 Lucas L. Lintermans
Lucas L. Lintermans Coen A. Stegeman
Coen A. Stegeman Peter Heeringa
Peter Heeringa Wayel H. Abdulahad
Wayel H. Abdulahad- 1Department of Rheumatology and Clinical Immunology, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
- 2Department of Nephrology, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
- 3Department of Pathology and Medical Biology, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
Inflammation of the human vasculature is a manifestation of many different diseases ranging from systemic autoimmune diseases to chronic inflammatory diseases, in which multiple types of immune cells are involved. For both autoimmune diseases and chronic inflammatory diseases several observations support a key role for T lymphocytes in these disease pathologies, but the underlying mechanisms are poorly understood. Previous studies in several autoimmune diseases have demonstrated a significant role for a specific subset of CD4+ T cells termed effector memory T (TEM) cells. This expanded population of TEM cells may contribute to tissue injury and disease progression. These cells exert multiple pro-inflammatory functions through the release of effector cytokines. Many of these cytokines have been detected in the inflammatory lesions and participate in the vasculitic reaction, contributing to recruitment of macrophages, neutrophils, dendritic cells, natural killer cells, B cells, and T cells. In addition, functional impairment of regulatory T cells paralyzes anti-inflammatory effects in vasculitic disorders. Interestingly, activation of TEM cells is uniquely dependent on the voltage-gated potassium Kv1.3 channel providing an anchor for specific drug targeting. In this review, we focus on the CD4+ T cells in the context of vascular inflammation and describe the evidence supporting the role of different T cell subsets in vascular inflammation. Selective targeting of pathogenic TEM cells might enable a more tailored therapeutic approach that avoids unwanted adverse side effects of generalized immunosuppression by modulating the effector functions of T cell responses to inhibit the development of vascular inflammation.
Introduction
Vasculitides comprises a group of rare diseases, characterized by inflammation of the blood vessel walls. The clinical manifestations are dependent upon the localization, the type of vessel involved as well as the nature of the inflammatory process. Vasculitis constitutes, in most cases, as a primary autoimmune disorder, but can also be secondary to other conditions. The underlying conditions to secondary vasculitis are infectious diseases, connective tissue disorders, or hypersensitivity disorders. In general, primary vasculitides are systemic diseases with variable clinical manifestations making it difficult to classify. According to the latest Chapel Hill Consensus Conference, primary systemic vasculitides can be divided into seven main entities of which three are most common; large vessel vasculitis, medium vessel vasculitis (MVV), and small vessel vasculitis (SVV) (1). The group of large vessel vasculitides (LVV) affects the aorta and its major branches. The two major variants of LVV are giant cell arteritis (GCA) and Takayasu’s arteritis (TA). MVV is vasculitis that predominantly affects medium arteries defined as the main visceral arteries and their branches. The two major categories are polyarteritis nodosa (PAN) and Kawasaki disease (KD). SVV is divided into anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) and immune complex SVV (2). AAV is characterized by necrotizing vasculitis with few or no immune deposits that predominantly affects small vessels, which lead to systemic organ damage. AAV are associated with the presence of circulating ANCA that are directed against proteinase-3 (PR3) or myeloperoxidase (MPO), proteins in the cytoplasmic granules of neutrophils. This group of systemic vasculitis includes granulomatosis with polyangiitis (GPA), primarily associated with antibodies to PR3–ANCA and microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA), both principally associated with antibodies to MPO–ANCA.
Besides autoimmune disorders related to vascular inflammation, a more common chronic vascular inflammatory disease is atherosclerosis. Clinical evidence indicates that patients suffering from large and medium-sized vessel vasculitis show accelerated atherosclerosis (3). In SVV, this relation is less well defined. However, many patients with SVV carry several risk factors (e.g., impaired renal function, persistent proteinuria, and increased level of C-reactive protein) that contribute to the acceleration of the atherosclerotic process (3, 4). Enhanced oxidation processes, persistently activated T cells and reduced numbers of regulatory T (TREG) cells are among the many pathophysiological factors that play a role in the acceleration of atherogenesis (5). Both vasculitis and atherosclerosis, although in nature different forms of chronic conditions, reveal similarities in T cell repertoire that occur within the process of vascular inflammation.
This review provides an overview of the role of adaptive immune mechanisms in vascular inflammation focusing on the T lymphocytes in particular. The main emphasis will be on the role of effector memory T (TEM) cells in vasculitis (i.e., AAV and atherosclerosis) and the potential therapeutic interventions for modulating the activity of these cells.
T Lymphocytes: Key Participants in Vascular Inflammation
T cells are recruited to the vessel wall in conjunction with macrophages, but in lesser quantity. In the blood vessel wall or tissues, T cell responses are initiated by signals generated via the association of TCR complexes with specific peptide–MHC protein complexes on the surface of antigen-presenting cells (APCs) and through signals provided by co-stimulators expressed on APCs. The responses to antigen and co-stimulators include synthesis of pro-inflammatory mediators (e.g., IFN-γ) cellular proliferation, differentiation into effector and memory cells, and performance of effector functions. These initial events further amplify the inflammatory response, aggravating disease progression. Different T cell subsets exist that can influence vascular inflammation in various ways. In the last decade, substantial progress has been made in the characterization of T cell mediated responses in vascular inflammation.
T Cell Involvement in AAV and Atherosclerosis
In AAV it has been postulated that ANCA in vivo bind to surface expressed auto-antigens (PR3 or MPO) on primed neutrophils, which subsequently activates the neutrophils (6). These activated neutrophils enhance neutrophil degranulation and the release of cytotoxic products that promote endothelial cells damage leading to vascular inflammation and injury (6). This initial inflammatory response mediated by the innate immune system creates a pro-inflammatory (micro)environment to attract cells from the adaptive immune system. In the case of autoimmune mediated vascular pathologies, like AAV, loss of self-tolerance, and continuous antigen presentation also contributes to the involvement of the adaptive immune system. The contribution of T cell mediated immune responses in vascular inflammation is most likely because infiltrating T cells are detected in inflammatory lesions observed in the microvascular bed of kidney, lung, and in nasal biopsies from AAV patients (7–11). In accordance with these findings, soluble T cell activation markers [soluble interleukin-2-receptor (sIL-2R) and soluble CD30] are elevated in plasma or serum and have been shown to be associated with disease activity in AAV (12–15). Also, ANCA antigen specific T cells have been detected in AAV (16, 17). Moreover, the IgG subclass distribution of ANCA, predominantly consisting of IgG1 and IgG4 implies isotype switching of ANCA for which T cells are required (18). Importantly, Ruth et al. demonstrated a pivotal role of T cells in the expression of crescentic glomerulonephritis (19). They induced experimental anti-MPO-associated crescentic glomerulonephritis by immunizing C57BL/6 mice with human MPO followed by subsequent challenge with anti-glomerular basement membrane (anti-GBM) antibodies. Mice depleted of T cells at the time of administration of anti-GBM antibodies developed significantly less glomerular crescent formation and displayed less cell influx in glomeruli compared with control mice. Interestingly, specific T cell depleting therapies with anti-CD52 antibodies (Alemtuzumab) or anti-thymocyte globulin can induce remission in refractory AAV patients (20, 21).
Atherosclerosis is considered a chronic inflammatory disease, characterized by a slowly progressing passive lipid accumulation in large and medium-sized blood vessels that ultimately leads to the formation of plaques. Both innate and adaptive immunity are involved in this process. Ait-Oufella et al. recently reviewed the role of the adaptive immune response in atherosclerosis and discussed the role of dendritic cells (DCs) in the control of T cell involvement in atherosclerosis (5). Classically, DCs accumulate in the atherosclerotic plaque through direct chemokine mediated recruitment. DCs take up (atherosclerotic-specific) antigens such as ApoB100 and LDL and become activated and mature. Subsequently, DCs migrate to draining lymph nodes, where they can present antigens to naïve T cells. After activation, these T cells develop into effector cells, clonally expand and enter the bloodstream. When effector T cells are recruited into atherosclerotic plaques they are reactivated by antigens presented by local macrophages and DCs, boosting the immune response. In human atherosclerotic lesions, the ratio of macrophages to T cell has been reported to be approximately 10:1, thus T cells are not as abundant as macrophages. However, because T cells are activated in the lesions resulting in the production of pro-atherogenic mediators, they can importantly contribute to lesion growth and disease aggravation. The first evidence of T cell involvement in atherosclerosis came with the demonstration that MHC class II positive cells and T cell cytokines (e.g., IFN-γ) are expressed in human atherosclerotic plaques (22). Later, the presence of T cells was observed in atherosclerotic plaques in humans (23, 24) and mice (25, 26). These observations only demonstrated the association of T cell with atherosclerosis but did not revealed the role of T cells in atherogenesis. However, Zhou et al. demonstrated a specific role of T cells in atherogenesis using an animal model of atherosclerosis. They showed that transfer of CD4+ T cells into ApoE−/− mice crossed with immunodeficient mice (scid/scid mice) fully reversed the atheroprotection provided by T and B cell deficiency (27).
Taken together, these observations indicate that T cell mediated immunity is an important contributor to the pathogenesis of vascular diseases such as AAV and atherosclerosis. In line with this, different T cell populations have been identified in vascular inflammation as will be discussed below. Figure 1 presents a proposed mechanism of the T cell mediated vascular inflammatory process.
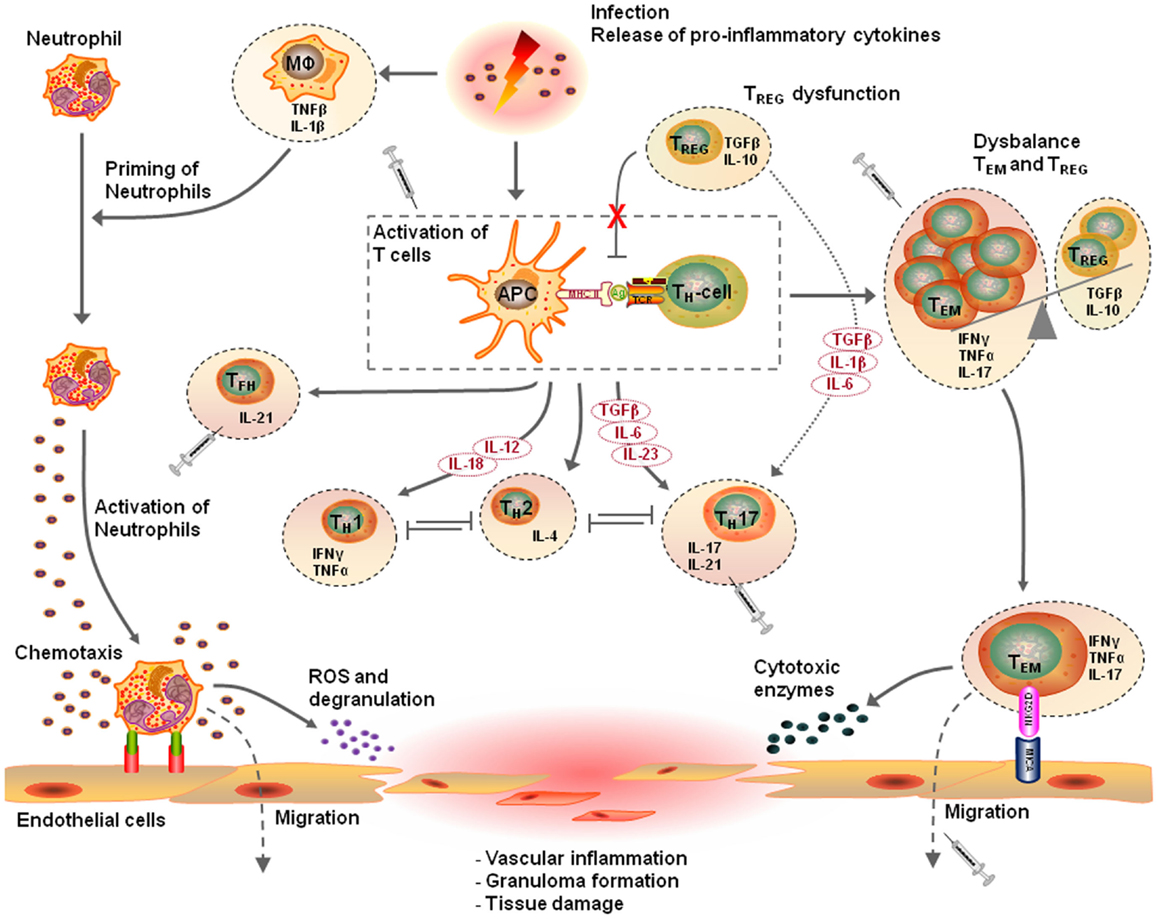
Figure 1. Proposed pathophysiological mechanism of T cell mediated vascular inflammation. Vascular inflammation is initiated by a pro-inflammatory trigger such as an infection. Release of pro-inflammatory cytokines causes priming of neutrophils, up-regulation of adhesion molecules on endothelial cells, and an expansion of circulation effector T cells. Activation of primed neutrophils enhances vessel wall adherence and the transmigration capacity of the neutrophils. Production of reactive oxygen species and degranulation of fully activated primed neutrophils causes damage to vascular endothelial cells. This acute injury together with pro-inflammatory triggers elicits an innate inflammatory response that recruits T lymphocytes, which replace the neutrophils and either resolves or mediate the development of vasculitis. In this pro-inflammatory environment, the innate immune system with antigen-presenting cells (APCs) and T cells start to mediate the inflammatory response. Distinct cytokine patterns in combination with a defect in regulatory T (TREG) cell function or frequency results in expansion of effector memory T (TEM) cells. The dysbalance in the homeostasis of TREG cells and TEM cells, results in additional releases of pro-inflammatory cytokines promoting neutrophil priming and persistent activation of TEM cells. Expanded circulating TEM cells upregulate their killer immunoglobulin-like receptor (NKG2D) and interact with their ligand major histocompatibility complex class-I chain-related molecule A (MICA) on vascular endothelial cells. This event results in the migration of TEM cells into target tissues, drive granuloma formation leading to tissues destruction in a perforin-dependent, and granzyme-dependent way, ending up in vasculitis. The T cell driven vascular inflammatory response is a multistep process and has different therapeutic possibilities. For this purpose, selective TEM cell modulation might be beneficial to regulate the TEM cell activity, proliferation, and migration. Other therapeutic options are modulation of T cell activation by interfering with co-stimulatory molecules, depletion of T cells, inhibition of T cell migration, or neutralizing secreted pro-inflammatory cytokines (This figure was created using Visi ScienceSlides® Software).
T Helper Cells in Vascular Inflammation
Aberrant T helper (TH) cell polarization has been described in patients with vascular diseases. The involvement of different TH cells subsets in the pathogenesis of vascular disease has been suggested to depend on disease activity/stage and whether the disease is localized or systemic.
In AAV, analysis of patients sera for soluble markers associated with either TH1 cells (IFN-γ, sCD26) or TH2 cells (IL-4, IL-5, IL-10, IL-13, sCD23, and sCD30) revealed a shift toward a TH2-type response in patients with active generalized disease, whereas a TH1-type response is predominant in patients with localized disease (28, 29). Consistent with these observations, analysis of nasal granulomatous lesions from AAV patients demonstrated a relative increase of cells expressing TH1-associated markers such as IFN-γ and CD26 during localized disease, whereas the TH2-associated marker IL-4 was found in generalized AAV (11). In addition, Lamprecht et al. compared chemokine receptors on peripheral blood-derived T cells. The inducible inflammatory TH1-type chemokine receptor CCR5 was more prominent in the granulomatous lesions of AAV patients (30).
Similar to AAV, a study on cytokine expression in advanced human atherosclerotic plaques confirmed the dominance of pro-inflammatory TH1 cytokines (IFN-γ, TNF-α, and IL-2) (31). Genetic deficiency in IFN-γ or its receptor in ApoE−/− mice reduced atherosclerotic lesion formation and enhanced plaque stability (32), whereas exogenously administered IFN-γ enhanced atherosclerosis in ApoE−/− mice (33). Intriguingly, it seems that the protective effect of IFN-γ deficiency is restricted to male ApoE−/− mice (34). In addition, several studies revealed that intervention in IL-12 or IL-18 gene, or receptor function was found to reduce plaque development in mouse models of atherosclerosis (35–37). Furthermore, administration of these cytokines accelerated disease progression (38, 39). Collectively, these data point toward a pro-inflammatory TH1 response in atherosclerosis. However, the role of TH2 immune responses in atherosclerosis is controversial. IL-4, the signature cytokine of the TH2 lineage, is not frequently observed in human atherosclerotic plaques (31). Moreover, experimental studies examining the involvement of TH2 cells are contradictory, some showing pro-atherosclerotic effects (36, 40), whereas others show no or athero-protective effects (41, 42).
Overall, the balance between TH1 and TH2 cells plays a key role in the development of vascular inflammation. Interestingly, in the last decade TH17 cells have emerged as a new CD4+ T cell subset characterized by secretion of IL-17A and other cytokines including IL-17F, IL-21, and IL-22. These cells are considered another major pathogenic effector subset involved in the development of inflammatory and autoimmune diseases (43).
IL-17 has been reported to promote the release of the pro-inflammatory cytokines IL-1β and TNF-α from macrophages (44), which are essential for priming and activation of neutrophils. Furthermore, this pro-inflammatory milieu induces CXC chemokine release (45) and up-regulation of endothelial adhesion molecules (46) responsible for the recruitment of neutrophils to the site of inflammation (47). These pro-inflammatory events suggest that IL-17 may directly contribute to the acute vascular inflammatory response in AAV. Convincing experimental evidence that support this notion comes from several studies. Hoshino et al. demonstrated that neutrophils produce IL-17A and IL-23 in response to MPO–ANCA creating local conditions to promote TH17-mediated autoimmunity (48). In addition, Gan et al. showed that immunization of C57BL/6 mice with murine MPO resulted in MPO-specific dermal delayed type hypersensitivity and systemic IL-17A production (49). Upon injection of low-dose anti-GBM antibodies these mice developed glomerulonephritis. In contrast, IL-17A deficient mice were nearly completely protected from disease induction due to reduced neutrophil recruitment and MPO deposition (49). Consistent with this finding, Odobasic et al. demonstrated that IL-17A contributes to early glomerular injury, but it paradoxically, attenuates the severity of fully established crescentic disease by limiting the TH1 responses (50). They used a mouse model of crescentic anti-GBM glomerulonephritis assessing the renal injury and immune responses in IL-17A−/− and in wild-type (WT) mice. Crescentic glomerulonephritis was enhanced in IL-17A−/− mice, with increased glomerular T cell accumulation and augmented TH1 responses (50). In contrast, mice lacking IL-12(p35), the key TH1-promoting cytokine, had decreased TH1 responses and increased TH17 responses and developed less severe crescentic glomerulonephritis than WT animals (50). Thus, they provided evidence that TH1 responses mediate severe crescentic injury and that TH1 and TH17 cells counter regulate each other during disease development in this model. In line with the in vivo observation, our group observed a skewing toward TH17 cells following in vitro stimulation of peripheral blood samples of AAV patients (51). Moreover, it has been shown that CD4+CD45RClow cells (T cells with a memory phenotype) are a source of IL-17 in AAV patients (52). These observations were corroborated by Nogueira et al., demonstrating significant elevated levels of serum IL-17A and its associated upstream cytokine IL-23 in acute AAV patients (53). Additionally, auto-antigen-specific IL-17 producing cells were significantly elevated in patients during disease convalescence compared to healthy controls (53). Moreover, increased frequencies of circulating TH17 cells have been observed in various forms of vasculitis (GCA, EGPA, and Behçet disease) and correlated with disease activity (54–58).
A possible explanation for the involvement of TH17 cells in AAV lies within the major physiological role of TH17 cells. Physiologically, TH17 cells are important in the defense against fungi and bacterial infections [e.g., Staphylococcus aureus (S.aureus) infections] by activating neutrophils through the production of IL-17 and IL-17F. It has been shown that peptidoglycans and superantigens of S. aureus might have an immunomodulatory effect on DCs by imprinting of a strong TH17 polarization capacity (59). Furthermore, S. aureus α-toxin was shown to induce IL-17A secretion in CD4+ T cells (60). In addition, Zielinski et al. demonstrated that S. aureus specific TH17 cells produced IL-17 and surprisingly could produce IL-10 upon restimulation (61). Intriguingly, chronic nasal carriage of S. aureus has been found to be an important risk factor for disease relapse in AAV patients (62). This suggests that carriage of S. aureus may drive the TH17 responses in AAV.
The role of TH17 cells in atherosclerosis remains controversial. It has been demonstrated that TH17 cells and IL-17 accumulate in atherosclerotic lesion of both mice and humans, but both atherogenic as well as athero-protective effects of IL-17 have been reported (63–67). Studies in ApoE−/− mice genetically deficient for IL-17 or treated with anti-IL-17A antibodies demonstrated that absence or depletion of IL-17 attenuated development of atherosclerosis (65, 68). Also, Ldlr−/− mice transplanted with bone marrow from mice deficient in IL-17 receptor showed smaller atherosclerotic lesions (63). In patients, IL-17A expressing T cells were detected in atherosclerotic lesions and increased IL-17 expression in these lesions has been shown to be associated with increased inflammation and plaque vulnerability (67). In contrast to the pathogenic role of TH17 cells, Taleb et al. found a protective role for TH17 cells in atherosclerosis. Using Ldlr−/− mice deficient for suppressor of cytokine signaling 3 (SOCS3), a suppressor of signaling from IL-17, showed less disease development (64). In the same study, administration of an anti-IL-17A antibody accelerated atherosclerosis, indicating a protective role for TH17 cells (64).
A possible explanation for these contradictory observations may be that IL-17 is not only produced by T cells. The presence of different IL-17 isoforms (IL-17A, -E, and -F) in human atherosclerotic plaques revealed that the IL-17 family cytokines were expressed by various cells of the immune system (e.g., neutrophils) depending on the stage of the atherosclerotic plaque (66). Furthermore, not only immune cell are targets of IL-17. It has been demonstrated that endothelial cells and smooth muscle cells are likely to be IL-17E-responsive, given the expression of IL-17 receptor components on these cells and transient activation of ERK1/2 upon stimulation with recombinant IL-17E (66). Thus, this indicates a complex contribution of IL-17 in atherogenesis depending on the isoform and phases of atherosclerosis.
Beside IL-17, TH17 cells can also produce IL-21, a cytokine that is produced primarily by T follicular helper (TFH) cells. IL-21 is required for B cell class switching, antibody production (69), and induces differentiation of B cells toward plasma cells by synergizing with B cell activating factor (BAFF) (70, 71). The role of IL-21 has been demonstrated in Behçet disease, a form of variable vessel vasculitis. Geri et al. demonstrated increased serum levels of IL-21 that correlated with the disease activity in patient with Behçet disease (58). In addition, they showed that IL-21 producing central memory CD4+ T cells positively correlated with TH17 responses and negatively correlated with FoxP3 TREG cells. Conversely, blockade of IL-21 with IL-21R-Fc fusion protein resorted the balance between TREG cells and TH17 cells by suppressing IL-17A production and increasing FoxP3 expression by CD4+ T cells (58). Interestingly, a significant increased population of IL-21 producing TFH cells was observed in the circulation of AAV patients (72). In addition, IL-21 was shown to enhance the production of cytotoxic products such as granzyme B and perforin, by CD8+ T cells and natural killer (NK) cells (73). Based on the studies in various forms of vasculitis it is therefore conceivable that IL-21 together with IL-17 plays a critical role in the pathogenesis of AAV.
In atherosclerosis, no major studies have been conducted to date to investigate the role of TFH cells, but IL-21 may be involved in tissue damage. There is evidence that IL-21 acts directly on gut epithelial cells to induce the production of macrophage inflammatory protein-3α (MIP-3α), a chemokine that attracts both TH1 and TH17 cells to inflamed tissues (74). Given that endothelial cells are known to produce MIP-3α, it is possible that IL-21 enhances the migration and accumulation of TH1 and TH17 cells into the vascular wall in both vasculitis and atherosclerosis resulting in inflammation.
Regulatory T Cells in Vascular Inflammation
The actions of TH cells can be balanced by TREG cells, a subpopulation that is characterized by their ability to suppress a variety of physiological and pathological immune responses and prevent autoimmunity (75). TREG cells are characterized by their expression of forkhead/winged helix transcription factor (FoxP3) that is required for their development and function (76). Defects in TREG function or reduced numbers of TREG cells have been described in several autoimmune disorders and chronic inflammatory disorders associated with vascular inflammation (77). To date different research groups reported controversial results regarding the frequency of TREG cells in AAV patients compared to healthy controls. However, a consistent finding has been that these studies all reported impaired functionality of circulating TREG cells (78–80). It has been found that the suppressive function of TREG cells was defective in GPA patients compared to healthy controls (78). However, the GPA patients showed a significant increase of memory FoxP3+CD25high TREG cells. Consistent with this finding, Klapa et al. demonstrated an increased number of FoxP3+ T cells as well as phenotypical and functional alteration of TREG cells in GPA patients (79). They reported an increased number of interferon receptor I-positive TREG cells in the peripheral blood of GPA patients. In addition, they showed that IFN-α exaggerates functional TREG impairment ex vivo in response to the auto-antigen PR3 (79). Furthermore, Morgan et al. also reported altered TREG function in GPA patients (80). They observed that TREG cells from healthy controls and from ANCA-negative patients were able to suppress T cell proliferation to PR3, whereas TREG cells from PR3-ANCA-positive patients failed to suppress this antigen specific response (80). Dysfunction of TREG cells is thus believed to play a role in the development of GPA. In contrast, TREG function in MPA patients was comparable to that in healthy controls although FoxP3 levels were diminished, suggesting that in MPA a numerical deficiency of TREG cells exists (81). Additionally, Saito et al. demonstrated that the proportion of TREG cells in the peripheral blood reflects the relapse or remission status of EGPA patients. They observed that FoxP3-expressing cells and IL-10 producing TREG cells were detected in lower frequencies in patients with a relapse compared to patient in remission (82). However, the suppressive function of TREG cells in EGPA patients still needs to be investigated. All together there are some inconsistent observations regarding the number and/or frequencies of TREG cells in AAV patients. These differences might be due to variations in the methodology and gating strategies for the TREG cells between the different studies. However, in all studies impaired functionality of the TREG subset has been demonstrated indicating that TREG cells from AAV patients are not able to suppress proliferation of other TH cell subsets.
The terminally differentiated TREG cells are not defined entirely by FoxP3 expression, and the FoxP3+ T cell population is heterogeneous, consisting of a committed TREG lineage and an uncommitted subpopulation with developmental plasticity (83). It has been reported that human TREG cells can convert into pro-inflammatory IL-17 producing T cells depending on a specific cytokine environment (81–83). Both TH cell subsets (i.e. TREG and TH17 cells) may develop from the same precursors under distinct cytokine conditions, and a subset of IL-17-producing CD4+FoxP3+ TREG cells can be generated upon polarization by pro-inflammatory cytokines such as IL-6, which is crucial in orchestrating the balance of TREG and TH17 cells (84–86). It has been shown that the combination of transforming growth factor-beta (TGF-β) and IL-6 treatment can synergistically promote FoxP3 degradation (87), and induce the transcription of ROR-γt, which in turn participates in the induction of IL-17 expression and mediates the skewing toward a TH17 cell phenotype.
Besides the local cytokine environment that orchestrates the balance of TREG and TH17 cells, the functional stability of FoxP3 might influence the developmental pathway. Post-translational modifications can transiently alter the functionality of transcription factors, and there is evidence that FoxP3 can be regulated via acetylation. For example, hyperacetylation of FoxP3 increases the stability of FoxP3 and treatment with histone deacetylases inhibitors results in increased numbers and functional TREG cells (88). Indeed, Koenen et al. demonstrated that histone deacetylases inhibitors suppresses the conversion from TREG to TH17 cells (89). In addition, different isoforms of FoxP3 have been investigated in human TREG that have been shown to affect TREG function and lineage commitment. More specifically, the full length isoform FoxP3 interacts with ROR-γt and inhibits the expression of genes that define the TH17 lineage, whereas the isoform lacking exon 2, FoxP3Δ2 fails to inhibit ROR-γt. Upon stimulation in an inflammatory environment these non-functional TREG convert into IL-17 producing effector T cells. Based on these findings, our previous described non-functional TREG cells in AAV patients may lack their suppressive function due to the up-regulation of FoxP3Δ2 that fails to inhibit ROR-γt mediated IL-17 transcription. Indeed, Free et al., demonstrated that TREG cells from patients with active AAV disproportionately used FoxP3Δ2, which might alter TREG cell function (90).
In atherosclerosis, several studies have demonstrated a protective effect of TREG cells. FoxP3+ T cells have been found in atherosclerotic plaques of humans, although in low numbers (91). In mice, the TREG cytokine products IL-10 and TGF-β, have been demonstrated to induce potent anti-atherosclerotic activities. Genetic inactivation or blockade of IL-10 and TGF-β with neutralizing antibodies aggravated atherosclerosis in mice (92, 93). Depletion of TREG cells directly addressed the protective role of these cells in atherosclerosis. Significant aggravation of atherosclerosis was observed in Ldlr−/− mice with reduced TREG cell numbers, achieved either by deletion of CD80/86 or CD28, inducible T cell co-stimulators, or upon treatment with CD25-depleting antibodies (94, 95).
Thus, the interplay and imbalances between different TH cells are important in the pathogenesis of vascular inflammatory diseases (Table 1). An imbalance in TH1/TH2 toward the TH1 response promotes the development of vascular inflammation, whereas skewing toward prominent TH2 and TREG responses is anti-inflammatory and results in a reduction of vascular inflammation. In AAV, TH17 cells are considered to be pathogenic but how TH17 cell affect inflammation in atherosclerosis still needs to be determined.
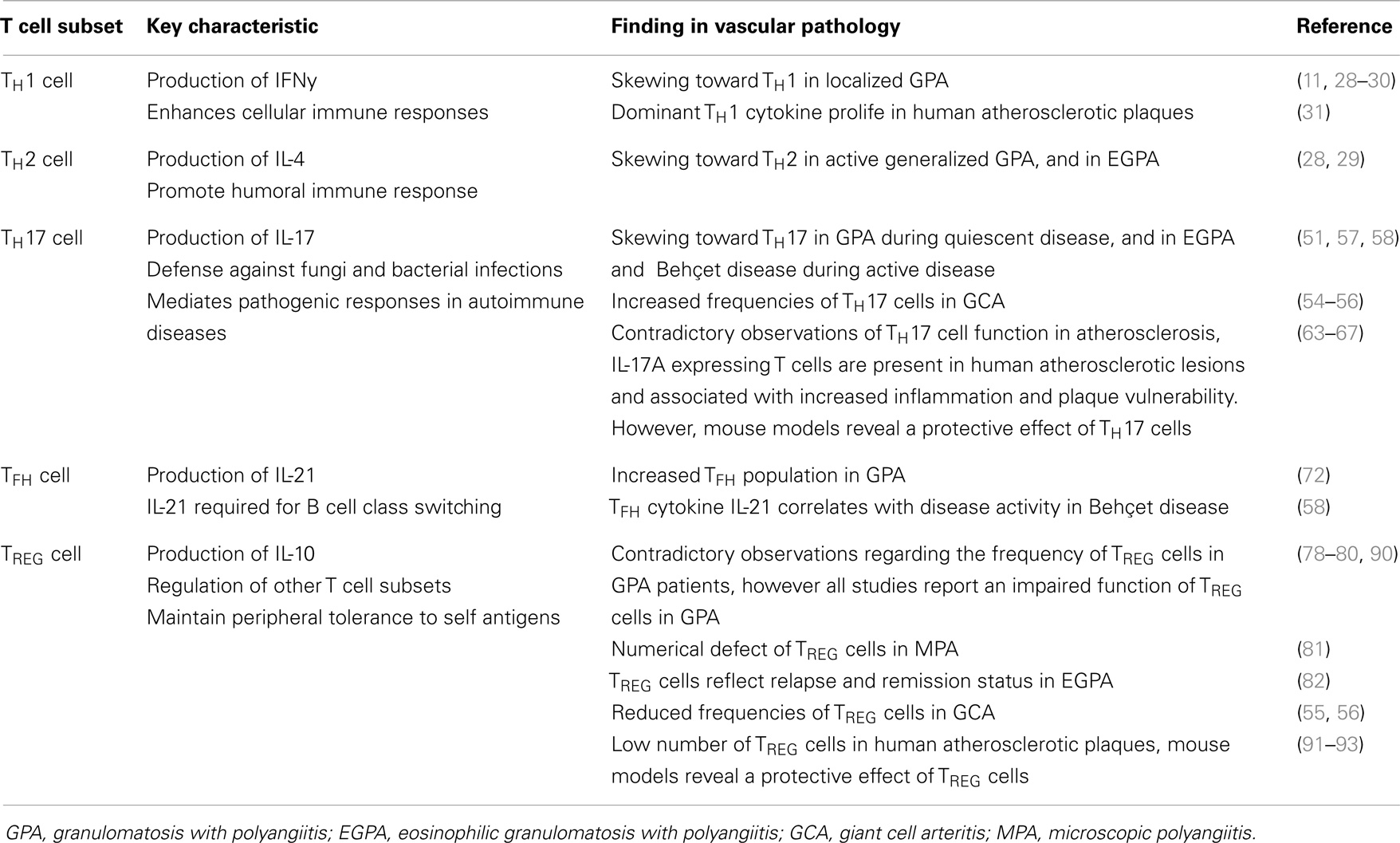
Table 1. T cell subsets associated with vascular pathologies.
Involvement of CD4+ T Effector Memory Cells
As mentioned above, several observations support the involvement of CD4+ TH cells in the pathogenesis of vascular inflammatory diseases like AAV and atherosclerosis. In line with these observations, an expanded population of CD4+ T cells lacking the co-stimulatory molecule CD28 was observed in peripheral blood and in inflammatory lesions of AAV patients (10, 96). Furthermore, these CD4+CD28− T cells are a major source of IFN-γ and TNF-α, display up-regulation of the T cell differentiation marker CD57 and show cytoplasmic perforin expression, indicating the cytotoxic potential of these cells (10).
Consistent with these findings, our group observed a significant increase in the frequency of circulating CD4+ TEM cells in the peripheral blood of AAV patients in remission (97). Subsequently, it was found that the number of these circulating CD4+ TEM cells decrease during active disease when compared with the number during complete remission (97). We proposed that these CD4+ TEM cells migrate toward inflamed tissues (97). In accordance, infiltrating T cells found in granulomas within lung and/or kidney tissues resemble mainly the CD4+ T cell memory phenotype. A remarkable increase in CD4+ TEM cells in the urinary sediment with a concomitant decrease of circulating CD4+ TEM cells in patients with active renal involvement strongly suggests migration of CD4+ TEM cells during active renal disease into the affected organs (98). This finding might reflect the role of CD4+ TEM cells in renal injury during active disease. In line with these findings, is the observation that CD4+ TEM cells expressing CD134 are expanded in peripheral blood of AAV patients (99). It has been reported that the ligand for CD134 (CD134L) is expressed on endothelial cells (100) and ligation of CD134 contributes to T cell migration and tissue infiltration through its interaction with CD134L on vascular endothelial cells (101). Furthermore, CD134 expressing T cells have been detected in the inflammatory lesions of AAV patients (99), supporting the hypothesis that CD4+ TEM migrate to inflamed areas. The fact that CD4+ TEM cells migrate to the kidney during active disease suggests that a specific stimulus is being expressed by the (micro)vascular bed of the kidney, which attracts these cells.
In atherosclerotic disease, analysis of CD4+ T cell subsets (i.e., naïve T cells, TCM, and TEM cells) revealed that CD4+ TEM cells are key players in disease pathogenesis (102). It has been shown that the frequency of circulating TEM cells was significantly increased in Ldlr−/− and ApoE−/− mice compared to control C57BL/6 mice and also correlated with the extent of atherosclerotic lesions (102). In line with these observations, Almanzar et al. demonstrated that T cells isolated from early atherosclerotic lesions are mostly CD4+ TEM cells (24). Subsequently, the intralesional atherosclerotic CD4+ T cells produce high amounts of the pro-inflammatory cytokines IFN-γ and IL-17 (24), which suggest that memory T cells in atherosclerotic plaques are in a state of activation reflecting the pro-inflammatory cytokine production.
At the functional level, CD4+ TEM cells have been shown to mimic features of NK cell including surface expression of the NK group 2 member D (NKG2D) and cytotoxic potential (103, 104). NKG2D is an activating C-type lectin-like receptor, which differs from other NKG2 family members as it apparently lacks an antagonist and substitutes for CD28-mediated co-stimulatory signaling in CD28− TEM cells (104). One of the NKG2D ligands in human is the major histocompatibility complex class-I chain-related molecule A (MICA). MICA is usually absent on normal cells, but expressed upon cellular stress on target cells such as fibroblasts, epithelial cells, and endothelial cells (104). The expression of MICA on the surface of the endothelium makes this polymorphic molecule a potential target in vasculitis. In rheumatoid arthritis (RA), an other chronic and systemic autoimmune disorder, an unusual CD4+CD28−NKG2D+ population was detected in peripheral blood and synovial tissue (105). Furthermore, the NKG2D ligand MICA is dramatically upregulated in RA synoviocytes and is capable of activating autoreactive T cells in an NKG2D-dependent manner (105). In addition, it has been shown in patients with Crohn’s disease, a chronic inflammatory disorder, that CD4+NKG2D+ TEM cells can kill target cells that express MICA via NKG2D–MICA interaction (106). These findings in RA and Crohn’s disease translate very well to AAV. In AAV, it has been found that NKG2D was anomalously expressed on circulating CD4+CD28− TEM cells (107). Furthermore, it has been demonstrated that NKG2D, MICA, and IL-15 are simultaneously expressed in granulomatous lesions in AAV patients. Importantly, it was reported that survival, expansion, and cytotoxic properties of CD4+NKG2D+ T cells were dependent on IL-15 signaling in AAV (108). Therefore, it is tempting to speculate that combined IL-15 and MICA expression contributes to the killing mechanisms of CD4+NKG2D+ T cells in vessel inflammation and disease progression in AAV.
In atherosclerosis, Xia et al. reported that immune activation resulting from NKG2D–ligand interaction promotes atherosclerosis (109). They observed soluble MICA in sera and upregulated MICA expression in atherosclerotic plaques of patients with type 2 diabetes mellitus. Moreover, they investigated the role of NKG2D in atherosclerosis using ApoE−/− mice genetically deficient for NKG2D or treated with anti-NKG2D antibodies. Preventing NKG2D–ligand interaction resulted in a dramatic reduction in plaque formation and suppressed systemic and local inflammation mediated by multiple immune cell types. Since this is the only study reported on NKG2D in relation with atherosclerosis development, further studies are needed to fully elucidate the role of NKG2D in the pathogenesis of atherosclerosis.
T Cell Directed Therapeutic Interventions
As described in this review, T cells and T cell migration are pivotal in vascular inflammatory diseases such as vasculitides and atherosclerosis. Therefore interfering with T cell activation, proliferation, and migration might be a beneficial approach to dampen the inflammatory response and cell-based therapy to modulate the T cell compartment may be a therapeutic option.
Regulatory T expansion may be of benefit to counterbalance persistent T cell activation in AAV. In this respect, treatment with low-dose IL-2 has been found to promote TREG recovery and clinical improvement in patients with autoimmune vasculitis (110). Interestingly, tocilizumab, a humanized anti-IL-6 receptor antibody used in the treatment of RA demonstrated that blocking IL-6 function affects the balance between TH17 and TREG cells favoring a more anti-inflammatory response (111). In addition, control of T cell activation might be an attractive therapeutic possibility. In this regard, blockage of the co-stimulatory pathway CD28/CD80 using CTLA-4 fusion proteins has successfully been used in RA (112) and shown to be well tolerated in a small open-label trial in GPA patients (113), which suggests this treatment as a possible therapeutic opportunity in AAV.
Modulation of other T cell subsets is also considered as a means for future therapies. For example, since TH17 cells contribute to inflammation and granuloma formation, this TH cell subset could be a novel therapeutic target for AAV. Depletion of TH17 cells by targeting specific surface proteins may be difficult as TH17 cells share many surface markers with other T cell subsets. Another therapeutic approach could be to specifically target its signature cytokine, IL-17, which would probably be more feasible. Indeed, neutralizing IL-17 by IL-17A specific antibodies or administration of soluble IL-17 receptors reduces inflammation in animal models of atherosclerosis (49, 65, 68, 114). Several IL-17A blockers, including the anti-IL-17A monoclonal antibodies secukinumab and ixekizumab, and the anti-IL-17 receptor subunit A monoclonal antibody brodalumab have been evaluated in clinical trials (115–117), and shown to induce clinically relevant responses in patients. Besides IL-17, IL-21 also seems an interesting target in the treatment of autoimmune mediated vascular inflammation. Manipulation of IL-21 levels may have desirable therapeutic consequences as it might reduce the recruitment of inflammatory TH1 and TH17 cells to inflammatory lesions preventing tissue damage and inhibit expansion of autoreactive B cells. Recently, phase I clinical trials using an IL-21-specific monoclonal antibody have been completed for RA (NCT01208506 and EudraCT-2011-005376-42, www.clinicaltrial.gov) but terminated for SLE (NCT01689025, www.clinicaltrial.gov). Neutralization of IL-17 or IL-21 could therefore represent also novel therapeutic approaches for patients with AAV. However, experiments using animal models of AAV and clinical trials need to elucidate the therapeutic potential of neutralizing IL-17 and IL-21 in AAV. It is important to note that interfering with the cytokine environment might also cause disturbances in the developmental pathways of the different T cell lineages. Since there is a tight interplay between different T cell lineages such as the TREG and TH17 cells, one should be cautious in modulating the cytokine environments. Besides targeting pro-inflammatory cytokines like IL-17 or IL-21, one can also consider to target effector T cells, which are predominantly responsible for the production of these cytokines.
Effector Memory CD4+ T Cells as Therapeutic Targets
According to aforementioned evidence in AAV and atherosclerosis, CD4+ TEM cells are considered to play a pivotal role in the pathogenesis of vascular inflammation and therefore, may serve as a potential therapeutic target. Selective targeting of CD4+ TEM cells without impairing other parts of the humoral or cellular immune system could be a major step forward in the treatment of chronic and/or autoimmune mediated vascular inflammation disorders.
The capacity of CD4+ TEM cells to interact with target cells via NKG2D–MICA interaction and attack them by releasing cytolytic enzymes has been demonstrated (104, 106). Therefore, NKG2D expressed on pathogenic TEM cells could be an interesting target to inhibit the pathogenic effects of TEM cells. Interestingly, interference with NKG2D signaling using anti-NKG2D antibodies has shown beneficial effects when administered early in two different mouse models. Blockade of NKG2D prevented autoimmune diabetes in non-obese diabetic mice (118) and attenuated transfer-induced colitis in SCID mice (119). However, no clinical trials on the blockade of NKG2D or its ligands in autoimmune diseases have been conducted.
Besides interfering with the NKG2D–MICA interaction, biophysical analyses revealed that ion channels expressed by immune cells perform functions vital for cellular homeostasis and T cell activation [reviewed by Ref. (120)]. In particular, human T cells express two types of potassium channels (voltage-gate potassium Kv1.3 channel; Kv1.3 and Ca2+-activated potassium KCa3.1 channel; KCa3.1) that play a major role in their activation. Interestingly, Kv1.3 channels and the KCa3.1 channels are expressed on T cells in a distinct pattern that depends on the state of activation as well as on the state of differentiation of the given T lymphocyte subset (121). It has been shown that Kv1.3 channels are highly expressed in CD4+ TEM cells (~1500 channels per cell), whereas naïve and TCM cells express lower levels of Kv1.3 channels (~250 channels per cell) (122). Therefore, Kv1.3 channels may serve as an attractive target for specific immunomodulation in TEM cell mediated chronic or autoimmune diseases. Hu et al. have demonstrated that genetic silencing of Kv1.3 in human CD4+ T cells results in selective expansion of TCM cells and the disappearance of TEM cells after multiple rounds of stimulation with anti-CD3/CD28 in vitro, suggesting that Kv1.3 is essential for maintaining the TEM pool (123). Indeed, selective blocking of Kv1.3 channels inhibits Ca2+ signaling, pro-inflammatory cytokine production, and proliferation of CD4+ TEM cells in vitro, with little or no effects on CD4+ naïve and TCM cells (124). Furthermore, it has been shown that specific Kv1.3 blockade suppressed TEM cell motility in inflamed tissues, but had no effect on homing to or motility in lymph nodes of naïve and TCM in vivo (125). In addition, Kv1.3 blockers ameliorate disease development in animal models of multiple sclerosis, RA, T1DM, and contact dermatitis without compromising the protective immune responses to acute infections (124, 126, 127). Noteworthy, Gocke et al. demonstrated that genetic deletion of Kv1.3 biases T cells toward an immunoregulatory phenotype and renders mice resistant to experimental autoimmune encephalomyelitis (128). They showed that Kv1.3 is required for expression of pro-inflammatory cytokines IFN-γ and IL-17, whereas its absence led to increased IL-10 production (128). Thus, it is tempting to speculate that pharmacological blockade or genetic suppression of Kv1.3 channels can be employed as a means to skew CD4+ T cell differentiation toward a regulatory phenotype, which might be beneficial for autoimmune mediated vascular diseases in general. Importantly, Kv1.3 blockers have a good safety prolife in rodents and primates and do not compromise the protective immune response to acute viral (Influenza) or bacterial (Chlamydia) infections (124, 125, 127).
Taken together, these studies demonstrated that specific blockade of Kv1.3 channels on TEM cells suppresses the pathogenicity of TEM cells by inhibition of their activation, proliferation and migration. Recently, it has been shown that expression of T cell Kv1.3 channels correlated with disease activity in ulcerative colitis (129). Therefore, Kv1.3 channels on CD4+ TEM cells in chronic or autoimmune inflammatory diseases could constitute a novel pharmacological target in immunomodulation therapies and at the same time may serve as a marker for disease activity.
Conclusion
Vascular inflammation can be driven by chronic inflammatory disorders or by autoimmune mediated diseases. In this inflammation process, there is a tight interplay between the innate and adaptive immune system. However, as described in this review, substantial evidence point to an important role for cell mediated adaptive immune responses in the pathogenesis of vascular inflammation. In particular, T cells in chronic or autoimmune mediated vascular inflammation show several functional abnormalities. The T cell compartment shows dysregulation in TH subsets, including an imbalance between TH1 and TH2 cells, skewing toward TH17 response and defective functions of TREG cells. However, the importance of TH2 and TH17 cells in atherogenesis is controversial. The underlying mechanisms responsible for orchestrating the aberrations within the T cell compartment are not completely understood. However, multiple studies indicate an interplay between TREG cells and TH17 cells. It has been suggested that in the context of an inflammatory environment TREG cells convert into IL-17 producing cells. Furthermore, TREG cells clearly have a protective effect in experimental models of atherosclerosis. In AAV, TREG cells are often quantitatively or functional defective. It has been suggested that a defect in TREG cell function, may also contribute to the expansion of CD4+ TEM cell population and migration of these cells to inflamed sites. Indeed, observations in AAV and atherosclerosis support TEM cell involvement in vascular inflammation that in part contributes to tissue damage. The persistent activation of TEM cells results in a selective up-regulation of Kv1.3 channels. These potassium channels play a key role in TEM cell homeostasis, proliferation, and activation. Therefore, they seem to be a highly interesting target for immunomodulation of TEM cells without compromising other subsets of the T cell compartment.
Currently efforts are being made to develop biological agents that can modulate the different T cell compartments specifically. In the case of autoimmune mediated vascular diseases such strategies could first be used for the prevention of disease flares. Considering that expansion of TREG cells might be inadequate to control inflammatory responses, regulating TEM cells would probably be the most effective approach. This approach may diminish effector responses and convert these into a more disease regulating response. To develop such therapeutic strategies further studies on the basic immunological properties of TEM and TREG cells in especially humans are needed. Investigation of the functional characteristics of TEM cells in the pathogenesis of vascular inflammatory diseases and selective targeting of these cells will enable their application for the treatment of TEM cell mediated vascular diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the Dutch Arthritis Foundation (12-2-407). Dr. Wayel H. Abdulahad has received funding from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no 261382.
References
1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum (2013) 65(1):1–11. doi: 10.1002/art.37715
2. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med (1997) 337(21):1512–23. doi:10.1056/NEJM199711203372106
3. Cohen Tervaert JW. Cardiovascular disease due to accelerated atherosclerosis in systemic vasculitides. Best Pract Res Clin Rheumatol (2013) 27(1):33–44. doi:10.1016/j.berh.2012.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. de Leeuw K, Sanders JS, Stegeman C, Smit A, Kallenberg CG, Bijl M. Accelerated atherosclerosis in patients with Wegener’s granulomatosis. Ann Rheum Dis (2005) 64(5):753–9. doi:10.1136/ard.2004.029033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Ait-Oufella H, Sage AP, Mallat Z, Tedgui A. Adaptive (T and B cells) immunity and control by dendritic cells in atherosclerosis. Circ Res (2014) 114(10):1640–60. doi:10.1161/CIRCRESAHA.114.302761
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Kallenberg CG, Heeringa P, Stegeman CA. Mechanisms of disease: pathogenesis and treatment of ANCA-associated vasculitides. Nat Clin Pract Rheumatol (2006) 2(12):661–70. doi:10.1038/ncprheum0355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Gephardt GN, Ahmad M, Tubbs RR. Pulmonary vasculitis (Wegener’s granulomatosis). Immunohistochemical study of T and B cell markers. Am J Med (1983) 74(4):700–4. doi:10.1016/0002-9343(83)91030-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Weidner S, Carl M, Riess R, Rupprecht HD. Histologic analysis of renal leukocyte infiltration in antineutrophil cytoplasmic antibody-associated vasculitis: importance of monocyte and neutrophil infiltration in tissue damage. Arthritis Rheum (2004) 50(11):3651–7. doi:10.1002/art.20607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Lamprecht P, Moosig F, Csernok E, Seitzer U, Schnabel A, Mueller A, et al. CD28 negative T cells are enriched in granulomatous lesions of the respiratory tract in Wegener’s granulomatosis. Thorax (2001) 56(10):751–7. doi:10.1136/thorax.56.10.751
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Komocsi A, Lamprecht P, Csernok E, Mueller A, Holl-Ulrich K, Seitzer U, et al. Peripheral blood and granuloma CD4(+)CD28(-) T cells are a major source of interferon-gamma and tumor necrosis factor-alpha in Wegener’s granulomatosis. Am J Pathol (2002) 160(5):1717–24. doi:10.1016/S0002-9440(10)61118-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Muller A, Trabandt A, Gloeckner-Hofmann K, Seitzer U, Csernok E, Schonermarck U, et al. Localized Wegener’s granulomatosis: predominance of CD26 and IFN-gamma expression. J Pathol (2000) 192(1):113–20. doi:10.1002/1096-9896(2000)9999:9999<::AID-PATH656>3.0.CO;2-M
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Schmitt WH, Heesen C, Csernok E, Rautmann A, Gross WL. Elevated serum levels of soluble interleukin-2 receptor in patients with Wegener’s granulomatosis. Association with disease activity. Arthritis Rheum (1992) 35(9):1088–96. doi:10.1002/art.1780350914
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Stegeman CA, Tervaert JW, Huitema MG, Kallenberg CG. Serum markers of T cell activation in relapses of Wegener’s granulomatosis. Clin Exp Immunol (1993) 91(3):415–20. doi:10.1111/j.1365-2249.1993.tb05918.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Popa ER, Stegeman CA, Bos NA, Kallenberg CG, Tervaert JW. Differential B- and T-cell activation in Wegener’s granulomatosis. J Allergy Clin Immunol (1999) 103(5 Pt 1):885–94. doi:10.1016/S0091-6749(99)70434-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Sanders JS, Huitma MG, Kallenberg CG, Stegeman CA. Plasma levels of soluble interleukin 2 receptor, soluble CD30, interleukin 10 and B cell activator of the tumour necrosis factor family during follow-up in vasculitis associated with proteinase 3-antineutrophil cytoplasmic antibodies: associations with disease activity and relapse. Ann Rheum Dis (2006) 65(11):1484–9. doi:10.1136/ard.2005.046219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Brouwer E, Stegeman CA, Huitema MG, Limburg PC, Kallenberg CG. T cell reactivity to proteinase 3 and myeloperoxidase in patients with Wegener’s granulomatosis (WG). Clin Exp Immunol (1994) 98(3):448–53. doi:10.1111/j.1365-2249.1994.tb05511.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Seta N, Tajima M, Kobayashi S, Kawakami Y, Hashimoto H, Kuwana M. Autoreactive T-cell responses to myeloperoxidase in patients with antineutrophil cytoplasmic antibody-associated vasculitis and in healthy individuals. Mod Rheumatol (2008) 18(6):593–600. doi:10.1007/s10165-008-0109-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Brouwer E, Tervaert JW, Horst G, Huitema MG, van der Giessen M, Limburg PC, et al. Predominance of IgG1 and IgG4 subclasses of anti-neutrophil cytoplasmic autoantibodies (ANCA) in patients with Wegener’s granulomatosis and clinically related disorders. Clin Exp Immunol (1991) 83(3):379–86. doi:10.1111/j.1365-2249.1991.tb05647.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Ruth AJ, Kitching AR, Kwan RY, Odobasic D, Ooi JD, Timoshanko JR, et al. Anti-neutrophil cytoplasmic antibodies and effector CD4+ cells play nonredundant roles in anti-myeloperoxidase crescentic glomerulonephritis. J Am Soc Nephrol (2006) 17(7):1940–9. doi:10.1681/ASN.2006020108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Lockwood CM, Thiru S, Isaacs JD, Hale G, Waldmann H. Long-term remission of intractable systemic vasculitis with monoclonal antibody therapy. Lancet (1993) 341(8861):1620–2. doi:10.1016/0140-6736(93)90759-A
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Schmitt WH, Hagen EC, Neumann I, Nowack R, Flores-Suarez LF, van der Woude FJ, et al. Treatment of refractory Wegener’s granulomatosis with antithymocyte globulin (ATG): an open study in 15 patients. Kidney Int (2004) 65(4):1440–8. doi:10.1111/j.1523-1755.2004.00534.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Jonasson L, Holm J, Skalli O, Gabbiani G, Hansson GK. Expression of class II transplantation antigen on vascular smooth muscle cells in human atherosclerosis. J Clin Invest (1985) 76(1):125–31. doi:10.1172/JCI111934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis (1986) 6(2):131–8. doi:10.1161/01.ATV.6.2.131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Almanzar G, Ollinger R, Leuenberger J, Onestingel E, Rantner B, Zehm S, et al. Autoreactive HSP60 epitope-specific T-cells in early human atherosclerotic lesions. J Autoimmun (2012) 39(4):441–50. doi:10.1016/j.jaut.2012.07.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Roselaar SE, Kakkanathu PX, Daugherty A. Lymphocyte populations in atherosclerotic lesions of apoE-/- and LDL receptor-/- mice. Decreasing density with disease progression. Arterioscler Thromb Vasc Biol (1996) 16(8):1013–8. doi:10.1161/01.ATV.16.8.1013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Zhou X, Stemme S, Hansson GK. Evidence for a local immune response in atherosclerosis. CD4+ T cells infiltrate lesions of apolipoprotein-E-deficient mice. Am J Pathol (1996) 149(2):359–66.
27. Zhou X, Nicoletti A, Elhage R, Hansson GK. Transfer of CD4(+) T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation (2000) 102(24):2919–22. doi:10.1161/01.CIR.102.24.2919
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Wang G, Hansen H, Tatsis E, Csernok E, Lemke H, Gross WL. High plasma levels of the soluble form of CD30 activation molecule reflect disease activity in patients with Wegener’s granulomatosis. Am J Med (1997) 102(6):517–23. doi:10.1016/S0002-9343(97)00049-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Schonermarck U, Csernok E, Trabandt A, Hansen H, Gross WL. Circulating cytokines and soluble CD23, CD26 and CD30 in ANCA-associated vasculitides. Clin Exp Rheumatol (2000) 18(4):457–63.
30. Lamprecht P, Bruhl H, Erdmann A, Holl-Ulrich K, Csernok E, Seitzer U, et al. Differences in CCR5 expression on peripheral blood CD4+CD28- T-cells and in granulomatous lesions between localized and generalized Wegener’s granulomatosis. Clin Immunol (2003) 108(1):1–7. doi:10.1016/S1521-6616(03)00121-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Frostegard J, Ulfgren AK, Nyberg P, Hedin U, Swedenborg J, Andersson U, et al. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis (1999) 145(1):33–43. doi:10.1016/S0021-9150(99)00011-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest (1997) 99(11):2752–61. doi:10.1172/JCI119465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E-/- mice. Am J Pathol (2000) 157(6):1819–24. doi:10.1016/S0002-9440(10)64820-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Whitman SC, Ravisankar P, Daugherty A. IFN-gamma deficiency exerts gender-specific effects on atherogenesis in apolipoprotein E-/- mice. J Interferon Cytokine Res (2002) 22(6):661–70. doi:10.1089/10799900260100141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Elhage R, Jawien J, Rudling M, Ljunggren HG, Takeda K, Akira S, et al. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc Res (2003) 59(1):234–40. doi:10.1016/S0008-6363(03)00343-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol (2003) 163(3):1117–25. doi:10.1016/S0002-9440(10)63471-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Hauer AD, Uyttenhove C, de Vos P, Stroobant V, Renauld JC, van Berkel TJ, et al. Blockade of interleukin-12 function by protein vaccination attenuates atherosclerosis. Circulation (2005) 112(7):1054–62. doi:10.1161/CIRCULATIONAHA.104.533463
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Lee TS, Yen HC, Pan CC, Chau LY. The role of interleukin 12 in the development of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol (1999) 19(3):734–42. doi:10.1161/01.ATV.19.3.734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Whitman SC, Ravisankar P, Daugherty A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(-/-) mice through release of interferon-gamma. Circ Res (2002) 90(2):E34–8. doi:10.1161/hh0202.105292
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. King VL, Szilvassy SJ, Daugherty A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor-/- mice. Arterioscler Thromb Vasc Biol (2002) 22(3):456–61. doi:10.1161/hq0302.104905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. King VL, Cassis LA, Daugherty A. Interleukin-4 does not influence development of hypercholesterolemia or angiotensin II-induced atherosclerotic lesions in mice. Am J Pathol (2007) 171(6):2040–7. doi:10.2353/ajpath.2007.060857
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Huber SA, Sakkinen P, David C, Newell MK, Tracy RP. T helper-cell phenotype regulates atherosclerosis in mice under conditions of mild hypercholesterolemia. Circulation (2001) 103(21):2610–6. doi:10.1161/01.CIR.103.21.2610
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Ooi JD, Kitching AR, Holdsworth SR. Review: T helper 17 cells: their role in glomerulonephritis. Nephrology (Carlton) (2010) 15(5):513–21. doi:10.1111/j.1440-1797.2010.01343.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol (1998) 160(7):3513–21.
45. Laan M, Cui ZH, Hoshino H, Lotvall J, Sjostrand M, Gruenert DC, et al. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol (1999) 162(4):2347–52.
46. Panettieri RA Jr., Lazaar AL, Pure E, Albelda SM. Activation of cAMP-dependent pathways in human airway smooth muscle cells inhibits TNF-alpha-induced ICAM-1 and VCAM-1 expression and T lymphocyte adhesion. J Immunol (1995) 154(5):2358–65.
47. Hoshino H, Laan M, Sjostrand M, Lotvall J, Skoogh BE, Linden A. Increased elastase and myeloperoxidase activity associated with neutrophil recruitment by IL-17 in airways in vivo. J Allergy Clin Immunol (2000) 105(1 Pt 1):143–9. doi:10.1016/S0091-6749(00)90189-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Hoshino A, Nagao T, Nagi-Miura N, Ohno N, Yasuhara M, Yamamoto K, et al. MPO-ANCA induces IL-17 production by activated neutrophils in vitro via classical complement pathway-dependent manner. J Autoimmun (2008) 31(1):79–89. doi:10.1016/j.jaut.2008.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Gan PY, Steinmetz OM, Tan DS, O’Sullivan KM, Ooi JD, Iwakura Y, et al. Th17 cells promote autoimmune anti-myeloperoxidase glomerulonephritis. J Am Soc Nephrol (2010) 21(6):925–31. doi:10.1681/ASN.2009070763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Odobasic D, Gan PY, Summers SA, Semple TJ, Muljadi RC, Iwakura Y, et al. Interleukin-17A promotes early but attenuates established disease in crescentic glomerulonephritis in mice. Am J Pathol (2011) 179(3):1188–98. doi:10.1016/j.ajpath.2011.05.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Abdulahad WH, Stegeman CA, Limburg PC, Kallenberg CG. Skewed distribution of Th17 lymphocytes in patients with Wegener’s granulomatosis in remission. Arthritis Rheum (2008) 58(7):2196–205. doi:10.1002/art.23557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Ordonez L, Bernard I, L’faqihi-Olive FE, Tervaert JW, Damoiseaux J, Saoudi A. CD45RC isoform expression identifies functionally distinct T cell subsets differentially distributed between healthy individuals and AAV patients. PLoS One (2009) 4(4):e5287. doi:10.1371/journal.pone.0005287
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Nogueira E, Hamour S, Sawant D, Henderson S, Mansfield N, Chavele KM, et al. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol Dial Transplant (2010) 25(7):2209–17. doi:10.1093/ndt/gfp783
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation (2010) 121(7):906–15. doi:10.1161/CIRCULATIONAHA.109.872903
55. Terrier B, Geri G, Chaara W, Allenbach Y, Rosenzwajg M, Costedoat-Chalumeau N, et al. Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheum (2012) 64(6):2001–11. doi:10.1002/art.34327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Samson M, Audia S, Fraszczak J, Trad M, Ornetti P, Lakomy D, et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum (2012) 64(11):3788–98. doi:10.1002/art.34647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Saito H, Tsurikisawa N, Tsuburai T, Oshikata C, Akiyama K. Cytokine production profile of CD4+ T cells from patients with active Churg-Strauss syndrome tends toward Th17. Int Arch Allergy Immunol (2009) 149(Suppl 1):61–5. doi:10.1159/000210656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Geri G, Terrier B, Rosenzwajg M, Wechsler B, Touzot M, Seilhean D, et al. Critical role of IL-21 in modulating TH17 and regulatory T cells in Behcet disease. J Allergy Clin Immunol (2011) 128(3):655–64. doi:10.1016/j.jaci.2011.05.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Ivanov S, Bozinovski S, Bossios A, Valadi H, Vlahos R, Malmhall C, et al. Functional relevance of the IL-23-IL-17 axis in lungs in vivo. Am J Respir Cell Mol Biol (2007) 36(4):442–51. doi:10.1165/rcmb.2006-0020OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Niebuhr M, Gathmann M, Scharonow H, Mamerow D, Mommert S, Balaji H, et al. Staphylococcal alpha-toxin is a strong inducer of interleukin-17 in humans. Infect Immun (2011) 79(4):1615–22. doi:10.1128/IAI.00958-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, et al. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature (2012) 484(7395):514–8. doi:10.1038/nature10957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Stegeman CA, Tervaert JW, Sluiter WJ, Manson WL, de Jong PE, Kallenberg CG. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med (1994) 120(1):12–7. doi:10.7326/0003-4819-120-1-199401010-00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. van Es T, van Puijvelde GH, Ramos OH, Segers FM, Joosten LA, van den Berg WB, et al. Attenuated atherosclerosis upon IL-17R signaling disruption in LDLr deficient mice. Biochem Biophys Res Commun (2009) 388(2):261–5. doi:10.1016/j.bbrc.2009.07.152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Taleb S, Romain M, Ramkhelawon B, Uyttenhove C, Pasterkamp G, Herbin O, et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med (2009) 206(10):2067–77. doi:10.1084/jem.20090545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Smith E, Prasad KM, Butcher M, Dobrian A, Kolls JK, Ley K, et al. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation (2010) 121(15):1746–55. doi:10.1161/CIRCULATIONAHA.109.924886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. de Boer OJ, van der Meer JJ, Teeling P, van der Loos CM, Idu MM, van Maldegem F, et al. Differential expression of interleukin-17 family cytokines in intact and complicated human atherosclerotic plaques. J Pathol (2010) 220(4):499–508. doi:10.1002/path.2667
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Erbel C, Dengler TJ, Wangler S, Lasitschka F, Bea F, Wambsganss N, et al. Expression of IL-17A in human atherosclerotic lesions is associated with increased inflammation and plaque vulnerability. Basic Res Cardiol (2011) 106(1):125–34. doi:10.1007/s00395-010-0135-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Erbel C, Chen L, Bea F, Wangler S, Celik S, Lasitschka F, et al. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J Immunol (2009) 183(12):8167–75. doi:10.4049/jimmunol.0901126
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Fazilleau N, Mark L, McHeyzer-Williams LJ, McHeyzer-Williams MG. Follicular helper T cells: lineage and location. Immunity (2009) 30(3):324–35. doi:10.1016/j.immuni.2009.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Ettinger R, Sims GP, Robbins R, Withers D, Fischer RT, Grammer AC, et al. IL-21 and BAFF/BLyS synergize in stimulating plasma cell differentiation from a unique population of human splenic memory B cells. J Immunol (2007) 178(5):2872–82. doi:10.4049/jimmunol.178.5.2872
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Karnell JL, Ettinger R. The interplay of IL-21 and BAFF in the formation and maintenance of human B cell memory. Front Immunol (2012) 3:2. doi:10.3389/fimmu.2012.00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Abdulahad WH, Lepse N, Stegeman CA, Huitema MG, Doornbos-van der Meer B, Tadema HH, et al. Increased frequency of circulating IL-21 producing Th-cells in patients with granulomatosis with polyangiitis. Arthritis Res Ther (2013) 15(3):R70. doi:10.1186/ar4247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Ebert EC. Interleukin 21 up-regulates perforin-mediated cytotoxic activity of human intra-epithelial lymphocytes. Immunology (2009) 127(2):206–15. doi:10.1111/j.1365-2567.2008.02941.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Caruso R, Fina D, Peluso I, Stolfi C, Fantini MC, Gioia V, et al. A functional role for interleukin-21 in promoting the synthesis of the T-cell chemoattractant, MIP-3alpha, by gut epithelial cells. Gastroenterology (2007) 132(1):166–75. doi:10.1053/j.gastro.2006.09.053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol (2014) 14(3):154–65. doi:10.1038/nri3605
76. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi:10.1038/ni904
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Valencia X, Lipsky PE. CD4+CD25+FoxP3+ regulatory T cells in autoimmune diseases. Nat Clin Pract Rheumatol (2007) 3(11):619–26. doi:10.1038/ncprheum0624
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Abdulahad WH, Stegeman CA, van der Geld YM, Doornbos-van der Meer B, Limburg PC, Kallenberg CG. Functional defect of circulating regulatory CD4+ T cells in patients with Wegener’s granulomatosis in remission. Arthritis Rheum (2007) 56(6):2080–91. doi:10.1002/art.22692
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Klapa S, Mueller A, Csernok E, Fagin U, Klenerman P, Holl-Ulrich K, et al. Lower numbers of FoxP3 and CCR4 co-expressing cells in an elevated subpopulation of CD4+CD25high regulatory T cells from Wegener’s granulomatosis. Clin Exp Rheumatol (2010) 28(1 Suppl 57):72–80.
80. Morgan MD, Day CJ, Piper KP, Khan N, Harper L, Moss PA, et al. Patients with Wegener’s granulomatosis demonstrate a relative deficiency and functional impairment of T-regulatory cells. Immunology (2010) 130(1):64–73. doi:10.1111/j.1365-2567.2009.03213.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Chavele KM, Shukla D, Keteepe-Arachi T, Seidel JA, Fuchs D, Pusey CD, et al. Regulation of myeloperoxidase-specific T cell responses during disease remission in antineutrophil cytoplasmic antibody-associated vasculitis: the role of Treg cells and tryptophan degradation. Arthritis Rheum (2010) 62(5):1539–48. doi:10.1002/art.27403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Saito H, Tsurikisawa N, Tsuburai T, Oshikata C, Akiyama K. The proportion of regulatory T cells in the peripheral blood reflects the relapse or remission status of patients with Churg-Strauss syndrome. Int Arch Allergy Immunol (2011) 155(Suppl 1):46–52. doi:10.1159/000327265
83. Abdulahad WH, Boots AM, Kallenberg CG. FoxP3+ CD4+ T cells in systemic autoimmune diseases: the delicate balance between true regulatory T cells and effector Th-17 cells. Rheumatology (Oxford) (2011) 50(4):646–56. doi:10.1093/rheumatology/keq328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature (2008) 453(7192):236–40. doi:10.1038/nature06878
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441(7090):235–8. doi:10.1038/nature04753
86. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441(7090):231–4. doi:10.1038/nature04754
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Gao Z, Gao Y, Li Z, Chen Z, Lu D, Tsun A, et al. Synergy between IL-6 and TGF-beta signaling promotes FOXP3 degradation. Int J Clin Exp Pathol (2012) 5(7):626–33.
88. van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YY, Beekman JM, van Beekum O, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood (2010) 115(5):965–74. doi:10.1182/blood-2009-02-207118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood (2008) 112(6):2340–52. doi:10.1182/blood-2008-01-133967
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Free ME, Bunch DO, McGregor JA, Jones BE, Berg EA, Hogan SL, et al. Patients with antineutrophil cytoplasmic antibody-associated vasculitis have defective treg cell function exacerbated by the presence of a suppression-resistant effector cell population. Arthritis Rheum (2013) 65(7):1922–33. doi:10.1002/art.37959
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. de Boer OJ, van der Meer JJ, Teeling P, van der Loos CM, van der Wal AC. Low numbers of FOXP3 positive regulatory T cells are present in all developmental stages of human atherosclerotic lesions. PLoS One (2007) 2(8):e779. doi:10.1371/journal.pone.0000779
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Mallat Z, Besnard S, Duriez M, Deleuze V, Emmanuel F, Bureau MF, et al. Protective role of interleukin-10 in atherosclerosis. Circ Res (1999) 85(8):e17–24. doi:10.1161/01.RES.85.8.e17
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Mallat Z, Gojova A, Marchiol-Fournigault C, Esposito B, Kamate C, Merval R, et al. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ Res (2001) 89(10):930–4. doi:10.1161/hh2201.099415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Gotsman I, Grabie N, Gupta R, Dacosta R, MacConmara M, Lederer J, et al. Impaired regulatory T-cell response and enhanced atherosclerosis in the absence of inducible costimulatory molecule. Circulation (2006) 114(19):2047–55. doi:10.1161/CIRCULATIONAHA.106.633263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med (2006) 12(2):178–80. doi:10.1038/nm1343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Moosig F, Csernok E, Wang G, Gross WL. Costimulatory molecules in Wegener’s granulomatosis (WG): lack of expression of CD28 and preferential up-regulation of its ligands B7-1 (CD80) and B7-2 (CD86) on T cells. Clin Exp Immunol (1998) 114(1):113–8. doi:10.1046/j.1365-2249.1998.00695.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Abdulahad WH, van der Geld YM, Stegeman CA, Kallenberg CG. Persistent expansion of CD4+ effector memory T cells in Wegener’s granulomatosis. Kidney Int (2006) 70(5):938–47. doi:10.1038/sj.ki.5001670
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Abdulahad WH, Kallenberg CG, Limburg PC, Stegeman CA. Urinary CD4+ effector memory T cells reflect renal disease activity in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum (2009) 60(9):2830–8. doi:10.1002/art.24747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Wilde B, Dolff S, Cai X, Specker C, Becker J, Totsch M, et al. CD4+CD25+ T-cell populations expressing CD134 and GITR are associated with disease activity in patients with Wegener’s granulomatosis. Nephrol Dial Transplant (2009) 24(1):161–71. doi:10.1093/ndt/gfn461
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Mestas J, Crampton SP, Hori T, Hughes CC. Endothelial cell co-stimulation through OX40 augments and prolongs T cell cytokine synthesis by stabilization of cytokine mRNA. Int Immunol (2005) 17(6):737–47. doi:10.1093/intimm/dxh255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Imura A, Hori T, Imada K, Ishikawa T, Tanaka Y, Maeda M, et al. The human OX40/gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. J Exp Med (1996) 183(5):2185–95. doi:10.1084/jem.183.5.2185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Ammirati E, Cianflone D, Vecchio V, Banfi M, Vermi AC, De Metrio M, et al. Effector memory T cells are associated with atherosclerosis in humans and animal models. J Am Heart Assoc (2012) 1(1):27–41. doi:10.1161/JAHA.111.000125
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Appay V. The physiological role of cytotoxic CD4(+) T-cells: the holy grail? Clin Exp Immunol (2004) 138(1):10–3. doi:10.1111/j.1365-2249.2004.02605.x
104. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285(5428):727–9. doi:10.1126/science.285.5428.727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci U S A (2003) 100(16):9452–7. doi:10.1073/pnas.1632807100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Allez M, Tieng V, Nakazawa A, Treton X, Pacault V, Dulphy N, et al. CD4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology (2007) 132(7):2346–58. doi:10.1053/j.gastro.2007.03.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Capraru D, Muller A, Csernok E, Gross WL, Holl-Ulrich K, Northfield J, et al. Expansion of circulating NKG2D+ effector memory T-cells and expression of NKG2D-ligand MIC in granulomaous lesions in Wegener’s granulomatosis. Clin Immunol (2008) 127(2):144–50. doi:10.1016/j.clim.2007.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. de Menthon M, Lambert M, Guiard E, Tognarelli S, Bienvenu B, Karras A, et al. Excessive interleukin-15 transpresentation endows NKG2D+CD4+ T cells with innate-like capacity to lyse vascular endothelium in granulomatosis with polyangiitis (Wegener’s). Arthritis Rheum (2011) 63(7):2116–26. doi:10.1002/art.30355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Xia M, Guerra N, Sukhova GK, Yang K, Miller CK, Shi GP, et al. Immune activation resulting from NKG2D/ligand interaction promotes atherosclerosis. Circulation (2011) 124(25):2933–43. doi:10.1161/CIRCULATIONAHA.111.034850
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med (2011) 365(22):2067–77. doi:10.1056/NEJMoa1105143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Samson M, Audia S, Janikashvili N, Ciudad M, Trad M, Fraszczak J, et al. Brief report: inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum (2012) 64(8):2499–503. doi:10.1002/art.34477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Vincenti F. Costimulation blockade in autoimmunity and transplantation. J Allergy Clin Immunol (2008) 121(2):299–306. doi:10.1016/j.jaci.2008.01.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Langford CA, Monach PA, Specks U, Seo P, Cuthbertson D, McAlear CA, et al. Vasculitis clinical research consortium. An open-label trial of abatacept (CTLA4-IG) in non-severe relapsing granulomatosis with polyangiitis (Wegener’s). Ann Rheum Dis (2014) 73(7):1376–9. doi:10.1136/annrheumdis-2013-204164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Gao Q, Jiang Y, Ma T, Zhu F, Gao F, Zhang P, et al. A critical function of Th17 proinflammatory cells in the development of atherosclerotic plaque in mice. J Immunol (2010) 185(10):5820–7. doi:10.4049/jimmunol.1000116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Tse MT. IL-17 antibodies gain momentum. Nat Rev Drug Discov (2013) 12(11):815–6. doi:10.1038/nrd4152
116. Kellner H. Targeting interleukin-17 in patients with active rheumatoid arthritis: rationale and clinical potential. Ther Adv Musculoskelet Dis (2013) 5(3):141–52. doi:10.1177/1759720X13485328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Brown G, Malakouti M, Wang E, Koo JY, Levin E. Anti-IL-17 phase II data for psoriasis: a review. J Dermatolog Treat (2014). doi:10.3109/09546634.2013.878448
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, et al. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity (2004) 20(6):757–67. doi:10.1016/j.immuni.2004.05.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Kjellev S, Haase C, Lundsgaard D, Urso B, Tornehave D, Markholst H. Inhibition of NKG2D receptor function by antibody therapy attenuates transfer-induced colitis in SCID mice. Eur J Immunol (2007) 37(5):1397–406. doi:10.1002/eji.200636473
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Cahalan MD, Chandy KG. The functional network of ion channels in T lymphocytes. Immunol Rev (2009) 231(1):59–87. doi:10.1111/j.1600-065X.2009.00816.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci (2004) 25(5):280–9. doi:10.1016/j.tips.2004.03.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Wulff H, Calabresi PA, Allie R, Yun S, Pennington M, Beeton C, et al. The voltage-gated Kv1.3 K(+) channel in effector memory T cells as new target for MS. J Clin Invest (2003) 111(11):1703–13. doi:10.1172/JCI16921
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Hu L, Pennington M, Jiang Q, Whartenby KA, Calabresi PA. Characterization of the functional properties of the voltage-gated potassium channel Kv1.3 in human CD4+ T lymphocytes. J Immunol (2007) 179(7):4563–70. doi:10.4049/jimmunol.179.7.4563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington MW, et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci U S A (2006) 103(46):17414–9. doi:10.1073/pnas.0605136103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Matheu MP, Beeton C, Garcia A, Chi V, Rangaraju S, Safrina O, et al. Imaging of effector memory T cells during a delayed-type hypersensitivity reaction and suppression by Kv1.3 channel block. Immunity (2008) 29(4):602–14. doi:10.1016/j.immuni.2008.07.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Beeton C, Wulff H, Barbaria J, Clot-Faybesse O, Pennington M, Bernard D, et al. Selective blockade of T lymphocyte K(+) channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proc Natl Acad Sci U S A (2001) 98(24):13942–7. doi:10.1073/pnas.241497298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Beeton C, Pennington MW, Wulff H, Singh S, Nugent D, Crossley G, et al. Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases. Mol Pharmacol (2005) 67(4):1369–81. doi:10.1124/mol.104.008193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Gocke AR, Lebson LA, Grishkan IV, Hu L, Nguyen HM, Whartenby KA, et al. Kv1.3 deletion biases T cells toward an immunoregulatory phenotype and renders mice resistant to autoimmune encephalomyelitis. J Immunol (2012) 188(12):5877–86. doi:10.4049/jimmunol.1103095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Koch Hansen L, Sevelsted-Moller L, Rabjerg M, Larsen D, Hansen TP, Klinge L, et al. Expression of T-cell K1.3 potassium channel correlates with pro-inflammatory cytokines and disease activity in ulcerative colitis. J Crohns Colitis (2014). doi:10.1016/j.crohns.2014.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: vascular inflammation, ANCA-associated vasculitis, atherosclerosis, T lymphocytes, effector memory T cells, Kv1.3 channels
Citation: Lintermans LL, Stegeman CA, Heeringa P and Abdulahad WH (2014) T cells in vascular inflammatory diseases. Front. Immunol. 5:504. doi: 10.3389/fimmu.2014.00504
Received: 18 July 2014; Accepted: 28 September 2014;
Published online: 14 October 2014.
Edited by:
Giuseppe Alvise Ramirez, Università Vita-Salute San Raffaele, ItalyReviewed by:
Gaurav K. Gupta, Harvard Medical School, USAFrédéric Velard, Université de Reims Champagne-Ardenne, France
Copyright: © 2014 Lintermans, Stegeman, Heeringa and Abdulahad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucas L. Lintermans, Department of Rheumatology and Clinical Immunology, University of Groningen, University Medical Center Groningen, IPC AA21, Hanzeplein 1, Groningen 9713 GZ, Netherlands e-mail:bC5sLmxpbnRlcm1hbnNAdW1jZy5ubA==