 Giovanni Piotti
Giovanni Piotti Alessandra Palmisano
Alessandra Palmisano Umberto Maggiore
Umberto Maggiore Carlo Buzio
Carlo Buzio
- Kidney and Pancreas Transplantation Unit, Department of Clinical Medicine, Nephrology and Health Sciences, University Hospital of Parma, Parma, Italy
This review of clinical and experimental studies aims at analyzing the interplay between graft endothelium and host immune system in renal transplantation, and how it affects the survival of the graft. Graft endothelium is indeed the first barrier between self and non-self that is encountered by host lymphocytes upon reperfusion of vascularized solid transplants. Endothelial cells (EC) express all the major sets of antigens (Ag) that elicit host immune response, and therefore represent a preferential target in organ rejection. Some of the Ag expressed by EC are target of the antibody-mediated response, such as the AB0 blood group system, the human leukocyte antigens (HLA), and MHC class I related chain A antigens (MICA) systems, and the endothelial cell-restricted Ag; for each of these systems, the mechanisms of interaction and damage of both preformed and de novo donor-specific antibodies are reviewed along with their impact on renal graft survival. Moreover, the rejection process can force injured EC to expose cryptic self-Ag, toward which an autoimmune response mounts, overlapping to the allo-immune response in the damaging of the graft. Not only are EC a passive target of the host immune response but also an active player in lymphocyte activation; therefore, their interaction with allogenic T-cells is analyzed on the basis of experimental in vitro and in vivo studies, according to the patterns of expression of the HLA class I and II and the co-stimulatory molecules specific for cytotoxic and helper T-cells. Finally, as the response that follows transplantation has proven to be not necessarily destructive, the factors that foster graft endothelium functioning in spite of rejection, and how they could be therapeutically harnessed to promote long-term graft acceptance, are described: accommodation that is resistance of EC to donor-specific antibodies, and endothelial cell ability to induce Foxp3+ regulatory T-cells, that are crucial mediators of tolerance.
Introduction
Over the last few decades, the practice of kidney transplantation has improved in many areas up to becoming the optimal treatment for end-stage renal disease (1). However, despite brilliantly achieving a 95% 1-year survival, long-term outcomes have not benefited from such improvements and remain unsatisfying (2). One of the major causes of late graft loss is occurrence of antibody-mediated rejection (ABMR), which current immunosuppressive regimens have mostly proven to be unable to cure (3). Another issue is the development of accelerated cardiovascular disease, which is due to the side effects inherent in the immunosuppressive drugs and, along with opportunistic infections and malignancies, represents the first cause of recipient death (4).
Allograft endothelium is the first barrier between self and non-self in vascularized solid-organ transplantation, and preservation of its integrity and functions is mandatory to ensure a prolonged survival of the graft (5). As endothelial cells (EC) express a number of antigens (Ag) that are visible by the immune system of a genetically disparate individual, donor endothelium is invariably recognized by the host immune system, and therefore, it is the first and preferential target of the allo-immune response that follows organ transplantation without an adequate immunosuppression (6).
Both naturally occurring and induced allo-antibodies directed to the Ag expressed on the membrane of EC are commonly found in renal recipients, and such antibodies, being capable of fixing the complement and damaging the tissues, are detrimental for the correct functioning of the endothelium (7). Moreover EC, besides being target of antibody-mediated response, can directly interact with allogenic T-cells by displaying not only the major histocompatibility complex (MHC) antigens but also adequate co-stimulatory molecules and adhesion proteins on their surface (8).
Nevertheless, the host immune response that follows recognition of EC allo-Ag is not necessarily destructive, in spite of graft rejection, accommodation, where not active tolerance, may operationally establish, thus fostering the endothelium to fulfill its functions (5). Endothelial regulation of blood flow and vessel permeability is paramount not only for the survival of any vascularized allograft but also for the specific depurative activities of the kidneys.
This review aims at analyzing the interplay between allograft endothelium and host immune system, and how differential unfolding of this interplay may ultimately affect the fate of the graft. A particular attention will be paid to the factors that, at the endothelial level, contribute to tipping the balance in favor of graft acceptance rather than rejection.
Discussion
Antibody-Mediated Immune Response toward Allograft Endothelial Cells
The importance of donor-specific antibodies in causing allograft rejection has progressively been uncovered, to the extent that a humoral theory of transplantation has been formulated in juxtaposition to the cellular one (9). Here, all the sets of Ag expressed on human EC that are relevant for kidney transplantation will be discussed (Table 1), along with the mechanisms of damage and accommodation.
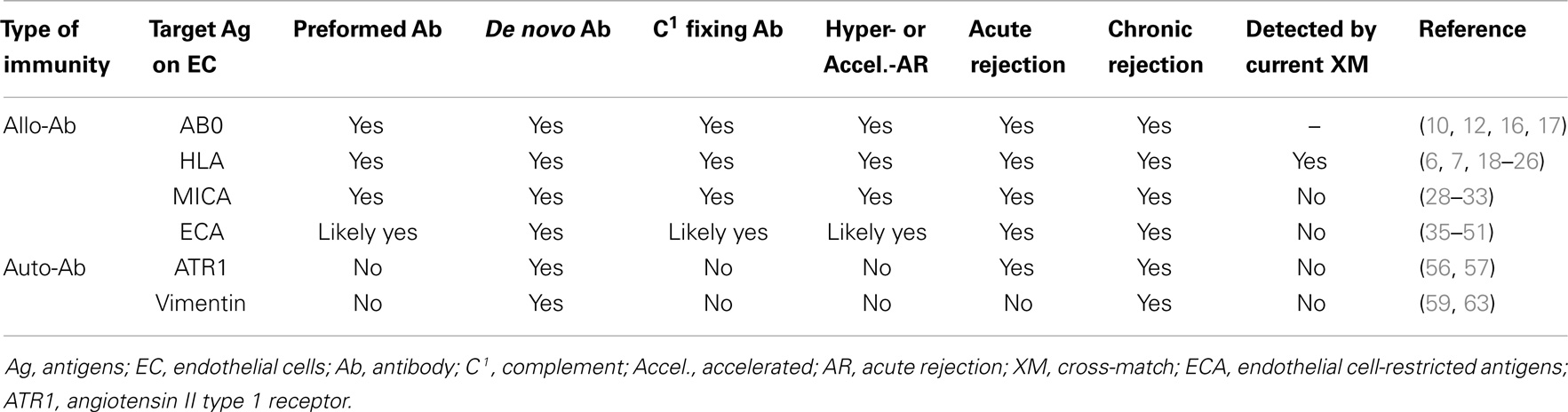
Table 1. Antibody-mediated immune response toward allograft endothelial cells.
Endothelial Cell Antigens Target of Allo-Antibodies
AB0 blood group antigens
Endothelial cells highly express the AB0 blood group antigens on their surface (10). Such Ag are carbohydrates linked to glycoproteins and glycolipids and are targets of specific Ab (isoagglutinins), which occur naturally in people lacking the A and/or B antigens (11). Isoagglutinins, upon binding to A/B incompatible EC, cause hyperacute or accelerated acute graft rejection (12). AB0 compatibility has, therefore, been required for successful cadaveric transplantation; however, being the allelic variability of this system little, it does not represent a barrier for allocation of deceased donor organs. On the other hand, due to the allelic frequencies, any two individuals have roughly a 35% probability of being AB0 incompatible (AB0i), and this is an actual limitation to living renal donations (11). A first breach to the absolute requirement for AB0 compatibility emerged soon after the recognition of AB0 Ag as a barrier for solid-organ transplantation; the analysis of the outcomes showed that acceptable results were only obtained when renal grafts from A2 donors had been transplanted to non-A, i.e., 0 or B, recipients (12, 13). This donor-recipient combination proved somehow permissive because of the scarce expression of A2 Ag on EC, and the consequent low titers of anti-A2 Ab in non-A2 recipients (13). A second breach was the good results reached with AB0i heart transplants in children, who are known to express lower amount of A/B Ag and produce less Ab compared to adults (14). Finally, in 1981, the report of a successful rescue treatment for a mistakenly performed AB0i renal transplantation was published (15); the concept of removal of the isoagglutinins with plasmapheresis in order to avoid hyperacute rejection laid the basis to current practice that aims at reducing anti-A/B Ab titers before and soon after transplantation as a strategy to overcome AB0 barrier (16). Since then, kidney transplantation from AB0i living donor has become a routine practice in many transplantation centers (17).
Human leukocyte antigens
Major histocompatibility complex antigens, also known as human leukocyte antigens (HLA) in human beings, are highly polymorphic surface proteins whose principal function is to display Ag to T-cells for recognition and activation. Two different classes of HLA molecules exist: class I molecules are constitutively expressed by all cell types and present Ag to CD8+ cytotoxic T-cells; class II molecules, that present Ag to CD4+ helper T-cells, are commonly restricted to professional antigen-presenting cells (APC) such as dendritic cells, but, upon stimulation, can be induced onto other cell types. Human EC highly express class I and, albeit at lesser extent, also class II HLA molecules, which can be further enhanced by appropriate stimuli of inflammatory and immunologic origin (6, 7).
Apart from presenting Ag to lymphocytes, HLA molecules can themselves be recognized by an allogenic immune system, and anti-HLA Ab are produced following immunizing events like pregnancies, blood transfusions, and organ transplantation (18). Preformed anti-HLA donor-specific Ab (DSA), i.e., DSA present prior to transplantation, have long been known to be responsible for hyperacute or accelerated acute graft rejection, which is determined by mechanisms similar to those for AB0i transplantation performed without an adequate desensitization (19). Nevertheless, due to the extreme polymorphism of HLA genes, donor-recipient matching is an exceptional occurrence; therefore, unlike bone marrow transplantation, most of the kidney transplants are performed across the HLA barrier, for which a profound immunosuppression is lifelong required. In order to eliminate the risk of hyperacute rejection, laboratory cross-matching techniques have been developed, and routinely applied, to identify preformed anti-HLA Ab in the serum of recipients before transplantation; a positive donor-recipient HLA cross-match would currently halt the organ allocation in the absence of adequate desensitization (20). Moreover, anti-HLA DSA can occur de novo after transplantation mainly as a consequence of suboptimal immunosuppression or scarce adherence to the therapy (21). De novo DSA, particularly if complement-fixing Ab, is pathogenic for both acute and chronic rejection of the allograft (22); and the presence of such Ab in recipient’s serum has prospectively been linked to graft failure in several studies (23). The EC of graft peritubular capillaries (PTC) are the preferred targets of DSA so much so that microvascular inflammation is a required criterion for histopathologic diagnosis of ABMR (24), which is further underpinned by the presence of deposits of the complement fragment C4d on PTC endothelium (25, 26), and of circulating DSA (24).
MHC class I related chain A antigens
The description of sporadic cases of hyperacute or accelerated acute rejection of non-AB0i kidneys in recipients lacking anti-HLA DSA have urged researchers to investigate further sets of allo-antigens that might be relevant for transplantation (27). MHC class I related chain A antigens (MICA) are surface glycoproteins encoded by highly polymorphic genes located on chromosome 6 within the region of MHC genes (28). MICA, whose function is related to immune surveillance, are expressed by different types of cells including EC, but importantly neither T nor B lymphocytes; thus, current standard cross-match procedures are unable to detect anti-MICA antibodies (28). MICA have proven to be the target of complement-fixing allo-Ab that can cause hyperacute, acute, and chronic ABMR (28–32); the presence of anti-MICA DSA negatively impact short-term and long-term graft survival, albeit at lesser extent compared to the effect of anti-HLA DSA (30). Endothelium damage, microvascular inflammation, and C4d deposition on PTC endothelium are the hallmark also of ABMR mediated through anti-MICA DSA (33).
Non-HLA nor-MICA endothelium-restricted antigens
Along with MICA, other non-HLA systems are thought to add to the gamut of the traditional transplantation Ag (34). Indeed, ABMR may exceptionally occur following renal transplantation between non-AB0i HLA-identical siblings, who usually also share MICA, being them in linkage disequilibrium with the HLA genes (28).
Between 1997 and 2005, endothelium-restricted antigens (EA), expressed neither by lymphocytes nor by monocytes, were proposed as possible targets of pathogenic Ab in renal recipients who had experienced acute ABMR without any obvious DSA (35–40). Following these results, the suggestion to adopt newer cross-matching techniques that would investigate the presence of these anti-endothelial cell antibodies (AECA) in the recipient’s serum before transplantation has become stronger (41–46). Meanwhile, the first studies have come out and shown an association between circulating AECA and acute rejection, chronic rejection, poor renal graft survival, and transplant glomerulopathy (47–51).
As for the identity of EA, it is still ill defined, despite the application of proteomic, protein microarrays, and transcriptome measures (52). The most relevant information we have is that EA are expressed only by activated or damaged EC (53). This observation has lead some authors to hypothesize that the EA, or at least some of them, might actually be self-molecules rather than allo-antigens, and AECA would be auto-Ab that arise following the exposure of these cryptic self-Ag on EC primarily hit by host immune response, and they would cooperate to graft destruction with allo-immunity (54, 55). Two examples of self-Ag displayed on EC and targeted by host immune response following transplantation are angiotensin II type 1 receptor and vimentin.
Angiotensin II type 1 receptor. In 2005, Dragun et al. linked the presence of anti-angiotensin II type 1 receptor (AT1R) Ab to acute rejection of non-AB0i kidneys in 16 recipients without anti-HLA or anti-MICA DSA (56). Anti-AT1R Ab were studied because malignant hypertension was part of the clinical picture in all the rejecting patients, thus somehow resembling preeclampsia, a condition the researchers had already linked to the presence of such Abs (57). AT1R is present on EC and vascular smooth muscle cells and, upon ligation of angiotensin II, elicits transduction of secondary signals that contribute to the regulation of body liquids and blood pressure. Anti-AT1R Ab are agonistic non-complement-fixing immunoglobulins that promote malignant hypertension by over-activating AT1R (47); moreover, anti-AT1R Ab can induce microvascular inflammation and coagulation by stimulating NF-kB pathway and tissue factor expression by EC (58). In accordance with these data, graft biopsies from the renal recipients in Dragun’s study lacked C4d deposition, but revealed the presence of endoarteritic lesions, fibrinoid necrosis, and thrombi. The authors finally provided evidence of the pathogenic role of anti-AT1R Ab as their removal with plasmapheresis and selective blockade of AT1R with losartan significantly improved graft survival (56). The origin of such Abs is not clear yet, but, as no polymorphism of the AT1R gene has been identified, they could be auto-Ab occurring due to molecular mimicry or to anomalous presentation of over-expressed AT1R on damaged EC (58).
Vimentin. Vimentin is a cytoskeleton intermediate filament protein present within the cytosol of cells of mesenchymal origin, such as EC, fibroblasts, and leukocytes. Anti-vimentin auto-Abs (AVA) are described in a number of autoimmune diseases (59). In organ transplantation, exposition of vimentin isoforms on apoptotic EC, irreparably damaged by the allo-immune response, results in break of self-tolerance, emergence of active vimentin-specific CD8+ T-cells, and production of AVA (59, 60). AVA have been found in heart as well as kidney transplantation (61); albeit capable of fixing the complement and activating platelets (62), AVA alone are not pathogenic, they instead contribute with the allo-immune response to cause the vascular lesions typical of chronic rejection, and to accelerate the progression of atherosclerotic lesions (63).
Focus on the Mechanisms of Rejection toward Graft Endothelial Cells
Nature and abundance of the allo-Ag expressed on EC, and type and titers of the DSA are the determinants of the intensity of host antibody-mediated immune response toward the allograft (Figure 1). This evidence has both theoretical and practice consequences. First, AB0 Ag, carbohydrates in nature, are less immunogenic than HLA molecules that are proteins, and indeed desensitization procedures for AB0i recipients are less intense and obtain better outcomes than those for anti-HLA immunized patients (16, 20). Second, A1 Ag elicit a more powerful response in kidney recipients of non-A blood group compared to A2 Ag due to minor expression of the latter on EC (13). Third, preformed DSA cause a more acute and severe rejection than de novo DSA, because they are more rapidly and abundantly produced by memory B-cells without any need of helper T-cells, and are more harmful for the graft (64). Fourth, complement-fixing DSA are associated with a poorer prognosis compared to non-fixing Abs (65). Finally, DSA concentration is critical for survival of both AB0i and HLAi renal transplants, with higher titers linked to more rejection episodes and shorter graft survival (20, 66).
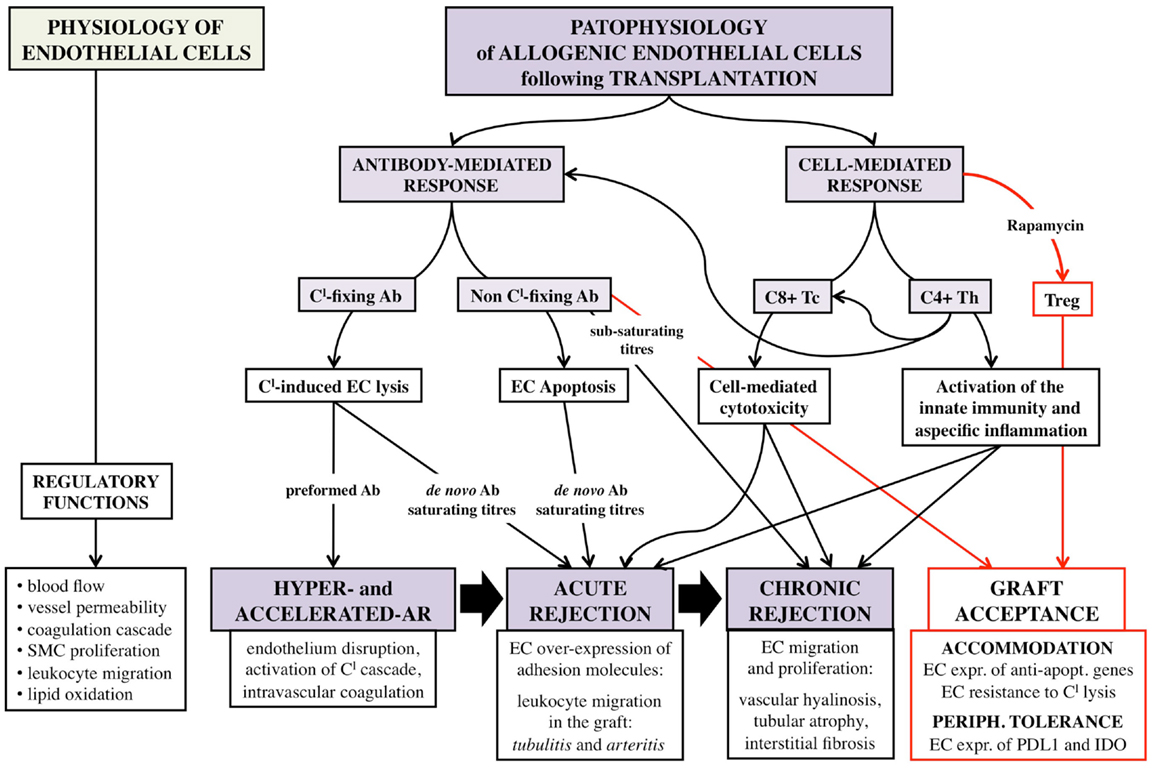
Figure 1. Schematic representation of the complex interaction between allogenic endothelial cells and host immune system following vascularized solid-organ transplantation. EC, endothelial cells; Tc, CD8+ cytotoxic T-cells; Th, CD4+ helper T-cells; Treg, CD4+ CD25+ Foxp3+ regulatory T-cells; SMC, smooth muscle cells; AR, acute rejection; Ab, antibody; Cl, complement; expr., expression; apopt., apoptotic.
Preformed DSA can cause hyperacute or accelerated acute rejection within minutes from the revascularization by binding to EC and fixing the complement; antibody-mediated complement activation extensively damages the endothelium integrity and initiates intravascular coagulation cascade that results in vessel thrombosis and tissue infarction (67).
Acute ABMR is a severe, albeit less catastrophic, event characterized by deposition of complement-fixing Abs on graft endothelium, mainly on the PTC endothelium, without initial activation of the coagulation (3). Upon DSA binding, activated EC increase the display on their surface membrane of MHC molecules, which further amplify the allo-immune specific response (65). Moreover, anti-HLA Ab ligation forces targeted EC to release prothrombotic mediators, like von Willebrand Factor (VWF), and to express more adhesion molecules, which foster platelets aggregation and leukocytes invasion of the graft (68). Graft invasion by T-cells is sustained by the enhanced expression on EC of vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and endothelium leukocyte adhesion molecule-1 (ELAM-1) (69), adhesion molecules, which can be induced by the inflammatory mediators interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), and interferon-γ (INF-γ), produced by the lured leukocytes (70–72). The invasion by host lymphocytes and inflammatory cells of functional structures of the graft, such as the tubuli and the PTC, and the development of small vessel occlusion due to thrombi formation and cell accumulation determine acute deterioration of graft function (3).
Alternatively, acute episodes self-limit and relapse thereafter repeatedly, thus resulting in chronic injuries that lead to transplant atherosclerosis, tubular atrophy, and interstitial fibrosis, the hallmark of chronic rejection (24). Transplant atherosclerosis in particular is sustained by the acquisition by EC of proliferative capabilities. Upon DSA binding to HLA molecule, EC are driven to express growth factor receptors, such as the fibroblast growth factor receptor (FGF-R) (73), and to re-arrange filaments of the cytoskeleton, such as forming stress fibers (74) and recruiting integrin-ß4 (75). Cytoskeleton rearrangements confer EC the capability of reacting to appropriate stimuli by proliferating. The transduction of integrin-ß4-dependent signals activates several cytoplasmatic kinases that ultimately result in the stimulation of the mechanistic target of rapamycin (mTOR) pathway (76); mTOR activation promotes progression of cell cycle from G1 phase to S phase and ultimately induces EC division, such a proliferation is reinforced by the susceptibility to FGF of EC expressing FGF-R (73).
Accommodation
Accommodation was originally the term used to describe the acquisition by EC of resistance to ABMR of AB0i renal grafts following reappearance of anti-A/B donor Ab (77). Nowadays, much interest is dedicated at understanding the mechanisms of accommodation and whether they could also be therapeutically harnessed to prevent transplant damage from anti-HLA DSA, that unlike anti-A/B Ab, are currently thought to invariably lead to allograft ABMR (78).
A small but interesting study investigated the behavior of EC in hyper-immune desensitized recipients who experienced reappearance of anti-HLA Abs following renal transplantation (79). None of the seven enrolled patients had hyperacute ABMR but three lost the graft due to various immunological damages, and three of the four remaining patients suffered curable acute rejection or transplant glomerulopathy. Analysis of the graft biopsies revealed increased expression of anti-apoptotic Bcl-xl gene in glomerular and peritubular capillary EC. Furthermore, in vitro incubation of human EC with sub-saturating concentrations of anti-HLA Ab eluted from the patients decreased ICAM-1 expression and provided resistance to complement-mediated cell lysis (79).
These preliminary results have been corroborated by further studies according to which sub-saturating anti-HLA Ab can induce EC expression of the anti-apoptotic genes Bcl-2 and Bcl-xL, whereas saturating titers induce EC apoptosis (Figure 1) (80–82).
Despite these encouraging results, a full knowledge of the accommodation process is far from been achieved and many more studies are needed in order to establish adequate protocol to desensitize hyper-immune recipients and safely perform transplantation in such population.
Cell-Mediated Immune Response toward Allograft Endothelial Cells
As said, EC have all the properties required to drive direct activation of allogenic T-cells that is the pivotal step in all the forms of rejection non-mediated by preformed DSA (Figure 1) (8).
The rejection process has long been thought to be induced, at least initially, by donor professional APC that, upon migration into host secondary lymphoid organs, would present allo-Ag to T-cells. Nevertheless, human EC, which are not professional APC, have proven to be able to directly activate T-cells (Table 2); human EC provide “signal 1” as, unlike porcine and rodent EC, they robustly express HLA molecules, and in particular small vessel and capillary human EC constitutively express both class I and II HLA molecules (8). They also provide “signal 2” by expressing the co-stimulatory molecules required for an effective Ag presentation (8). Direct activation of T-cells by EC is of particular importance because while donor professional APC are destined to fade over time, EC, whose survival is linked to that of the allograft, can potentially ignite acute rejection at any time following transplantation (83).

Table 2. Cell-mediated immune response toward allograft endothelial cells.
Endothelial Cells and CD8+ Cytotoxic T-Cells
In vitro mixed lymphocyte reaction (MLR) experiments using EC as stimulators of allogenic CD8+ sorted naive T-cells have confirmed that EC are able to behave as professional APC (84).
CD8+ cytotoxic T-cells (Tc), so co-cultured with EC, respond proliferating and acquiring an effector phenotype defined by higher expression of perforin and production of IL-2 and INF-γ, which in turn enhance EC expression of HLA class I and II molecules (84).
With the use of blocking monoclonal Ab (mAb), the essential signals for Tc activation have been identified in the HLA-A and B class I molecules on EC that are target of the T-cell receptor (TCR) and CD8 co-receptor, as well as the co-stimulatory molecule CD80 (B7-I) on EC and its ligand CD28 on T-cells (85). In vivo experiments have confirmed that CD8+ T-cell direct activation by non-hematopoietic cells, such as EC, leads to graft rejection in a murine model of class I restricted heart allo-grafts transplanted into CD4-depleted recipients devoided of secondary lymphoid organs (86). Finally, the finding of vimentin-specific autoreactive CD8+ T-cells in heart recipients have shown that transplantation cellular response, as for the humoral response, may spread from being directed to allo-antigens to autoimmunity (60).
Endothelial Cells and CD4+ Helper T-Cells
Accumulating evidence has convincingly clarified that human EC of microvascular origin can directly activate CD4+ helper T-cells (Th) (8, 87–89). Th are central mediators of allo-immunity as they provide help for allo-Ab production, they arm cytotoxic T-cells, and drive innate unspecific inflammatory response (8). Human EC constitutively express MHC class II molecules, HLA-DR, DP, and DQ, albeit at lesser extent compared to class I molecules. These class II molecules, which are recognized by TCR and bound with the help of CD4 co-receptor, are “signal 1” for Th direct activation. EC can also provide “signal 2” specific for Th; they indeed display ICAM-1 (CD54) and lymphocyte function-associated antigen-3 (LFA3) (CD58), which are bound by LFA1 (CD11a/18) and LFA2 (CD2) on T-cells. As shown in vitro, following EC provision of signals 1 and 2, Th start to proliferate and acquire an effector phenotype characterized by the induction of the co-stimulatory molecule CD40L (90) and of the adhesion molecules that favor trans-endothelium migration (91). Depending on local cytokine microenvironment, resting CD4+ T-cells differentiate into different Th subsets. Along with Th1 and Th2 subsets, whose ability to mediate rejection is well known (92), also Th17, which are implicated in a number of autoimmune diseases, can emerge guided by activated EC that provide the critical cytokine IL-6 in the presence, under inflammatory conditions, of transforming growth factor-ß (TGF-ß) (93). This is of particular interest as not only Th17 have recently proven capable of causing allograft rejection (94) but also they and CD4+ CD25+ Foxp3+ regulatory T-cells (Treg) seem to keep reciprocally at bay. Since Treg, that are crucial cells for transplantation tolerance, require TGF-ß but not IL-6 for their induction, it has been hypothesized that EC could also mediate the induction of peripheral Treg.
Endothelial Cells and Regulatory T-Cells
CD4+ CD25+ Treg are a well-defined subset of CD4+ T-cells identified by the expression of the master transcription factor Foxp3. They are crucial regulators of the immune response; natural Treg of thymic origin prevent autoimmune diseases, while peripherally induced Treg actively regulate transplantation tolerance (95). Treg act at a tissue level where they influence APC ability of presenting Ag to conventional T-cells (Tconv), which in turn become either anergic or regulatory cells themselves. Therefore, empowering Treg at the expense of Tconv can induce a state of local immune privilege that promotes long-term graft survival (95).
A few papers have investigated the ability of EC to interact with Treg. INF-γ-stimulated EC have proven to be capable of inducing Treg when co-cultured with allogenic CD4+ T-cells (96); but more importantly, similar results have been obtained when EC were pre-treated with the clinically available immunosuppressant rapamycin (Rapa), which exerts its functions by inhibiting the mTOR pathway (97). Expansion of Treg results from the conversion of naive CD4+ cells into Foxp3+ cells, and depends on cell–cell contact and the local microenvironment (98); not only Rapa reduces the display of VCAM-1 in EC (99) but also forces the expression of the inhibitory molecules programed death ligand-1 (PDL-1) and indoleamine 2,3-dioxygenase (IDO), which are crucial for Treg induction (96–98). Finally, EC pre-treated with Rapa produce less IL-6 (97), which is instead required for the expansion of Th17 but not that of Treg (93). EC-induced Treg are functionally active as they can effectively suppress the proliferative response of Ag-stimulated CD8+ T-cells. On the other side, Treg can influence EC behavior; Treg release TGF-ß that downregulate the expression on EC of the adhesion molecules CD62E and CD62P (respectively, E- and P-selectin), thus limiting transmigration of Tc and reducing local inflammation (100).
Conclusion
Graft endothelium is the first barrier between self and non-self in transplantation of vascularized solid organs, such as kidney transplants. Indeed, upon organ reperfusion, host lymphocytes initially encounter graft EC, which express all the most relevant antigens in transplantation immunobiology. Such antigens are invariably recognized by the host immune system, and toward them antibody-mediated and cell-mediated immune responses mount. Moreover, not only are EC a passive target of the host immune system but they also are an active player in the recruitment and activation of allogenic lymphocytes, and in the invasion of graft tissues by them. The ability of EC to directly activate allogenic naive T-cells deserves to be highlighted as EC represent a long-term source of allo-antigens and can potentially induce graft rejection at any time post-transplantation. Therefore, the fate of renal transplants largely depends on how such interplay between graft endothelium and host immune system unfolds: on one side, the activation of the immune response may lead to all the forms of graft rejection, from hyperacute to chronic, and to the deterioration of graft function primarily because of damages to the endothelium integrity. On the other side, even in the presence of circulating donor-specific antibodies directed to antigens expressed on donor EC, graft endothelium may thrive and fulfill its functions of regulating blood flow and vessel permeability, which are crucial not only for graft survival but also for the depurative activities of the kidney.
Current immunosuppressive drugs have been developed with the aim of targeting host immune response, be it depleting or blocking T-cells and B-cells or halting the complement cascade; however, given the central role of EC in modulating the allo-immune response, graft endothelium may represent a preferential target of newer immunosuppressive protocols aimed at promoting long-term graft acceptance by reducing EC immunogenicity and antigen presentation, while favoring their survival. The class of immunosuppressants mTOR inhibitors has been more and more utilized in the clinic for its pro-tolerogenic and anti-neoplastic activities; the pro-tolerogenic effects of the mTOR inhibitor rapamycin have proven to rely at least in part on its ability to condition antigen presentation by EC so that regulatory T-cells emerge at the expense of rejecting conventional T-cells. Although scientific data lack, it is conceivable that also the latest immunosuppressant introduced in the clinic, belatacept, may prevent graft rejection by limiting EC ability to activate T-cells (101); belatacept is a CTLA-4 fusion protein with the human IgG Fc, which blocks “signal 2,” by binding to the co-stimulatory molecules CD80 and CD86 that are expressed not only on the APC but also on the surface of EC.
As therapeutically blocking EC from presenting antigens prevents direct activation of T-cells and, in turn, induction of de novo donor-specific antibodies, developing such immunosuppressive strategy could become the answer to the issue of late ABMR, toward which current immunosuppressants are blunt, and could contribute to the improvement of long-term transplantation outcomes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sayegh MH, Carpenter CB. Transplantation 50 years later - progress, challenges, and promises. N Engl J Med (2004) 351:2761–6. doi:10.1056/NEJMon043418
2. U.S. Renal Data System. USRDS 2011 Annual Data Report: Atlas of End-Stage Renal Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (2011).
3. Colvin RB, Smith RN. Antibody-mediated organ-allograft rejection. Nat Rev Immunol (2005) 5(10):807–17. doi:10.1038/nri1702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Hill GS, Nochy D, Loupy A. Accelerated arteriosclerosis: a form of transplant arteriopathy. Curr Opin Organ Transplant (2010) 15(1):11–5. doi:10.1097/MOT.0b013e3283342684
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol (2007) 7(10):803–15. doi:10.1038/nri2171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Rose ML. editor. The immune response to endothelial cells. Transplant-Associated Coronary Artery Vasculopathy, Chapter 3. Austin, TX: Landes Bioscience, Georgetown Texas USA; Eurekah.com (2001). p. 71–89.
7. Glotz D, Lucchiari N, Pegaz-Fiornet B, Suberbielle-Boissel C. Endothelial cells as targets of allograft rejection. Transplantation (2006) 82(1 Suppl):S19–21. doi:10.1097/01.tp.0000231348.55262.5a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Rose ML. Endothelial cells as antigen-presenting cells: role in human transplant rejection. Cell Mol Life Sci (1998) 54:965–78. doi:10.1007/s000180050226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Terasaki PI. Humoral theory of transplantation. Am J Transplant (2003) 3(6):665–73. doi:10.1034/j.1600-6143.2003.00135.x
10. Rydberg L. AB0-incompatibility in solid organ transplantation. Transfus Med (2001) 11:325–42. doi:10.1046/j.1365-3148.2001.00313.x
11. Warner PR, Nester TA. AB0-incompatible solid-organ transplantation. Am J Clin Pathol (2006) 125(Suppl 1):S87–94. doi:10.1309/8W4X9H6F8FTLCGYX
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Starzl TE, Marchioro TL, Holmes JH, Hermann G, Brittain RS, Stonington H, et al. Renal homografts in patients with major donor-recipient blood group incompatibilities. Surgery (1964) 55:195–200.
13. Nelson PW, Landreneau MD, Luger AM, Pierce GE, Ross G, Shield CF III, et al. Ten-year experience in transplantation of A2 kidneys into B and 0 recipients. Transplantation (1998) 65:256–60. doi:10.1097/00007890-199801270-00020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Dipchand AI, Pollock BarZiv SM, Manlhiot C, West LJ, VanderVliet M, McCrindle BW. Equivalent outcomes for pediatric heart transplantation recipients: ABO-blood group incompatible versus ABO-compatible. Am J Transplant (2010) 10(2):389–97. doi:10.1111/j.1600-6143.2009.02934.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Slapak M, Naik RB, Lee HA. Renal transplant in a patient with major donor-recipient blood group incompatibility: reversal of acute rejection by the use of modified plasmapheresis. Transplantation (1981) 31:4–7. doi:10.1097/00007890-198101000-00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Flint SM, Walker RG, Hogan C, Haeusler MN, Robertson A, Francis DM, et al. Successful ABO-incompatible kidney transplantation with antibody removal and standard immunosuppression. Am J Transplant (2011) 11(5):1016–24. doi:10.1111/j.1600-6143.2011.03464.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Montgomery RA, Locke JE, King KE, Segev DL, Warren DS, Kraus ES, et al. ABO incompatible renal transplantation: a paradigm ready for broad implementation. Transplantation (2009) 87(8):1246–55. doi:10.1097/TP.0b013e31819f2024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Hung SY, Lin TM, Chang MY, Wang HH, Lee YC, Ho LC, et al. Risk factors of sensitization to human leukocyte antigen in end-stage renal disease patients. Hum Immunol (2014) 75(6):531–5. doi:10.1016/j.humimm.2014.02.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Morris PJ, Mickey MR, Singal DP, Terasaki PI. Serotyping for homotransplantation. XXII, Specificity of cytotoxic antibodies developing after renal transplantation. Br Med J (1969) I:758–9. doi:10.1136/bmj.1.5646.758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Montgomery RA, Lonze BE, King KE, Kraus ES, Kucirka LM, Locke JE, et al. Desensitization in HLA-incompatible kidney recipients and survival. N Engl J Med (2011) 365(4):318–26. doi:10.1056/NEJMoa1012376
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Sellarés J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis1 B, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and non-adherence. Am J Transplant (2012) 12(2):388–99. doi:10.1111/j.1600-6143.2011.03840.x
22. Lee PC, Terasaki PI, Takemoto SK, Lee PH, Hung CJ, Chen YL, et al. All chronic rejection failures of kidney transplants were preceded by the development of HLA antibodies. Transplantation (2002) 74(8):1192–4. doi:10.1097/00007890-200210270-00025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Loupy A, Lefaucheur C, Vernerey D, Prugger C, Duong vanHuyen JP, Mooney N, et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med (2013) 369:1215–26. doi:10.1056/NEJMoa1302506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Haas M, Sis B, Racusen LC, Solez K, Glotz D, Colvin RB, et al. Banff 2013 meeting report: inclusion of c4d-negative antibody-mediated rejection and antibody-associated arterial lesions. Am J Transplant (2014) 14(2):272–83. doi:10.1111/ajt.12590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Feucht HE, Felber E, Gokel MJ, Hillebrand G, Nattermann U, Brockmeyer C, et al. Vascular deposition of complement-split products in kidney allografts with cell-mediated rejection. Clin Exp Immunol (1991) 86(3):464–70. doi:10.1111/j.1365-2249.1991.tb02954.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Kluth-Pepper B, Schneeberger H, Lederer SR, Albert E, Land W, Feucht HE. Impact of humoral alloreactivity early after transplantation on the long-term survival of renal allografts. Kidney Int (2001) 59:334–41. doi:10.1046/j.1523-1755.2001.00495.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Opelz G. Non-HLA transplantation immunity revealed by lymphocytotoxic antibodies. Lancet (2005) 365:1570–6. doi:10.1016/S0140-6736(05)66458-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Sumitran-Holgersson S, Wilczek HE, Holgersson J, Söderström K. Identification of the nonclassical HLA molecules, MICA, as targets for humoral immunity associated with irreversible rejection of kidney allografts. Transplantation (2002) 74(2):268–77. doi:10.1097/00007890-200207270-00019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Mizutani K, Terasaki P, Rosen A, Esquenazi V, Miller J, Shih RN, et al. Serial ten-year follow-up of HLA and MICA antibody production prior to kidney graft failure. Am J Transplant (2005) 5(9):2265–72. doi:10.1111/j.1600-6143.2005.01016.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Terasaki PI, Ozawa M, Castro R. Four-year follow-up of a prospective trial of HLA and MICA antibodies on kidney graft survival. Am J Transplant (2007) 7(2):408–15. doi:10.1111/j.1600-6143.2006.01644.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Zou Y, Stastny P, Süsal C, Döhler B, Opelz G. Antibodies against MICA antigens and kidney-transplant rejection. N Engl J Med (2007) 357(13):1293–300. doi:10.1056/NEJMoa067160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Cox ST, Stephens HA, Fernando R, Karasu A, Harber M, Howie AJ, et al. Major histocompatibility complex class I-related chain A allele mismatching, antibodies, and rejection in renal transplantation. Hum Immunol (2011) 72(10):827–34. doi:10.1016/j.humimm.2011.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Narayan S, Tsai EW, Zhang Q, Wallace WD, Reed EF, Ettenger RB. Acute rejection associated with donor-specific anti-MICA antibody in a highly sensitized pediatric renal transplant recipient. Pediatr Transplant (2011) 15(1):E1–7. doi:10.1111/j.1399-3046.2010.01407.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Grafft CA, Cornell LD, Gloor JM, Cosio FG, Gandhi MJ, Dean PG, et al. Antibody-mediated rejection following transplantation from an HLA-identical sibling. Nephrol Dial Transplant (2010) 25(1):307–10. doi:10.1093/ndt/gfp526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Sumitran-Karuppan S, Tyden G, Reinholt F, Berg U, Moller E. Hyperacute rejections of two consecutive renal allografts and early loss of the third transplant caused by non-HLA antibodies specific for endothelial cells. Transpl Immunol (1997) 5(4):321–7. doi:10.1016/S0966-3274(97)80016-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Perrey C, Brenchley PE, Johnson RW, Martin S. An association between antibodies specific for endothelial cells and renal transplant failure. Transpl Immunol (1998) 6(2):101–6. doi:10.1016/S0966-3274(98)80024-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Shin YS, Yang CW, Ahn HJ, Park CW, Jin DC, Kim YS, et al. Clinical significance of anti-endothelial cell antibody in renal transplant recipients. Korean J Intern Med (2001) 16(1):24–9.
38. Grandtnerová B, Laca L, Jahnová E, Horváthová M, Baláz V, Hovoricová B, et al. Hyperacute rejection of living related kidney graft caused by IgG endothelial specific antibodies with a negative monocyte cross-match. Ann Transplant (2002) 7(4):52–4.
39. Le Bas-Bernardet S, Hourmant M, Coupel S, Bignon JD, Soulillou JP, Charreau B. Non-HLA-type endothelial cell reactive alloantibodies in pre-transplant sera of kidney recipients trigger apoptosis. Am J Transplant (2003) 3(2):167–77. doi:10.1034/j.1600-6143.2003.00021.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Sun Q, Liu Z, Yin G, Chen H, Chen J, Li L. Detectable circulating antiendothelial cell antibodies in renal allograft recipients with C4d-positive acute rejection: a report of three cases. Transplantation (2005) 79(12):1759–62. doi:10.1097/01.TP.0000163290.19788.E7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Rodríguez PC, Arroyave IH, Mejía G, García LF. Detection of alloantibodies against non-HLA antigens in kidney transplantation by flow cytometry. Clin Transplant (2000) 14(5):472–8. doi:10.1034/j.1399-0012.2000.140505.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Pontes LF, Carvalho L, Stumbo AC, Porto LC. Detection and localization of non-HLA-ABC antigenic sites relevant to kidney rejection on endothelial cells. J Immunol Methods (2001) 251(1–2):73–80. doi:10.1016/S0022-1759(01)00309-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Breimer ME, Rydberg L, Jackson AM, Lucas DP, Zachary AA, Melancon JK, et al. Multicenter evaluation of a novel endothelial cell crossmatch test in kidney transplantation. Transplantation (2009) 87(4):549–56. doi:10.1097/TP.0b013e3181949d4e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Alheim M, Johansson SM, Hauzenberger D, Grufman P, Holgersson J. A flow cytometric crossmatch test for simultaneous detection of antibodies against donor lymphocytes and endothelial precursor cells. Tissue Antigens (2010) 75(3):269–77. doi:10.1111/j.1399-0039.2009.01439.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Jackson AM, Lucas DP, Melancon JK, Desai NM. Clinical relevance and IgG subclass determination of non-HLA antibodies identified using endothelial cell precursors isolated from donor blood. Transplantation (2011) 92(1):54–60. doi:10.1097/TP.0b013e31821b60e9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Canet E, Devallière J, Gérard N, Karam G, Giral M, Charreau B, et al. Profiling posttransplant circulating antibodies in kidney transplantation using donor endothelial cells. Transplantation (2012) 93(3):257–64. doi:10.1097/TP.0b013e31823ec0ec
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Ronda C, Borba SC, Ferreira SC, Glotz D, Ianhez LE, Rodrigues H, et al. Non-human leukocyte antigen antibodies reactive with endothelial cells could be involved in early loss of renal allografts. Transplant Proc (2011) 43(4):1345–8. doi:10.1016/j.transproceed.2011.03.059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Sun Q, Liu Z, Chen J, Chen H, Wen J, Cheng D, et al. Circulating anti-endothelial cell antibodies are associated with poor outcome in renal allograft recipients with acute rejection. Clin J Am Soc Nephrol (2008) 3(5):1479–86. doi:10.2215/CJN.04451007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Sigdel TK, Li L, Tran TQ, Khatri P, Naesens M, Sansanwal P, et al. Non-HLA antibodies to immunogenic epitopes predict the evolution of chronic renal allograft injury. J Am Soc Nephrol (2012) 23(4):750–63. doi:10.1681/ASN.2011060596
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Dinavahi R, George A, Tretin A, Akalin E, Ames S, Bromberg JS, et al. Antibodies reactive to non-HLA antigens in transplant glomerulopathy. J Am Soc Nephrol (2011) 22(6):1168–78. doi:10.1681/ASN.2010111183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Hernandez NM, Casselbrant A, Joshi M, Johansson BR, Sumitran-Holgersson S. Antibodies to kidney endothelial cells contribute to a “leaky” glomerular barrier in patients with chronic kidney diseases. Am J Physiol Renal Physiol (2012) 302(7):F884–94. doi:10.1152/ajprenal.00250.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Li L, Wadia P, Chen R, Kambham N, Naesens M, Sigdel TK, et al. Identifying compartment-specific non-HLA targets after renal transplantation by integrating transcriptome and “antibodyome” measures. Proc Natl Acad Sci U S A (2009) 106(11):4148–53. doi:10.1073/pnas.0900563106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Saini D, Weber J, Ramachandran S, Phelan D, Tiriveedhi V, Liu M, et al. Alloimmunity-induced autoimmunity as a potential mechanism in the pathogenesis of chronic rejection of human lung allografts. J Heart Lung Transplant (2011) 30(6):624–31. doi:10.1016/j.healun.2011.01.708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Zhang Q, Reed EF. Non-MHC antigenic targets of the humoral immune response in transplantation. Curr Opin Immunol (2010) 22(5):682–8. doi:10.1016/j.coi.2010.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Xu Y, Chester AH, Hariri B, McCormack A, Sarathchandra P, Rose ML. The indirect alloimmune response causes microvascular endothelial dysfunction-a possible role for alloantibody. Transplantation (2010) 90(11):1157–64. doi:10.1097/TP.0b013e3181fa9480
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Dragun D, Müller DN, Bräsen JH, Fritsche L, Nieminen-Kelhä M, Dechend R, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med (2005) 352(6):558–69. doi:10.1056/NEJMoa035717
57. Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jüpner A, et al. Patients with preeclampsia develop agonistic antibodies against the angiotensin AT1 receptor. J Clin Invest (1999) 103:945–52. doi:10.1172/JCI4106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Reinsmoen NL, Lai CH, Heidecke H, Haas M, Cao K, Ong G, et al. Anti-angiotensin type 1 receptor antibodies associated with antibody mediated rejection in donor HLA antibody negative patients. Transplantation (2010) 90(12):1473–7. doi:10.1097/TP.0b013e3181fd97f1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Rose ML. Role of anti-vimentin antibodies in allograft rejection. Human Immunol (2013) 74:1459–62. doi:10.1016/j.humimm.2013.06.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Barber LD, Whitelegg A, Madrigal JA, Banner NR, Rose ML. Detection of vimentin-specific autoreactive CD8+ T cells in cardiac transplant patients. Transplantation (2004) 77(10):1604–9. doi:10.1097/01.TP.0000129068.03900.25
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Carter V, Shenton BK, Jaques B, Turner D, Talbot D, Gupta A, et al. Vimentin antibodies: a non-HLA antibody as a potential risk factor in renal transplantation. Transplant Proc (2005) 37(2):654–7. doi:10.1016/j.transproceed.2004.12.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Leong HS, Mahesh BM, Day JR, Smith JD, McCormack AD, Ghimire G, et al. Vimentin autoantibodies induce platelet activation and formation of platelet-leukocyte conjugates via platelet-activating factor. J Leukoc Biol (2008) 83(2):263–71. doi:10.1189/jlb.0607339
63. Mahesh B, Leong HS, Nair KS, McCormack A, Sarathchandra P, Rose ML. Autoimmunity to vimentin potentiates graft vasculopathy in murine cardiac allografts. Transplantation (2010) 90(1):4–13. doi:10.1097/TP.0b013e3181dfa694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Snanoudj R, Candon S, Legendre C. Targeting B cells in sensitized kidney transplant patients: state of the art and future perspectives. Curr Opin Organ Transplant (2010) 15(6):709–15. doi:10.1097/MOT.0b013e3283402cf4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Zhang X, Reed EF. Effect of antibodies on endothelium. Am J Transplant (2009) 9:2459–65. doi:10.1111/j.1600-6143.2009.02819.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Tobian AA, Shirey RS, Montgomery RA, Cai W, Haas M, Ness PM, et al. AB0 anitbody titer and risk of antibody-mediated rejection in AB0-incompatible renal transplantation. Am J Transplant (2010) 10:1247–53. doi:10.1111/j.1600-6143.2010.03103.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. KissmeyerNielsen F, Olsen S, Petersen VP, Fjeldborg O. Hyperacute rejection of kidney allografts, associated with preexisting humoral antibodies against donor cells. Lancet (1966) 2(7465):6625.
68. Yamakuchi M, Kirkiles-Smith NC, Ferlito M, Cameron SJ, Bao C, Fox-Talbot K, et al. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proc Natl Acad Sci U S A (2007) 104(4):1301–6. doi:10.1073/pnas.0602035104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Fuggle SV, Sanderson JB, Gray DW, Richardson A, Morris PJ. Variation in expression of endothelial adhesion molecules in pre transplant and transplanted kidneys correlation with intra graft events. Transplantation (1993) 55(1):11723.
70. Cavender DE, Haskard DO, Joseph B, Ziff M. Interleukin 1 increases the binding of human B and T lymphocytes to endothelial cell monolayer. J Immunol (1986) 136(1):203–7.
71. Cavender DE, Saegusa Y, Ziff M. Stimulation of endothelial cell binding of human lymphocytes by tumor necrosis factor. J Immunol (1987) 139(6):1855–60.
72. Yu CL, Haskard D, Cavender DE, Johnson AR, Ziff M. Human gamma interferon increases the binding of T lymphocytes to endothelial cell monolayer. Clin Exp Immunol (1985) 62(1):554–60.
73. Harris PE, Bian H, Reed EF. Induction of high affinity fibroblast growth factor receptor expression and proliferation in human endothelial cells by anti-HLA antibodies: a possible mechanism for transplant atherosclerosis. J Immun (1997) 159:5697–704.
74. Ziegler ME, Souda P, Jin Y, Whitelegge JP, Reed EF. Characterization of the endothelial cell cytoskeleton following HLA class I ligation. PLoS One (2012) 7(1):e29472. doi:10.1371/journal.pone.0029472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Zhang X, Rozengurt E, Reed EF. HLA class I molecules partner with Integrin ß4 to stimulate endothelial cell proliferation and migration. Sci Signal (2014) 3(149):1–25. doi:10.1126/scisignal.2001158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Jindra PT, Jin Y, Rozengurt E, Reed EF. HLA class I antibody-mediated endothelial cell proliferation via the mTOR pathway. J Immunol (2008) 180:2357–66. doi:10.4049/jimmunol.180.4.2357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Chopek MW, Simmons RL, Platt JL. ABO-incompatible kidney transplantation: initial immunopathologic evaluation. Transplant Proc (1987) 19(6):4553–7.
78. Rose ML, West LJ. Accommodation: does it apply to human leukocyte antigens? Transplantation (2012) 93(3):244–6. doi:10.1097/TP.0b013e31823f468c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Salama AD, Delikouras A, Pusey CD, Cook HT, Bhangal G, Lechler RI, et al. Transplant accommodation in highly sensitized patients: a potential role for Bc – xL and allo.antibody. Am J Transplant (2001) 1:260–9. doi:10.1034/j.1600-6143.2001.001003260.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Narayanan K, Jaramillo A, Phelan DL, Mohanakumar T. Pre-exposure to sub-saturating concentrations of HLA class I antibodies confers resistance to endothelial cells against antibody complement mediated lysis by regulating Bad through the phosphatidylinositol 3-kinase/Akt pathway. Eur J Immunol (2004) 34:2303–12. doi:10.1002/eji.200324843
81. Jindra PT, Zhang X, Mulder A, Claas F, Veale J, Jin YP, et al. Anti-HLA antibodies can induce endothelial cell survival or proliferation depending on their concentration. Transplantation (2006) 82:S33–5. doi:10.1097/01.tp.0000231447.34240.3c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Jindra PT, Hsueh A, Hong L, Gjertson D, Shen XD, Gao F, et al. Anti-MHC class I antibody activation of proliferation and survival signaling in murine cardiac allografts. J Immunol (2008) 180(4):2214–24. doi:10.4049/jimmunol.180.4.2214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Pober JS, Orosz CG, Rose ML, Savage CO. Can graft endothelial cells initiate a host anti-graft immune response? Transplantation (1996) 61(3):343–9. doi:10.1097/00007890-199602150-00001
84. Epperson DE, Pober JS. Antigen-presenting function of human endothelial cells. Direct activation of resting CD8 T cells. J Immunol (1994) 153(12):5402–12.
85. Kreisel D, Krupnick AS, Balsara KR, Riha M, Gelman AE, Popma SH, et al. Mouse vascular endothelium activates CD8+ T lymphocytes in a B7-dependent fashion. J Immunol (2002) 169:6154–61. doi:10.4049/jimmunol.169.11.6154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Kreisel D, Krupnick AS, Gelman AE, Engels FH, Popma SH, Krasinskas AM, et al. Non-hematopoietic allograft cells directly activate CD8+ T cells and trigger acute rejection: an alternative mechanism of allorecognition. Nat Med (2002) 8(3):233–9. doi:10.1038/nm0302-233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Savage C, Hughes CC, Pepinsky RB, Wallner BP, Freedman AS, Pober JS. Endothelial cell lymphocyte function-associated antigen-3 and an unidentified ligand act in concert to provide costimulation to human peripheral blood CD4+ T cells. Cell Immunol (1991) 137(1):15063. doi:10.1016/0008-8749(91)90065-J
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Adams PW, Lee HS, Waldman WJ, Sedmak DD, Morgan CJ, Ward JS, et al. Alloantigenicity of human endothelial cells. 1. Frequency and phenotype of human T helper lymphocytes that can react to allogeneic endothelial cells. J Immunol (1992) 148(12):375360.
89. Page C, Thompson C, Yacoub M, Rose M. Human endothelial stimulation of allogeneic T cells via a CTLA4 independent pathway. Transpl Immunol (1994) 2(4):3427. doi:10.1016/0966-3274(94)90013-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Murakami K, Ma W, Fuleihan R, Pober JS. Human endothelial cells augment early CD40 ligand expression in activated CD41 T Cells through LFA-3-mediated stabilization of mRNA. J Immunol (1999) 163:2667–73.
91. Marelli-Berg FM, Frasca L, Weng L, Lombardi G, Lechler RI. Antigen recognition influences transendothelial migration of CD4+ T Cells. J Immunol (1999) 162:696–703.
92. Zelenika D, Adams E, Mellor A, Simpson E, Chandler P, Stockinger B, et al. Rejection of H-Y disparate skin grafts by monospecific CD4+ Th1 and Th2 cells: no requirement for CD8+ T cells or B cells. J Immunol (1998) 161(4):1868–74.
93. Taflin C, Favier B, Baudhuin J, Savenay A, Hemon P, Bensussan A, et al. Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc Natl Acad Sci U S A (2011) 108(7):2891–6. doi:10.1073/pnas.1011811108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Agorogiannis EI, Regateiro FS, Howie D, Waldmann H, Cobbold SP. Th17 cells induce a distinct graft rejection response that does not require IL-17A. Am J Transplant (2012) 12(4):835–45. doi:10.1111/j.1600-6143.2011.03971.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Kendal AR, Chen Y, Regateiro FS, Ma J, Adams E, Cobbold SP, et al. Sustained suppression by Foxp3+ regulatory T cells is vital for infectious transplantation tolerance. J Exp Med (2011) 208(10):2043–53. doi:10.1084/jem.20110767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Thebault P, Condamine T, Heslan M, Hill M, Bernard I, Saoudi A, et al. Role of IFNc in allograft tolerance mediated by CD4+CD25+ regulatory T cells by induction of IDO in endothelial cells. Am J Transplant (2007) 7:2472–82. doi:10.1111/j.1600-6143.2007.01960.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Wang C, Yi T, Qin L, Maldonado RA, von Andrian UH, Kulkarni S, et al. Rapamycin-treated human endothelial cells preferentially activate allogeneic regulatory T cells. J Clin Invest (2013) 123(4):1677–93. doi:10.1172/JCI66204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Krupnick AS, Gelman AE, Barchet W, Richardson S, Kreisel FH, Turka LA, et al. Cutting edge: murine vascular endothelium activates and induces the generation of allogeneic CD4 25 Foxp3 regulatory T Cells. J Immunol (2005) 175:6265–70. doi:10.4049/jimmunol.175.10.6265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Wang C, Qin L, Manes TD, Kirkiles-Smith NC, Tellides G, Pober JS. Rapamycin antagonizes TNF induction of VCAM-1 on endothelial cells by inhibiting mTORC2. J Exp Med (2014) 211(3):395–404. doi:10.1084/jem.20131125
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Maganto-García E, Bu DX, Tarrio ML, Alcaide P, Newton G, Griffin GK, et al. Foxp3+-inducible regulatory T cells suppress endothelial activation and leukocyte recruitment. J Immunol (2011) 187:3521–9. doi:10.4049/jimmunol.1003947
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Rostaing L, Vincenti F, Grinyó J, Rice KM, Bresnahan B, Steinberg S, et al. Long-term belatacept exposure maintains efficacy and safety at 5 years: results from the long-term extension of the benefit study. Am J Transplant (2013) 13(11):2875–83. doi:10.1111/ajt.12460
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: endothelial cell antigens, angiotensin II type 1 receptor, vimentin, accommodation, regulatory T-cells, renal transplantation, antibody-mediated rejection, mTOR inhibitors
Citation: Piotti G, Palmisano A, Maggiore U and Buzio C (2014) Vascular endothelium as a target of immune response in renal transplant rejection. Front. Immunol. 5:505. doi: 10.3389/fimmu.2014.00505
Received: 26 July 2014; Accepted: 28 September 2014;
Published online: 21 October 2014.
Edited by:
Giuseppe Alvise Ramirez, Università Vita-Salute San Raffaele, ItalyReviewed by:
Kottarappat N. Dileepan, The University of Kansas Medical Center, USABo Shen, University of Illinois at Chicago, USA
Copyright: © 2014 Piotti, Palmisano, Maggiore and Buzio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanni Piotti, Kidney and Pancreas Transplantation Unit, Department of Clinical Medicine, Nephrology and Health Sciences, University Hospital of Parma, Via Gramsci 14, 43121 Parma, Italy e-mail:Z3Bpb3R0aUB5YWhvby5pdA==