is known that certain toxicants contain fractions that may inhibit the AHR or cytochrome P450 enzymes, and certain weak AHR agonists actually inhibit activation by other agonists that may be found in different fractions of a given exposure (43). This implies that in clinically relevant mixtures of chemicals, the overall response could be one of antagonism of the AHR and worsening of skin inflammation.
AHR Meets Immunology: Friend or Foe?
So does the relationship between the AHR and the immune system make us healthier and more able to withstand interactions with the environment, or leave us at risk for pathologic manipulation of the immune system in response to external and internal signals? We assert that the AHR does make us healthier, and allows us to survive in the environment that currently exists. The relationship of the endogenous ligand kynurenine and the AHR allows a feedback loop that dampens an immune response to prevent too much inflammation and autoimmunity, allowing a response to be self-limited. Exogenous ligands serve as “danger” signals that alert the immune system to toxic insults from the environment, allowing immune cells to play an early role in containing damage done by these exposures, helping repair local barriers, and containing local endogenous infectious exposures. At the same time, there are examples where disease processes have taken advantage of this connection to increase pathology. This scenario, where normal physiology or “protective” responses become overwhelmed and lead to increased severity of illness, is well documented in immune responses to pathology, including systemic inflammatory response syndrome in response to infection, multiple forms of autoimmunity, and resistance of cancer to its own destruction. As we better understand the mechanisms and signals behind the role of the AHR in the immune system, we will be able to manipulate this receptor to treat or prevent diseases as diverse as cancer, autoimmunity, and transplant rejection.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Denison MS, Pandini A, Nagy SR, Baldwin EP, Bonati L. Ligand binding and activation of the Ah receptor. Chem Biol Interact (2002) 141(1–2):3–24. doi:10.1016/S0009-2797(02)00062-5
2. Mason GG. Dioxin-receptor ligands in urban air and vehicle exhaust. Environ Health Perspect (1994) 102(Suppl 4):111–6. doi:10.2307/3432226
3. Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol (2003) 43:309–34. doi:10.1146/annurev.pharmtox.43.100901.135828
4. Kitamura M, Kasai A. Cigarette smoke as a trigger for the dioxin receptor-mediated signaling pathway. Cancer Lett (2007) 252(2):184–94. doi:10.1016/j.canlet.2006.11.015
5. Hahn ME. Biomarkers and bioassays for detecting dioxin-like compounds in the marine environment. Sci Total Environ (2002) 289(1–3):49–69. doi:10.1016/S0048-9697(01)01016-6
6. Vorderstrasse BA, Kerkvliet NI. 2,3,7,8-Tetrachlorodibenzo-p-dioxin affects the number and function of murine splenic dendritic cells and their expression of accessory molecules. Toxicol Appl Pharmacol (2001) 171(2):117–25. doi:10.1006/taap.2000.9119
7. Funatake CJ, Marshall NB, Steppan LB, Mourich DV, Kerkvliet NI. Cutting edge: activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+ CD25+ cells with characteristics of regulatory T cells. J Immunol (2005) 175(7):4184–8. doi:10.4049/jimmunol.175.7.4184
8. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453(7191):65–71. doi:10.1038/nature06880
9. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links T(H)17-cell-mediated autoimmunity to environmental toxins. Nature (2008) 453(7191):106–9. doi:10.1038/nature06881
10. Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol (2010) 11(9):854–61. doi:10.1038/ni.1912
11. Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A (2010) 107(48):20768–73. doi:10.1073/pnas.1009201107
12. Nakahama T, Kimura A, Nguyen NT, Chinen I, Hanieh H, Nohara K, et al. Aryl hydrocarbon receptor deficiency in T cells suppresses the development of collagen-induced arthritis. Proc Natl Acad Sci U S A (2011) 108(34):14222–7. doi:10.1073/pnas.1111786108
13. Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol (2008) 21(1):102–16. doi:10.1021/tx7001965
14. Walisser JA, Bunger MK, Glover E, Bradfield CA. Gestational exposure of Ahr and Arnt hypomorphs to dioxin rescues vascular development. Proc Natl Acad Sci U S A (2004) 101(47):16677–82. doi:10.1073/pnas.0404379101
15. Walisser JA, Bunger MK, Glover E, Harstad EB, Bradfield CA. Patent ductus venosus and dioxin resistance in mice harboring a hypomorphic Arnt allele. J Biol Chem (2004) 279(16):16326–31. doi:10.1074/jbc.M400784200
16. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science (1998) 281(5380):1191–3. doi:10.1126/science.281.5380.1191
17. Mellor AL, Munn DH. Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol Today (1999) 20(10):469–73. doi:10.1016/S0167-5699(99)01520-0
18. Belladonna ML, Grohmann U, Guidetti P, Volpi C, Bianchi R, Fioretti MC, et al. Kynurenine pathway enzymes in dendritic cells initiate tolerogenesis in the absence of functional IDO. J Immunol (2006) 177(1):130–7. doi:10.4049/jimmunol.177.1.130
19. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol (2006) 176(11):6752–61. doi:10.4049/jimmunol.176.11.6752
20. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol (2010) 185(6):3190–8. doi:10.4049/jimmunol.0903670
21. Duarte JH, Di Meglio P, Hirota K, Ahlfors H, Stockinger B. Differential influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PLoS One (2013) 8(11):e79819. doi:10.1371/journal.pone.0079819
22. Di Meglio P, Duarte JH, Ahlfors H, Owens ND, Li Y, Villanova F, et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity (2014) 40(6):989–1001. doi:10.1016/j.immuni.2014.04.019
23. Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A (2008) 105(28):9721–6. doi:10.1073/pnas.0804231105
24. Alam MS, Maekawa Y, Kitamura A, Tanigaki K, Yoshimoto T, Kishihara K, et al. Notch signaling drives IL-22 secretion in CD4+ T cells by stimulating the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A (2010) 107(13):5943–8. doi:10.1073/pnas.0911755107
25. Ramirez JM, Brembilla NC, Sorg O, Chicheportiche R, Matthes T, Dayer JM, et al. Activation of the aryl hydrocarbon receptor reveals distinct requirements for IL-22 and IL-17 production by human T helper cells. Eur J Immunol (2010) 40(9):2450–9. doi:10.1002/eji.201040461
26. Pan H, Hong F, Radaeva S, Gao B. Hydrodynamic gene delivery of interleukin-22 protects the mouse liver from concanavalin A-, carbon tetrachloride-, and Fas ligand-induced injury via activation of STAT3. Cell Mol Immunol (2004) 1(1):43–9.
27. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity (2004) 21(2):241–54. doi:10.1016/j.immuni.2004.07.007
28. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity (2007) 27(4):647–59. doi:10.1016/j.immuni.2007.07.023
29. van Voorhis M, Knopp S, Julliard W, Fechner JH, Zhang X, Schauer JJ, et al. Exposure to atmospheric particulate matter enhances Th17 polarization through the aryl hydrocarbon receptor. PLoS One (2013) 8(12):e82545. doi:10.1371/journal.pone.0082545
30. Kumar P, Rajasekaran K, Palmer JM, Thakar MS, Malarkannan S. IL-22: an evolutionary missing-link authenticating the role of the immune system in tissue regeneration. J Cancer (2013) 4(1):57–65. doi:10.7150/jca.5048
31. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature (2011) 478(7368):197–203. doi:10.1038/nature10491
32. Liu Z, Wu X, Zhang F, Han L, Bao G, He X, et al. AhR expression is increased in hepatocellular carcinoma. J Mol Histol (2013) 44(4):455–61. doi:10.1007/s10735-013-9481-z
33. Selmi C, Leung PS, Sherr DH, Diaz M, Nyland JF, Monestier M, et al. Mechanisms of environmental influence on human autoimmunity: a National Institute of Environmental Health Sciences expert panel workshop. J Autoimmun (2012) 39(4):272–84. doi:10.1016/j.jaut.2012.05.007
34. van Voorhis M, Fechner JH, Zhang X, Mezrich JD. The aryl hydrocarbon receptor: a novel target for immunomodulation in organ transplantation. Transplantation (2013) 95(8):983–90. doi:10.1097/TP.0b013e31827a3d1d
35. Nawrot TS, Vos R, Jacobs L, Verleden SE, Wauters S, Mertens V, et al. The impact of traffic air pollution on bronchiolitis obliterans syndrome and mortality after lung transplantation. Thorax (2011) 66(9):748–54. doi:10.1136/thx.2010.155192
36. Verleden SE, Scheers H, Nawrot TS, Vos R, Fierens F, Geenens R, et al. Lymphocytic bronchiolitis after lung transplantation is associated with daily changes in air pollution. Am J Transplant (2012) 12(7):1831–8. doi:10.1111/j.1600-6143.2012.04134.x
37. Burlingham WJ, Love RB, Jankowska-Gan E, Haynes LD, Xu Q, Bobadilla JL, et al. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest (2007) 117(11):3498–506. doi:10.1172/JCI28031
38. Vanaudenaerde BM, De Vleeschauwer SI, Vos R, Meyts I, Bullens DM, Reynders V, et al. The role of the IL23/IL17 axis in bronchiolitis obliterans syndrome after lung transplantation. Am J Transplant (2008) 8(9):1911–20. doi:10.1111/j.1600-6143.2008.02321.x
39. Fan L, Benson HL, Vittal R, Mickler EA, Presson R, Fisher AJ, et al. Neutralizing IL-17 prevents obliterative bronchiolitis in murine orthotopic lung transplantation. Am J Transplant (2011) 11(5):911–22. doi:10.1111/j.1600-6143.2011.03482.x
40. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science (2011) 334(6062):1561–5. doi:10.1126/science.1214914
41. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol (2012) 13(2):144–51. doi:10.1038/ni.2187
42. Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell (2011) 147(3):629–40. doi:10.1016/j.cell.2011.09.025
Keywords: indoleamine 2,3-dioxygenase, Th17 Cells, Treg cells, immunomodulation, kynurenine, aryl hydrocarbon receptor
Citation: Julliard W, Fechner JH and Mezrich JD (2014) The aryl hydrocarbon receptor meets immunology: friend or foe? A little of both. Front. Immunol. 5:458. doi: 10.3389/fimmu.2014.00458
Received: 31 July 2014; Accepted: 08 September 2014;
Published online: 02 October 2014.
Edited by:
Ursula Grohmann, University of Perugia, ItalyReviewed by:
Lauren A. Zenewicz, The University of Oklahoma Health Sciences Center, USAUrsula Grohmann, University of Perugia, Italy
Copyright: © 2014 Julliard, Fechner and Mezrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joshua D. Mezrich, Division of Transplantation, Department of Surgery, University of Wisconsin School of Medicine and Public Health, 600 Highland Avenue, Madison, WI 53792, USA e-mail:bWV6cmljaEBzdXJnZXJ5Lndpc2MuZWR1