 Bin Zhang
Bin Zhang Yijun Yin
Yijun Yin Ruenn Chai Lai
Ruenn Chai Lai Sai Kiang Lim
Sai Kiang Lim- 1Exosome and Secreted Nano-vesicle Group, A*STAR Institute of Medical Biology, Singapore
- 2Department of Surgery, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
Extracellular vesicle or EV is a term that encompasses all classes of secreted lipid membrane vesicles. Despite being scientific novelties, EVs are gaining importance as a mediator of important physiological and pathological intercellular activities possibly through the transfer of their cargo of protein and RNA between cells. In particular, exosomes, the currently best characterized EVs have been notable for their in vitro and in vivo immunomodulatory activities. Exosomes are nanometer-sized endosome-derived vesicles secreted by many cell types and their immunomodulatory potential is independent of their cell source. Besides immune cells such as dendritic cells, macrophages, and T cells, cancer and stem cells also secrete immunologically active exosomes that could influence both physiological and pathological processes. The immunological activities of exosomes affect both innate and adaptive immunity and include antigen presentation, T cell activation, T cell polarization to regulatory T cells, immune suppression, and anti-inflammation. As such, exosomes carry much immunotherapeutic potential as a therapeutic agent and a therapeutic target.
Introduction
Extracellular vesicles (EVs) are increasingly implicated as a major mode of intercellular communication. Most cell types are known to secrete EVs which are essentially bi-lipid membrane vesicles carrying a complex cargo of proteins and RNAs. These EVs can be taken up by other cell types thereby transferring proteins and RNAs from one cell to another.
There are many classes of EVs such as exosomes, microvesicles, ectosomes, membrane particles, exosome-like vesicles and apoptotic bodies, and they could be distinguished by their biogenesis pathway, size, flotation density on a sucrose gradient, lipid composition, sedimentation force, and protein cargo [reviewed in Ref. (1, 2)]. However, many of these differentiating parameters such as size, flotation density on a sucrose gradient, lipid composition, sedimentation force, and protein cargo are not discrete values that are exclusive to a specific class of EVs. Consequently, classification of EVs has been challenging. Exosomes are presently the best characterized EVs. They are defined as membrane vesicles of 50–100 nm diameter containing proteins, RNAs, and lipids (3–8). They are secreted by cells when endosomal membranes invaginate inward to form multivesicular bodies (MVBs) and the MVs fused with the plasma membrane. This endosomal biogenesis is a distinctive feature of exosomes, and is presently known to be unique to exosomes and not any of the other classes of EVs. As endocytosis is most active at specialized microdomains in plasma membrane such as lipid rafts, exosomes such as mesenchymal stem cell (MSC) exosomes have membranes enriched in elements of lipid rafts such as GM1 gangliosides and transferrin receptors (9). However, ascertaining the biogenesis of EVs is technically challenging and not always practical, and consequently, the term “exosomes” have been used generically to describe any EVs that share some of the biophysical or biochemical parameters of exosomes without validating their biogenesis. Hence, the term EVs and exosomes in this review will be used synonymously.
Many cell types are known to secrete EVs, and they include epithelial cells (10, 11), fibroblasts (12), erythrocytes (13, 14), platelets (15), mast cells (16), tumor cells (17–19), stem cells (20–22), and immune cells such as dendritic cells (DCs) (23, 24), monocytes (25, 26), macrophages (27, 28), NK cells (29, 30), B lymphocytes (31, 32), and T lymphocytes (33, 34). However, the physiological functions of EVs remain tenuous. Part of this could be attributed to the lack of a definitive criterion to purify, characterize, and classify the classes of EVs unambiguously.
Exosomes were first thought to be a cellular means for the disposal of redundant proteins by groups studying reticulocyte maturation (35–37), but they are now generally viewed as mediators of intercellular communication through the transfer of biologically active materials. However, with the present lack of clarity in defining the classes of EVs, the role of EVs or exosomes as mediators of intercellular communication, and their effects on biological or physiological processes remains a conundrum. For example, do cells secrete or even have the capacity to produce all classes of EVs? Do cells secrete more than one class of EVs at any one time? Are secretion or production of EVs and their cargo regulated? It is likely that our present day understanding of EV functions is an amalgam of the diametrically different functions of different EV classes, leading to a confusing and sometimes contradictory perception of EV or exosome functions. One of the earliest reported physiological targets of exosome-mediated intercellular communication is the immune system. B lymphocytes were the first immune cell type reported to secrete vesicles and these vesicles express abundant major histocompatibility complex (MHC) Class I and II molecules, B7.1 (CD80) and B7.2 (CD86) co-stimulatory molecules, and ICAM-1 (CD54) adhesion molecules. Consistent with the presence of these proteins, which are important in antigen presentation, the vesicles could activate CD4+ T cells suggesting that they mediate antigen presentation by lymphocytes (38). Subsequently, other immune cells such as DCs and mast cells were also found to secrete EVs/exosomes (39, 40). Incidentally, non-immune cells have also been reported to modulate the immune system by exosomes. For example, cancer cells are known to modulate the immune system to facilitate growth and metastasis. One of the earliest reports described the downregulation of surface NKG2D expression on NK cells and CD8+ T cells by exosomes from cancer cells or mesothelioma patients (41). We have also demonstrated that exosomes from MSCs modulate the immune system through TLRs and induce expansion of regulatory T cells (Tregs) (22). Together, these studies demonstrate the immense immunotherapeutic potential of exosomes produced by both immune and non-immune cells and also highlight the divergent effects of EVs/exosomes on the immune system. While this divergence could be due to a difference in cell source of the EVs/exosomes, other parameters such as the classes of EVs and the methods of isolation are equally important.
Biogenesis and Preparation of EVs
Among the EVs, exosomes are best defined in terms of their biogenesis and consequently, are the best characterized EVs. Exosomes are small membrane vesicles about 50–100 nm in diameter that are secreted by cells into the extracellular compartment when MVBs in the cells fuse with the plasma membrane [reviewed in Ref. (42)]. MVBs in turn are formed by membrane invagination of late endosomes. Therefore, all cells that could generate MVBs have the potential to produce exosomes. As the biogenesis of exosomes from endosomes involves a reverse budding process, exosome membranes have the same directional orientation as cells with the cytosol inside the luminal space, and extracellular domains of membrane proteins exposed at external surface. The release of exosomes through fusion of MVBs with the plasma membrane has been documented by electron microscopy while the endosomal origin of exosomes are supported by the enrichment of endosome-associated proteins such as the Rab proteins, ALIX, TSG101, or endocytic proteins such as transferring receptors, clathrins (3, 9, 43, 44) (Figure 1).
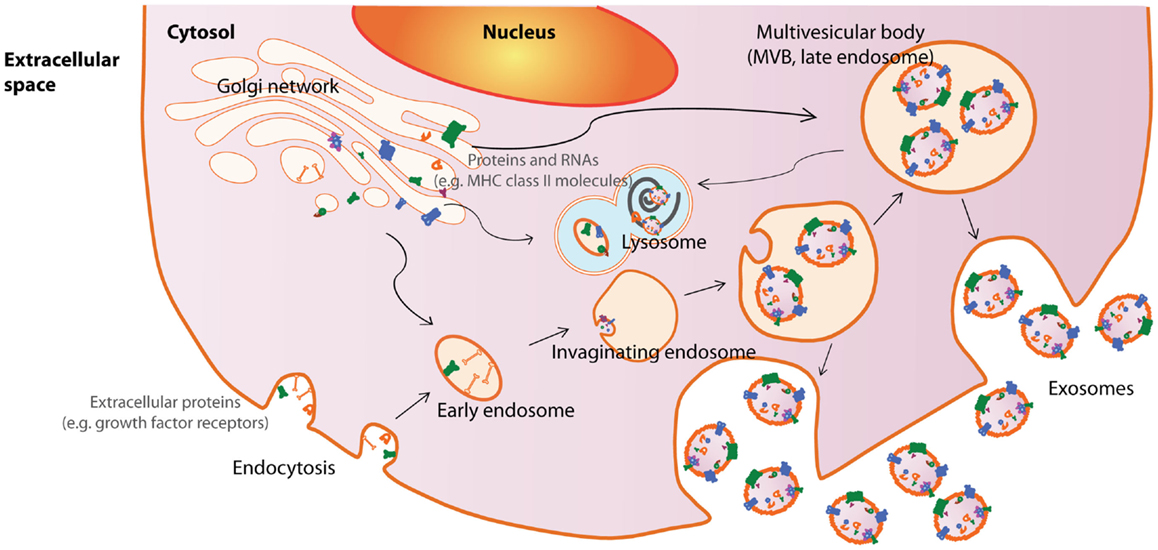
Figure 1. Exosome biogenesis and secretion. Exosomes are released by cells when intracellular organelles called multivesicular bodies (MVBs) fuse with the plasma membrane. MVBs are formed by invaginations of late endosomes, which contained molecules from the Golgi (e.g., MHC class II molecules) or the cell surface (e.g., growth factor receptors). Consequently, exosomes contain cytosolic materials and are enriched in endosome-associated protein markers such as the the Rab proteins, ALIX, TSG101, and MHC class II molecules or endocytic proteins, such as transferrin receptors and clathrins. This figure was modified from Lai et al. (169).
Since EVs are released to the extracellular space, these vesicles are routinely purified from cell culture supernatants or biological fluids. EV purification from cell-conditioned culture media classically involves a series of differential centrifugations to remove dead cells and large debris, and then a final high speed ultracentrifugation of about 100,000 × g for at least 1 h to pellet EVs (37, 45, 46). As exosomes have a characteristic flotation density of 1.13–1.21 g/ml, an additional equilibrium density gradient centrifugation is often used to enhance the purity of the exosome preparation. However, ultracentrifugation has poor scalability and is thus not amenable to large scale manufacturing processes. Another common method is to isolate EVs or exosomes by their size as in size exclusion chromatography (47, 48). To enhance the resolution of size exclusion chromatography, our laboratory routinely uses high-performance liquid chromatography (HPLC) to purify exosomes from MSC-conditioned medium (49). Other methods of isolation include immunoaffinity chromatography where exosomes are captured using antibody that recognizes a marker enriched on exosomes such as MHC class II molecules for antigen-presenting cells (APC)-derived exosomes (50), A33 (5), or EpCAM (51) for exosomes from tumor cell line, or CD63, which could theoretically be used for exosomes from all sources (52, 53). However, these isolation techniques frequently require biologically harsh conditions, such as low pH or high salt concentration, to release the bound EVs such that the biological activity of the EVs would be compromised. Like the other isolation techniques, these methods may not differentiate between different classes of EVs or between EVs and large protein complexes. Despite the challenges in isolating specific classes of EVs, the isolation of exosomes for use in clinical testing has already been described (54, 55). The isolation process usually includes a filtration step to isolate large complexes followed by equilibrium density gradient centrifugation to isolate complexes according to density.
EVs and Immunity
Vertebrates have two arms of immune systems, namely the innate and adaptive immune systems. The innate immune system is an evolutionarily conserved immune system found in all multi-cellular organisms while the adaptive immune system is found only in vertebrates (56). The innate immune system is generally the first line of defense against microbial pathogens or tissue damage and mediates inflammation. It is activated through a limited set of germline-encoded receptors that recognizes pathogen-associated molecular patterns (PAMPs) from infectious agents, or damage-associated molecular patterns (DAMPs) releasing from dying cells (57, 58). These germline-encoded receptors are often referred to as pattern recognition receptors (PRRs). In mammals, Toll-like receptors (TLRs) are the best characterized examples. In addition, other receptors such as nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and some C-type lectin receptors (CLRs) also recognize specific components of microbes and are therefore considered as innate immune receptors (59, 60). In contrast, antigen receptors in the adaptive immune system are not germline-encoded but generated through somatic hypermutations (61). Hence the adaptive immune system has an immensely wider repertoire of antigen receptors compared to the limited set of receptors for PAMPs or DAMPs in the innate immune system. As such, the adaptive immune system also has the potential to fine tune its antigen recognition to a high degree of specificity and avidity. Induction of adaptive immunity depends on antigen recognition by antigen receptors on adaptive immune cells followed by subsequent activation and clonal expansion of cells carrying the appropriate antigen-specific receptors (62). The two main cell types in adaptive immune system are T and B cells. Unlike B cells, naïve T cells do not recognize antigens and their capacity to recognize antigens have to be activated through a process known as antigen presentation. During antigen presentation, APCs such as DCs or macrophages internalize foreign antigens, process, and load the processed antigens onto MHC I and MHC II molecules for presentation to naïve CD8+ and CD4+ T cells, respectively. Together with co-stimulatory molecules such as CD80 and CD86, the antigen-MHC class I or II complex on APCs activates T cells and imparts the memory of the antigen to the T cells (63–66).
Antigen presentation was generally assumed to be a cell–cell interaction until it was discovered that Epstein–Barr virus-transformed B lymphocytes secreted exosomes carrying antigenic peptide bound-MHC class II dimers. These exosomes could induce antigen-specific MHC class II-restricted T cell response (Figure 2A) (38). Subsequently, DCs were also found to secrete exosomes with MHC class I and II, and T cell co-stimulatory molecules. When pulsed with tumor peptides, DCs secrete exosomes that could activate cytotoxic T lymphocytes and suppress tumor growth in a T cell-dependent manner (Figure 2B) (23). It was further observed that allogeneic and autologous exosomes were equally efficient in eliciting anti-tumor protection (67). Since then, exosomes have been reported to play an extensive role in antigen presentation. Exosomes purified from ascites of tumor patients (68) or culture medium of tumor cell lines (17, 69) have been shown to carry tumor antigens. When pulsed with these exosomes, DCs endocytose the exosomes and present the exosome-associated tumor antigens on their MHC molecules to activate tumor-specific cytotoxic T lymphocytes (Figure 3) (17, 69–71). However, the role of exosomes in antigen presentation is not restricted to the conveyance of antigens to APCs for T cell activation. Some exosomes, particularly those derived from mature DCs are known to carry MHC II–peptide complexes and they could activate T lymphocytes directly (Figure 2B). For example, DC-derived exosomes could activate CD8+ cytotoxic T lymphocytes clones by themselves (72–74). However, it has been reported that such activation is generally inefficient compared to activation in the presence of DCs (75).
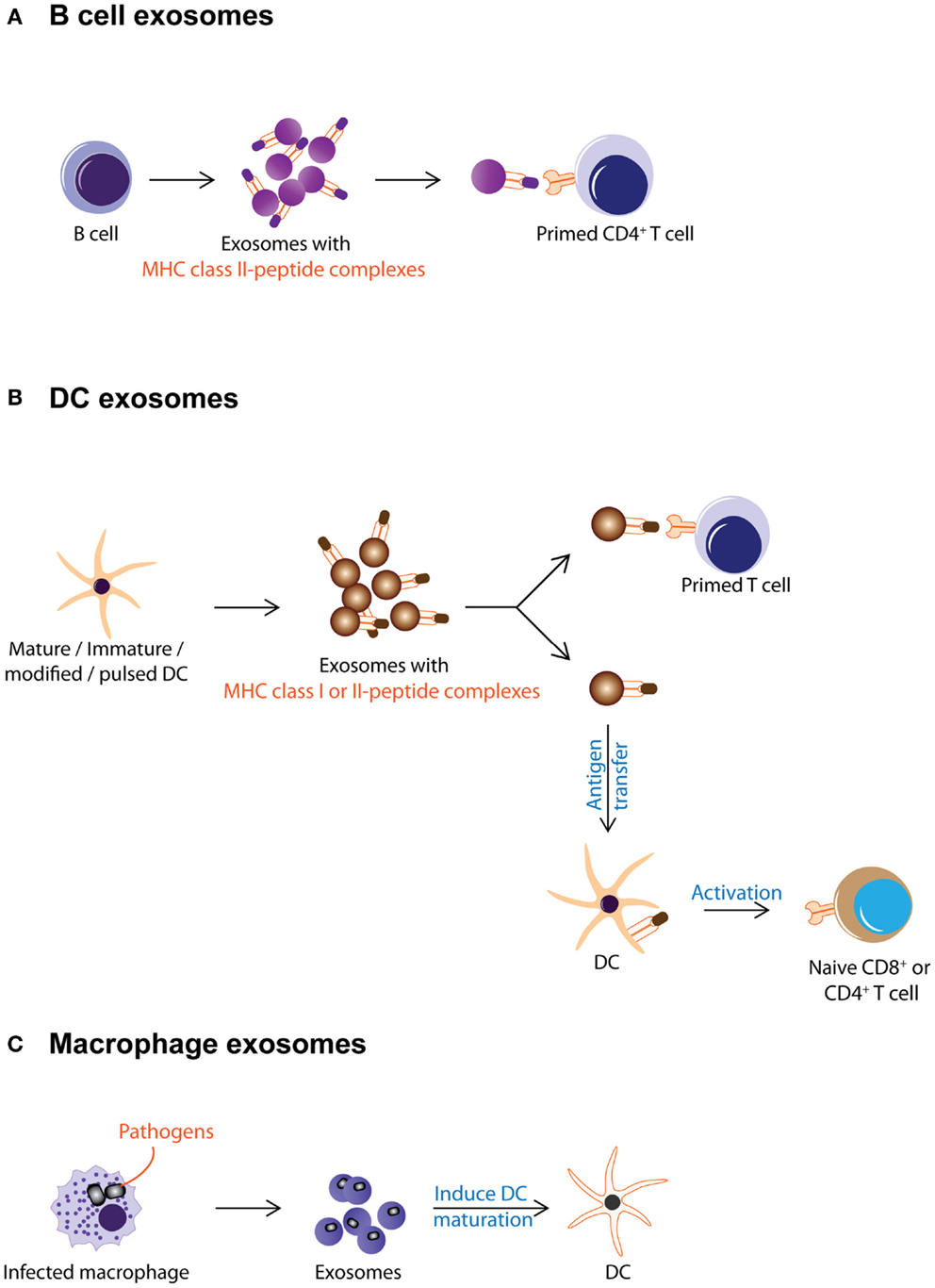
Figure 2. Exosomes from immune cells are shown. This figure summarizes the different known activities of exosomes from immune cells. (A) B cell secretes exosomes carrying MHC class II–peptide complexes as a mode of antigen presentation to primed CD4+ T cell. (B) DC-derived exosomes carry MHC class I or II-peptide complexes that can be either directly recognized by pre-activated CD4+ or CD8+ T cells or captured and presented by DCs to activate naïve T cells. (C) Macrophages infected with pathogens secrete exosomes with pathogen antigens. These exosomes induce maturation of DCs and promote secretion of pro-inflammatory cytokines.
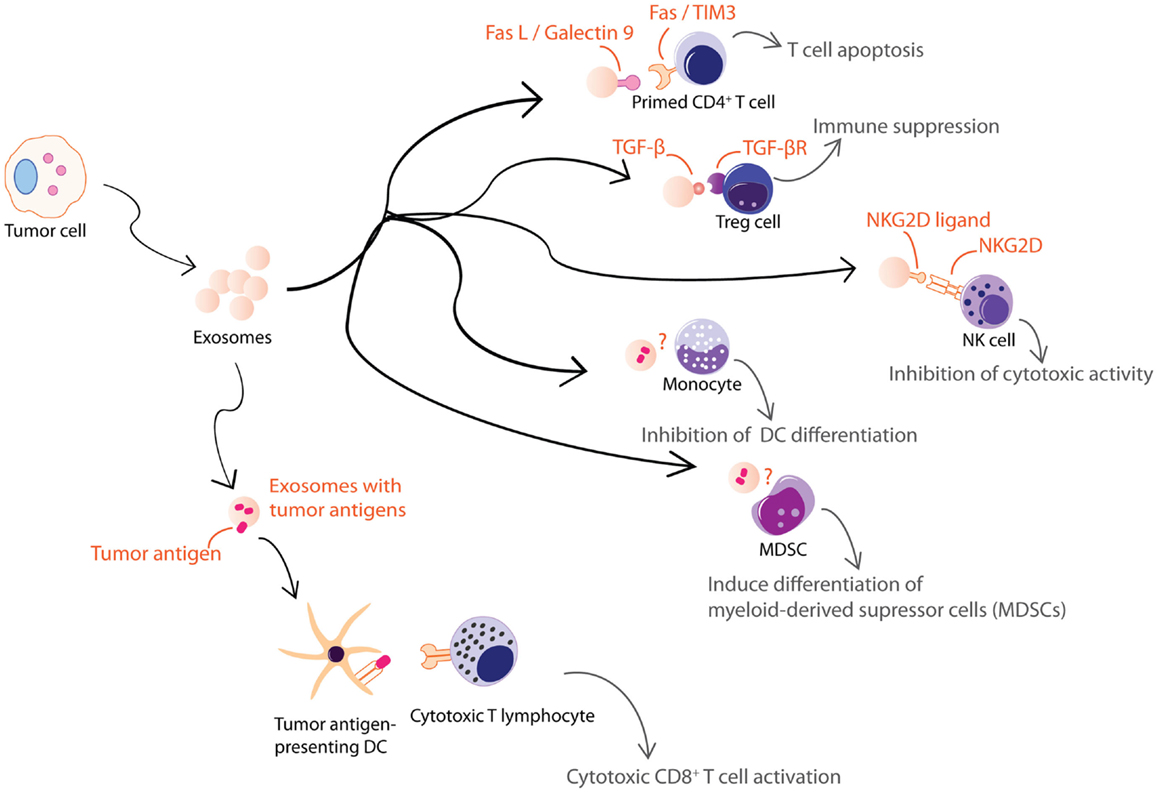
Figure 3. Exosomes from tumor cells are shown. Tumor cell-derived exosomes can be either immune suppressive or activating. These exosomes exert some of their immune suppressive activities on T cells through FasL, galectin-9, TGFβ, or NKG2D ligand carried by the exosomes. Others inhibit differentiation of DCs or MDSCs through unknown mechanisms. However, tumor cell-derived exosomes also carry tumor antigens that could elicit an anti-tumor response.
The antigen-presenting capacity of exosomes from DCs depends on the maturation state of the DCs with less exosomes being produced as the cells mature (76–79). Unlike exosomes from immature DCs, exosomes from mature DCs carry MHC class II and co-stimulatory molecules such as B7.2, ICAM-1, but not MFG-E8 on their membrane (70, 80–84). As such, they can interact directly with T cells to activate the immune system and eradicate tumor or bacteria or virus (23, 85, 86). However, these MHC class II-expressing exosomes have also been implicated in immune suppression and prolonged survival of allografts. When administered to rats prior to heart allograft transplantation, MHC class II-expressing exosomes from donor DCs prolong graft survival with a significant concomitant decrease in recipient CD4+ T cells and increased anti-donor MHC class II alloantibody production suggesting that these exosomes can modulate immune response by either inducing tolerance or immune reactivity (Figure 2B) (87).
On the other hand, exosomes from immature DCs while produced in greater abundance, are 50- to 100-fold less potent in T cell stimulation (82). This attenuated potency has been attributed to the absence of MHC class II and co-stimulatory molecules such as B7.2, ICAM-1 on their membrane (70, 80–84). Instead of T cell stimulation, these exosomes help distribute antigens to other APCs for antigen presentation and subsequent immune modulation (Figure 2B) (79). Additionally, exosomes from immature DCs carry MFG-E8, which binds phosphatidylserine and opsonizes apoptotic cells for phagocytosis (88), possibly enhancing the antigen presentation function of immature DC exosomes.
Exosomes secreted by immature DCs could be either immune suppressive or activating. Exosomes derived from immature, gene-modified, or interleukin (IL)-10-treated DCs have been shown to induce immune suppression in murine models of transplantation and auto-immune disease (89–97). At the same time, when immature DCs were pulsed with pathogen-associated antigens such as Toxoplasma gondii antigen, diphtheria toxin, or Eimeria tenella antigens, they produce exosomes that confer immune protection against these pathogens (23, 67, 85, 86, 98–100). Incidentally, this immune protection is known to include polyclonal activation of B cells as well (101). Thus exosomes from immature DCs elicit the immune suppression or immune activation (Figure 2B) [reviewed in Ref. (90, 102, 103)].
This capacity of DC-derived exosome to suppress or activate the immune system indicates that exosome is an important component of the complex immune network in mediating a steady state between immune tolerance and rejection. It also serves an underpinning rationale for the use of DC-derived exosomes in vaccination trials to treat melanoma and non-small cell lung cancer patients (102).
Besides DCs, which secrete exosomes targeting mainly the adaptive immunity (104), other cell types when challenged also secrete immunologically active exosomes targeting primarily but not exclusively the innate immunity. Macrophages when infected by pathogens such as mycobacterium or toxoplasma, release EVs containing PAMPs that induce secretion of pro-inflammatory cytokines and recruitment of immune cells (Figure 2C) (105–107). These EVs have also been implicated in the progression of auto-immune diseases. For example, it was observed that fibroblasts from synovial fluid of patients with rheumatoid arthritis secreted EVs carrying active membrane-bound TNF-α can inhibit T cell activation-induced cell death (AICD) (108) and EVs present in the bronchoalveolar fluid of patients with sarcoidosis also displayed pro-inflammatory activities (109).
Tumor cells also secrete immunologically active EVs. Some of these EVs are pro-inflammatory while others are immune suppressive (Figure 3). Heat-shocked tumor cells secrete EVs that promote NK cell activity (110), secretion of TNF-α by macrophages (111), and stimulate T cell activation (112, 113). Anticancer drug-treated carcinoma cells release exosomes with heat shock proteins (HSPs) that elicit effective NK cell anti-tumor responses (114) while exosomes from ascites of gastric cancer patients induce a tumor-specific CTL response (115). On the other hand, EVs from tumor cell lines or tumor-bearing patients were found to carry various immunosuppressive molecules that when tested in vitro, induced T cell apoptosis via Fas ligand (FasL) (48, 116–118) and galectin-9 (119, 120), inhibited IL-2-induced T cell proliferation (121), and/or promote differentiation into Tregs (122, 123). They also decreased NK cell activity (41, 124, 125), inhibited myeloid precursor differentiation into DCs (126), and promoted the differentiation of myeloid-derived supressor cells (MDSCs) (127–130). Hence, tumor cells secrete EVs that could both inhibit tumor growth by eliciting anti-tumor immune responses (131) and promote tumor growth by inhibiting anti-tumor immunity or enhancing angiogenesis and/or metastases (132).
Aside from pathological situations, exosomes are also used to modulate immune activity to support normal physiological processes. Like tumor EVs, normal tissues also produce EVs that are functionally dichotomous (Figure 4). During early pregnancy, trophoblast secretes exosomes to recruit and educate monocytes to initiate a pro-inflammatory microenvironment essential for embryo implantation, angiogenesis, and stromal remodeling associated with early pregnancy (133). These exosomes induce macrophages to synthesize and release pro-inflammatory cytokines, including IL-1β via exosome-associated fibronectin (134). In contrast to the trophoblast, other tissues such as the prostate and placenta also secrete EVs to attenuate immune activity and facilitate normal physiological activity such as fertilization and pregnancy. It has been proposed that EVs in semen and secretion from placental explants mediate protection of the sperms and fetus from immune attack by the host tissues via NKG2D ligands, which reduce cytotoxicity of NK and CD8+ T cells (135, 136). EVs have also been implicated in pregnancy-associated immune suppression via the expression FasL, which is known to induce T cell anergy (137, 138). Other tissues that secrete EVs to attenuate inflammatory response include EVs secreted by intestinal epithelium to promote oral tolerance in rat (139) and EVs in milk and colostrum that exert immunosuppressive effects on T cells (140).
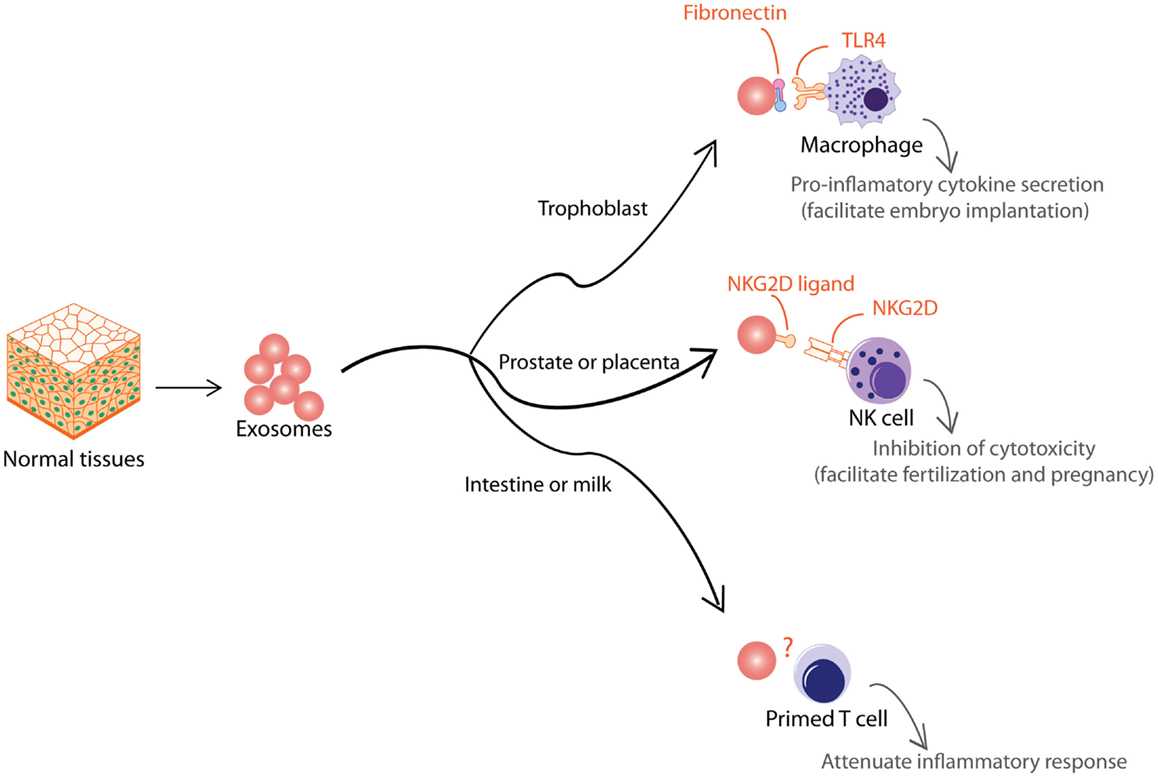
Figure 4. Exosomes from normal tissues are shown. Normal tissue-derived exosomes exert multiple immune-modulatory activities to support normal physiological processes such as embryo implantation, fertilization, pregnancy, and oral tolerance.
Therapeutic Stem Cells and Exosomes
Mesenchymal stem cell, a widely distributed stromal support cell in body, is the most widely used stem cell type in clinical trials today with more than 306 trials1 to treat many diseases including many immune diseases such as multiple sclerosis (141) and auto-immune rheumatologic disorders (142). MSCs have been reported to exert both suppressive and regulatory effects on autologous and allogeneic adaptive and innate immune cells (143) such as inhibiting mitogen-activated T cell proliferation (144–148), inducing an anti-inflammatory tolerant phenotype in immune cells (149) and inhibit B cell proliferation (150). These effects provide a rationale for the approval of MSC in the treatment of pediatric graft-versus-host disease (GVHD) in Canada and New Zealand, and in its testing against immune diseases such as Crohn’s disease (151) and type 1 diabetes (152). However, the molecular mechanism underpinning these immune activities remains unresolved. It is observed that the efficacy of MSCs against severe GVHD (153) does not impair graft versus-leukemia (GVL) reaction (154) or suppress T cell proliferation (155). Increasingly, it was proposed that MSC secretes factors such as interferon-γ (IFNγ), TGF-β, PGE2, HLA-G, IL-10, and indoleamine 2,3-dioxygenase (IDO) to expand Tregs, which in turn, attenuate GVHD [reviewed in Ref. (156)]. However over the years, each of these secreted factors could not by themselves adequately account for the immunomodulatory activity of MSCs [reviewed in Ref. (157)]. For example, the immunomodulatory activity of MSCs was not compromised by a lack of IDO production caused by either defective IFN(γ) receptor 1 or IDO inhibitors (158) thus eliminating interferon-γ (IFNγ) (159–161) and IDO (162) as the secreted candidates for MSC immune activity.
The molecular mechanism underpinning MSC-mediated immunoregulation is likely to be complex and mediated by the synergism of several rather than single molecules. Hence, exosomes or EVs, which carry a multitude of molecules, are ideal secreted agents to mediate the immune-modulatory activity of MSCs. MSCs were first reported to secrete microvesicles in 2009 (163) and exosomes in 2010 (49). Some features of the immunomodulatory property of MSCs have been replicated by purified MSC exosomes. In a mouse model of myocardial ischemia/reperfusion injury, MSC exosomes attenuate injury induced inflammatory response as evidenced by reduced white blood cell count and tissue infiltration of neutrophils (164). We have also shown that MSC exosomes delay the rejection of allogeneic skin grafts in mice with a concomitant increase in Tregs (22) and inhibit complement-mediated lysis of sheep red blood cells in a CD59-dependent manner (165).
EVs or Exosomes as Immunotherapeutic Agents
The extensive implication of EVs in modulating diverse aspects of the immune system to either enhance or suppress immune activity makes EVs attractive candidate immunotherapeutics. While this functional dichotomy increases the versatility of EVs as therapeutic agents, it also increases the risk of unpredictable adverse outcomes. To mitigate this risk, it is therefore critical that EVs with the appropriate functional properties are isolated for therapy. As discussed earlier, the capacity of EVs to enhance or suppress immune activity depends on the cell source, state of the cell source, e.g., maturity, and the type of EV. Hence a minimum requisite for the development of EVs as immunotherapeutic agents is a physical and biological characterization of each of the different EV types produced by a cell in a specific physiological state such that the EV type with appropriate functional properties can be isolated. Unfortunately, this is a highly intractable issue that is endemic in the EV research community. As discussed in the Section “Introduction,” there is presently a lack of clarity in defining the classes of EVs, the role of EVs or exosomes as mediators of intercellular communication. There is presently a concerted effort within the EV research community2 to first define and standardize the nomenclature for EVs and the EV isolation.
In spite of these challenges, the prospect of EV-based immunotherapy remains highly promising. Unlike cell-based immunotherapy, EV being a non-viable cellular product has many advantages. In cellular immunotherapy, the need to preserve cell viability increases a layer of complexity to its manufacture, storage, transport, and transplantation. In contrast, EVs are biologics and are thus more amenable to a strictly regulated and monitored manufacturing process. This will essentially translate into better qualified and safer off-the-shelf products that could be delivered to patients in a timely manner. The administration of EVs instead of cells would also alleviate many of the risks associated with viable cellular therapy. The use of viable replicating cells as therapeutic agents carries unique safety risks and challenges as the biological potency of the agent may persist or amplify even after the disease has been resolved, leading to adverse immune dysfunctions such as increased cancer risk and increased susceptibility to infection or auto-immune disease. In addition, the process of administering cells while generally safe (166) could cause complications such as occlusions in the distal microvasculature, as cells are generally much larger than the usual therapeutic small molecules or biologics (167). Furthermore, the differentiation potential of cells could generate inappropriate and potentially deleterious cell types. For example, the osteogenic and chondrogenic potential of MSC has raised safety concerns as a high frequency (51.2%) of ossifications and/or calcifications in tissues has been reported in some animal studies (168). In conclusion, the benefits of an EV-based immunotherapy vis-a-vis the risks of a cell-based immunotherapy provide a compelling rationale to develop an EV-based immunotherapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^http://www.clinicaltrials.gov/; accessed March 2013
- ^http://www.isev.org
References
1. Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol (2009) 9:581–93. doi: 10.1038/nri2567
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Duijvesz D, Luider T, Bangma CH, Jenster G. Exosomes as biomarker treasure chests for prostate cancer. Eur Urol (2011) 59:823–31. doi:10.1016/j.eururo.2010.12.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Escola JM, Kleijmeer MJ, Stoorvogel W, Griffith JM, Yoshie O, Geuze HJ. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem (1998) 273:20121–7. doi:10.1074/jbc.273.32.20121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Mathivanan S, Simpson RJ. ExoCarta: a compendium of exosomal proteins and RNA. Proteomics (2009) 9:4997–5000. doi:10.1002/pmic.200900351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Simpson RJ, Jensen SS, Lim JW. Proteomic profiling of exosomes: current perspectives. Proteomics (2008) 8:4083–99. doi:10.1002/pmic.200800109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Duijvesz D, Burnum-Johnson KE, Gritsenko MA, Hoogland AM, Vredenbregt-van den Berg MS, Willemsen R, et al. Proteomic profiling of exosomes leads to the identification of novel biomarkers for prostate cancer. PLoS One (2013) 8:e82589. doi:10.1371/journal.pone.0082589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Laulagnier K, Motta C, Hamdi S, Roy S, Fauvelle F, Pageaux JF, et al. Mast cell- and dendritic cell-derived exosomes display a specific lipid composition and an unusual membrane organization. Biochem J (2004) 380:161–71. doi:10.1042/BJ20031594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Subra C, Laulagnier K, Perret B, Record M. Exosome lipidomics unravels lipid sorting at the level of multivesicular bodies. Biochimie (2007) 89:205–12. doi:10.1016/j.biochi.2006.10.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Tan SS, Yin Y, Lee T, Lai RC, Yeo RW, Zhang B, et al. Therapeutic MSC exosomes are derived from lipid raft microdomains in the plasma membrane. J Extracell Vesicles (2013):2. doi:10.3402/jev.v2i0.22614
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. van Niel G, Raposo G, Candalh C, Boussac M, Hershberg R, Cerf-Bensussan N, et al. Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology (2001) 121:337–49. doi:10.1053/gast.2001.26263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Lin XP, Almqvist N, Telemo E. Human small intestinal epithelial cells constitutively express the key elements for antigen processing and the production of exosomes. Blood Cells Mol Dis (2005) 35:122–8. doi:10.1016/j.bcmd.2005.05.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Ji H, Erfani N, Tauro BJ, Kapp EA, Zhu HJ, Moritz RL, et al. Difference gel electrophoresis analysis of Ras-transformed fibroblast cell-derived exosomes. Electrophoresis (2008) 29:2660–71. doi:10.1002/elps.200800015
13. Johnstone RM, Bianchini A, Teng K. Reticulocyte maturation and exosome release: transferrin receptor containing exosomes shows multiple plasma membrane functions. Blood (1989) 74:1844–51.
14. Geminard C, de Gassart A, Vidal M. Reticulocyte maturation: mitoptosis and exosome release. Biocell (2002) 26:205–15.
15. Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood (1999) 94:3791–9.
16. Raposo G, Tenza D, Mecheri S, Peronet R, Bonnerot C, Desaymard C. Accumulation of major histocompatibility complex class II molecules in mast cell secretory granules and their release upon degranulation. Mol Biol Cell (1997) 8:2631–45. doi:10.1091/mbc.8.12.2631
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med (2001) 7:297–303. doi:10.1038/85438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Mahaweni NM, Kaijen-Lambers ME, Dekkers J, Aerts JG, Hegmans JP. Tumour-derived exosomes as antigen delivery carriers in dendritic cell-based immunotherapy for malignant mesothelioma. J Extracell Vesicles (2013):2. doi:10.3402/jev.v2i0.22492
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Atay S, Godwin AK. Tumor-derived exosomes: a message delivery system for tumor progression. Commun Integr Biol (2014) 7:e28231. doi:10.4161/cib.28231
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Biancone L, Bruno S, Deregibus MC, Tetta C, Camussi G. Therapeutic potential of mesenchymal stem cell-derived microvesicles. Nephrol Dial Transplant (2012) 27:3037–42. doi:10.1093/ndt/gfs168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Lee JK, Park SR, Jung BK, Jeon YK, Lee YS, Kim MK, et al. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS One (2013) 8:e84256. doi:10.1371/journal.pone.0084256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Zhang B, Yin Y, Lai RC, Tan SS, Choo AB, Lim SK. Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev (2014). doi:10.1089/scd.2013.0479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med (1998) 4:594–600. doi:10.1038/nm0598-594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Sobo-Vujanovic A, Munich S, Vujanovic NL. Dendritic-cell exosomes cross-present toll-like receptor-ligands and activate bystander dendritic cells. Cell Immunol (2014) 289:119–27. doi:10.1016/j.cellimm.2014.03.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Ekstrom K, Omar O, Graneli C, Wang X, Vazirisani F, Thomsen P. Monocyte exosomes stimulate the osteogenic gene expression of mesenchymal stem cells. PLoS One (2013) 8:e75227. doi:10.1371/journal.pone.0075227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Akao Y, Iio A, Itoh T, Noguchi S, Itoh Y, Ohtsuki Y, et al. Microvesicle-mediated RNA molecule delivery system using monocytes/macrophages. Mol Ther (2011) 19:395–9. doi:10.1038/mt.2010.254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. McDonald MK, Tian Y, Qureshi RA, Gormley M, Ertel A, Gao R, et al. Functional significance of macrophage-derived exosomes in inflammation and pain. Pain (2014) 155:1527–39. doi:10.1016/j.pain.2014.04.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Lee HD, Kim YH, Kim DS. Exosomes derived from human macrophages suppress endothelial cell migration by controlling integrin trafficking. Eur J Immunol (2014) 44:1156–69. doi:10.1002/eji.201343660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Fais S. NK cell-released exosomes: natural nanobullets against tumors. Oncoimmunology (2013) 2:e22337. doi:10.4161/onci.22337
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Lugini L, Cecchetti S, Huber V, Luciani F, Macchia G, Spadaro F, et al. Immune surveillance properties of human NK cell-derived exosomes. J Immunol (2012) 189:2833–42. doi:10.4049/jimmunol.1101988
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Buschow SI, van Balkom BW, Aalberts M, Heck AJ, Wauben M, Stoorvogel W. MHC class II-associated proteins in B-cell exosomes and potential functional implications for exosome biogenesis. Immunol Cell Biol (2010) 88:851–6. doi:10.1038/icb.2010.64
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. McLellan AD. Exosome release by primary B cells. Crit Rev Immunol (2009) 29:203–17. doi:10.1615/CritRevImmunol.v29.i3.20
33. Zhang H, Xie Y, Li W, Chibbar R, Xiong S, Xiang J. CD4(+) T cell-released exosomes inhibit CD8(+) cytotoxic T-lymphocyte responses and antitumor immunity. Cell Mol Immunol (2011) 8:23–30. doi:10.1038/cmi.2010.59
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Yu X, Huang C, Song B, Xiao Y, Fang M, Feng J, et al. CD4+CD25+ regulatory T cells-derived exosomes prolonged kidney allograft survival in a rat model. Cell Immunol (2013) 285:62–8. doi:10.1016/j.cellimm.2013.06.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Harding C, Heuser J, Stahl P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol (1983) 97:329–39. doi:10.1083/jcb.97.2.329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Pan BT, Teng K, Wu C, Adam M, Johnstone RM. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J Cell Biol (1985) 101:942–8. doi:10.1083/jcb.101.3.942
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem (1987) 262:9412–20.
38. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med (1996) 183:1161–72. doi:10.1084/jem.183.3.1161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Bodey B, Bodey B Jr, Kaiser HE. Dendritic type, accessory cells within the mammalian thymic microenvironment. Antigen presentation in the dendritic neuro-endocrine-immune cellular network. In vivo (1997) 11:351–70.
40. Skokos D, Le Panse S, Villa I, Rousselle JC, Peronet R, David B, et al. Mast cell-dependent B and T lymphocyte activation is mediated by the secretion of immunologically active exosomes. J Immunol (2001) 166:868–76. doi:10.4049/jimmunol.166.2.868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z. Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol (2008) 180:7249–58. doi:10.4049/jimmunol.180.11.7249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Yeo RW, Lai RC, Zhang B, Tan SS, Yin Y, Teh BJ, et al. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv Drug Deliv Rev (2013) 65:336–41. doi:10.1016/j.addr.2012.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Denzer K, Kleijmeer MJ, Heijnen HF, Stoorvogel W, Geuze HJ. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci (2000) 113(Pt 19):3365–74.
44. Raiborg C, Rusten TE, Stenmark H. Protein sorting into multivesicular endosomes. Curr Opin Cell Biol (2003) 15:446–55. doi:10.1016/S0955-0674(03)00080-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol (2006) 3:22. doi:10.1002/0471143030.cb0322s30
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Davis JQ, Dansereau D, Johnstone RM, Bennett V. Selective externalization of an ATP-binding protein structurally related to the clathrin-uncoating ATPase/heat shock protein in vesicles containing terminal transferrin receptors during reticulocyte maturation. J Biol Chem (1986) 261:15368–71.
47. Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, Whiteside TL. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res (2005) 11:1010–20.
48. Taylor DD, Gercel-Taylor C, Lyons KS, Stanson J, Whiteside TL. T-cell apoptosis and suppression of T-cell receptor/CD3-zeta by Fas ligand-containing membrane vesicles shed from ovarian tumors. Clin Cancer Res (2003) 9:5113–9.
49. Lai RC, Arslan F, Lee MM, Sze NS, Choo A, Chen TS, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res (2010) 4:214–22. doi:10.1016/j.scr.2009.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Clayton A, Court J, Navabi H, Adams M, Mason MD, Hobot JA, et al. Analysis of antigen presenting cell derived exosomes, based on immuno-magnetic isolation and flow cytometry. J Immunol Methods (2001) 247:163–74. doi:10.1016/S0022-1759(00)00321-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Taylor DD, Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol (2008) 110:13–21. doi:10.1016/j.ygyno.2008.04.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Chen C, Skog J, Hsu CH, Lessard RT, Balaj L, Wurdinger T, et al. Microfluidic isolation and transcriptome analysis of serum microvesicles. Lab Chip (2010) 10:505–11. doi:10.1039/b916199f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol (2010) 12:19–30. doi:10.1038/ncb2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Navabi H, Croston D, Hobot J, Clayton A, Zitvogel L, Jasani B, et al. Preparation of human ovarian cancer ascites-derived exosomes for a clinical trial. Blood Cells Mol Dis (2005) 35:149–52. doi:10.1016/j.bcmd.2005.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Lamparski HG, Metha-Damani A, Yao JY, Patel S, Hsu DH, Ruegg C, et al. Production and characterization of clinical grade exosomes derived from dendritic cells. J Immunol Methods (2002) 270:211–26. doi:10.1016/S0022-1759(02)00330-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Sirisinha S. Evolutionary insights into the origin of innate and adaptive immune systems: different shades of grey. Asian Pac J Allergy Immunol (2014) 32:3–15.
57. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol (2007) 81:1–5. doi:10.1189/jlb.0306164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22:240–73. doi:10.1128/CMR.00046-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Olive C. Pattern recognition receptors: sentinels in innate immunity and targets of new vaccine adjuvants. Expert Rev Vaccines (2012) 11:237–56. doi:10.1586/erv.11.189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol (2011) 30:16–34. doi:10.3109/08830185.2010.529976
61. Vivier E, Malissen B. Innate and adaptive immunity: specificities and signaling hierarchies revisited. Nat Immunol (2005) 6:17–21. doi:10.1038/ni1153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Schenten D, Medzhitov R. The control of adaptive immune responses by the innate immune system. Adv Immunol (2011) 109:87–124. doi:10.1016/B978-0-12-387664-5.00003-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Medzhitov R, Janeway CA Jr. Innate immune recognition and control of adaptive immune responses. Semin Immunol (1998) 10:351–3. doi:10.1006/smim.1998.0136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Chesnut RW, Grey HM. Antigen presenting cells and mechanisms of antigen presentation. Crit Rev Immunol (1985) 5:263–316.
65. Katz DR. Antigen presentation, antigen-presenting cells and antigen processing. Curr Opin Immunol (1988) 1:213–9. doi:10.1016/0952-7915(88)90004-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Thery C, Amigorena S. The cell biology of antigen presentation in dendritic cells. Curr Opin Immunol (2001) 13:45–51. doi:10.1016/S0952-7915(00)00180-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Chaput N, Schartz NE, Andre F, Taieb J, Novault S, Bonnaventure P, et al. Exosomes as potent cell-free peptide-based vaccine. II. exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J Immunol (2004) 172:2137–46. doi:10.4049/jimmunol.172.4.2137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Andre F, Schartz NE, Movassagh M, Flament C, Pautier P, Morice P, et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet (2002) 360:295–305. doi:10.1016/S0140-6736(02)09552-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Napoletano C, Rughetti A, Landi R, Pinto D, Bellati F, Rahimi H, et al. Immunogenicity of allo-vesicle carrying ERBB2 tumor antigen for dendritic cell-based anti-tumor immunotherapy. Int J Immunopathol Pharmacol (2009) 22:647–58.
70. Thery C, Duban L, Segura E, Veron P, Lantz O, Amigorena S. Indirect activation of naive CD4+ T cells by dendritic cell-derived exosomes. Nat Immunol (2002) 3:1156–62. doi:10.1038/ni854
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood (2004) 104:3257–66. doi:10.1182/blood-2004-03-0824
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Admyre C, Johansson SM, Paulie S, Gabrielsson S. Direct exosome stimulation of peripheral human T cells detected by ELISPOT. Eur J Immunol (2006) 36:1772–81. doi:10.1002/eji.200535615
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Luketic L, Delanghe J, Sobol PT, Yang P, Frotten E, Mossman KL, et al. Antigen presentation by exosomes released from peptide-pulsed dendritic cells is not suppressed by the presence of active CTL. J Immunol (2007) 179:5024–32. doi:10.4049/jimmunol.179.8.5024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Admyre C, Bohle B, Johansson SM, Focke-Tejkl M, Valenta R, Scheynius A, et al. B cell-derived exosomes can present allergen peptides and activate allergen-specific T cells to proliferate and produce TH2-like cytokines. J Allergy Clin Immunol (2007) 120:1418–24. doi:10.1016/j.jaci.2007.06.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Vincent-Schneider H, Stumptner-Cuvelette P, Lankar D, Pain S, Raposo G, Benaroch P, et al. Exosomes bearing HLA-DR1 molecules need dendritic cells to efficiently stimulate specific T cells. Int Immunol (2002) 14:713–22. doi:10.1093/intimm/dxf048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Morva A, Lemoine S, Achour A, Pers JO, Youinou P, Jamin C. Maturation and function of human dendritic cells are regulated by B lymphocytes. Blood (2012) 119:106–14. doi:10.1182/blood-2011-06-360768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Pletinckx K, Dohler A, Pavlovic V, Lutz MB. Role of dendritic cell maturity/costimulation for generation, homeostasis, and suppressive activity of regulatory T cells. Front Immunol (2011) 2:39. doi:10.3389/fimmu.2011.00039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Zanoni I, Granucci F. The regulatory role of dendritic cells in the induction and maintenance of T-cell tolerance. Autoimmunity (2011) 44:23–32. doi:10.3109/08916931003782148
79. Thery C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, et al. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol (1999) 147:599–610. doi:10.1083/jcb.147.3.599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Utsugi-Kobukai S, Fujimaki H, Hotta C, Nakazawa M, Minami M. MHC class I-mediated exogenous antigen presentation by exosomes secreted from immature and mature bone marrow derived dendritic cells. Immunol Lett (2003) 89:125–31. doi:10.1016/S0165-2478(03)00128-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Segura E, Nicco C, Lombard B, Veron P, Raposo G, Batteux F, et al. ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood (2005) 106:216–23. doi:10.1182/blood-2005-01-0220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Segura E, Amigorena S, Thery C. Mature dendritic cells secrete exosomes with strong ability to induce antigen-specific effector immune responses. Blood Cells Mol Dis (2005) 35:89–93. doi:10.1016/j.bcmd.2005.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Montecalvo A, Shufesky WJ, Stolz DB, Sullivan MG, Wang Z, Divito SJ, et al. Exosomes as a short-range mechanism to spread alloantigen between dendritic cells during T cell allorecognition. J Immunol (2008) 180:3081–90. doi:10.4049/jimmunol.180.5.3081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Muntasell A, Berger AC, Roche PA. T cell-induced secretion of MHC class II-peptide complexes on B cell exosomes. EMBO J (2007) 26:4263–72. doi:10.1038/sj.emboj.7601842
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Aline F, Bout D, Amigorena S, Roingeard P, Dimier-Poisson I. Toxoplasma gondii antigen-pulsed-dendritic cell-derived exosomes induce a protective immune response against T. gondii infection. Infect Immun (2004) 72:4127–37. doi:10.1128/IAI.72.7.4127-4137.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Del Cacho E, Gallego M, Lee SH, Lillehoj HS, Quilez J, Lillehoj EP, et al. Induction of protective immunity against Eimeria tenella infection using antigen-loaded dendritic cells (DC) and DC-derived exosomes. Vaccine (2011) 29:3818–25. doi:10.1016/j.vaccine.2011.03.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Peche H, Heslan M, Usal C, Amigorena S, Cuturi MC. Presentation of donor major histocompatibility complex antigens by bone marrow dendritic cell-derived exosomes modulates allograft rejection. Transplantation (2003) 76:1503–10. doi:10.1097/01.TP.0000092494.75313.38
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature (2002) 417:182–7. doi:10.1038/417182a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Yang X, Meng S, Jiang H, Zhu C, Wu W. Exosomes derived from immature bone marrow dendritic cells induce tolerogenicity of intestinal transplantation in rats. J Surg Res (2011) 171:826–32. doi:10.1016/j.jss.2010.05.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Li X, Li JJ, Yang JY, Wang DS, Zhao W, Song WJ, et al. Tolerance induction by exosomes from immature dendritic cells and rapamycin in a mouse cardiac allograft model. PLoS One (2012) 7:e44045. doi:10.1371/journal.pone.0044045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Bianco NR, Kim SH, Ruffner MA, Robbins PD. Therapeutic effect of exosomes from indoleamine 2,3-dioxygenase-positive dendritic cells in collagen-induced arthritis and delayed-type hypersensitivity disease models. Arthritis Rheum (2009) 60:380–9. doi:10.1002/art.24229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Kim SH, Bianco N, Menon R, Lechman ER, Shufesky WJ, Morelli AE, et al. Exosomes derived from genetically modified DC expressing FasL are anti-inflammatory and immunosuppressive. Mol Ther (2006) 13:289–300. doi:10.1016/j.ymthe.2005.09.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Kim SH, Bianco NR, Shufesky WJ, Morelli AE, Robbins PD. Effective treatment of inflammatory disease models with exosomes derived from dendritic cells genetically modified to express IL-4. J Immunol (2007) 179:2242–9. doi:10.4049/jimmunol.179.4.2242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Ruffner MA, Kim SH, Bianco NR, Francisco LM, Sharpe AH, Robbins PD. B7-1/2, but not PD-L1/2 molecules, are required on IL-10-treated tolerogenic DC and DC-derived exosomes for in vivo function. Eur J Immunol (2009) 39:3084–90. doi:10.1002/eji.200939407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Kim SH, Lechman ER, Bianco N, Menon R, Keravala A, Nash J, et al. Exosomes derived from IL-10-treated dendritic cells can suppress inflammation and collagen-induced arthritis. J Immunol (2005) 174:6440–8. doi:10.4049/jimmunol.174.10.6440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Cai Z, Zhang W, Yang F, Yu L, Yu Z, Pan J, et al. Immunosuppressive exosomes from TGF-beta1 gene-modified dendritic cells attenuate Th17-mediated inflammatory autoimmune disease by inducing regulatory T cells. Cell Res (2012) 22:607–10. doi:10.1038/cr.2011.196
97. Quah BJ, O’Neill HC. Maturation of function in dendritic cells for tolerance and immunity. J Cell Mol Med (2005) 9:643–54. doi:10.1111/j.1582-4934.2005.tb00494.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Beauvillain C, Juste MO, Dion S, Pierre J, Dimier-Poisson I. Exosomes are an effective vaccine against congenital toxoplasmosis in mice. Vaccine (2009) 27:1750–7. doi:10.1016/j.vaccine.2009.01.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Colino J, Snapper CM. Exosomes from bone marrow dendritic cells pulsed with diphtheria toxoid preferentially induce type 1 antigen-specific IgG responses in naive recipients in the absence of free antigen. J Immunol (2006) 177:3757–62. doi:10.4049/jimmunol.177.6.3757
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Qazi KR, Gehrmann U, Domange Jordo E, Karlsson MC, Gabrielsson S. Antigen-loaded exosomes alone induce Th1-type memory through a B-cell-dependent mechanism. Blood (2009) 113:2673–83. doi:10.1182/blood-2008-04-153536
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Quah BJ, O’Neill HC. Mycoplasma contaminants present in exosome preparations induce polyclonal B cell responses. J Leukoc Biol (2007) 82:1070–82. doi:10.1189/jlb.0507277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Viaud S, Thery C, Ploix S, Tursz T, Lapierre V, Lantz O, et al. Dendritic cell-derived exosomes for cancer immunotherapy: what’s next? Cancer Res (2010) 70:1281–5. doi:10.1158/0008-5472.CAN-09-3276
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Chaput N, Thery C. Exosomes: immune properties and potential clinical implementations. Semin Immunopathol (2011) 33:419–40. doi:10.1007/s00281-010-0233-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Quah BJ, O’Neill HC. The immunogenicity of dendritic cell-derived exosomes. Blood Cells Mol Dis (2005) 35:94–110. doi:10.1016/j.bcmd.2005.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Bhatnagar S, Schorey JS. Exosomes released from infected macrophages contain Mycobacterium avium glycopeptidolipids and are proinflammatory. J Biol Chem (2007) 282:25779–89. doi:10.1074/jbc.M702277200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Bhatnagar S, Shinagawa K, Castellino FJ, Schorey JS. Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood (2007) 110:3234–44. doi:10.1182/blood-2007-03-079152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Anand PK, Anand E, Bleck CK, Anes E, Griffiths G. Exosomal Hsp70 induces a pro-inflammatory response to foreign particles including mycobacteria. PLoS One (2010) 5:e10136. doi:10.1371/journal.pone.0010136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Zhang HG, Liu C, Su K, Yu S, Zhang L, Zhang S, et al. A membrane form of TNF-alpha presented by exosomes delays T cell activation-induced cell death. J Immunol (2006) 176:7385–93. doi:10.4049/jimmunol.176.12.7385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Qazi KR, Torregrosa Paredes P, Dahlberg B, Grunewald J, Eklund A, Gabrielsson S. Proinflammatory exosomes in bronchoalveolar lavage fluid of patients with sarcoidosis. Thorax (2010) 65:1016–24. doi:10.1136/thx.2009.132027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, et al. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res (2005) 65:5238–47. doi:10.1158/0008-5472.CAN-04-3804
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Vega VL, Rodriguez-Silva M, Frey T, Gehrmann M, Diaz JC, Steinem C, et al. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J Immunol (2008) 180:4299–307. doi:10.4049/jimmunol.180.6.4299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Dai S, Wan T, Wang B, Zhou X, Xiu F, Chen T, et al. More efficient induction of HLA-A*0201-restricted and carcinoembryonic antigen (CEA)-specific CTL response by immunization with exosomes prepared from heat-stressed CEA-positive tumor cells. Clin Cancer Res (2005) 11:7554–63. doi:10.1158/1078-0432.CCR-05-0810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Chen W, Wang J, Shao C, Liu S, Yu Y, Wang Q, et al. Efficient induction of antitumor T cell immunity by exosomes derived from heat-shocked lymphoma cells. Eur J Immunol (2006) 36:1598–607. doi:10.1002/eji.200535501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Lv LH, Wan YL, Lin Y, Zhang W, Yang M, Li GL, et al. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J Biol Chem (2012) 287:15874–85. doi:10.1074/jbc.M112.340588
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Zhong H, Yang Y, Ma S, Xiu F, Cai Z, Zhao H, et al. Induction of a tumour-specific CTL response by exosomes isolated from heat-treated malignant ascites of gastric cancer patients. Int J Hyperthermia (2011) 27:604–11. doi:10.3109/02656736.2011.564598
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P, et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med (2002) 195:1303–16. doi:10.1084/jem.20011624
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Czystowska M, Han J, Szczepanski MJ, Szajnik M, Quadrini K, Brandwein H, et al. IRX-2, a novel immunotherapeutic, protects human T cells from tumor-induced cell death. Cell Death Differ (2009) 16:708–18. doi:10.1038/cdd.2008.197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Huber V, Fais S, Iero M, Lugini L, Canese P, Squarcina P, et al. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: role in immune escape. Gastroenterology (2005) 128:1796–804. doi:10.1053/j.gastro.2005.03.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Klibi J, Niki T, Riedel A, Pioche-Durieu C, Souquere S, Rubinstein E, et al. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood (2009) 113:1957–66. doi:10.1182/blood-2008-02-142596
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Keryer-Bibens C, Pioche-Durieu C, Villemant C, Souquere S, Nishi N, Hirashima M, et al. Exosomes released by EBV-infected nasopharyngeal carcinoma cells convey the viral latent membrane protein 1 and the immunomodulatory protein galectin 9. BMC Cancer (2006) 6:283. doi:10.1186/1471-2407-6-283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Clayton A, Mitchell JP, Court J, Mason MD, Tabi Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res (2007) 67:7458–66. doi:10.1158/0008-5472.CAN-06-3456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Szajnik M, Czystowska M, Szczepanski MJ, Mandapathil M, Whiteside TL. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS One (2010) 5:e11469. doi:10.1371/journal.pone.0011469
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, Whiteside TL. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J Immunol (2009) 183:3720–30. doi:10.4049/jimmunol.0900970
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Liu C, Yu S, Zinn K, Wang J, Zhang L, Jia Y, et al. Murine mammary carcinoma exosomes promote tumor growth by suppression of NK cell function. J Immunol (2006) 176:1375–85. doi:10.4049/jimmunol.176.3.1375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Ashiru O, Boutet P, Fernandez-Messina L, Aguera-Gonzalez S, Skepper JN, Vales-Gomez M, et al. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res (2010) 70:481–9. doi:10.1158/0008-5472.CAN-09-1688
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Yu S, Liu C, Su K, Wang J, Liu Y, Zhang L, et al. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J Immunol (2007) 178:6867–75. doi:10.4049/jimmunol.178.11.6867
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, et al. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-beta-mediated suppressive activity on T lymphocytes. Cancer Res (2006) 66:9290–8. doi:10.1158/0008-5472.CAN-06-1819
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Liu Y, Xiang X, Zhuang X, Zhang S, Liu C, Cheng Z, et al. Contribution of MyD88 to the tumor exosome-mediated induction of myeloid derived suppressor cells. Am J Pathol (2010) 176:2490–9. doi:10.2353/ajpath.2010.090777
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest (2010) 120:457–71. doi:10.1172/JCI40483
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Xiang X, Liu Y, Zhuang X, Zhang S, Michalek S, Taylor DD, et al. TLR2-mediated expansion of MDSCs is dependent on the source of tumor exosomes. Am J Pathol (2010) 177:1606–10. doi:10.2353/ajpath.2010.100245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Zeelenberg IS, Ostrowski M, Krumeich S, Bobrie A, Jancic C, Boissonnas A, et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res (2008) 68:1228–35. doi:10.1158/0008-5472.CAN-07-3163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Ichim TE, Zhong Z, Kaushal S, Zheng X, Ren X, Hao X, et al. Exosomes as a tumor immune escape mechanism: possible therapeutic implications. J Transl Med (2008) 6:37. doi:10.1186/1479-5876-6-37
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Atay S, Gercel-Taylor C, Suttles J, Mor G, Taylor DD. Trophoblast-derived exosomes mediate monocyte recruitment and differentiation. Am J Reprod Immunol (2011) 65:65–77. doi:10.1111/j.1600-0897.2010.00880.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Atay S, Gercel-Taylor C, Taylor DD. Human trophoblast-derived exosomal fibronectin induces pro-inflammatory IL-1beta production by macrophages. Am J Reprod Immunol (2011) 66:259–69. doi:10.1111/j.1600-0897.2011.00995.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Tarazona R, Delgado E, Guarnizo MC, Roncero RG, Morgado S, Sanchez-Correa B, et al. Human prostasomes express CD48 and interfere with NK cell function. Immunobiology (2011) 216:41–6. doi:10.1016/j.imbio.2010.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Hedlund M, Stenqvist AC, Nagaeva O, Kjellberg L, Wulff M, Baranov V, et al. Human placenta expresses and secretes NKG2D ligands via exosomes that down-modulate the cognate receptor expression: evidence for immunosuppressive function. J Immunol (2009) 183:340–51. doi:10.4049/jimmunol.0803477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Taylor DD, Akyol S, Gercel-Taylor C. Pregnancy-associated exosomes and their modulation of T cell signaling. J Immunol (2006) 176:1534–42. doi:10.4049/jimmunol.176.3.1534
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Williams JL, Gatson NN, Smith KM, Almad A, McTigue DM, Whitacre CC. Serum exosomes in pregnancy-associated immune modulation and neuroprotection during CNS autoimmunity. Clin Immunol (2013) 149:236–43. doi:10.1016/j.clim.2013.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Ostman S, Taube M, Telemo E. Tolerosome-induced oral tolerance is MHC dependent. Immunology (2005) 116:464–76. doi:10.1111/j.1365-2567.2005.02245.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Admyre C, Johansson SM, Qazi KR, Filen JJ, Lahesmaa R, Norman M, et al. Exosomes with immune modulatory features are present in human breast milk. J Immunol (2007) 179:1969–78. doi:10.4049/jimmunol.179.3.1969
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Uccelli A, Laroni A, Freedman MS. Mesenchymal stem cells for the treatment of multiple sclerosis and other neurological diseases. Lancet Neurol (2011) 10:649–56. doi:10.1016/S1474-4422(11)70121-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Mascarenhas S, Avalos B, Ardoin SP. An update on stem cell transplantation in autoimmune rheumatologic disorders. Curr Allergy Asthma Rep (2012) 12:530–40. doi:10.1007/s11882-012-0298-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Marigo I, Dazzi F. The immunomodulatory properties of mesenchymal stem cells. Semin Immunopathol (2011) 33:593–602. doi:10.1007/s00281-011-0267-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Potian JA, Aviv H, Ponzio NM, Harrison JS, Rameshwar P. Veto-like activity of mesenchymal stem cells: functional discrimination between cellular responses to alloantigens and recall antigens. J Immunol (2003) 171:3426–34. doi:10.4049/jimmunol.171.7.3426
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation (2003) 75:389–97. doi:10.1097/01.TP.0000045055.63901.A9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Bartholomew A, Sturgeon C, Siatskas M, Ferrer K, McIntosh K, Patil S, et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol (2002) 30:42–8. doi:10.1016/S0301-472X(01)00769-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood (2002) 99:3838–43. doi:10.1182/blood.V99.10.3838
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Le Blanc K, Tammik L, Sundberg B, Haynesworth SE, Ringden O. Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand J Immunol (2003) 57:11–20. doi:10.1046/j.1365-3083.2003.01176.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood (2005) 105:1815–22. doi:10.1182/blood-2004-04-1559
150. Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, et al. Human mesenchymal stem cells modulate B-cell functions. Blood (2006) 107:367–72. doi:10.1182/blood-2005-07-2657
151. Newman RE, Yoo D, LeRoux MA, Danilkovitch-Miagkova A. Treatment of inflammatory diseases with mesenchymal stem cells. Inflamm Allergy Drug Targets (2009) 8:110–23. doi:10.2174/187152809788462635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Wu H, Mahato RI. Mesenchymal stem cell-based therapy for type 1 diabetes. Discov Med (2014) 17:139–43.
153. Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet (2008) 371:1579–86. doi:10.1016/S0140-6736(08)60690-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Sato K, Ozaki K, Mori M, Muroi K, Ozawa K. Mesenchymal stromal cells for graft-versus-host disease: basic aspects and clinical outcomes. J Clin Exp Hematop (2010) 50:79–89. doi:10.3960/jslrt.50.79
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. von Bahr L, Sundberg B, Lonnies L, Sander B, Karbach H, Hagglund H, et al. Long-term complications, immunologic effects, and role of passage for outcome in mesenchymal stromal cell therapy. Biol Blood Marrow Transplant (2012) 18:557–64. doi:10.1016/j.bbmt.2011.07.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Burr SP, Dazzi F, Garden OA. Mesenchymal stromal cells and regulatory T cells: the Yin and Yang of peripheral tolerance? Immunol Cell Biol (2013) 91:12–8. doi:10.1038/icb.2012.60
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Ghannam S, Bouffi C, Djouad F, Jorgensen C, Noel D. Immunosuppression by mesenchymal stem cells: mechanisms and clinical applications. Stem Cell Res Ther (2010) 1:2. doi:10.1186/scrt2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Gieseke F, Schütt B, Viebahn S, Koscielniak E, Friedrich W, Handgretinger R, et al. Human multipotent mesenchymal stromal cells inhibit proliferation of PBMCs independently of IFNγR1 signaling and IDO expression. Blood (2007) 110:2197–200. doi:10.1182/blood-2007-04-083162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Rasmusson I, Ringden O, Sundberg B, Le Blanc K. Mesenchymal stem cells inhibit lymphocyte proliferation by mitogens and alloantigens by different mechanisms. Exp Cell Res (2005) 305:33–41. doi:10.1016/j.yexcr.2004.12.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Kang HS, Habib M, Chan J, Abavana C, Potian JA, Ponzio NM, et al. A paradoxical role for IFN-γ in the immune properties of mesenchymal stem cells during viral challenge. Exp Hematol (2005) 33:796–803. doi:10.1016/j.exphem.2005.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Krampera M. Role for interferon-γ in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells (2006) 24:386–98. doi:10.1634/stemcells.2005-0008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Meisel R, Zibert A, Laryea M, Gobel U, Daubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood (2004) 103:4619–21. doi:10.1182/blood-2003-11-3909
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol (2009) 20:1053–67. doi:10.1681/ASN.2008070798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor EN, et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res (2013) 10:301–12. doi:10.1016/j.scr.2013.01.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Lai RC, Yeo RW, Tan SS, Zhang B, Yin Y, Sze NS, et al. Mesenchymal stem cell exosomes: the future msc-based therapy? In: Chase LG, Vemuri MC, editors. Mesenchymal Stem Cell Therapy. New York: Humana Press (2012). pp. 39–61. doi:10.1007/978-1-62703-200-1_3
166. Poh KK, Sperry E, Young RG, Freyman T, Barringhaus KG, Thompson CA. Repeated direct endomyocardial transplantation of allogeneic mesenchymal stem cells: safety of a high dose, “off-the-shelf”, cellular cardiomyoplasty strategy. Int J Cardiol (2007) 117:360–4. doi:10.1016/j.ijcard.2006.04.092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Furlani D, Ugurlucan M, Ong L, Bieback K, Pittermann E, Westien I, et al. Is the intravascular administration of mesenchymal stem cells safe? Mesenchymal stem cells and intravital microscopy. Microvasc Res (2009) 77:370–6. doi:10.1016/j.mvr.2009.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Breitbach M, Bostani T, Roell W, Xia Y, Dewald O, Nygren JM, et al. Potential risks of bone marrow cell transplantation into infarcted hearts. Blood (2007) 110:1362–9. doi:10.1182/blood-2006-12-063412
Keywords: extracellular vesicles, exosomes, immunomodulation, innate immunity, adaptive immunity, immunotherapy
Citation: Zhang B, Yin Y, Lai RC and Lim SK (2014) Immunotherapeutic potential of extracellular vesicles. Front. Immunol. 5:518. doi: 10.3389/fimmu.2014.00518
Received: 30 July 2014; Accepted: 04 October 2014;
Published online: 22 October 2014.
Edited by:
Francesc E. Borras, Institut d’Investigació en Ciències de la Salut Germans Trias i Pujol, SpainReviewed by:
Gregory B. Lesinski, The Ohio State University Comprehensive Cancer Center, USABegoña Pérez-Cabezas, Instituto de Biologia Molecular e Celular, Portugal
Copyright: © 2014 Zhang, Yin, Lai and Lim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sai Kiang Lim, Exosome and Secreted Nano-vesicle Group, A*STAR Institute of Medical Biology, 8A Biomedical Grove, #05-05 Immunos, 138648 Singapore e-mail:c2Fpa2lhbmcubGltQGltYi5hLXN0YXIuZWR1LnNn