 Monica Borghi1
Monica Borghi1 Giorgia Renga1
Giorgia Renga1 Matteo Puccetti2
Matteo Puccetti2 Vasileios Oikonomou1
Vasileios Oikonomou1 Melissa Palmieri1
Melissa Palmieri1 Claudia Galosi1Andrea Bartoli1
Claudia Galosi1Andrea Bartoli1 Luigina Romani1*
Luigina Romani1*- 1Pathology Section, Department of Experimental Medicine, University of Perugia, Perugia, Italy
- 2Polo GGB, Perugia, Italy
Fungal diseases represent an important paradigm in immunology since they can result from either the lack of recognition or over-activation of the inflammatory response. Current understanding of the pathophysiology underlying fungal infections and diseases highlights the multiple cell populations and cell-signaling pathways involved in these conditions. A systems biology approach that integrates investigations of immunity at the systems-level is required to generate novel insights into this complexity and to decipher the dynamics of the host–fungus interaction. It is becoming clear that a three-way interaction between the host, microbiota, and fungi dictates the types of host–fungus relationship. Tryptophan metabolism helps support this interaction, being exploited by the mammalian host and commensals to increase fitness in response to fungi via resistance and tolerance mechanisms of antifungal immunity. The cellular and molecular mechanisms that provide immune homeostasis with the fungal biota and its possible rupture in fungal infections and diseases will be discussed within the expanding role of antifungal Th cell responses.
Fungal Infections and Diseases in the Metagenomics Era: A Reappraisal
Fungi can interact with their hosts (plants, animals, or human beings) in multiple ways, establishing symbiotic, commensal, or pathogenic relationships. Most fungi, such as Aspergillus fumigatus and Cryptococcus neoformans, and the thermally dimorphic fungi are ubiquitous in the environment, and human beings are exposed by inhaling spores or small yeast cells. In addition, more than 400 species of fungi associated with human beings have been identified (1). In this case, co-evolution of commensals, such as Pneumocystis jirovecii, Malassezia spp., and Candida albicans, with their mammalian hosts implicates the existence of sophisticated mechanisms to antagonize immunity in order to survive. Once considered pathogenic microbes, the commensal fungal microbiota is now an important component of the human intestinal ecosystem. Indeed, despite the intimate contact of fungi with the human host, fungal diseases in immunocompetent hosts are fairly uncommon, indicating that low-virulence fungi have evolved particular adaptation mechanisms that allow them to persist relatively unnoticed by the immune system (2). This “peaceful” coexistence may digress into overt disease under conditions of immune deregulation, such as in primary immunodeficiency human immunodeficiency virus infection and as a result of immunosuppressive therapies (2). In addition, invasive fungal diseases continue to be a serious problem in patients with hematologic disorders, solid, and hematopoietic organ transplantation as well as in non-high-risk, sensu strictu, patients, such as patients with Mycobacterium tubercolosis infection, hyper IgE syndrome, and anti-TNF-alpha therapy (3).
The increasing understanding of the importance of the microbiota in shaping the host immune and metabolic activity has rendered fungal interactions with the host and its microbiome more complex than previously appreciated (4) (Box 1). Indeed, the complex interactions between fungal and bacterial commensals, either directly or through the participation of the host immune system, all impact on the pathophysiology of a number of inflammatory disease that, in turn, may lead to secondary fungal infections (5, 6). Evidence is accumulating to support the exciting concept that the interaction between different biomes and between the host and the mycobiome are critical in the pathogenesis of fungal infections and other human diseases (1, 7, 8). Here, we will discuss recent findings on host- and microbial-dependent mechanisms of immune homeostasis with the fungal biota and its possible rupture in fungal infections and diseases.
Box 1. The mycobiome at the host/microbiome interface.
The development of culture-independent methods has expanded our knowledge of the mycobiomes found in different body sites, their interface with other biomes, and their association with human health and diseases (1). Alterations in the mycobiome are frequently reported to be associated with various diseases such as cystic fibrosis (9), inflammatory bowel diseases (6, 10, 11), atopic dermatitis (12), or mucocutaneous candidiasis (13). However, it remains to be elucidated whether this variation is primary or secondary to an imbalanced bacterial microbiome. Indeed, interactions of fungi with bacteria in vitro have been described [reviewed in Ref. (6)] as well as the clinical relevance of these interactions (14), such as the occurrence of intractable candidiasis in association with antibiotic-induced dysbiosis (15) and of mixed fungal–bacterial species in biofilms (14). Fungal–bacterial interactions can be antagonistic, synergistic, or symbiotic; regardless, they influence the physiological characteristics and survival of either one partner and, consequently, impact on host immune reactivity. Variations in the mycobiome can also be secondary to dysregulated host immune reactivity. The traditional view of a single direction by which bacteria stimulates the immune system, leading to inflammation or autoimmune disorders, has been challenged by a more complex view; the gut immune system does not simply protect from pathogens, but is actively involved in the maintenance of a rich and healthy community of gut bacteria (16). Faults in the immune regulation lead to changes in the bacterial community that in turn feed back into the immune system. Similar to the microbiome, the host/mycobiome interactions also lead to mutual influences. Not only is the host affecting the mycobiome composition and variations, by means of genotype, physiology, immune system, and lifestyle, but also the fungal microbiota may contribute to the balance of inflammation and tolerance at local mucosal surfaces and at distal sites (17).
Resistance and Tolerance Mechanisms of Antifungal Immunity
As the immune system has evolved to accommodate colonization by symbiotic microbes while retaining the capacity to oppose their infectivity, a fine balance between pro- and anti-inflammatory signals is a prerequisite for a stable host/fungal relationship, the disruption of which may lead to pathological consequences. Indeed, despite the occurrence of severe fungal infections in immunocompromised patients, clinical evidence indicates that fungal diseases also occur in the setting of a heightened inflammatory response, in which immunity occurs at the expense of host damage and pathogen eradication (18). A number of fungal diseases are critical examples of such bidirectional influences between infection and immune-related pathology, a condition that highlights the bipolar nature of the inflammatory process in infection. Early inflammation prevents or limits infection, but an uncontrolled response may eventually oppose disease eradication. This conceptual principle is best exemplified by the occurrence of severe fungal infections in patients with chronic granulomatous disease (19), cystic fibrosis (20), or with immune reconstitution inflammatory syndrome (IRIS) (21), an entity characterized by local and systemic inflammatory reactions that can result in quiescent or latent infections manifesting as opportunistic mycoses. Chronic mucocutaneous candidiasis (CMC) and chronic disseminated candidiasis also belongs to the spectrum of fungus-related IRIS (22). Thus, an immune response that limits both fungal infectivity and host collateral damage is required to maintain a homeostatic environment (23). This dual role has recently been accommodated within the conceptual framework of a two-component antifungal immune response, i.e., resistance – the ability to limit fungal burden – and tolerance – the ability to limit the host damage caused by either the immune response or other mechanisms (2). Resistance is meant to reduce pathogen burden through innate and adaptive immune mechanisms, whereas a plethora of tolerance mechanisms, despite less known relative to resistance mechanisms, protect the host from immune- or pathogen-induced damage (24).
Mechanisms of Antifungal Resistance
Innate immune mechanisms are used by the host to respond to a range of fungal pathogens in an acute and conserved fashion. The constitutive mechanisms of innate defense are present at sites of continuous interaction with fungi and include the barrier function of body surfaces and the mucosal epithelial surfaces of the respiratory, gastrointestinal, and genitourinary tracts. Microbial antagonism, defensins, collectins, and the complement system realize the strict fungus specificity of the constitutive mechanisms and provide opsonic recognition. Multiple cell populations and cell-signaling pathways are involved in the antigen-independent recognition of fungi by PRRs (2, 25). Both murine and human studies have confirmed the association of susceptibility to fungal infections and diseases with genetic deficiency of selected PRRs (2). Because PRRs not only mediate downstream intracellular events related to fungal clearance but also participate in activation of adaptive immunity, deficiencies on innate immune genes also reverberate on the type and quality of the adaptive immune response, including effector CD4+ T helper (Th), regulatory T (Treg), and CD8+ T-cells (2, 25–27).
Dendritic Cells
It is well established that the adaptive immune response, in particular that of T-cells, plays a pivotal role in antifungal host defense (2, 25). Dendritic cells (DCs) play a key role in promoting T-cell differentiation and responses to ubiquitous or commensal fungi. Studies have shown that lung DCs can transport fungal antigens to the draining lymph nodes (28, 29), where they orchestrate T-cell activation and differentiation into effector cells. Through elaboration of distinct sets of cytokines and other mediators, DCs have the unique ability to elicit a robust T-cell response that can be either tolerogenic or pro-inflammatory in nature, based on anatomical location and local metabolic environment. The whole-genome transcriptional analysis of DCs stimulated with fungi evidenced the presence of peculiar transcriptional programs governing the recognition of fungi (30).
These include common signaling pathways involving Syk kinase, Card9 and NF-κB downstream CLRs and ERK kinase, PI3K/Akt downstream TLRs for Th1/Th2/Th17 priming by conventional, inflammatory DCs, as well as p38/TRIF/STAT3 for Treg priming by plasmacytoid DCs (2, 31). In a mutual interaction, the host and the fungus control each other to avoid potential harmful inflammatory response. The ability of a given DC subset to respond with flexible activating programs and activation of distinct intracellular signaling pathways to the different PRR/fungal molecules’ combinations confers unexpected plasticity to the DC system and pivotally contributes in shaping adaptive Th cells responses in infection and vaccination. The capacity of DCs to initiate different adaptive antifungal immune responses also depends upon specialization and cooperation between DC subsets (32). The multiple, functionally distinct, receptor/signaling pathways in DCs, ultimately affecting the local Th/Treg balance, are likely successfully exploited by fungi from commensalism to infection (33).
Th1 Cells
CD4+ Th cells exist in a variety of epigenetic states that determine their function, phenotype, and capacity for persistence, and form long-term immune memory (34). Well-balanced Th1 and Th17 cell responses are crucial in antifungal immunity and facilitate phagocytic clearance of fungal recognition, mainly through release of cytokines such as TNF-α, IFN-γ, and IL-17A and IL-17F (Table 1). These cytokines stimulate the disparate antifungal effector functions of phagocytes, as well as the generation of optimal T-cell-dependent immunity (2, 25). A dominant Th1 response correlates with the expression of protective immunity to fungi (2, 35) and vaccines (36, 37). Through the production of the signature cytokine IFN-γ and help for opsonizing antibodies, the activation of Th1 cells is instrumental in the optimal activation of phagocytes at sites of infection. Therefore, the failure to deliver activating signals to effector phagocytes may predispose patients to overwhelming infections, limit the therapeutic efficacy of antifungals and antibodies, and favor fungal persistency (2). Patients who are deficient in IL-12Rβ are susceptible to CMC, which is frequently recurrent or persistent (38), as well as to deep paracoccidioidomycosis (39).
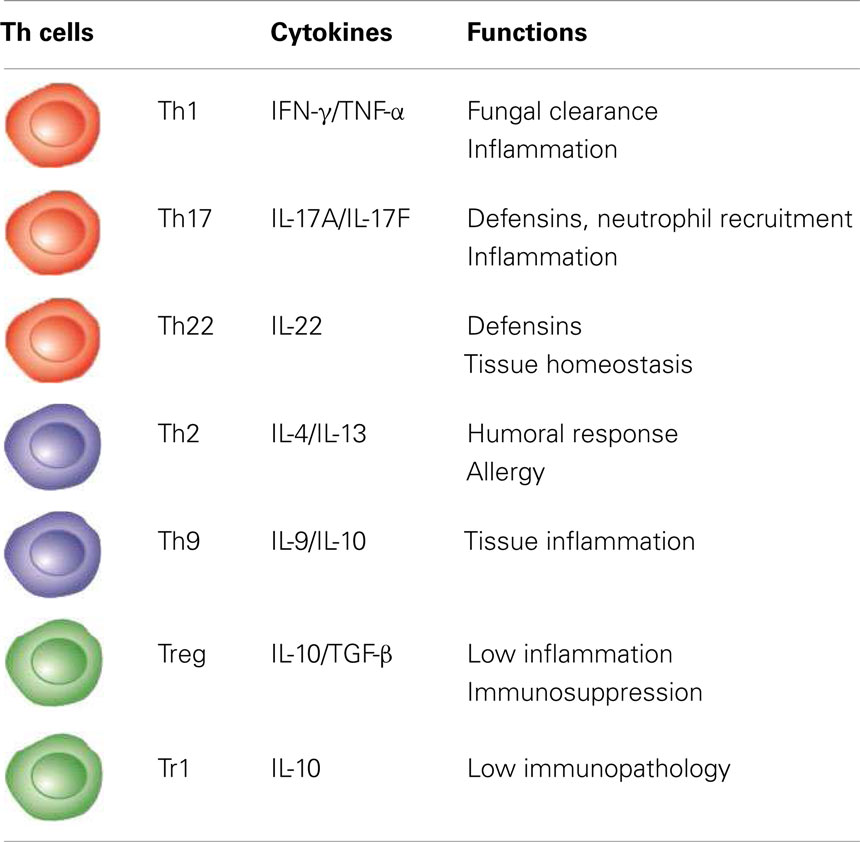
Table 1. CD4+ Th cell subsets in fungal infections.
Th17 Cells
Th17 are present in the human T-cell memory repertoire to fungi (2) and inborn errors of human IL-17 immunity underlie susceptibility to CMC (40) in which both Th17 (41) and Th1 (38, 42, 43) responses are defective. Combined deficiency of the Th1 and Th17 pathway predisposes to fungal diseases (44, 45), thus emphasizing the important role played by both pathways in resistance against fungi. This could be explained with the notion that Th17 cells, although found early during the initiation of an immune response, are involved in a broad range of Th1-, Th2- and Treg-dominated immune responses (2, 46). In terms of effector functions, the ability of IL-17A to mobilize neutrophils and induce defensins may contribute to a prompt and efficient control of the infection at the different body sites. In respiratory fungal infections, Th17 cell are dispensable for resistance to the primary infection caused by A. fumigatus (47), but are required for vaccine-induced immunity against systemic mycoses endemic to North America (48). Thus, both Th17 and Th1 (27) cells are required for vaccine immunity to respiratory fungal pathogens.
It is intriguing that Th17 responses are down regulated by C. albicans (49). Regardless of the contribution of this phenomenon to infection or commensalism, this finding suggests that Th17 responses are finely tuned by fungi, as the failure to downregulate Th17 may eventually result in chronic inflammation and failure to resolve the infection (47, 50). The mechanisms that linked inflammation to chronic infection have been credited to the offending potential of IL-17A that, although promoting neutrophil recruitment, impeded the timely restriction of neutrophil inflammatory potential (51) while directly promoting fungal virulence (52). Thus, the Th17 pathway could be involved in the immunopathogenesis of chronic fungal diseases where persistent fungal antigens may maintain immunological dysreactivity. This may happen in autoimmune polyendocrine syndrome type 1 patients (APS-1) and Aire-deficient mice (53) where an excessive Th17 reactivity was observed. This finding apparently conflicts with the presence of autoantibodies against IL-22, IL-17A, and IL-17F observed in these patients (54, 55). Although correlated to susceptibility to CMC, these antibodies were also present in patients without CMC. In addition, despite the presence of antibodies to type I IFN, APS-I patients do not appear prone to recurrent viral infections. It has instead been shown that autoantibodies to pro-inflammatory cytokines may act as beneficial autoimmunity in their ability to dampen pro-inflammatory mediators and restrict self-destructive immunity (56).
Th2 Cells
IL-4 and IL-13 act as the most potent proximal signal for commitment to Th2 reactivity that, by dampening protective Th1 responses and promoting the alternative pathway of macrophage activation, favors fungal persistence, allergy, and disease relapse. Limiting IL-4 production restores antifungal resistance (2) (Table 1). In atopic subjects and neonates, the suppressed DTH response to fungi is associated with elevated levels of antifungal IgE, IgA, and IgG. In CF patients, heightened Th2 reactivity associates with allergic bronchopulmonary aspergillosis and is sensitive to vitamin 3 (57). However, alternatively activated macrophages may have a protective role in defense against some respiratory fungi (58, 59) and Th2-dependent humoral immunity may afford some protection, in part by promoting Th1 immunity (60) and by altering fungal gene expression and intracellular trafficking (61–63). The efficacy of certain vaccines that elicit protective antibody strongly indicates that antibody responses can make a decisive contribution to host defense to fungi (61).
Th9 Cells
The realization that Th effectors can produce various other cytokines alone or in combination in patterns not fitting the preconceived definitions of Th1/Th2 or Th17 subsets has led to the description of additional Th cell lineages, including Th9 and Th22. Initially thought to be a Th2-specific cytokine by virtue of its role in the pathogenesis of asthma, IgE class switch recombination, and resolution of parasitic infections, IL-9 is now considered to be the product of a distinct Th subset, the Th9 (64). Despite its relationship with other subsets, such as Th2, Th17, and Treg cells, Th9 cell subset can mediate tumor immunity and participate in autoimmune and allergic inflammation. Recently, human memory Th9 cells were found to be skin tropic or skin resident. Human Th9 cells co-expressed TNF-α and granzyme B, lacked coproduction of Th1/Th2/Th17 cytokines, and many were specific for C. albicans. IL-9 production preceded the upregulation of other inflammatory cytokines, such as IFN-γ, IL-13, and IL-17. IL-9-producing T-cells were increased in the skin lesions of psoriasis, suggesting that these cells may contribute to human inflammatory skin disease in the presence of Candida (65). Recent findings demonstrated that IL-9 is predominantly produced in vivo by a novel subset of innate lymphoid cells termed ILC2 (66). It has been proposed that IL-9 might have a regulatory and prosurvival function for many lymphoid and myeloid cells (67). Our recent evidence suggests that different types of ILCs are defective in IL-9-deficient mice infected with either C. albicans or A. fumigatus, and this profoundly affects the outcome of either infection and the associated pathology (unpublished observations) (Table 1).
Th22 Cells
Th22 cells producing only IL-22 but neither IFN-γ nor IL-17A have been identified in human beings (68). They are induced in the presence of TNF-α and IL-6 and require ligation of aryl hydrocarbon receptor (AhR). Th22 cells via IL-22 influence the function of mesenchymal and epithelial cells and have been implicated in the dermatopathology of psoriasis and atopic dermatitis (69, 70). Memory C. albicans-specific IL-22+ CD4+ cells are present in human beings and defective in patients with CMC (71). Recent evidence indicates that IL-22 may play a crucial role in the innate immune resistance and local protection in mucocutaneous fungal diseases (72–74). Through the exploitation of primitive anti-fungal defense mechanisms, IL-22 was crucially involved in the control of Candida growth at mucosal sites in conditions of Th1 and Th17 deficiency (72, 74). Produced by ILC3 cells expressing AhR, IL-22 directly targeted gut epithelial cells to induce STAT3 phosphorylation and the release of S100A8 and S100A9 peptides known to have anti-candidal activity and anti-inflammatory effects (72, 74). Thus, due to dominant-negative mutations of STAT3, patients with autosomal dominant hyper-IgE syndrome have a defective Th17 (41) that is likely amplified on ECs where STAT3 mutation compromises the IL-22 effects. IL-22 also mediates antifungal resistance and epithelial protection in experimental and human vulvovaginal candidiasis (VVC) as well as in recurrent VVC (RVVC). In RVVC, functional genetic variants in IL22 genes were found to be associated with heightened resistance to RVVC, and they correlated with increased local expression of IL-22 (74). Thus, IL-22+ cells, employing ancient effector mechanisms of immunity, may represent a primitive mechanism of resistance against fungi under a condition of limited inflammation (Table 1). The fact that IL-22 production in the gut is driven by commensals (see below) also provides novel mechanistic insights on how antibiotic-related dysbiosis may predispose to candidiasis (75).
Mechanisms of Tolerance
Treg Cells
The exposure to fungi requires the generation of a controlled immune response in the host that recognizes and controls them, limits collateral damage to self-tissues, and restores a homeostatic environment. A number of clinical observations suggest an inverse relationship between IFN-γ and IL-10 production in patients with fungal infections. High levels of IL-10, negatively affecting IFN-γ production, are detected in chronic candidal diseases, in the severe form of endemic mycoses, and in neutropenic patients with aspergillosis. Thus, high levels of IL-10 have been linked to susceptibility to fungal infections (76). However, given its prominent effect on resolution of inflammation, IL-10 production may be a consequence, rather than the cause, of the infection. This predicts that, in the case of chronic fungal infections dominated by non-resolving, persisting inflammation, IL-10 produced by Treg cells acts as homeostatic host-driven response to keep inflammation under control. Treg cells with anti-inflammatory activity have been described in fungal infections of both mice and human beings (2, 25). In experimental fungal infections, inflammatory immunity and immune tolerance in the respiratory or the gastrointestinal mucosa were all controlled by the coordinate activation of different Treg cell subsets, exerting a fine control over effector components of innate and adaptive immunity. Seen in this context, the Treg/IL-10 axis is a dangerous necessity, the failure of which may lead to detrimental inflammation. However, as the Treg responses may handicap the efficacy of protective immunity, the consequence of Treg activity is less damage to the host but also fungal persistence and immunosuppression, eventually (Table 1). Thus, by controlling the quality and magnitude and effector innate and adaptive responses, the spectrum of Treg cell activities may go from “protective tolerance,” defined as a host’s response that ensures survival of the host in a trade-off between sterilizing immunity and its negative regulation limiting pathogen elimination to overt immunosuppression. Taking a step further, this suggests that the interaction between fungi and the host immune status may determine their position from commensals to pathogens, and this position can change continuously. The salutary effects of Treg cells may go beyond their anti-inflammatory properties, to include the polarization of protective Th17 cells (46).
Tr1 cells
T regulatory Type 1 (Tr1) cells are adaptive Treg cells characterized by the ability to secrete high levels of IL-10. Since their discovery, Tr1 cells have been proven to be important in maintaining immunological homeostasis and preventing T-cell-mediated diseases. Tr1 cells suppress T- and DC-dependent responses primarily via the secretion of IL-10 and TGF-β, release of granzyme B and perforin, and by disrupting the metabolic state of T effector cells. Tr1 cells have been demonstrated to have a role in infectious diseases, autoimmunity, and transplant rejection in different pre-clinical disease models and in patients (77). It has recently been shown that Tr1 cells play a distinct, yet complementary role, in response to A. fumigatus in human beings and mice. Tr1 cells specific for an epitope derived from the cell wall glucanase Crf-1 of A. fumigatus (Crf-1/p41) were identified in healthy human beings and mice after vaccination with Crf-1/p41+ zymosan. These cells produced high amounts of interleukin IL-10 and suppressed the expansion of antigen-specific T-cells in vitro and in vivo, thus limiting immunopathology (Table 1). In vivo differentiation of Tr1 cells was dependent on the presence of AhR, c-Maf, and IL-27. In comparison to Tr1 cells, Foxp3+ induced Treg that recognize the same epitope were induced in an interferon gamma-type inflammatory environment and more potently suppressed innate immune cell activities. These data provide evidence that Tr1 cells are involved in the maintenance of antifungal immune homeostasis, and most likely play a distinct, yet complementary, role compared with Foxp3+ Treg cells (78).
Tryptophan Metabolism
The enzyme indoleamine 2,3-dioxygenase 1 (IDO1) and its downstream catabolites sustain the delicate balance between Th1/Th17 pathways and Treg cells, by providing the host with adequate protective immune mechanisms without necessarily eliminating the pathogen or causing undesirable tissue damage (79). As a result of their ability to induce differentiation of Treg cells and inhibit Th17 cells, IDO1 is critical to cell lineage commitment in experimental fungal infections and contributes to the overall outcome of inflammation, allergy, and Th17-driven inflammation in these infections. Under these circumstances, the Th17 pathway, by inhibiting tryptophan catabolism, may instead favor pathology and provides evidence accommodating the apparently paradoxical association of chronic inflammation with fungal disease (19). IDO1 is a “metabolic” enzyme conserved through the past 600 million years of evolution. Initially recognized in infection because of antimicrobial activity (“tryptophan starvation” of intracellular parasites), IDO1 is now widely recognized as suppressor of acute inflammatory responses and regulator of mammalian immune homeostasis (79). Not surprising, IDO1 may represent an evasion mechanism for microbes that establish commensalism or chronic infection (79). In their capacity to induce Tregs and inhibit Th17, IDO1-expressing DCs and epithelial cells and kynurenines revealed an unexpected potential in the control of inflammation, allergy, and Th17-driven inflammation in these infections (51, 80).
Microbiota Regulation of Resistance and Tolerance to Fungi
Commensal-driven mucosal responses are upregulated in IDO1 deficiency (81) and IL-22 responses are upregulated in conditions of defective adaptive immunity (72) and IDO deficiency (75). AhR is a ligand-activated transcription factor that mediates IL-22 production (82). A variety of indole derivatives act as endogenous ligands for AhR (83) and are generated through conversion from dietary tryptophan by commensal intestinal microbes (84). Recent evidence has shown that AhR is involved in the (patho)physiology of skin including the regulation of skin pigmentation, photocarcinogenesis, and skin inflammation (85, 86). Of interest, the ability of Malassezia-derived indoles to activate AhR correlated with local immunoregulation (87) and pathogenicity in seborrheic dermatitis (88). Similarly, metabolomics has revealed that bioactive indoles with Ahr agonist activity are also present in mice with candidiasis (75). Thus, the trpyptophan metabolism pathway is exploited by commensals and the mammalian host to increase fitness in response to fungi via induction of resistance and tolerance at the skin and mucosal surface. The new findings support a model in which the IL-22 axis controls the initial fungal growth (i.e., resistance) and epithelial cells homeostasis likely exploiting primitive anti-fungal effector defense mechanisms. In contrast, the exploitation of the IFN-γ/IDO 1 axis for functional specialization of antifungal regulatory mechanisms (i.e., protective tolerance) may have allowed the fungal microbiota to co-evolutes with the mammalian immune system, survives in conditions of high-threat inflammation, and prevents dysregulated immunity (79). The two pathways, although non-redundant, are reciprocally regulated and compensate each other in the relative absence of either one (72), consistent with the theme that adaptive immunity depends on innate immunity but innate immunity requires adaptive regulation. This finding not only helps to explain the association of fungal infections with dysbiosis but also points to the essential help the microbiota may provide in fungal colonization and pathogenicity in immunodeficient patients.
Conclusion
Vertebrates have co-evolved with microorganisms resulting in a symbiotic relationship, which plays an important role in shaping host immunity. However, intestinal inflammation also dictates the composition of gut-associated microbial communities (89), a finding indicating the reciprocal influence of the microbiota and the mammalian immune status. The mycobiome is not an exception to the rule. The activation of different Th cells with distinct effector and immunoregulatory functions may impact differently on the local mycobiome composition. Indeed, the findings that fungi oppositely react to IFN-γ (90) or IL-17A (52), in terms of growth and virulence, suggest that the local Th environment may contribute to the diversity of the mycobiome at different body sites. Ultimately, fungi have evolved a contingency-based system during co-evolution to adapt to host immunity and persist in an inflammatory host environment. In turn, this feeds back into the host immune fitness. For instance, manipulation of the regulatory network of the host by the fungal microbiota, resulting in the activation of Treg-dependent immune tolerance, is a mechanism to ensure fungal survival and commensalism at different body sites, as well as local immune tolerance (76, 91, 92). Thus, challenging existing paradigms with new perspectives from the crosstalk between fungi, the immune system, and the microbiota will eventually lead toward the development of multi-pronged therapeutic approaches for mucosal and systemic fungal diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AhR, aryl hydrocarbon receptor; APS-1, autoimmune polyendocrine syndrome type 1 patients; CF, cystic fibrosis; CLR, C-type lectin receptors; CMC, chronic mucocutaneous candidiasis; DCs, dendritic cells; DTH, delayed type hypersensitivity; IDO1, indoleamine 2,3-dioxygenase 1; ILC3, innate lymphoid cells 3; IRIS, immune reconstitution inflammatory syndrome; PRRs, pattern recognition receptors; RVVC, recurrent VVC; Th, T helper; Treg, regulatory T-cells; VVC, human vulvovaginal candidiasis.
Acknowledgments
This work is supported by the Specific Targeted Research Project FUNMETA (ERC-2011-AdG-293714), project FFC#22/2014, and project HYPOXINFECT no. Z100CD11A1 to Luigina Romani. The authors thank Dr. Cristina Massi-Benedetti for editorial assistance.
References
1. Cui L, Morris A, Ghedin E. The human mycobiome in health and disease. Genome Med (2013) 5:63. doi: 10.1186/gm467
3. Segal BH, Herbrecht R, Stevens DA, Ostrosky-Zeichner L, Sobel J, Viscoli C, et al. Defining responses to therapy and study outcomes in clinical trials of invasive fungal diseases: Mycoses Study Group and European Organization for Research and Treatment of Cancer consensus criteria. Clin Infect Dis (2008) 47:674–83. doi:10.1086/590566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Pflughoeft KJ, Versalovic J. Human microbiome in health and disease. Annu Rev Pathol (2012) 7:99–122. doi:10.1146/annurev-pathol-011811-132421
5. Paulino LC, Tseng CH, Strober BE, Blaser MJ. Molecular analysis of fungal microbiota in samples from healthy human skin and psoriatic lesions. J Clin Microbiol (2006) 44:2933–41. doi:10.1128/JCM.00785-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Wang ZK, Yang YS, Stefka AT, Sun G, Peng LH. Review article: fungal microbiota and digestive diseases. Aliment Pharmacol Ther (2014) 39:751–66. doi:10.1111/apt.12665
7. Huffnagle GB, Noverr MC. The emerging world of the fungal microbiome. Trends Microbiol (2013) 21:334–41. doi:10.1016/j.tim.2013.04.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Underhill DM, Iliev ID. The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol (2014) 14:405–16. doi:10.1038/nri3684
9. Delhaes L, Monchy S, Frealle E, Hubans C, Salleron J, Leroy S, et al. The airway microbiota in cystic fibrosis: a complex fungal and bacterial community – implications for therapeutic management. PLoS One (2012) 7:e36313. doi:10.1371/journal.pone.0036313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Ott SJ, Kuhbacher T, Musfeldt M, Rosenstiel P, Hellmig S, Rehman A, et al. Fungi and inflammatory bowel diseases: alterations of composition and diversity. Scand J Gastroenterol (2008) 43:831–41. doi:10.1080/00365520801935434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Li Q, Wang C, Tang C, He Q, Li N, Li J. Dysbiosis of gut fungal microbiota is associated with mucosal inflammation in Crohn’s disease. J Clin Gastroenterol (2014) 48:513–23. doi:10.1097/MCG.0000000000000035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Zhang E, Tanaka T, Tajima M, Tsuboi R, Nishikawa A, Sugita T. Characterization of the skin fungal microbiota in patients with atopic dermatitis and in healthy subjects. Microbiol Immunol (2011) 55:625–32. doi:10.1111/j.1348-0421.2011.00364.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Smeekens SP, Huttenhower C, Riza A, van de Veerdonk FL, Zeeuwen PL, Schalkwijk J, et al. Skin microbiome imbalance in patients with STAT1/STAT3 defects impairs innate host defense responses. J Innate Immun (2014) 6:253–62. doi:10.1159/000351912
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Peleg AY, Hogan DA, Mylonakis E. Medically important bacterial-fungal interactions. Nat Rev Microbiol (2010) 8:340–9. doi:10.1038/nrmicro2313
15. Krause R, Schwab E, Bachhiesl D, Daxbock F, Wenisch C, Krejs GJ, et al. Role of Candida in antibiotic-associated diarrhea. J Infect Dis (2001) 184:1065–9. doi:10.1086/323550
16. Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y, et al. Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity (2014) 41:152–65. doi:10.1016/j.immuni.2014.05.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Noverr MC, Huffnagle GB. Does the microbiota regulate immune responses outside the gut? Trends Microbiol (2004) 12:562–8. doi:10.1016/j.tim.2004.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Perfect JR. The impact of the host on fungal infections. Am J Med (2012) 125:S39–51. doi:10.1016/j.amjmed.2011.10.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Romani L, Fallarino F, De Luca A, Montagnoli C, D’Angelo C, Zelante T, et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature (2008) 451:211–5. doi:10.1038/nature06471
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Iannitti RG, Carvalho A, Cunha C, De Luca A, Giovannini G, Casagrande A, et al. Th17/Treg imbalance in murine cystic fibrosis is linked to indoleamine 2,3-dioxygenase deficiency but corrected by kynurenines. Am J Respir Crit Care Med (2013) 187:609–20. doi:10.1164/rccm.201207-1346OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Singh N, Perfect JR. Immune reconstitution syndrome associated with opportunistic mycoses. Lancet Infect Dis (2007) 7:395–401. doi:10.1016/S1473-3099(07)70085-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Legrand F, Lecuit M, Dupont B, Bellaton E, Huerre M, Rohrlich PS, et al. Adjuvant corticosteroid therapy for chronic disseminated candidiasis. Clin Infect Dis (2008) 46:696–702. doi:10.1086/527390
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Casadevall A, Pirofski LA. The damage-response framework of microbial pathogenesis. Nat Rev Microbiol (2003) 1:17–24. doi:10.1038/nrmicro732
24. Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol (2010) 10:170–81. doi:10.1038/nri2711
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Wuthrich M, Deepe GS Jr, Klein B. Adaptive immunity to fungi. Annu Rev Immunol (2012) 30:115–48. doi:10.1146/annurev-immunol-020711-074958
26. Carvalho A, De Luca A, Bozza S, Cunha C, D’Angelo C, Moretti S, et al. TLR3 essentially promotes protective class I-restricted memory CD8(+) T-cell responses to Aspergillus fumigatus in hematopoietic transplanted patients. Blood (2012) 119:967–77. doi:10.1182/blood-2011-06-362582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. De Luca A, Iannitti RG, Bozza S, Beau R, Casagrande A, D’Angelo C, et al. CD4(+) T cell vaccination overcomes defective cross-presentation of fungal antigens in a mouse model of chronic granulomatous disease. J Clin Invest (2012) 122:1816–31. doi:10.1172/JCI60862
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Bozza S, Gaziano R, Spreca A, Bacci A, Montagnoli C, Di Francesco P, et al. Dendritic cells transport conidia and hyphae of Aspergillus fumigatus from the airways to the draining lymph nodes and initiate disparate Th responses to the fungus. J Immunol (2002) 168:1362–71. doi:10.4049/jimmunol.168.3.1362
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Ersland K, Wuthrich M, Klein BS. Dynamic interplay among monocyte-derived, dermal, and resident lymph node dendritic cells during the generation of vaccine immunity to fungi. Cell Host Microbe (2010) 7:474–87. doi:10.1016/j.chom.2010.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Rizzetto L, De Filippo C, Rivero D, Riccadonna S, Beltrame L, Cavalieri D. Systems biology of host-mycobiota interactions: dissecting dectin-1 and dectin-2 signalling in immune cells with DC-ATLAS. Immunobiology (2013) 218:1428–37. doi:10.1016/j.imbio.2013.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Gringhuis SI, Den Dunnen J, Litjens M, Van Der Vlist M, Wevers B, Bruijns SC, et al. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol (2009) 10:203–13. doi:10.1038/ni.1692
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Roy RM, Klein BS. Dendritic cells in antifungal immunity and vaccine design. Cell Host Microbe (2012) 11:436–46. doi:10.1016/j.chom.2012.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Romani L, Bistoni F, Puccetti P. Fungi, dendritic cells and receptors: a host perspective of fungal virulence. Trends Microbiol (2002) 10:508–14. doi:10.1016/S0966-842X(02)02460-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood (2013) 121:2402–14. doi:10.1182/blood-2012-09-378653
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Jarvis JN, Casazza JP, Stone HH, Meintjes G, Lawn SD, Levitz SM, et al. The phenotype of the Cryptococcus-specific CD4+ memory T-cell response is associated with disease severity and outcome in HIV-associated cryptococcal meningitis. J Infect Dis (2013) 207:1817–28. doi:10.1093/infdis/jit099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. de Oliveira LL, Coltri KC, Cardoso CR, Roque-Barreira MC, Panunto-Castelo A. T helper 1-inducing adjuvant protects against experimental paracoccidioidomycosis. PLoS Negl Trop Dis (2008) 2:e183. doi:10.1371/journal.pntd.0000183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Spellberg B, Ibrahim AS, Lin L, Avanesian V, Fu Y, Lipke P, et al. Antibody titer threshold predicts anti-candidal vaccine efficacy even though the mechanism of protection is induction of cell-mediated immunity. J Infect Dis (2008) 197:967–71. doi:10.1086/529204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Ouederni M, Sanal O, Ikinciogullari A, Tezcan I, Dogu F, Sologuren I, et al. Clinical features of candidiasis in patients with inherited interleukin 12 receptor beta1 deficiency. Clin Infect Dis (2014) 58:204–13. doi:10.1093/cid/cit722
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Moraes-Vasconcelos D, Grumach AS, Yamaguti A, Andrade ME, Fieschi C, De Beaucoudrey L, et al. Paracoccidioides brasiliensis disseminated disease in a patient with inherited deficiency in the beta1 subunit of the interleukin (IL)-12/IL-23 receptor. Clin Infect Dis (2005) 41:e31–7. doi:10.1086/432119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Puel A, Cypowyj S, Marodi L, Abel L, Picard C, Casanova JL. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol (2012) 12:616–22. doi:10.1097/ACI.0b013e328358cc0b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature (2008) 452:773–6. doi:10.1038/nature06764
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Eyerich K, Rombold S, Foerster S, Behrendt H, Hofmann H, Ring J, et al. Altered, but not diminished specific T cell response in chronic mucocutaneous candidiasis patients. Arch Dermatol Res (2007) 299:475–81. doi:10.1007/s00403-007-0792-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Ryan KR, Hong M, Arkwright PD, Gennery AR, Costigan C, Dominguez M, et al. Impaired dendritic cell maturation and cytokine production in patients with chronic mucocutanous candidiasis with or without APECED. Clin Exp Immunol (2008) 154:406–14. doi:10.1111/j.1365-2249.2008.03778.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med (2011) 365:54–61. doi:10.1056/NEJMoa1100102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Vinh DC. Insights into human antifungal immunity from primary immunodeficiencies. Lancet Infect Dis (2011) 11:780–92. doi:10.1016/S1473-3099(11)70217-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Pandiyan P, Conti HR, Zheng L, Peterson AC, Mathern DR, Hernandez-Santos N, et al. CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 cell infection model. Immunity (2011) 34:422–34. doi:10.1016/j.immuni.2011.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Zelante T, De Luca A, Bonifazi P, Montagnoli C, Bozza S, Moretti S, et al. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol (2007) 37:2695–706. doi:10.1002/eji.200737409
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Wuthrich M, Gern B, Hung CY, Ersland K, Rocco N, Pick-Jacobs J, et al. Vaccine-induced protection against 3 systemic mycoses endemic to North America requires Th17 cells in mice. J Clin Invest (2011) 121:554–68. doi:10.1172/JCI43984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Cheng SC, van de Veerdonk F, Smeekens S, Joosten LA, Van Der Meer JW, Kullberg BJ, et al. Candida albicans dampens host defense by downregulating IL-17 production. J Immunol (2010) 185:2450–7. doi:10.4049/jimmunol.1000756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Loures FV, Pina A, Felonato M, Calich VL. TLR2 is a negative regulator of Th17 cells and tissue pathology in a pulmonary model of fungal infection. J Immunol (2009) 183:1279–90. doi:10.4049/jimmunol.0801599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Romani L, Zelante T, De Luca A, Fallarino F, Puccetti P. IL-17 and therapeutic kynurenines in pathogenic inflammation to fungi. J Immunol (2008) 180:5157–62. doi:10.4049/jimmunol.180.8.5157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Zelante T, Iannitti RG, De Luca A, Arroyo J, Blanco N, Servillo G, et al. Sensing of mammalian IL-17A regulates fungal adaptation and virulence. Nat Commun (2012) 3:683. doi:10.1038/ncomms1685
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Ahlgren KM, Moretti S, Ardesjö Lundgren B, Karlsson I, Åhlin E, Norling A, et al. Increased IL-17A secretion in response to Candida albicans in autoimmune polyendocrine syndrome type 1 and its animal model. Eur J Immunol (2011) 41:235–45. doi:10.1002/eji.200939883
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med (2010) 207:299–308. doi:10.1084/jem.20091669
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med (2010) 207:291–7. doi:10.1084/jem.20091983
56. Wildbaum G, Nahir MA, Karin N. Beneficial autoimmunity to proinflammatory mediators restrains the consequences of self-destructive immunity. Immunity (2003) 19:679–88. doi:10.1016/S1074-7613(03)00291-7
57. Kreindler JL, Steele C, Nguyen N, Chan YR, Pilewski JM, Alcorn JF, et al. Vitamin D3 attenuates Th2 responses to Aspergillus fumigatus mounted by CD4+ T cells from cystic fibrosis patients with allergic bronchopulmonary aspergillosis. J Clin Invest (2010) 120:3242–54. doi:10.1172/JCI42388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Bhatia S, Fei M, Yarlagadda M, Qi Z, Akira S, Saijo S, et al. Rapid host defense against Aspergillus fumigatus involves alveolar macrophages with a predominance of alternatively activated phenotype. PLoS One (2011) 6:e15943. doi:10.1371/journal.pone.0015943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Nelson MP, Christmann BS, Werner JL, Metz AE, Trevor JL, Lowell CA, et al. IL-33 and M2a alveolar macrophages promote lung defense against the atypical fungal pathogen Pneumocystis murina. J Immunol (2011) 186:2372–81. doi:10.4049/jimmunol.1002558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Guimaraes AJ, Frases S, Gomez FJ, Zancope-Oliveira RM, Nosanchuk JD. Monoclonal antibodies to heat shock protein 60 alter the pathogenesis of Histoplasma capsulatum. Infect Immun (2009) 77:1357–67. doi:10.1128/IAI.01443-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Casadevall A, Pirofski LA. A reappraisal of humoral immunity based on mechanisms of antibody-mediated protection against intracellular pathogens. Adv Immunol (2006) 91:1–44. doi:10.1016/S0065-2776(06)91001-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. McClelland EE, Nicola AM, Prados-Rosales R, Casadevall A. Ab binding alters gene expression in Cryptococcus neoformans and directly modulates fungal metabolism. J Clin Invest (2010) 120:1355–61. doi:10.1172/JCI38322
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Subramaniam KS, Datta K, Quintero E, Manix C, Marks MS, Pirofski LA. The absence of serum IgM enhances the susceptibility of mice to pulmonary challenge with Cryptococcus neoformans. J Immunol (2010) 184:5755–67. doi:10.4049/jimmunol.0901638
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Kaplan MH. Th9 cells: differentiation and disease. Immunol Rev (2013) 252:104–15. doi:10.1111/imr.12028
65. Schlapbach C, Gehad A, Yang C, Watanabe R, Guenova E, Teague JE, et al. Human TH9 cells are skin-tropic and have autocrine and paracrine proinflammatory capacity. Sci Transl Med (2014) 6:219ra218. doi:10.1126/scitranslmed.3007828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol (2011) 12:1071–7. doi:10.1038/ni.2133
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Wilhelm C, Turner JE, Van Snick J, Stockinger B. The many lives of IL-9: a question of survival? Nat Immunol (2012) 13:637–41. doi:10.1038/ni.2303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Trifari S, Spits H. IL-22-producing CD4+ T cells: middle-men between the immune system and its environment. Eur J Immunol (2010) 40:2369–71. doi:10.1002/eji.201040848
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol (2009) 10:857–63. doi:10.1038/ni.1767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Kagami S, Rizzo HL, Lee JJ, Koguchi Y, Blauvelt A. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol (2010) 130:1373–83. doi:10.1038/jid.2009.399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Liu Y, Yang B, Zhou M, Li L, Zhou H, Zhang J, et al. Memory IL-22-producing CD4+ T cells specific for Candida albicans are present in humans. Eur J Immunol (2009) 39:1472–9. doi:10.1002/eji.200838811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. De Luca A, Zelante T, D’Angelo C, Zagarella S, Fallarino F, Spreca A, et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol (2010) 3:361–73. doi:10.1038/mi.2010.22
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Zelante T, Iannitti R, De Luca A, Romani L. IL-22 in antifungal immunity. Eur J Immunol (2011) 41:270–5. doi:10.1002/eji.201041246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. De Luca A, Carvalho A, Cunha C, Iannitti RG, Pitzurra L, Giovannini G, et al. IL-22 and IDO1 affect immunity and tolerance to murine and human vaginal candidiasis. PLoS Pathog (2013) 9:e1003486. doi:10.1371/journal.ppat.1003486
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity (2013) 39:372–85. doi:10.1016/j.immuni.2013.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Romani L, Puccetti P. Protective tolerance to fungi: the role of IL-10 and tryptophan catabolism. Trends Microbiol (2006) 14:183–9. doi:10.1016/j.tim.2006.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr Top Microbiol Immunol (2014) 380:39–68. doi:10.1007/978-3-662-43492-5_3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Bedke T, Iannitti RG, De Luca A, Giovannini G, Fallarino F, Berges C, et al. Distinct and complementary roles for Aspergillus fumigatus-specific Tr1 and Foxp3 regulatory T cells in humans and mice. Immunol Cell Biol (2014) 92:659–70. doi:10.1038/icb.2014.34
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Zelante T, Fallarino F, Bistoni F, Puccetti P, Romani L. Indoleamine 2,3-dioxygenase in infection: the paradox of an evasive strategy that benefits the host. Microbes Infect (2009) 11:133–41. doi:10.1016/j.micinf.2008.10.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Grohmann U, Volpi C, Fallarino F, Bozza S, Bianchi R, Vacca C, et al. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med (2007) 13:579–86. doi:10.1038/nm1563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Harrington L, Srikanth CV, Antony R, Rhee SJ, Mellor AL, Shi HN, et al. Deficiency of indoleamine 2,3-dioxygenase enhances commensal-induced antibody responses and protects against Citrobacter rodentium-induced colitis. Infect Immun (2008) 76:3045–53. doi:10.1128/IAI.00193-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol (2009) 10:864–71. doi:10.1038/ni.1770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Heath-Pagliuso S, Rogers WJ, Tullis K, Seidel SD, Cenijn PH, Brouwer A, et al. Activation of the Ah receptor by tryptophan and tryptophan metabolites. Biochemistry (1998) 37:11508–15. doi:10.1021/bi980087p
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc Natl Acad Sci U S A (1991) 88:9543–7. doi:10.1073/pnas.88.21.9543
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Esser C, Bargen I, Weighardt H, Haarmann-Stemmann T, Krutmann J. Functions of the aryl hydrocarbon receptor in the skin. Semin Immunopathol (2013) 35:677–91. doi:10.1007/s00281-013-0394-4
86. Di Meglio P, Duarte JH, Ahlfors H, Owens ND, Li Y, Villanova F, et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity (2014) 40:989–1001. doi:10.1016/j.immuni.2014.04.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Vlachos C, Schulte BM, Magiatis P, Adema GJ, Gaitanis G. Malassezia-derived indoles activate the aryl hydrocarbon receptor and inhibit toll-like receptor-induced maturation in monocyte-derived dendritic cells. Br J Dermatol (2012) 167:496–505. doi:10.1111/j.1365-2133.2012.11014.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Gaitanis G, Magiatis P, Stathopoulou K, Bassukas ID, Alexopoulos EC, Velegraki A, et al. AhR ligands, malassezin, and indolo[3,2-b]carbazole are selectively produced by Malassezia furfur strains isolated from seborrheic dermatitis. J Invest Dermatol (2008) 128:1620–5. doi:10.1038/sj.jid.5701252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Faber F, Baumler AJ. The impact of intestinal inflammation on the nutritional environment of the gut microbiota. Immunol Lett (2014) S0165-2478(14):83–82. doi:10.1016/j.imlet.2014.04.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Kalo-Klein A, Witkin SS. Prostaglandin E2 enhances and gamma interferon inhibits germ tube formation in Candida albicans. Infect Immun (1990) 58:260–2.
91. Bonifazi P, Zelante T, D’Angelo C, De Luca A, Moretti S, Bozza S, et al. Balancing inflammation and tolerance in vivo through dendritic cells by the commensal Candida albicans. Mucosal Immunol (2009) 2:362–74. doi:10.1038/mi.2009.17
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Bonifazi P, D’Angelo C, Zagarella S, Zelante T, Bozza S, De Luca A, et al. Intranasally delivered siRNA targeting PI3K/Akt/mTOR inflammatory pathways protects from aspergillosis. Mucosal Immunol (2010) 3:193–205. doi:10.1038/mi.2009.130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Th cell subsets, immunity, tolerance, fungi
Citation: Borghi M, Renga G, Puccetti M, Oikonomou V, Palmieri M, Galosi C, Bartoli A and Romani L (2014) Antifungal Th immunity: growing up in family. Front. Immunol. 5:506. doi: 10.3389/fimmu.2014.00506
Received: 22 August 2014; Paper pending published: 15 September 2014;
Accepted: 28 September 2014; Published online: 15 October 2014.
Edited by:
Dragana Jankovic, National Institutes of Health, USAReviewed by:
Edward John Collins, The University of North Carolina at Chapel Hill, USACosima T. Baldari, University of Siena, Italy
Copyright: © 2014 Borghi, Renga, Puccetti, Oikonomou, Palmieri, Galosi, Bartoli and Romani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luigina Romani, Pathology Section, Department of Experimental Medicine, University of Perugia, Polo Unico Sant’Andrea delle Fratte, Perugia 06132, Italy e-mail:bHVpZ2luYS5yb21hbmlAdW5pcGcuaXQ=