 Kate A. Redgrove
Kate A. Redgrove Eileen A. McLaughlin
Eileen A. McLaughlin- 1Priority Research Centre in Reproductive Biology and Chemical Biology, University of Newcastle, Callaghan, NSW, Australia
- 2School of Environmental and Life Science, University of Newcastle, Callaghan, NSW, Australia
Chlamydia trachomatis (CT) is the most prevalent bacterial sexually transmitted infection in the world, with more than 100 million cases reported annually. While there have been extensive studies into the adverse effects that CT infection has on the female genital tract, and on the subsequent ability of these women to conceive, studies into the consequences on male fertility have been limited and controversial. This is in part due to the asymptomatic nature of the infection, where it is estimated that 50% of men with Chlamydia fail to show any symptoms. It is accepted, however, that acute and/or persistent CT infection is the causative agent for conditions such as urethritis, epididymitis, epididymo-orchitis, and potentially prostatitis. As with most infections, the immune system plays a fundamental role in the body’s attempts to eradicate the infection. The first and most important immune response to Chlamydia infection is a local one, whereby immune cells such as leukocytes are recruited to the site of infections, and subsequently secrete pro-inflammatory cytokines and chemokines such as interferon gamma. Immune cells also work to initiate and potentiate chronic inflammation through the production of reactive oxygen species (ROS), and the release of molecules with degradative properties including defensins, elastase, collagenase, cathespins, and lysozyme. This long-term inflammation can lead to cell proliferation (a possible precursor to cancer), tissue remodeling, and scarring, as well as being linked to the onset of autoimmune responses in genetically disposed individuals. This review will focus on the ability of the immune system to recognize and clear acute and persistent chlamydial infections in the male genital tract, and on the paradoxical damage that chronic inflammation resulting from the infection can cause on the reproductive health of the individual.
Introduction
Sexually transmitted infections (STIs) are a major public health problem in most parts of the world, and are responsible for a number of acute illnesses, infertility, long-term disability, and premature death, in addition to contributing to an increase in the spread of HIV. It is estimated that $90–160 million is spent annually by the Australian health care system as a direct cost of Chlamydia treatment; this includes both initial treatment with antibiotics by a GP, and the more severe outcomes of untreated infection (1, 2). Perhaps the highest cost is associated with individuals who have troubles conceiving as result of infection and turn to assisted reproductive technologies (ART). In 2011 in Australia alone, a total of 61,158 ART treatment cycles were performed at a cost >$500 million (3). While there has been an increase in the promotion of Chlamydia prevention and screening programs, the largest barrier to reducing the rates of infection lies with the limited knowledge that people between the ages of 16 and 24 years possess, concerning the consequences, symptoms, prevalence, screening recommendations, testing procedures, and treatment of Chlamydia infection. It is, therefore, becoming essential that identification and treatment of Chlamydia infection is instigated before irreversible tissue damage occurs. Underpinning a successful public health program would be the development of a novel and effective chlamydial vaccine for young men. However, in order to undertake this, a thorough understanding of the intricate and often paradoxical immune response to Chlamydia infection in male reproductive tissues is needed. This review will highlight the known impacts that acute and chronic Chlamydia infection has on the male reproductive tract, as well as outlining some of the mechanisms that underlie the immune response in these unique tissues.
Chlamydia
Background and Life Cycle
Chlamydiae are obligate intracellular Gram-negative bacteria that are surrounded by a rigid cell wall. They are able to infect both human (Chlamydia trachomatis and Chlamydia pneumonia) and animals (Chlamydia muridarum, Chlamydia suis, Chlamydia abortus, Chlamydia pecorum, Chlamydia psittaci, and Chlamydia caviae) and depend entirely on the biosynthesis pathways of a host cell to multiply, as they are unable to synthesize essential nutrients (4, 5). For human C. trachomatis, there are 19 currently identified serotypes, determined based on their major outer membrane protein (MOMP) characteristics (6), with serotypes A, B, and C causing trachoma of the eye, serotypes D through to K infecting urogenital tissue (7), and serotype L being responsible for lymphogranuloma venereum (LGV), an infection of the lymphatics and lymph nodes (8, 9). Despite their common intracellular lifestyles, Chlamydia exhibits a range of hosts, as well as diversity in morphology, biological properties, and pathological consequences. The level of similarity for individual proteins encoded by C. trachomatis and C. pneumonia spans a wide spectrum (22–95% amino acid identity between orthologs from the two species) (10, 11).
Chlamydia exists in two developmental forms: the elementary body (EB), which is infectious, non-replicating, and extracellular. It also displays no metabolic activity; and the reticulate body (RB), which is non-infectious, replicating, and intracellular. Infection begins when the small (~0.2–0.3 μm) EB’s make contact with the epithelial cell surface. It has been proposed that number of receptor–ligand interactions take place at this point, after which the EB is endocytosed. The endocytic-vesicles are modified by the EB to prevent it from entering endocytic-lysosomal pathway and are then trafficked on cytoskeletal intermediate filaments to the endoplasmic reticulum/Golgi activity center. After arriving here, the transformation of the essentially non-metabolically active EB into the larger (~0.8 μm) metabolically active RB begins; the EB DNA is relaxed, signals for DNA, RNA, and protein synthesis are activated and RB cell division ensues. This intracellular Chlamydial micro-colony is now termed an inclusion, and after several hours of logarithmic RB growth, the inclusion expands. This, in conjunction with nutrient depletion and ATP scavenging from the infected host signals the transformation of the non-infectious RB back into the infectious EB, which are then exocytosed from the host cell to infect neighboring epithelial cells, in order to perpetuate the infection process (12–14) (Figure 1).
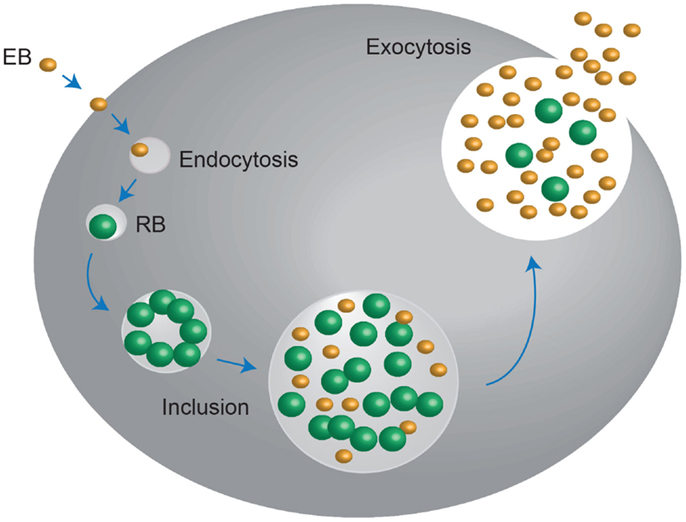
Figure 1. Chlamydia undergo a unique biphasic developmental cycle. The infectious form of Chlamydia, the elementary body (EB) enters into the host cell via endocytosis. Upon entry, the EB convert into the metabolically active, non-infectious reticulate body (RB), which replicates within a vaculolar compartment, termed the inclusion. Once the developmental cycle is almost complete, the RBs revert back into EBs, stimulating host cell lysis and release of the infectious EBs into the extracellular space. These EBs then move onto to infect new host cells. Adapted from Roan and Starnbach (346).
Infection/Interaction with Host Cells
Chlamydia are capable of invading the majority of cultured cells, which would suggest that the receptor(s) that facilitate the invasion are either ubiquitous, or that multiple receptors are used. However, the receptor–ligand interactions involved during chlamydial entry have proven to be elusive. This is in part due to the use of different species or strains of Chlamydia as well as different experimental procedures and parameters, making it difficult to draw comparisons between the multitudes of studies performed. It is thought that the binding may be a two-step process in some species, involving an initial, reversible, electrostatic interaction mediated by heparin-sulfate proteoglycans (HSPGs) (15–17), followed by high-affinity, irreversible binding to a secondary receptor (11). In addition to this, cleavage of the N-linked oligosaccharides, on the surface of C. trachomatis and C. pneumonia, inhibited attachment of the bacteria to a number of cell types, suggesting that glycan moieties of proteins expressed on the Chlamydia cell surface also participate in binding (18). Some of the proposed ligands that allow Chlamydia to attach to and infect host cells include the MOMP (16, 19), heat shock protein 70 (20), and glycosaminoglycans (GAGs) (15). Studies have also shown that lipopolysaccharide (LPS), one of the major components of the chlamydial cell surface, may play a role in the infectivity of Chlamydia into host cells, although the evidence suggests that the interaction may be complex and rely on more than one moiety (21). There has also recently been some interest generated by the entry of C. trachomatis into host cells via cholesterol-rich membrane domains, or lipid “rafts,” which appear to be serotype-dependent (22).
Pathophysiology – Males and Females
Approximately, 75% of C. trachomatis infections in women and up to 50% of those in men are asymptomatic (23, 24). Clinical manifestations of C. trachomatis infections in women include acute urethral syndrome, urethritis, bartholinitis, cervicitis, upper genital tract infection (including endometritis, salpingo-oophoritis, and pelvic inflammatory disease), perihepatitis, and reactive arthritis (23). In women, untreated C. trachomatis infection can lead to severe reproductive complications. Pelvic inflammatory disease is a particularly common complication of chlamydial infection with consequences including infertility, ectopic pregnancy, chronic pelvic pain, and miscarriage (25–30). In fact, it is estimated that up to two-thirds of cases of tubal-factor infertility and one-third of cases of ectopic pregnancy can be attributed to C. trachomatis infection (31). In addition to this, chlamydial infection during pregnancy is associated with a number of adverse outcomes, including preterm labor, premature rupture of amniotic membranes, low birth weight, neonatal death, and post-partum endometritis (32, 33). Chlamydial infection can also be transmitted to the infant during delivery (34). An infant born to a mother with an active infection has an estimated 50–75% risk of acquiring infection. Indeed, approximately 30–50% of infants born to chlamydial-positive mothers will have chlamydial conjunctivitis, and at least half of these infants will also display nasopharyngeal infection. Further to this, it is common in about 30% of cases that infant with nasopharyngeal infection will also develop chlamydial pneumonia (23).
Male Genital Tract Infection and Pathology
In men, C. trachomatis infection is known to be responsible for urethritis (35), epididymitis, epidiymo-orchitis (36–38), and it is becoming more widely accepted that it also acts as a causative agent for prostatitis (39–44), as well as causing an enlargement of seminal vesicles in the epididymis (37, 45–48), although the direct consequences of the C. trachomatis infection on the prostate and seminal vesicles remains unknown.
Urethritis
Urethritis is commonly defined as infection-induced inflammation of the urethra. The term is usually reserved for urethral inflammation caused by an STD and is normally characterized into gonococcal urethritis (GU), or non-gonococcal urethritis (NGU). Interestingly, many patients with urethritis, including approximately 25% of patients with NGU, are clinically asymptomatic (49). It is, therefore, unsurprising that C. trachomatis is a major cause of urethritis in men. Various studies have estimated that 30% of urethritis cases can be attributed to C. trachomatis infection (50). In addition to this, it is though that up to 42% of NGU cases may be caused by C. trachomatis (51). Importantly, C. trachomatis infection appears to be equally present in both symptomatic and asymptomatic urethral diseases (52, 53).
Epididymitis and epididymo-orchitis
Epididymitis and epididymo-orchitis are conditions exemplified by inflammation of the epididymis and testes, respectively, with or without infection. Indeed, according to the National Institute of Health, in the United States, where the population contains approximately 150 million males, 600,000 cases of epididymitis are recorded each year (38). The symptoms for epididymitis include pain, nodules, edema, urinary difficulties, fever, urethral discharge, and infertility (54). The condition can be classified based on the duration of symptoms as acute, sub-acute, or chronic. If the symptoms include pain and swelling, and desist within 6 weeks then the case is termed acute. If there is pain without swelling and the symptoms persist for longer than 3 months, then it is labeled as chronic (43). Epididymo-orchitis occurs when the inflammation from the epididymis spreads to the adjacent testicle (38). In men younger than 14 years and older than 35 years, epididymitis is generally caused by infection with a common urinary tract pathogen such as Escherichia coli. In those between 14 and 35 years, however, it is most commonly caused by sexually transmitted Neisseria gonorrheae or C. trachomatis (55, 56). Importantly, decreased sperm counts and decreased motility are often consequences of acute epididymo-orchitis (57), and this pathology is also associated with high rates of infertility (58).
Prostatitis
Prostatitis is a state of inflammation of the prostate, which can be described both with and without infection. Prostatitis syndromes can be divided into four different classifications: (I) acute, (II) chronic, (III) non-bacterial prostatitis, chronic pelvic pain syndrome (CPPS), and (IV) asymptomatic inflammatory prostatitis (59). Although acute and chronic prostatitis have a clear etiology and patients respond well to anti-microbial treatment, these types of prostatitis only encompass 10% of the cases seen in clinical practice (60). CPPS is the most common prostatitis syndrome, constituting 90–95% of cases. Patients with CPPS have no evidence of urinary tract infection, making it a common disease of unclear etiology (60). There have been a number of studies highlighting the prevalence of C. trachomatis infection in patients with prostatitis (9, 39, 40, 42, 44, 46, 61–66). The rates of prevalence, however, are variable between studies, and have been related back to differences in the types of samples analyzed, for example, urethral swab, first void urine, semen, or expressed prostate secretion. In addition to this, concerns have been raised about the reliability of the samples used in these studies. In particular, it has been postulated that bacterial isolation from urethral swabs, expressed prostatic secretions (EPS), and/or urine following prostatic massage, have the potential to be contaminated as a result of transiting through the urethra, thus limiting the interpretations that can be made from such tests (64, 67). However, a number of studies indicate that semen/EPS specimens are often positive for C. trachomatis in patients with negative urethral swabs (40, 63, 66, 68). Moreover, pure prostatic biopsies from CPPS have identified the presence of C. trachomatis in the absence of urethral infection (39, 42). It is thought that C. trachomatis infection of the prostate gland may cause inflammation and thus impair the normal functionality of the gland and impact on male fertility (69). As stated above, the literature concerning this issue is controversial, with some reports arguing in favor of a positive relationship between chronic prostatitis induced by C. trachomatis and altered semen quality (70–73), whereas other reports support the concept that no alterations in semen quality and male fertility are observed (46, 68, 74–79).
Seminal vesiculitis
Seminal vesiculitis is inflammation of the seminal vesicles, and is most often a secondary outcome of prostatitis, although it is also know to occur independently. It is still uncertain in human beings whether C. trachomatis can infect seminal vesicles and lead to inflammation and a specific pathology. This is mainly due to the lack of clinical symptoms and/or significant consequences that the infection produces (80). However, studies performed by Furuya and colleagues reported the presence of inflammation in the seminal vesicles of patients with acute epididymitis, and that C. trachomatis was the pathogen most frequently detected in the seminal vesicle fluid (81). In addition, vesiculitis-associated symptoms disappeared simultaneously with improvement in symptoms of epididymitis after anti-microbial treatment (81). These finding strongly suggest that seminal vesicles are involved in the urogenital inflammation process. Furthermore, some researchers have proposed that chlamydial epididymitis may originate from seminal vesiculitis (37). This is supported by a reported case of seminal vesiculitis appearing prior to acute epididymitis in a patient whose female partner had been diagnosed with chlamydial cervicitis (82). It has also been shown that patients with urethritis are more likely to have accompanying seminal vesiculitis (83).
Chlamydia and Male Infertility
While it is clear that the inflammation caused during chlamydial infection has a direct effect on the male reproductive tract itself, it remains largely controversial as to whether infection with Chlamydia has a dramatic effect on sperm quality and subsequent male infertility. Studies using electron microscopy have demonstrated an interaction between Chlamydia and sperm in biopsies taken from both testis and epididymis, in addition to semen samples (84, 85). In addition to this, a study using transmission electron microscopy showed that specific C. trachomatis serotypes (86). More conclusive evidence for negative effects of Chlamydia infection on male fertility is offered in a number studies that demonstrate that the EBs of C. trachomatis can lead to apoptosis of human sperm in vitro, via the activation of specific caspases (87–89). It is well known that this effect can be elicited by Chlamydia LPS (90, 91). Based on this evidence, it has been proposed that chlamydial LPS interacts with CD14 on the surface of the sperm leading to an increased production of reactive oxygen species (ROS), subsequently activating apoptotic caspases (92). Importantly, excessive generation of ROS has been correlated with an increase in sperm defects both in vitro (93) and in infertile men (94). In support of the detrimental effects of infection on male fertility, a number of studies have demonstrated that C. trachomatis infection correlates with reduced sperm motility (95–98), increased proportion of sperm abnormalities (99), a significant reduction in semen density, sperm morphology and viability (100), and an increase likelihood of leukocytospermia (98, 101). Co-infection with Chlamydia and Mycoplasma results in over threefold more sperm cells presenting with fragmented DNA than uninfected controls (98). In contrast to this, however, there have been multiple studies that suggest that positive markers for C. trachomatis infection are not associated with altered sperm parameters and quality (46, 68, 74–79, 102). A role for C. trachomatis in male factor infertility is also yet to be proven (24, 31). A number of studies have attempted to prove an association between infection and infertility. For example, Mosli and colleagues examined chlamydial incidence via direct immunofluorescence and culture of urethral swabs in age-matched partners of infertile couples (MPIC) and fertile controls, and demonstrated rates of Chlamydia infection to be 25 and 4%, respectively (103). However, it is difficult to compare these results with other studies performed due to the variance among the studies including the differences in patient demographics, the methods of Chlamydia diagnosis used and the samples that were examined (43).
It has been estimated that 5–10% of male factor infertility can be attributed to inflammatory or autoimmune responses, including orchitis, epididymitis, and epididymo-orchitis, as discussed above. However, an additional etiology caused by these immune responses is the production of anti-sperm antibody (ASA) formation. Indeed, ASA can be found in the seminal plasma or attached to the sperm surface in 5–12% of infertile men (104).These ASAs function to impair sperm fertilizing ability including affecting motility (105, 106), the ability to undergo a successful acrosome reaction (107), penetration of the cervical mucosa (108), binding to the zona pellucida (109), as well as sperm-oocyte fusion (110). These antibodies directed against sperm antigens can be found in the seminal fluid and seminal plasma in men, as well as in the follicular fluid in women. They can also be detected in the blood serum of both men and women (111). However, only those antibodies that are bound to the sperm are considered to have an effect on fertility (108, 112).
While all of the studies performed into the direct effects that Chlamydia infection may have on the fertility of untreated males, it is also important to consider the role that the immune response toward the bacterial infection may play toward creating the observed pathophysiology. While the biochemical environment, and immune response, of reproductive organs such as the prostate and the epididymis closely relate to those of mucosal tissues, the micro-environment that the developing sperm exist in within the testes is immunologically privileged. Indeed, developing sperm cells are produced long after the immune library has been established, and harbor specific antigens that cannot be found in any other organ, tissue, or cell within the body. As a consequence of this, these developing cells and the resultant spermatozoa are deemed to be “foreign” by the male immune system. However, the immune privileged environment, they experience within the testis and the epididymis aid in preventing an immune response (113, 114). In addition to this, foreign cells are able to exist within the testes without the induction of a large-scale immune response (115–117).
Testis as an Immune Privileged Site
Structure of the testis
The testes fulfill two major functions for male reproduction: the first is to produce morphological mature and functional spermatozoa, and the second is the production and controlled release of sex steroids (primarily androgens). The testis is compartmentalized both histologically and functionally into two distinct regions to accommodate the two separate functions. Spermatogenesis takes place in the seminiferous tubules, while androgens are synthesized in the Leydig cells in the interstitial compartment that is dispersed between the tubules. The seminiferous tubules are tightly coiled, and originate and terminate at the rete testis. Each tubule is surrounded by myoid pertitubulur tissue that, together with the Sertoli cells, provides structural support, and secretes the components of the basal membrane enclosing the seminiferous epithelium. The columnar Sertoli cells extend from the basal lamina toward the lumen of the tubules, and are responsible for the physical support of the developing germ cells, as well as providing essential nutrients and growth factors (Figure 2). The most prominent components of the interstitial space are the Leydig cells that work to produce steroid hormones. The interstitium also contains blood vessels and immune cells, such as macrophages, dendritic cells (DC), lymphocytes and increasingly with age, mast cells (118) (Figure 3). Also studies performed by Holstein and Davidoff have described the presence of large, flat fibroblastoid cells, which compartmentalize the microvessels, the Leydig cells and part of the seminiferous tubules (119). These cells, termed “co-cells” (abbreviated from connective tissue cells/compartmentalizing cells/covering cells), appear to produce extracellular matrix components such as decorin, vimentin, and fibroblast surface protein (120), and are typically found only in human testis. The variability in the extracellular matrix components observed is important for cell–cell interactions within the testis (121). Some of these proteins are able to bind to various types of growth factors, creating a reservoir from which the bioavailability of these growth factors can be modulated (122).
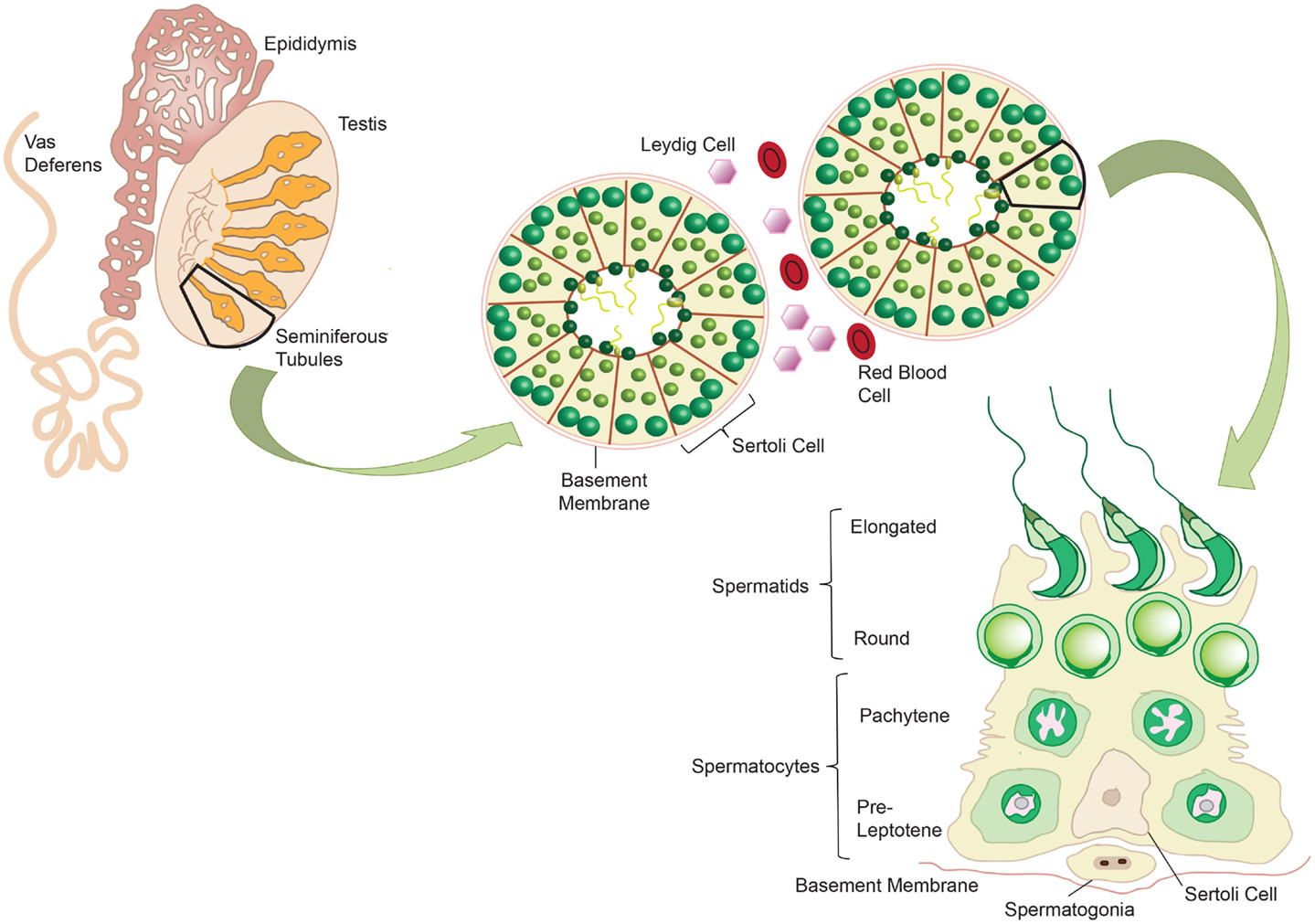
Figure 2. Mammalian spermatogenesis occurs within the seminiferous tubules of the testis, with development of the mature sperm cells occurring in a radial fashion from the basement membrane into the lumen. Spermatogonia reside on the basement membrane and undergo mitotic division to produce pre-leptotene spermatocytes. These primary spermatocytes undergo meiosis to give two pachytene spermatocytes, which in turn undergo meiosis to give round spermatids. These cells elongate and develop into mature spermatozoa. Sertoli cells support the germ cells as they develop, providing essential nutrients. Adapted from ‘Spermatogenesis Online’. Reproduction Data Systems 2011–2012. mcg.ustc.edu.cb/sdap1/spermgenes
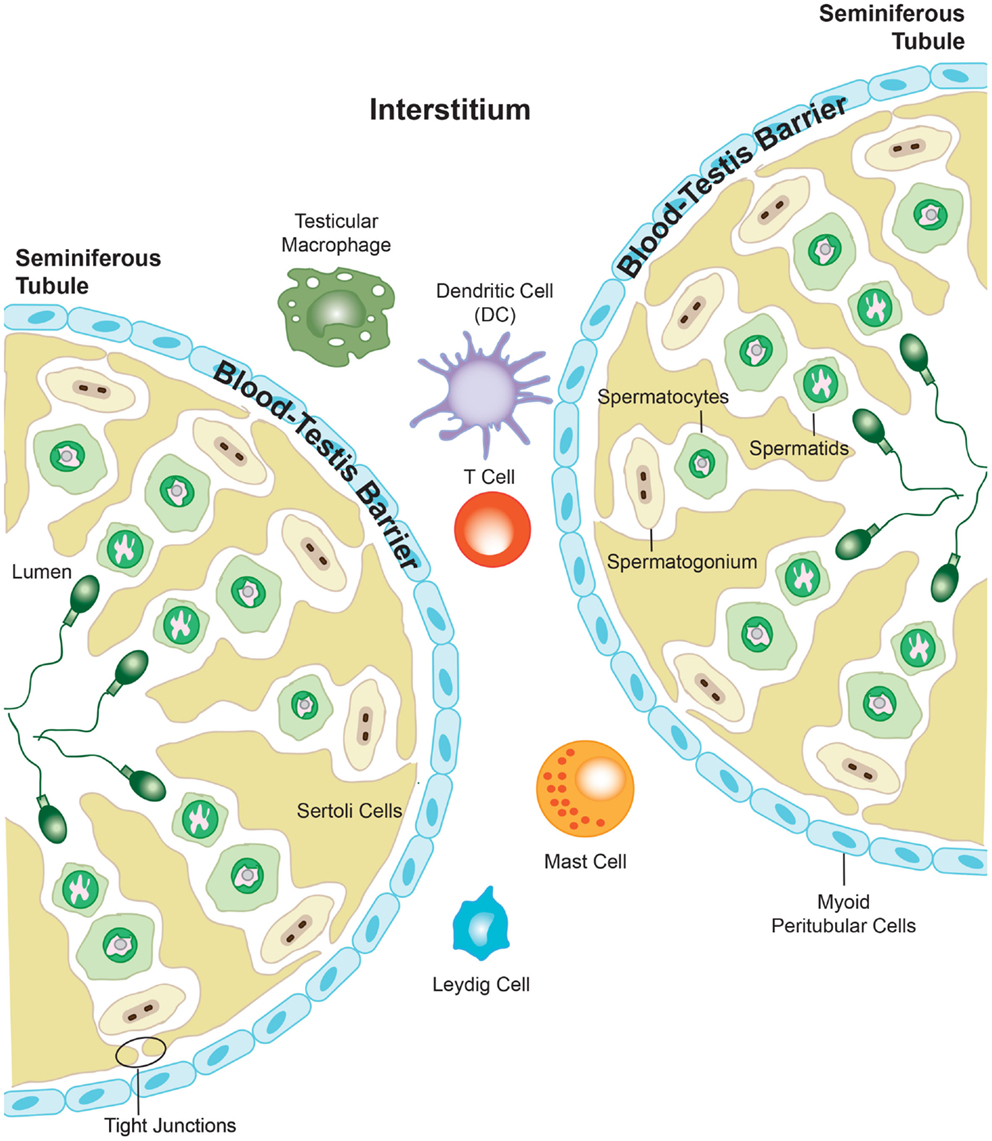
Figure 3. The presence of the blood–testis barrier creates a region of immune privilege within the testis. At the onset of puberty, developing sperm cells express novel antigens that the immune system would normally identify as “foreign.” However, segregation of antigens in the seminiferous tubules from the immune cells that are able to enter into the interstitial space of the testis prevents the body from eliciting an immune response against these vulnerable cells. Adapted from ‘Blood-Testis Barrier’. Immunopaedia.org 2010. www.immunopaedia.org.za/index.php?id+668
Blood–testis barrier
The blood–testis barrier is formed by tight junctions between neighboring Sertoli cells localized in the seminiferous epithelium, and function to restrict the movement of molecules through the intracellular spaces (Figure 3). The barrier divides the seminiferous tubule into two distinct compartments: the basal compartment, which contains spermatogonial stem cells and early stage spermatocytes, and the adluminal compartment containing meiotic pachytene and secondary spermatocytes, in addition to haploid spermatids. The importance of the blood–testis barrier in creating immune privilege was first demonstrated using Sertoli cell-depleted, androgen-receptor knockout mice. The integrity of the tight junctions that form the blood–testis barrier was compromised in these mice, and as such, immune molecules were able to pass into the adluminal compartment and mount an attack on the spermatocytes and spermatids held within. This lead to an arrest in spermatogenesis, and it was proposed to have downstream negative effects on fertility (123). However, it is well known that the blood–testis barrier alone cannot be wholly responsible for the immune privileged status that exists within the testis, as germ cell auto-antigens have been shown to be expressed in the basal compartment and in spermatogonia and early spermatocytes, which are not protected by the blood–testis barrier (124, 125). As described previously, the blood–testis barrier is incomplete in the rete testis, a location where large numbers of morphologically mature spermatozoa expressing newly adapted surface molecules, move toward the epididymis (126, 127). Furthermore, Head and Billingham demonstrated that allografts placed in the interstitial space outside the blood–testis barrier, could survive for an extended period without experiencing immune rejection (117). They concluded that mechanisms additional to the physical barrier must be in place to maintain immune privilege in the testis. The blood–testis barrier terminates at the rete testis, and subsequently, spermatozoa are no longer protected from immune attack, which is confirmed by the observation that various forms of autoimmune orchitis manifest first in the rete testis (126–129). Spermatozoa move from the rete testis into the epididymis, which consists of a long, convoluted duct, and gain the capacity for fertilization. The epididymis also contains a blood–epididymis barrier, although this barrier is more functionally related to those found in other epithelia, with fewer exclusively apical tight junctions (130–132). In contrast to the seminiferous epithelium, T-lymphocytes and macrophages are frequently found within the epididymis epithelium and in the lumen of the epididymal duct (133–135), suggesting that the epididymis operates within a different immune environment to that of the testis.
Immune Response
Innate Immunity
The first line of defense from chlamydial genital infection is the mucosal barrier of the genital tract. However, upon entering the mucosal lining and establishing a productive infection, it is the innate immune system that provides the next stage of defense against the bacteria. Although epithelial cells, which are the initial targets for Chlamydia infection, are not considered to be a part of the classical innate immune system, they are capable of initiating and sustaining innate immune responses (136). It is well known that C. trachomatis infection in both human beings and murine epithelial cells can induce the production of pro-inflammatory cytokines such as interleukin 1 (IL-1), Il-6, and tumor necrosis factor alpha (TNFα) (137, 138). In addition, secretion of chemokines such as Il-8 by infected cells can recruit classical innate immunity cells such as natural killer (NK) cells and DCs, which are abundant in the genital mucosa (139). However, as discussed above, the testis as an immune privileged site differs in the way the immune response develops against foreign pathogens, and thus warrants further exploration.
Neutrophils and natural killer cells
Neutrophils are the most predominant form of white blood cell, and have the dual functions of immune surveillance and in situ elimination of microorganisms (140). NK cells are classified as being cytotoxic lymphocytes that play a similar role to that of neutrophils (141). Importantly, NK cells and neutrophils are the first immune cells that are recruited to the site of chlamydial infection. It is thought that neutrophils work to reduce direct chlamydial infection and limit spreading, with human neutrophils being able to effectively inactivate C. trachomatis in vitro (142, 143). Additionally, mice that were neutrophil depleted had up to a 10-times greater burden of C. muridarum in the female genital tract than neutrophil-competent controls. However, both sets of mice were able to effectively eliminate the infection within the same period of time (144), which suggests that neutrophils are not critical for the resolution of the infection. As neutrophils are usually the first immune cells recruited to the site of infection, and are generally short-lived (145, 146), it is likely that the primary role for neutrophils is to reduce chlamydial infection and to limit it from spreading. In the context of the testes, the short-life span of the neutrophils is particularly important, as these cells are a major source of tissue-damaging cytokines, such as matrix metalloproteinase 9 (MMP9), during acute infection (147), and a prolonged life span for these cells may contribute to fibrosis and infertility. In support of this, recent evidence has emerged that indicates that C. trachomatis may delay neutrophil apoptosis, prolonging their life span (148). It remains unclear whether neutrophils carry out the same function in the clearance of Chlamydia from the immune privileged space of the testis, although their presence has been documented in the rat testis (149), and they have been shown to accumulate in the interstitium of mouse testis 9–12 h following exposure to E. coli (150). NK cells are more traditionally known for the role they play in viral infections and cancers. However, they have also been shown to be important in the early immune response and subsequent elimination of bacterial infections (151, 152), are their activity becomes enhanced in the presence of cytokines such as IL-12 and interferon γ (IFNγ) (Figure 3). While it has been shown the NK cells exist in the testis, their specific function remains unknown, although it is assumed that they undertake a traditional role in virus and tumor surveillance (153).
Dendritic cells
Dendritic cells are recognized as being the archetypal antigen presenting cells (APCs). DCs migrate as immature or precursor cells from the bone marrow into peripheral tissues, whereupon they receive activation signals associated with inflammation or pathogen invasion. Once activated, they migrate to the local lymph nodes, where they mature and phagocytose the antigen. Once internalized, the DCs degrade the components of the antigen and present their peptides to T-cells via major histocompatibility complex (MHC) receptors, which activate the T-cells to initiate a cell-mediated and/or humoral immune response (Figure 4). DCs also have the ability to tolerize T cells to antigens, thereby minimizing aggressive autoimmune responses (154). Immature DCs have increased capacity to internalize antigens, but low T cell stimulatory activity, while mature DCs reciprocally down-regulate their ability to endocytose antigens but have high functioning T lymphocyte stimulatory capabilities (155). In addition to this, mature DCs express surface T cell stimulatory molecules such as CD40, CD80, and CD86, as well as MHC class II molecules. They also produce bioactive IL-12 and TNFα, and display altered migratory behavior (156), and are ultimately potent stimulators of immune responses. In contrast to this, those DCs in a resting state have been implicated in the creation of self-tolerance (157), with the presentation of “self” antigens on the surface of DCs thought to play a significant role in the initiation of autoimmunity, and its progression toward autoimmune disease (158, 159). Further to this, as DCs are APCs, they are not capable of targeting specific antigens, but instead present a wide range of antigens on their cell surface, including what have been labeled as danger signals. The working hypothesis, or “danger model” suggests that stressed or damaged cells and tissues express and release heat shock proteins (Hsps) during injury caused by trauma, inflammation, pathogens, or toxins. Recently, it has been shown that these Hsps act as testicular autoantigens, and may provide a mechanism for how DCs in the testis participate in the activation of lymphocytes and the subsequent damage of testicular tissue (160).
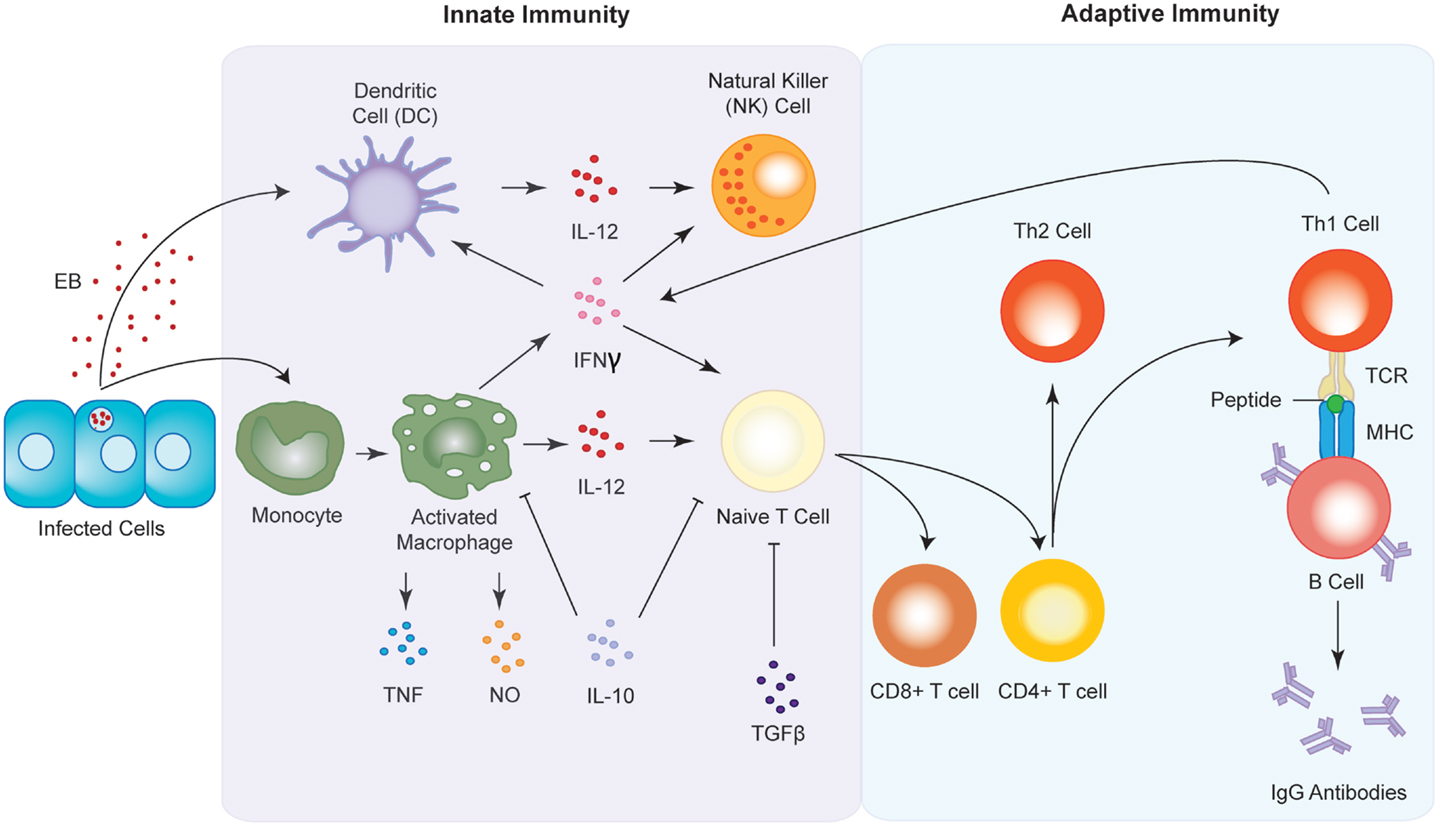
Figure 4. Innate and adaptive immune responses to Chlamydia infection. Upon infection, antigen presenting cells (APC) such as macrophages and dendritic cells are sequestered to the site of infection where they begin to release pro-inflammatory cytokines such as IFNγ and IL-12. The chemokines in turn activate natural killer (NK) cells and induce the maturation of T cells into either CD8+ or CD4+ T cells. CD4+ T cells go on to form either T-helper 1 (Th1) or T-helper 2 type (Th2) T cells. Th1 cell interact with B cells via the T cell receptor (TCR) and the major histocompatibility complex (MHC) to produce antibodies against the chlamydial infection.
Both MHC class I and II molecules are expressed in the interstitial tissue of testis, including on macrophages and Leydig cells. In addition, DCs also express MHC class II molecules in the testis (161). Interestingly, developing germ cells fail to express MHC antigens on their surface, perhaps giving an indication of how these cells avoid detection by CD4+ and CD8 + T cells (162–168). However, despite the importance they play in regulating the immune response in other tissues, very few studies have investigated DCs in the testis. Cells that express DC markers or possess DC-like morphology have been observed in the testes of mice (169, 170), rats (117), and human beings (165, 171). Although these markers are also expressed on macrophages, the positive identification of these cells as DCs remains difficult. Nevertheless, co-stimulatory molecules such as CD80 and CD86 are expressed in the testis of 14- to 22-week-old non-obese diabetic mice (172), as well as in the rat testis (161), which suggests that DC-dependent activation of T-lymphocytes via binding to their specific antigens is at least possible in rodent testis.
Sertoli cells
Sertoli cells are somatic cells that, as described above, provide essential support for developing sperm cells in the testis and have a critical role in the establishment and maintenance of immune privilege. A number of studies have demonstrated that Sertoli cells possess the ability to act as immunosuppressants (173), and are capable of being transplanted into a variety of tissues (174–176) and inducing immune tolerance (173). In addition to this, these cells are known to possess characteristics more traditionally displayed by immune cells, and thus are thought to play an essential role in the immune response within the testis. They are capable of phagocytosing apoptotic spermatogenic germ cells (177), producing anti-microbial proteins (178–180), and perhaps most importantly express pattern-recognition receptors (PRRs) such as Toll-like receptors (TLRs) (181–184). TLRs operate by recognizing pathogen-associated molecular patterns (PAMPs) and structural subunits, including those for microbial cell walls (e.g., peptidoglycan), cell membranes (e.g., LPS), and virulence proteins (e.g., flagellin) (185), that are absent in the host. Upon recognizing these PAMPs, the TLRs become activated and induce production of pro-inflammatory cytokines, as well as cell adhesion molecules that recruit macrophages, neutrophils, and NK cells, and invoke maturation of DCs. They also secrete specific anti-microbial products such as interferons and defensins (186). To date, 10 TLRs have been identified in human beings, and 13 in mice (187), and the roles that each TLR plays depend on which PAMPs it recognizes. TLR2, in conjunction with TLR1 and TLR6 recognize various bacterial components (peptidoglycans, lipopeptide, and lipoprotein of Gram-negative bacteria, and lipopeptide of mycoplasma) (188, 189). TLR3 recognizes the dsRNA that is produced by replicating viruses (190). TLR4 in association with the co-receptor CD14 and the extracellular molecule MD-2 recognizes LPS (191, 192). TLR5 recognizes a highly conserved structure that is specific to bacterial flagellin (193). The presence of TLR2 and TLR4 on Sertoli cells has been demonstrated in both prepubertal and adult testis, with TLR5 and TLR6 also detected at lower levels. Importantly, it has been shown that the presence of IFNγ or TLR agonists induces a significant increase in TLR2, TLR4, and TLR6 mRNA, and TLR2 protein becomes up-regulated (181). Further to this, stimulation with pro-inflammatory cytokines resulted in Sertoli cells being able to bind lymphocytes, as well as secrete IL-6, indicating the potential to promote inflammation in the testis (181).
Interferon γ
Interferon γ plays a functional role in both the innate and adaptive immune response (Figure 4). It has the dual responsibility of inhibiting the growth of Chlamydia (194), as well as being one of the main cytokines important for type 1 T-helper cell (Th1) immune response (195). IFNγ is a cytokine produced by NK cells, as well as CD4+ and CD8+ T-cells, in response to signals such as Interleukin 2 (IL-2), basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF). The importance of IFNγ in vivo has been shown through enhanced levels of bacterial infection in IFNγ−∖− or IFNγ-receptor−∖− mice, compared to controls (196–200). However, mice are able to resolve chlamydial infection in the absence of IFNγ, suggesting that while beneficial in the process of clearing chlamydial infection, it is not essential, and that there are T-and B-lymphocyte dependent mechanisms also playing roles. Indeed, studies using double-knockout mice null for both IFNγ (IFNγ−∖−) and T-and B-lymphocytes (RAG−∖−), demonstrated that when infected with C. pneumonia, the null mice displayed a higher rate of mortality, when compared with IFNγ−∖− and RAG−∖− mice single KO mice (201). IFNγ has been shown both in cell culture and in vivo models to provide protection against infection by C. muridarum and C. psittaci (202–204). The mechanism of action of this inhibitory effect is not the direct result of the IFNγ acting on the Chlamydia but instead due to alterations made to the host cell physiology and environment that impact on the ability of the Chlamydia to grow and replicate.
IFNγ affects human host cells in vitro by inducing the expression of indoleamine 2, 3-dioxygenase (IDO), an enzyme that catalyzes the initial step in the degradation of the amino acid tryptophan to N-formalkynurenine and kynurenine (205). IDO has been shown to mediate potent immunosuppression in classical immune responses, as well as fetal tolerance, tumor resistance, regulation of autoimmune responses, and maintenance of immune privilege in the epididymis (206–211). Depletion of exogenous intracellular tryptophan by IDO has been shown to starve the Chlamydia of an amino acid essential for its ability to differentiate into infections EBs (194, 203, 212). However, there are chlamydial species, which have successfully adapted to tryptophan starvation by transforming into a unique non-replicating but viable form (213). Following removal of IFNγ from the host cell, and subsequent resumption of tryptophan synthesis, these unique forms quickly differentiate back into infectious EBs and continue the infection. This cycle is known as a “persistent” infection, and is discussed in more detail in below.
Furthermore, it is well established in murine cells that IFNγ also activates inducible nitric oxide synthase (iNOS), an enzyme that catalyzes the production of anti-microbial reactive nitrogen intermediates, including nitric oxide (NO), from l-arginine (214). There have been a number of studies showing a correlation between the inhibition of intracellular chlamydial growth and the induction of NO secretion (214–219), suggesting that control of chlamydial infection in mice may involve the activation of the cytokine-iNOS system. In support of this, a recent study has linked IL-17A and IFNγ, suggesting that they work in a synergistic fashion to up-regulate iNOS, and subsequently NO production, in order to inhibit chlamydial growth.
Iron has also been shown to be important for Chlamydia survival (220–222). It is thought that the ability of IFNγ to down-regulate transferrin receptor (223–226), which regulates the import of iron into the cell, may limit the availability of the amount of intracellular iron available to the bacterium, thus limiting its growth. This has been shown to be true for other bacterial species such as Legionella pneumophila (225) and Salmonella typhimurium (227). In addition to this, IFNγ is able of enhancing the phagocytic capabilities of macrophages, and may promote the engulfment and subsequent elimination of Chlamydia (228, 229). It has been known for some time that IFNγ is produced by the Sertoli cells, peritubular cells and, at low levels, early spermatids within the testes (230). It has also been shown to influence testicular germ-cell tumors (TGCTs), paradoxically having either anti- or pro-apoptotic activity depending on the tumor type. Within these tumors, IFNγ bind to its receptor (IFNγ R) and activates the JAK/STAT (Janus kinase/signal transducer and activator of transcription) pathway. JAKs that have been activated by IFNγ phosphorylate STAT1 proteins, which are translocated to the nucleus, resulting in the transcriptional activation of specific target genes. Several of these genes have been identified, but the gene of note that is shown to be activated by IFNγ is that of interferon regulatory factor 1 (IRF-1) (231). IRF-1 mediates a diverse range of functions, including tumor suppression, myeloid differentiation, macrophage activation, antigen presentation, and T- and B-cell differentiation (232). In addition to this, studies by Kanzaki and colleagues has demonstrated the presence of IFNγR in rat Sertoli cells and that it also regulates the expression of the IRF-1 gene in the testis, potentially as a means of regulating the apoptosis if neoplastic germ cells (233). Importantly, interstitial macrophages also express high levels of IFNγ and TNFα during testicular inflammation, and that abnormal elevation of these cytokines has been associated with reduced fertility in inflamed testes (234). A recent study by Gao and colleagues further indicates that IFNγ works synergistically with TNFα, having pronounced disruptive effects on the blood–testis barrier and Sertoli-germ cell adhesion in the seminiferous epithelium, resulting in reduced fertility (235).
In the epididymis, however, it appears that IFNγ plays a minor role in the establishment and maintenance of the immune response. Although IDO is highly abundant in the epididymis, its expression is constitutive, and is IFNγ-independent. It is, therefore, thought that the epididymis is constantly in an inflammatory state, as IDO is considered to be a component of the early immune response to inflammation and infection (236).
Macrophages
Macrophages are phagocytic immune cells produced through the differentiation of monocytes and are central for the induction of the innate immune response. They are also APC, equipped with PRRs that aid in the recognition of various moieties from pathogens termed PAMPs, in addition to danger-associated molecular patterns (DAMPS) (237). During Chlamydia infection, macrophages migrate to the infected site (238), phagocytose the bacteria (239), and depending on the specific receptor-PAMP/DAMP match, induce various downstream effectors and pathways to produce pro-inflammatory cytokines (240, 241). Macrophages are activated by IFNγ, which is produced by CD4+ and CD8+ cells, and works to convert resting macrophages into potent cells with increased antigen presenting capacity, increased synthesis of pro-inflammatory cytokines and toxic mediators, and augmented complement-mediated phagocytosis (Figure 4). Further to this, the destruction of Chlamydia inside macrophages has been associated with autophagy, a process by which cells degrade cytoplasmic proteins and organelles (242–244). Studies have also demonstrated that macrophage autophagy can enhance antigen presentation to T cells (245). It is now well known that the macrophages make up the majority of the immune cells found within the testis (118, 246). Under normal conditions, macrophages, and indeed all other leukocytes, are found exclusively in the interstitial space. It is only under pathological conditions that macrophages are able to enter the germ cell compartment where they are capable of phagocytosing degenerating germ cells. In addition to possessing features common to all macrophages, testicular macrophages also appear to play an important role in male reproductive function. Indeed, it has been shown that macrophages within the testis exist in direct contact with Leydig cells, forming specialized contact sites known as digitations (247, 248). Furthermore, they are involved in Leydig cell development and the regulation of steroidogenesis in adults (249–253). The high levels of macrophages present within the testis are almost exclusively regulated by a Leydig-cell-mediated mechanism, with testosterone and macrophage-migration inhibitory factor (MIF) playing only minor roles, if any at all (254). The evidence for the close relationship between testicular macrophages and Leydig cells is represented by the loss of approximately 50% of these macrophages in Leydig-cell-depleted testis (255). These macrophages are also capable of influencing Sertoli cell function, and subsequent spermatogenesis through the release of soluble mediators (256).
It is well-established testicular macrophages are responsible for establishing and maintaining the immune response within the testis. Macrophages from normal tissues are APCs, with the ability to direct the outcome of T cell responses, and the expression of major histocompatibility (MHC) class II expression on the testicular macrophages suggests a similar role for these cells (118). However, the antigen presenting capabilities of testicular macrophages have yet to be fully elucidated, although it is known that they suppress both T cell responses in vitro (257) and in vivo (258). Interestingly, a number of studies have shown the testicular macrophages are significantly poorer at producing pro-inflammatory cytokines, as well as IL-1β and TNFα, although the production of prostaglandins and other cytokines are not inhibited (257, 259–261). However, despite this limitation in testicular macrophage function, the testis is still able to support an inflammatory response. Evidence suggests that testicular somatic cells such as Leydig cells, Sertoli cells, and peritubular cells, are capable of producing various pro-inflammatory mediators such as MIF, iNOS, and various isoforms of IL-1 (113, 262–268). In the rat testis, at least two subsets of macrophages have been identified, one which is recognized by the monoclonal antibody ED1, and the other which is a surface antigen recognized by the monoclonal antibody ED2 [reviewed by Hutson (269), Hedger (246), Hedger and Meinhardt (270), Hedger and Hales (113), and Hutson (248)]. While the ED2+ subset forms the majority of the macrophages expressed in the rat testis, a significant proportion of ED1+ ED2− cells are also present, representing about 15–20% of the population. These are assumed to be circulating “inflammatory” monocytes or recently arrived macrophage. While the ED2+ cells do not participate in the inflammatory response, it is clear that an influx of ED1+ monocytes during acute or chronic inflammation shift the cytokine balance toward and inflammatory response with the potential to overcome the immune privilege (271–273). Interestingly, it has been shown that in models of acute inflammation, the testis appears to possess a mechanism capable of counterbalancing the influx of ED1+ monocytes, as the increase observed is only temporary and resolved after 1–2 days (271). This mechanism is unable to counter-balance the inflammation caused by chronic infection, however, where the increase in macrophage number persists for much longer (274). In the mouse testis, a sub-population of macrophages has been identified that are capable of expressing high levels of TNFβ and appears to exhibit a tolergenic phenotype (275). In contrast to those identified in the rat testis, these macrophages have been recognized as having immunosuppressive qualities, as demonstrated by their inability to induce T-lymphocyte proliferation and their reduced antigen presenting activity (275).
Adaptive Immunity
Although the innate immune response is important as a first line of defense against Chlamydia infection, the adaptive response is necessary in limiting the spread of the infection, and in providing protection against recurrent infections. The adaptive, or acquired, immune response involves creating immunological memory after the initial response to the pathogen, which leads to an enhanced response in the case of subsequent exposure to the same pathogen. There are two cell types that regulate this immune response: T-lymphocytes and B-lymphocytes. Both cell types rely on their ability to distinguish between “self” and foreign antigens presented on the cell surface, a process that is regulated by the MHC. The B-lymphocytes, or B cells, regulate the humoral immune response, producing antibodies against foreign antigens. The T-lymphocytes, or T cells, regulate the cell-mediated immune response, and can be divided into sub-types. CD8+ T cells are also known as cytotoxic T cells and work to induce death in infected host cells. These cells predominately recognize foreign antigens presented on the cell surface by class I MHC. CD4+ T cells or helper T cells can be divided once again into two sub-types: Th1 cells, which induce the production of cytokines such as IFNγ. Release of IFNγ activates macrophages and induces B cells to make opsonizing and complement fixing antibodies. Th2 cell are characterized by the release of Interleukin 4 (IL-4), which in turn activate B cells to make non-cytolytic antibodies leading to humoral immunity. These cells recognize foreign antigens presented by class II MHC.
T cells
The involvement of T cells in the immune response against chlamydial infection was first demonstrated when Rank and colleagues observed that immunodeficient mice developed chronic C. muridarum infection following intravaginal inoculation, while their wild-type counterparts were able to clear the infection within 20 days (276). T cells unable to recognize pathogens or antigens without the help of APC, such as DC or macrophages. Once these cells phagocytose chlamydial EB, or infected host cells containing RBs, they degrade the chlamydial components and present the peptides via the MHC class I/II – antigen complexes. Studies in both human and mice have shown that both CD4+ and CD8+ cells are present at the site of chlamydial infection (238, 277–279). Both types of T-cell have been shown to recognize C. trachomatis antigens, including outer membrane protein 2 (OMP2) (280), polymorphic outer membrane protein D (POMP-D) (281), MOMP, heat shock protein 60 (hsp60) (282–284), chlamydial protease activating factor (CPAF) (285), pmpG, PmpF, and RpIF (286, 287). Upon C. trachomatis infection, CD4+ cells become activated, begin to proliferate and migrate to the genital mucosa (286, 288–292). These T-cells exhibit a characteristic Th1 response, secreting large amounts of IFNγ required to aid in clearing bacterial infection (290, 293). Previous studies have shown that infection of CD4−∖− mice with C. trachomatis results in higher infection load during primary infection, in addition to diminished protection from secondary infection (290). However, there has been controversy regarding the protective immunity that CD4+ T cell memory provides following C. trachomatis infection (290, 294). Indeed, one such study utilized antibodies to deplete CD4+ T cells and showed that previous infection with C. trachomatis does not induce strong protective immunity upon secondary infection, and that CD4+ T cells are not essential for clearance of infection (294). Conversely, there have been many studies highlighting the important of CD4+ T cells in providing protective immunity against bacterial infection (295, 296). More recently, a study compared the ability of wild-type mice and B cell-deficient mice to clear C. trachomatis genital infection, and demonstrated that CD4+ T cell immunity was essential for protective immunity to secondary infection (297). Evidence from Gondek and colleagues support this, whereby they demonstrated that infection of the upper genital tract with C. trachomatis induces a robust Chlamydia-specific CD4+ T cell response that is both necessary and sufficient to clear infection and provide protection against re-infection (298). The role of CD8+ T cells in aiding in clearing a bacterial infection was first described in studies using a mouse model whereby splenic CD8+ T cells could specifically lyse Chlamydia-infected fibroblasts. In addition to this, CD8+ cytotoxic T cells induced partial protection when adoptively transferred into infected mice (288, 299, 300). CD8+ cells control infection of intracellular pathogens through a number of mechanisms, including cytotoxicity via granule exocytosis, production of anti-microbial peptides, and production of cytokines and chemokines. It is assumed, based on these abilities that CD8+ T cells function by lysing infected cells and depriving the pathogen of its intracellular environment, as well as releasing inflammatory mediators that render developing bacteria non-infectious, and recruiting or activating other immune cells to limit the survival of the pathogen (301). Although both CD4+ and CD8+ T cells contribute protective immunity during Chlamydia infection, differences exist depending on the model of Chlamydia and the mode of infection studied. For example, depletion of CD8+ but not CD4+ T cells in immune mice diminishes immune protection upon challenge with C. psittaci (302).
In contrast, in a model of C. trachomatis infected mice, depletion of CD4+ T cells abrogates protection more significantly compared to a decrease in CD8+ T cells (303, 304). It is important to note that CD4+ T cells are often required for the induction and preservation of a functional CD8+ T cell response, and in their absence both CD4+ and CD8+ T cell effector functions are severely impaired (301). T cells comprise approximately 15% of the total leukocyte population in the interstitium of rodent testis (118, 305), with CD8+ cytotoxic T cells predominating over CD4+ T cells (306). Unfortunately, very little is known about the function that T-lymphocytes carry out in the testis although there is evidence that activated memory T cells, which would normally affect an immunological response, are instead targeted for destruction when they enter the testicular environment (307, 308).
B cells and antibodies
The importance of B cells and the antibodies they produce in mediating immunity against Chlamydia infection was demonstrated more than four decades ago, when it was observed that the presence of Chlamydia-specific antibodies correlated with reduced rates of infection in human beings (309, 310). Later, studies went on to demonstrate that monoclonal antibodies directed against the primary Chlamydia antigen MOMP could neutralize infection in vitro (311, 312), in addition to being moderately affective at providing immunity when passively administered to mice (313). Numerous Chlamydia proteins have also been shown to induce antigen-specific antibodies (314). There are several mechanisms by which B cells are able to modulate immunity during Chlamydia infection. The first and most predominate mechanism is through antibody-mediated neutralization, whereby the B cell produces specific antibodies directed against chlamydial peptides (315). Secondly, through antibody-dependent cellular cytotoxicity that targets cells that have antibodies attached to their surface for lysis (316). Finally, B cells aid in the formation of antibody–antigen complexes that bind to receptors on APC. These then enhance phagocytosis and antigen presentation to CD4+ T Cells (317). Until recently, it has been thought that B cells were predominantly important in controlling secondary chlamydial infection but their presence for clearing primary infection was not essential. This was based on evidence form Su and colleagues showing that B cell-deficient mice control Chlamydia infection as efficiently as wild-type mice but had delayed clearance of secondary infections (318, 319).
Latterly, it was observed that B cell-deficient mice depleted of CD4+ T cells are unable to control secondary chlamydial infection in contrast to mice who were devoid of CD4+ T cells alone were able to clear the secondary infection after only a slight delay (320). This has since been attributed to the ability of B cells to produce specific antibodies, as passive transfer of immune serum or anti-Chlamydia antibodies into B cell-deficient, CD4+ depleted mice rescues their ability to clear a secondary infection (321). Moreover, the susceptibility of these B cell-deficient, CD4+ depleted mice to Chlamydia infection suggests a relationship between CD4+ T cells and B cells in providing protective immunity. Further to this, a study by Li and colleagues proposes that it is the antibody-producing ability of the B cells, and not antigen presentation that is responsible for the containment of bacterial infection, as CD4+ T cell priming was markedly reduced in B cell-deficient mice, and Chlamydia were unable to disseminate as far compared with controls. Interestingly, these studies demonstrate a potential role for B cells in regulating local T cell activation and bacterial dissemination during primary Chlamydia infection (322). Under normal conditions, B cells are very rarely detected in rat testis (255), and as such the specific role that B cells and antibodies play in controlling bacterial infections in the testis remains unknown.
Persistence and Avoiding the Immune Response
Persistence is defined as a long-term association between Chlamydia and the host, in which the organism remains viable but in a culture-negative state. The exposure of Chlamydia to cellular and molecular stresses is the most common cause for the bacteria to enter into a state of persistence. This state is normally characterized by the existence of large pleomorphic RBs, termed aberrant bodies (ABs), which are non-infectious, viable, and thought to be clinically undetectable. One of the defining features of the ABs, as opposed to dying or dead RBs, is that the removal of the stressor often allows the Chlamydia to re-enter the normal developmental life cycle. The transition of RBs into ABs can be induced in vitro by treatment with antibiotics, deprivation of amino acids, heat shock, or pro-inflammatory cytokines (323). Treatment of Chlamydia with antibiotics, such as penicillin or erythromycin, arrests chlamydial development by blocking the conversion of RBs into EBs, as shown when McCoy cells infected with C. trachomatis were treated with erythromycin 12 h post-infection, and developed abnormally large RBs (324).
In addition to this, it was more recently observed that addition of ampicillin to culture media of C. pneumoniae-infected HeLa cells resulted in the formation of aberrant, giant RBs (325). Interestingly, when compared with C. trachomatis infection of the same cell type, the C. pneumonia took much longer to recover and re-enter a normal developmental cycle (325). The reason for this remains unclear, although it is intriguing as C. pneumoniae are normally recognized for being very efficient at reactivation following the removal of other types of stressors, such as tryptophan-depletion, or co-culture with IFNγ (see below) (326). It has been proposed that these studies give an indication toward the possibility that inadequate microbial therapy may allow Chlamydia to persist in vivo.
In contrast to the mechanism in place for inducing persistence with antibiotics, depletion of amino acids from the culture medium of Chlamydia-infected cells arrests the development of both the bacteria and the host cell until such a time as the amino acids are re-introduced back into the system (327). Interestingly though, re-introduction of the amino acids does not results in full recovery of Chlamydia development, with smaller particle sizes observed (328). While the progressive depletion of all amino acids causes abnormal development, the depletion of tryptophan is particularly important. Most species of Chlamydia regulate tryptophan for survival (329). As described previously, IFNγ induces the expression of the cellular tryptophan-degrading enzyme indoleamine 2,3-dioxygenase (IDO). In the majority of Chlamydia species, lack of this essential amino acid results in death. However, some species have instead adapted to this mode of starvation by reverting to a persistent state. Depletion of nutrients other than amino acids has also been shown to contribute toward inducing persistence, with examples including the removal of glucose from C. trachomatis in McCoy cells resulting in temporary loss of infectivity and an abnormal morphology comparable to that observed in amino acid depletion (330).
The form of nutrient-depletion that has been the best characterized, however, has been the depletion of iron. Exposure of C. trachomatis to iron chelators causes significant morphological changes that were distinct even from those observed in other persistence systems (331). Importantly, addition of iron-saturated transferrin is able to rescue the infectivity of the Chlamydiae. Exposure of Chlamydia in culture to moderate levels of the cytokine IFNγ induces the formation of large ABs (328, 332), as described in other persistence models although with different growth characteristics (333) and altered or decreased expression of predominant chlamydial proteins or constituents such as LPS, the 60 kDa outer membrane protein (OMP) and the MOMP (334). The mechanism of action is through the activation of IDO by IFNγ, resulting in tryptophan depletion (see above) (202). As with other models, the addition of tryptophan into the culture results in reactivation of the developmental cycle. Interestingly, observations recorded by Caldwell and colleagues suggest that each species of Chlamydia has its own pattern of resistance to the inhibitory effects of IFNγ, and this relates directly to polymorphisms in tryptophan synthesis genes (335). For example, ocular serovars of Chlamydia possess a non-functional tryptophan synthase and are consequently unable to produce tryptophan of their own accord, which would make them more susceptible to inhibition by IFNγ. Genital serovars, however, have a functional tryptophan synthase, and are capable of using indole as a substrate for tryptophan synthesis (335). Persistence has also been demonstrated in vivo, with electron microscopic visualization of morphologically aberrant Chlamydia forms in diseased tissue. Nanagara and colleagues showed that atypical, pleomorphic AB with poorly defined outer membranes dominated within infected fibroblasts and macrophages from patients with C. trachomatis-associated reactive arthritis (336). In addition, miniature C. trachomatis forms have been observed in total ejaculate and expressed prostate secretions from patients with chronic chlamydial prostatitis (337) and in the oviducts of mice experimentally infected with C. trachomatis (338). Importantly, there is also evidence for the ability of Chlamydia to become reactivated in vivo. A study performed by Dean and colleagues demonstrated the individuals experiencing recurrent infections possessed chlamydial isolates of the same genotype (339). Unfortunately, this study could not make allowance for recurrence from infected partners. However, an additional study did show a 10% recurrence of genital C. trachomatis infection in individuals that reported abstinence or complete condom use following treatment with antibiotics (340).
It is well understood that Chlamydia utilizes the state of persistence not only to continue its infectious life cycle but also as a means of evading the host immune response. However, Chlamydiae have also developed additional means of remaining undetected. One such survival strategy employed by both C. trachomatis and C. pneumonaie is to inhibit apoptosis of the host cells (341, 342), which ensures that host cell lysis does not occur prior to the completion of the bacterial developmental cycle. It is also thought that inhibition of apoptosis may limit the number of apoptotic infected cells available to APC, and allow infected cells to resist being killed by effector CD8+ T cells (301). Nevertheless, the influence that Chlamydia has on host cell apoptosis is complex and controversial, as studies have also shown Chlamydia is also able to induce apoptosis (343).
The induction of apoptosis is thought to occur in two ways: firstly through the expression of Chlamydia proteins associated with death domains, which may promote apoptosis through association with mammalian death receptors (344, 345). Although it appears counter-intuitive for Chlamydia to both induce and suppress apoptosis, it may help protect the bacterium from the immune response in vivo. As mentioned previously, it has been proposed that Chlamydia may restrict apoptosis in order to complete the developmental cycle, allowing RBs to convert back into infectious EBs. Once the developmental cycle nears completion, it may be beneficial for the bacterium to induce apoptosis in order to avoid necrosis, which is known to stimulate inflammation and subsequently enhance Chlamydia-specific immune response. It may also aid in the release of infectious EBs into the cytosol (346). In addition to this, it has been proposed that Chlamydia may down-regulate MHC class I and II expression on infected cells in order to evade T cell-mediated immune recognition. This is evidenced by the secretion of a chlamydial protease-like activity factor (CPAF) into the cytosol of C. trachomatis or C. pneumonaie infected cells, which then works to degrade specific host cell transcription factors that control the constitutive and IFNγ-inducible expression of MHC class I and II molecules, respectively (347–349). Down-regulation of MHC expression on infected cells has the potential to limit their recognition by CD4+ and CD8+ T cells, in addition to rendering the infected cells susceptible to lysis by NK cells. Evidence for Chlamydia’s ability to evade the immune system in vivo is indirect. It is clear that previous exposure of human beings to C. trachomatis does not provide robust immunity against re-infection, and this may be due to poor development of an adequate immune response (8). Studies performed in mice have demonstrated that Chlamydia-specific T cells do not appear to develop into memory T cells capable of mounting a robust recall response when compared with infection with well-known pathogen such as vaccinia (8).
Chronic Inflammation
Following chronic or recurrent genital tract infection with C. trachomatis, the chronic inflammation that develops can lead to tubal scarring, ectopic pregnancy, and infertility in women (350), as well as epididymitis, prostatitis, and orchitis in men (see above). As discussed above inflammation is primarily caused by the activation of macrophages during the immune response, resulting in the release of different mediators that perpetuate the pro-inflammatory response such as IL-1, TNFα, and prostaglandins. While this pathway is thought to result in the majority of the tissue damage associated with the pathology of Chlamydia infection, a number of other factors may also contribute to the etiology.
The first of which is the presence of ROS. ROS comprises several oxidizing, oxygen-containing species that are generated in a physiological system through one-electron-transfer reactions during metabolism, and include hydroxyl radical (OH−), superoxide anion radical and hydrogen peroxide (92). ROS are known mediators of the immune response, and work by promoting endothelial dysfunction through the oxidation of crucial signaling proteins (351). Importantly, testicular macrophages are known to produce ROS during infection and in response to LPS (352), and LPS is known to induce oxidative stress (353–355). The release of ROS by activate macrophages not only affects invading pathogens, but may also expose adjacent host tissue and cells to oxidative stress. It has been proposed that Leydig cells may be particularly susceptible to extracellular sources of ROS during immune responses due to their close proximity to interstitial testicular macrophages (356, 357). In addition to this, as ROS produced by C. trachomatis infection could be an important factor in the damage of sperm cells, it is thought that low molecular weight and enzymatic secretions of the male genital tract may play an active role in suppressing the deleterious effects of ROS, leading to the preservation of fertility (102, 358). ROS such as superoxide anion are able to rapidly combine with NO to form reactive nitrogen species (RNS). The RNS in turn induces nitrosative stress, which adds to the pro-inflammatory burden of ROS (359). In addition to this uncontrolled NO production alone has been implicated in chronic inflammation during Chlamydia infection (360, 361).
In addition to the presence of ROS, the presence of small anti-bacterial molecules in the male reproductive tract may also contribute to a sustained inflammatory response. Defensins are small, positively charged peptides that disrupt bacterial infection by forming multimeric pores in the pathogens membrane (186). A large majority of epithelial cells produce beta defensins, including those of the epididymis and testis, with a large number of epididymal-specific beta defensins being identified in both the mouse and rat (362–367). Most of these epididymal defensins are developmentally or hormonally regulated, with evidence that some might play a role in sperm maturation (362, 365). Normally, epithelial defensing production is stimulated by TLR activations and cytokines during inflammation, with studies showing that LPS induces mRNA expression of defensin and defensin-like spag11, as well as pro-inflammatory cytokines in the rat epididymal caput, cauda and testis (368). Importantly, it has been shown recently that stimulation of mouse macrophages with beta defensin 14 results in the synergistic and enhanced expression of pro-inflammatory cytokine and chemokines induced by TLR ligand re-stimulation (369). It is, therefore, possible that infection with Chlamydia may stimulate the up-regulation of beta defensins in the male reproductive tract, which may in turn activate mouse macrophages to produce an inflammatory response. Therefore, chronic, or recurrent chlamydial infection may lead to chronic inflammation, and subsequent tissue and cell damage.
Conclusion
Chlamydia infection is a public health concern, with the development of a vaccine capable of preventing infection without activating harmful immune responses the best solution for controlling this sexually transmitted disease. Unfortunately, as yet, no vaccine has been developed that has been able to block infection, highlighting the dynamic and complex nature of the immunobiological response that is mounted against the invading bacteria. Clearance of chlamydial infection requires the coordinated action of both the innate and adaptive immune systems, with multiple levels of cross-talk and redundancies in place. The problem is compounded even more in regions of the male reproductive tract, particularly the testis, whose blood–testis barrier allows the cells within to mature and function without risk of attack from the immune system. However this barrier, also prevents a normal immune response from occurring, and the underlying mechanisms of this response remain unclear. It is also unknown how the presence of the blood–testis barrier would affect the delivery of an effective vaccine. Therefore, research aiming to provide a greater understanding of the role and regulation of the immune response within the male reproductive tract would aid in the development of Chlamydial vaccine.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Nisyrios G. Should the Australian defence force screen for genital Chlamydia trachomatis infection? Aust Defence Force Health (2006) 7:20–1.
2. Wallesar S, Salkeld G, Donovan B. The cost-effectiveness of screening for genital Chlamydia trachomatis infection in Australia. Sex Health (2006) 3:225–34. doi:10.1071/SH06016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Macaldowies A, Wang YA, Chambers GM, Sullivan EA. In: AIHW editor. Assisted Reproductive Technologies in Australia and New Zealand 2010. Assisted Reproduction Technology Series (Cat no. PER55). Canberra, ACT: AIHW (2012).
4. Corsaro D, Greub G. Pathogenic potential of novel Chlamydiae and diagnostic approaches to infections due to these obligate intracellular bacteria. Clin Microbiol Rev (2006) 19:283–97. doi:10.1128/CMR.19.2.283-297.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Nunes A, Gomes JP. Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect Genet Evol (2014) 23C:49–64. doi:10.1016/j.meegid.2014.01.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Wang SP, Grayston JT. Three new serovars of Chlamydia trachomatis: Da, Ia, and L2a. J Infect Dis (1991) 163:403–5. doi:10.1093/infdis/163.2.403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Baud D, Regan L, Greub G. Emerging role of Chlamydia and Chlamydia-like organisms in adverse pregnancy outcomes. Curr Opin Infect Dis (2008) 21:70–6. doi:10.1097/QCO.0b013e3282f3e6a5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Brunham RC, Rey-Ladino J. Immunology of Chlamydia infection: implications for a Chlamydia trachomatis vaccine. Nat Rev Immunol (2005) 5:149–61. doi:10.1038/nri1551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Wagenlehner FM, Naber KG, Weidner W. Chlamydial infections and prostatitis in men. BJU Int (2006) 97:687–90. doi:10.1111/j.1464-410X.2006.06007.x
10. Kalman S, Mitchell W, Marathe R, Lammel C, Fan J, Hyman RW, et al. Comparative genomes of Chlamydia pneumoniae and C. trachomatis. Nat Genet (1999) 21:385–9. doi:10.1038/7716
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Dautry-Varsat A, Subtil A, Hackstadt T. Recent insights into the mechanisms of Chlamydia entry. Cell Microbiol (2005) 7:1714–22. doi:10.1111/j.1462-5822.2005.00627.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Belland RJ, Zhong G, Crane DD, Hogan D, Sturdevant D, Sharma J, et al. Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc Natl Acad Sci U S A (2003) 100:8478–83. doi:10.1073/pnas.1331135100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Abdelrahman YM, Belland RJ. The chlamydial developmental cycle. FEMS Microbiol Rev (2005) 29:949–59. doi:10.1016/j.femsre.2005.03.002
14. Wyrick PB. Chlamydia trachomatis persistence in vitro: an overview. J Infect Dis (2010) 201(Suppl 2):S88–95. doi:10.1086/652394
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Zhang JP, Stephens RS. Mechanism of C. trachomatis attachment to eukaryotic host cells. Cell (1992) 69:861–9. doi:10.1016/0092-8674(92)90296-O
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Su H, Raymond L, Rockey DD, Fischer E, Hackstadt T, Caldwell HD. A recombinant Chlamydia trachomatis major outer membrane protein binds to heparan sulfate receptors on epithelial cells. Proc Natl Acad Sci U S A (1996) 93:11143–8. doi:10.1073/pnas.93.20.11143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Davis CH, Wyrick PB. Differences in the association of Chlamydia trachomatis serovar E and serovar L2 with epithelial cells in vitro may reflect biological differences in vivo. Infect Immun (1997) 65:2914–24.
18. Kuo CC, Lee A, Campbell LA. Cleavage of the N-linked oligosaccharide from the surfaces of Chlamydia species affects attachment and infectivity of the organisms in human epithelial and endothelial cells. Infect Immun (2004) 72:6699–701. doi:10.1128/IAI.72.11.6699-6701.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Kuo C, Takahashi N, Swanson AF, Ozeki Y, Hakomori S. An N-linked high-mannose type oligosaccharide, expressed at the major outer membrane protein of Chlamydia trachomatis, mediates attachment and infectivity of the microorganism to HeLa cells. J Clin Invest (1996) 98:2813–8. doi:10.1172/JCI119109
20. Raulston JE, Davis CH, Schmiel DH, Morgan MW, Wyrick PB. Molecular characterization and outer membrane association of a Chlamydia trachomatis protein related to the hsp70 family of proteins. J Biol Chem (1993) 268:23139–47.
21. Fadel S, Eley A. Is lipopolysaccharide a factor in infectivity of Chlamydia trachomatis? J Med Microbiol (2008) 57:261–6. doi:10.1099/jmm.0.47237-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Stuart ES, Webley WC, Norkin LC. Lipid rafts, caveolae, caveolin-1, and entry by Chlamydiae into host cells. Exp Cell Res (2003) 287:67–78. doi:10.1016/S0014-4827(03)00059-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Stamm WE. Chlamydia trachomatis infections: progress and problems. J Infect Dis (1999) 179(Suppl 2):S380–3. doi:10.1086/513844
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Gonzales GF, Munoz G, Sanchez R, Henkel R, Gallegos-Avila G, Diaz-Gutierrez O, et al. Update on the impact of Chlamydia trachomatis infection on male fertility. Andrologia (2004) 36:1–23. doi:10.1046/j.0303-4569.2003.00594.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Westrom L, Joesoef R, Reynolds G, Hagdu A, Thompson SE. Pelvic inflammatory disease and fertility. A cohort study of 1,844 women with laparoscopically verified disease and 657 control women with normal laparoscopic results. Sex Transm Dis (1992) 19:185–92. doi:10.1097/00007435-199207000-00001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Baud D, Goy G, Jaton K, Osterheld MC, Blumer S, Borel N, et al. Role of Chlamydia trachomatis in miscarriage. Emerg Infect Dis (2011) 17:1630–5. doi:10.3201/eid1709.100865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Karaer A, Mert I, Cavkaytar S, Batioglu S. Serological investigation of the role of selected sexually transmitted infections in the aetiology of ectopic pregnancy. Eur J Contracept Reprod Health Care (2013) 18:68–74. doi:10.3109/13625187.2012.744818
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Kavanagh K, Wallace LA, Robertson C, Wilson P, Scoular A. Estimation of the risk of tubal factor infertility associated with genital chlamydial infection in women: a statistical modelling study. Int J Epidemiol (2013) 42:493–503. doi:10.1093/ije/dyt011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Mitchell C, Prabhu M. Pelvic inflammatory disease: current concepts in pathogenesis, diagnosis and treatment. Infect Dis Clin North Am (2013) 27:793–809. doi:10.1016/j.idc.2013.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Reekie J, Donovan B, Guy R, Hocking JS, Jorm L, Kaldor JM, et al. Hospitalisations for pelvic inflammatory disease temporally related to a diagnosis of Chlamydia or gonorrhoea: a retrospective cohort study. PLoS One (2014) 9:e94361. doi:10.1371/journal.pone.0094361
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Paavonen J, Eggert-Kruse W. Chlamydia trachomatis: impact on human reproduction. Hum Reprod Update (1999) 5:433–47. doi:10.1093/humupd/5.5.433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Andrews WW, Goldenberg RL, Mercer B, Iams J, Meis P, Moawad A, et al. The preterm prediction study: association of second-trimester genitourinary chlamydia infection with subsequent spontaneous preterm birth. Am J Obstet Gynecol (2000) 183:662–8. doi:10.1067/mob.2000.106556
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Mardh PA. Influence of infection with Chlamydia trachomatis on pregnancy outcome, infant health and life-long sequelae in infected offspring. Best Pract Res Clin Obstet Gynaecol (2002) 16:847–64. doi:10.1053/beog.2002.0329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Jain S. Perinatally acquired Chlamydia trachomatis associated morbidity in young infants. J Matern Fetal Med (1999) 8:130–3. doi:10.1002/(SICI)1520-6661(199905/06)8:3<130::AID-MFM11>3.0.CO;2-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Wagenlehner FM, Weidner W, Naber KG. Chlamydial infections in urology. World J Urol (2006) 24:4–12. doi:10.1007/s00345-005-0047-x
36. Mulcahy FM, Bignell CJ, Rajakumar R, Waugh MA, Hetherington JW, Ewing R, et al. Prevalence of chlamydial infection in acute epididymo-orchitis. Genitourin Med (1987) 63:16–8.
37. Krishnan R, Heal MR. Study of the seminal vesicles in acute epididymitis. Br J Urol (1991) 67:632–7. doi:10.1111/j.1464-410X.1991.tb15229.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Trojian TH, Lishnak TS, Heiman D. Epididymitis and orchitis: an overview. Am Fam Physician (2009) 79:583–7.
39. Toth M, Patton DL, Campbell LA, Carretta EI, Mouradian J, Toth A, et al. Detection of chlamydial antigenic material in ovarian, prostatic, ectopic pregnancy and semen samples of culture-negative subjects. Am J Reprod Immunol (2000) 43:218–22. doi:10.1111/j.8755-8920.2000.430406.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Skerk V, Schonwald S, Krhen I, Markovinovic L, Beus A, Kuzmanovic NS, et al. Aetiology of chronic prostatitis. Int J Antimicrob Agents (2002) 19:471–4. doi:10.1016/S0924-8579(02)00087-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Krieger JN. The problem with prostatitis. What do we know? What do we need to know? J Urol (2004) 172:432–3. doi:10.1097/01.ju.0000132864.57677.91
42. Krieger JN, Riley DE. Chronic prostatitis: Charlottesville to Seattle. J Urol (2004) 172:2557–60. doi:10.1097/01.ju.0000144291.05839.a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Cunningham KA, Beagley KW. Male genital tract chlamydial infection: implications for pathology and infertility. Biol Reprod (2008) 79:180–9. doi:10.1095/biolreprod.108.067835
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Ouzounova-Raykova V, Ouzounova I, Mitov IG. May Chlamydia trachomatis be an aetiological agent of chronic prostatic infection? Andrologia (2010) 42:176–81. doi:10.1111/j.1439-0272.2009.00973.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Furuya R, Takahashi S, Furuya S, Takeyama K, Masumori N, Tsukamoto T. Chlamydial seminal vesiculitis without symptomatic urethritis and epididymitis. Int J Urol (2006) 13:466–7. doi:10.1111/j.1442-2042.2006.01317.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Motrich RD, Cuffini C, Oberti JP, Maccioni M, Rivero VE. Chlamydia trachomatis occurrence and its impact on sperm quality in chronic prostatitis patients. J Infect (2006) 53:175–83. doi:10.1016/j.jinf.2005.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Mackern-Oberti JP, Motrich RD, Breser ML, Cejas H, Cuffini C, Maccioni M, et al. Male rodent genital tract infection with Chlamydia muridarum: persistence in the prostate gland that triggers self-immune reactions in genetically susceptible hosts. J Urol (2011) 186:1100–6. doi:10.1016/j.juro.2011.04.086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Motrich RD, Sanchez L, Maccioni M, Mackern-Oberti JP, Rivero VE. Male rat genital tract infection with Chlamydia muridarum has no significant consequence on male fertility. J Urol (2012) 187:1911–7. doi:10.1016/j.juro.2011.12.055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Gillespie CW, Manhart LE, Lowens MS, Golden MR. Asymptomatic urethritis is common and is associated with characteristics that suggest sexually transmitted etiology. Sex Transm Dis (2013) 40:271–4. doi:10.1097/OLQ.0b013e31827c9e42
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Wiggins RC, Holmes CH, Andersson M, Ibrahim F, Low N, Horner PJ. Quantifying leukocytes in first catch urine provides new insights into our understanding of symptomatic and asymptomatic urethritis. Int J STD AIDS (2006) 17:289–95. doi:10.1258/095646206776790268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Stamm WE, Batteiger BE, Mccormack WM, Totten PA, Sternlicht A, Kivel NM, et al. A randomized, double-blind study comparing single-dose rifalazil with single-dose azithromycin for the empirical treatment of nongonococcal urethritis in men. Sex Transm Dis (2007) 34:545–52. doi:10.1097/01.olq.0000253348.44308.8c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Tait IA, Hart CA. Chlamydia trachomatis in non-gonococcal urethritis patients and their heterosexual partners: routine testing by polymerase chain reaction. Sex Transm Infect (2002) 78:286–8. doi:10.1136/sti.78.4.286
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Takahashi S, Takeyama K, Kunishima Y, Takeda K, Suzuki N, Nishimura M, et al. Analysis of clinical manifestations of male patients with urethritis. J Infect Chemother (2006) 12:283–6. doi:10.1007/s10156-006-0466-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Hedger MP. Immunophysiology and pathology of inflammation in the testis and epididymis. J Androl (2011) 32:625–40. doi:10.2164/jandrol.111.012989
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Redfern TR, English PJ, Baumber CD, Mcghie D. The aetiology and management of acute epididymitis. Br J Surg (1984) 71:703–5. doi:10.1002/bjs.1800710921
56. Manavi K, Turner K, Scott GR, Stewart LH. Audit on the management of epididymo-orchitis by the Department of Urology in Edinburgh. Int J STD AIDS (2005) 16:386–7. doi:10.1258/0956462053888880
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Weidner W, Krause W, Ludwig M. Relevance of male accessory gland infection for subsequent fertility with special focus on prostatitis. Hum Reprod Update (1999) 5:421–32. doi:10.1093/humupd/5.5.421
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Ness RB, Markovic N, Carlson CL, Coughlin MT. Do men become infertile after having sexually transmitted urethritis? An epidemiologic examination. Fertil Steril (1997) 68:205–13. doi:10.1016/S0015-0282(97)81502-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Krieger JN, Nyberg L Jr, Nickel JC. NIH consensus definition and classification of prostatitis. JAMA (1999) 282:236–7. doi:10.1001/jama.282.3.236
60. Habermacher GM, Chason JT, Schaeffer AJ. Prostatitis/chronic pelvic pain syndrome. Annu Rev Med (2006) 57:195–206. doi:10.1146/annurev.med.57.011205.135654
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Shurbaji MS, Gupta PK, Myers J. Immunohistochemical demonstration of Chlamydial antigens in association with prostatitis. Mod Pathol (1988) 1:348–51.
62. Abdelatif OM, Chandler FW, Mcguire BS Jr. Chlamydia trachomatis in chronic abacterial prostatitis: demonstration by colorimetric in situ hybridization. Hum Pathol (1991) 22:41–4. doi:10.1016/0046-8177(91)90059-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Corradi G, Konkoly Thege M, Panovics J, Molnar G, Bodo A, Frang D. Is seminal fluid a suitable specimen for detecting chlamydial infection in men? Acta Microbiol Immunol Hung (1995) 42:389–94.
64. Weidner W, Diemer T, Huwe P, Rainer H, Ludwig M. The role of Chlamydia trachomatis in prostatitis. Int J Antimicrob Agents (2002) 19:466–70. doi:10.1016/S0924-8579(02)00094-8
65. Badalyan RR, Fanarjyan SV, Aghajanyan IG. Chlamydial and ureaplasmal infections in patients with nonbacterial chronic prostatitis. Andrologia (2003) 35:263–5. doi:10.1111/j.1439-0272.2003.tb00854.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Ostaszewska-Puchalska I, Zdrodowska-Stefanow B, Badyda J, Bulhak-Koziol V, Pucilo K, Darewicz B. Antichlamydial antibodies in the serum and expressed prostatic secretion in prostatitis. Arch Immunol Ther Exp (Warsz) (2004) 52:277–83.
67. Krause W, Bohring C. Male infertility and genital chlamydial infection: victim or perpetrator? Andrologia (2003) 35:209–16. doi:10.1046/j.1439-0272.2003.00561.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Gdoura R, Kchaou W, Chaari C, Znazen A, Keskes L, Rebai T, et al. Ureaplasma urealyticum, Ureaplasma parvum, Mycoplasma hominis and Mycoplasma genitalium infections and semen quality of infertile men. BMC Infect Dis (2007) 7:129. doi:10.1186/1471-2334-7-129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Lepor H, Wang B, Shapiro E. Relationship between prostatic epithelial volume and serum prostate-specific antigen levels. Urology (1994) 44:199–205. doi:10.1016/S0090-4295(94)80131-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Idahl A, Boman J, Kumlin U, Olofsson JI. Demonstration of Chlamydia trachomatis IgG antibodies in the male partner of the infertile couple is correlated with a reduced likelihood of achieving pregnancy. Hum Reprod (2004) 19:1121–6. doi:10.1093/humrep/deh155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Mazzoli S, Cai T, Addonisio P, Bechi A, Mondaini N, Bartoletti R. Chlamydia trachomatis infection is related to poor semen quality in young prostatitis patients. Eur Urol (2010) 57:708–14. doi:10.1016/j.eururo.2009.05.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. La Vignera S, Vicari E, Condorelli RA, D’Agata R, Calogero AE. Male accessory gland infection and sperm parameters (review). Int J Androl (2011) 34:e330–47. doi:10.1111/j.1365-2605.2011.01200.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Pajovic B, Radojevic N, Vukovic M, Stjepcevic A. Semen analysis before and after antibiotic treatment of asymptomatic Chlamydia- and Ureaplasma-related pyospermia. Andrologia (2013) 45:266–71. doi:10.1111/and.12004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Weidner W, Floren E, Zimmermann O, Thiele D, Ludwig M. Chlamydial antibodies in semen: search for “silent” chlamydial infections in asymptomatic andrological patients. Infection (1996) 24:309–13. doi:10.1007/BF01743366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Habermann B, Krause W. Altered sperm function or sperm antibodies are not associated with chlamydial antibodies in infertile men with leucocytospermia. J Eur Acad Dermatol Venereol (1999) 12:25–9. doi:10.1111/j.1468-3083.1999.tb00803.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Ochsendorf FR, Ozdemir K, Rabenau H, Fenner T, Oremek R, Milbradt R, et al. Chlamydia trachomatis and male infertility: chlamydia-IgA antibodies in seminal plasma are C. trachomatis specific and associated with an inflammatory response. J Eur Acad Dermatol Venereol (1999) 12:143–52. doi:10.1111/j.1468-3083.1999.tb01005.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Vigil P, Morales P, Tapia A, Riquelme R, Salgado AM. Chlamydia trachomatis infection in male partners of infertile couples: incidence and sperm function. Andrologia (2002) 34:155–61. doi:10.1046/j.1439-0272.2002.00472.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Eggert-Kruse W, Rohr G, Kunt B, Meyer A, Wondra J, Strowitzki T, et al. Prevalence of Chlamydia trachomatis in subfertile couples. Fertil Steril (2003) 80:660–3. doi:10.1016/S0015-0282(03)00761-1
79. de Barbeyrac B, Papaxanthos-Roche A, Mathieu C, Germain C, Brun JL, Gachet M, et al. Chlamydia trachomatis in subfertile couples undergoing an in vitro fertilization program: a prospective study. Eur J Obstet Gynecol Reprod Biol (2006) 129:46–53. doi:10.1016/j.ejogrb.2006.02.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Mackern-Oberti JP, Motrich RD, Breser ML, Sanchez LR, Cuffini C, Rivero VE. Chlamydia trachomatis infection of the male genital tract: an update. J Reprod Immunol (2013) 100:37–53. doi:10.1016/j.jri.2013.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Furuya R, Takahashi S, Furuya S, Kunishima Y, Takeyama K, Tsukamoto T. Is seminal vesiculitis a discrete disease entity? Clinical and microbiological study of seminal vesiculitis in patients with acute epididymitis. J Urol (2004) 171:1550–3. doi:10.1097/01.ju.0000116288.59223.e9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Furuya R, Takahashi S, Furuya S, Takeyama K, Tsukamoto T. A patient with seminal vesiculitis prior to acute chlamydial epididymitis. J Infect Chemother (2005) 11:250–2. doi:10.1007/s10156-005-0404-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Furuya R, Takahashi S, Furuya S, Saitoh N, Ogura H, Kurimura Y, et al. Is urethritis accompanied by seminal vesiculitis? Int J Urol (2009) 16:628–31. doi:10.1111/j.1442-2042.2009.02314.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Villegas H, Pinon M, Shor V, Karchmer S. Electron microscopy of Chlamydia trachomatis infection of the male genital tract. Arch Androl (1991) 27:117–26. doi:10.3109/01485019108987663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Erbengi T. Ultrastructural observations on the entry of Chlamydia trachomatis into human spermatozoa. Hum Reprod (1993) 8:416–21.
86. Wolner-Hanssen P, Mardh PA. In vitro tests of the adherence of Chlamydia trachomatis to human spermatozoa. Fertil Steril (1984) 42:102–7.
87. Hosseinzadeh S, Brewis IA, Eley A, Pacey AA. Co-incubation of human spermatozoa with Chlamydia trachomatis serovar E causes premature sperm death. Hum Reprod (2001) 16:293–9. doi:10.1093/humrep/16.2.293
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Eley A, Hosseinzadeh S, Hakimi H, Geary I, Pacey AA. Apoptosis of ejaculated human sperm is induced by co-incubation with Chlamydia trachomatis lipopolysaccharide. Hum Reprod (2005) 20:2601–7. doi:10.1093/humrep/dei082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Satta A, Stivala A, Garozzo A, Morello A, Perdichizzi A, Vicari E, et al. Experimental Chlamydia trachomatis infection causes apoptosis in human sperm. Hum Reprod (2006) 21:134–7. doi:10.1093/humrep/dei269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Galdiero F, Sommese L, Gorga F, Galdiero E, Rizzo A, Ajello M. Toxic effect on human spermatozoa by Chlamydia trachomatis purified lipopolysaccharide. FEMS Microbiol Lett (1994) 115:197–200. doi:10.1111/j.1574-6968.1994.tb06637.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Hosseinzadeh S, Pacey AA, Eley A. Chlamydia trachomatis-induced death of human spermatozoa is caused primarily by lipopolysaccharide. J Med Microbiol (2003) 52:193–200. doi:10.1099/jmm.0.04836-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Eley A, Pacey AA, Galdiero M, Galdiero M, Galdiero F. Can Chlamydia trachomatis directly damage your sperm? Lancet Infect Dis (2005) 5:53–7. doi:10.1016/S1473-3099(04)01254-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Aitken RJ, Buckingham DW, Brindle J, Gomez E, Baker HW, Irvine DS. Analysis of sperm movement in relation to the oxidative stress created by leukocytes in washed sperm preparations and seminal plasma. Hum Reprod (1995) 10:2061–71.
94. Aitken RJ, Baker MA, Sawyer D. Oxidative stress in the male germ line and its role in the aetiology of male infertility and genetic disease. Reprod Biomed Online (2003) 7:65–70. doi:10.1016/S1472-6483(10)61730-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Gdoura R, Keskes-Ammar L, Bouzid F, Eb F, Hammami A, Orfila J. Chlamydia trachomatis and male infertility in Tunisia. Eur J Contracept Reprod Health Care (2001) 6:102–7. doi:10.1080/713604209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Jakiel G, Robak-Cholubek D, Wieczorek P, Bokiniec M. Evaluation of some parameters of human semen with positive chlamydial reaction. Ann Univ Mariae Curie Sklodowska Med (2004) 59:61–4.
97. Bezold G, Politch JA, Kiviat NB, Kuypers JM, Wolff H, Anderson DJ. Prevalence of sexually transmissible pathogens in semen from asymptomatic male infertility patients with and without leukocytospermia. Fertil Steril (2007) 87:1087–97. doi:10.1016/j.fertnstert.2006.08.109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Gallegos G, Ramos B, Santiso R, Goyanes V, Gosalvez J, Fernandez JL. Sperm DNA fragmentation in infertile men with genitourinary infection by Chlamydia trachomatis and Mycoplasma. Fertil Steril (2008) 90:328–34. doi:10.1016/j.fertnstert.2007.06.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Custo GM, Lauro V, Saitto C, Frongillo RF. Chlamydial infection and male infertility: an epidemiological study. Arch Androl (1989) 23:243–8. doi:10.3109/01485018908986847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Cengiz T, Aydoganli L, Baykam M, Mungan NA, Tuncbilek E, Dincer M, et al. Chlamydial infections and male infertility. Int Urol Nephrol (1997) 29:687–93. doi:10.1007/BF02552187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Hosseinzadeh S, Eley A, Pacey AA. Semen quality of men with asymptomatic chlamydial infection. J Androl (2004) 25:104–9. doi:10.1002/j.1939-4640.tb02764.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Ochsendorf FR. Infections in the male genital tract and reactive oxygen species. Hum Reprod Update (1999) 5:399–420. doi:10.1093/humupd/5.5.399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Mosli HA, Gazzaz FS, Farsi HM, Abduljabar HS. Genital infection in males with idiopathic infertility. Ann Saudi Med (1996) 16:42–6.
104. Lenzi A, Gandini L, Lombardo F, Rago R, Paoli D, Dondero F. Antisperm antibody detection: 2. Clinical, biological, and statistical correlation between methods. Am J Reprod Immunol (1997) 38:224–30. doi:10.1111/j.1600-0897.1997.tb00303.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Barratt CL, Havelock LM, Harrison PE, Cooke ID. Antisperm antibodies are more prevalent in men with low sperm motility. Int J Androl (1989) 12:110–6. doi:10.1111/j.1365-2605.1989.tb01293.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Zouari R, De Almeida M, Rodrigues D, Jouannet P. Localization of antibodies on spermatozoa and sperm movement characteristics are good predictors of in vitro fertilization success in cases of male autoimmune infertility. Fertil Steril (1993) 59:606–12.
107. Tasdemir I, Tasdemir M, Fukuda J, Kodama H, Matsui T, Tanaka T. Sperm immobilization antibodies in infertile male sera decrease the acrosome reaction: a possible mechanism for immunologic infertility. J Assist Reprod Genet (1996) 13:413–6. doi:10.1007/BF02066174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Kremer J, Jager S. The significance of antisperm antibodies for sperm-cervical mucus interaction. Hum Reprod (1992) 7:781–4.
109. Francavilla F, Romano R, Santucci R, Marrone V, Properzi G, Ruvolo G. Interference of antisperm antibodies with the induction of the acrosome reaction by zona pellucida (ZP) and its relationship with the inhibition of ZP binding. Fertil Steril (1997) 67:1128–33. doi:10.1016/S0015-0282(97)81450-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Bronson RA, Cooper GW, Phillips DM. Effects of anti-sperm antibodies on human sperm ultrastructure and function. Hum Reprod (1989) 4:653–7.
111. Clarke GN, Stojanoff A, Cauchi MN, Johnston WI. The immunoglobulin class of antispermatozoal antibodies in serum. Am J Reprod Immunol Microbiol (1985) 7:143–7.
112. Eggert-Kruse W, Hofsass A, Haury E, Tilgen W, Gerhard I, Runnebaum B. Relationship between local anti-sperm antibodies and sperm-mucus interaction in vitro and in vivo. Hum Reprod (1991) 6:267–76.
113. Hedger M, Hales D. Immunolophysiology of the male reproductive tract. In: Knobil E, Neill J, editors. Physiology of Reproduction. Amsterdam: Academic Press (2006). p. 1195–286.
114. Guiton R, Henry-Berger J, Drevet J. The immunobiology of the mammalian epididymis: the black box is now open! Basic Clin Androl (2013) 23:1–10.
115. Whitmore WF, Gittes RF. Intratesticular grafts: the testis as an exceptional immunologically privileged site. Trans Am Assoc Genitourin Surg (1978) 70:76–80.
116. Head JR, Neaves WB, Billingham RE. Immune privilege in the testis. I. Basic parameters of allograft survival. Transplantation (1983) 36:423–31. doi:10.1097/00007890-198310000-00014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Head JR, Billingham RE. Immune privilege in the testis. II. Evaluation of potential local factors. Transplantation (1985) 40:269–75. doi:10.1097/00007890-198509000-00010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Hedger MP. Testicular leukocytes: what are they doing? Rev Reprod (1997) 2:38–47. doi:10.1530/ror.0.0020038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Holstein AF, Davidoff M. Compartmentalization of the intertubular space in the human testis. Adv Exp Med Biol (1997) 424:161–2. doi:10.1007/978-1-4615-5913-9_31
120. Ungefroren H, Ergun S, Krull NB, Holstein AF. Expression of the small proteoglycans biglycan and decorin in the adult human testis. Biol Reprod (1995) 52:1095–105. doi:10.1095/biolreprod52.5.1095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Dym M. Basement membrane regulation of Sertoli cells. Endocr Rev (1994) 15:102–15. doi:10.1210/edrv-15-1-102
122. Schlatt S, Meinhardt A, Nieschlag E. Paracrine regulation of cellular interactions in the testis: factors in search of a function. Eur J Endocrinol (1997) 137:107–17. doi:10.1530/eje.0.1370107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Meng J, Greenlee AR, Taub CJ, Braun RE. Sertoli cell-specific deletion of the androgen receptor compromises testicular immune privilege in mice. Biol Reprod (2011) 85:254–60. doi:10.1095/biolreprod.110.090621
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Yule TD, Montoya GD, Russell LD, Williams TM, Tung KS. Autoantigenic germ cells exist outside the blood testis barrier. J Immunol (1988) 141:1161–7.
125. Saari T, Jahnukainen K, Pollanen P. Autoantigenicity of the basal compartment of seminiferous tubules in the rat. J Reprod Immunol (1996) 31:65–79. doi:10.1016/0165-0378(96)00962-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Itoh M, Takeuchi Y, De Rooij D. Histopathology of the seminiferous tubules in mice injected with syngeneic testicular germ cells alone. Arch Androl (1995) 35:93–103. doi:10.3109/01485019508987859
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Itoh M, Terayama H, Naito M, Ogawa Y, Tainosho S. Tissue microcircumstances for leukocytic infiltration into the testis and epididymis in mice. J Reprod Immunol (2005) 67:57–67. doi:10.1016/j.jri.2005.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Hermo L, Dworkin J. Transitional cells at the junction of seminiferous tubules with the rete testis of the rat: their fine structure, endocytic activity, and basement membrane. Am J Anat (1988) 181:111–31. doi:10.1002/aja.1001810202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Itoh M, Xie Q, Miyamoto K, Takeuchi Y. F4/80-positive cells rapidly accumulate around tubuli recti and rete testis between 3 and 4 weeks of age in the mouse: an immunohistochemical study. Am J Reprod Immunol (1999) 42:321–6. doi:10.1111/j.1600-0897.1999.tb00108.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Friend DS, Gilula NB. Variations in tight and gap junctions in mammalian tissues. J Cell Biol (1972) 53:758–76. doi:10.1083/jcb.53.3.758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Cyr DG, Robaire B, Hermo L. Structure and turnover of junctional complexes between principal cells of the rat epididymis. Microsc Res Tech (1995) 30:54–66. doi:10.1002/jemt.1070300105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Hinton BT, Palladino MA. Epididymal epithelium: its contribution to the formation of a luminal fluid microenvironment. Microsc Res Tech (1995) 30:67–81. doi:10.1002/jemt.1070300106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Nashan D, Malorny U, Sorg C, Cooper T, Nieschlag E. Immuno-competent cells in the murine epididymis. Int J Androl (1989) 12:85–94. doi:10.1111/j.1365-2605.1989.tb01289.x
134. Nashan D, Cooper TG, Knuth UA, Schubeus P, Sorg C, Nieschlag E. Presence and distribution of leucocyte subsets in the murine epididymis after vasectomy. Int J Androl (1990) 13:39–49. doi:10.1111/j.1365-2605.1990.tb00958.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Flickinger CJ, Bush LA, Howards SS, Herr JC. Distribution of leukocytes in the epithelium and interstitium of four regions of the Lewis rat epididymis. Anat Rec (1997) 248:380–90. doi:10.1002/(SICI)1097-0185(199707)248:3<380::AID-AR11>3.3.CO;2-F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Quayle AJ. The innate and early immune response to pathogen challenge in the female genital tract and the pivotal role of epithelial cells. J Reprod Immunol (2002) 57:61–79. doi:10.1016/S0165-0378(02)00019-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Rasmussen SJ, Eckmann L, Quayle AJ, Shen L, Zhang YX, Anderson DJ, et al. Secretion of proinflammatory cytokines by epithelial cells in response to Chlamydia infection suggests a central role for epithelial cells in chlamydial pathogenesis. J Clin Invest (1997) 99:77–87. doi:10.1172/JCI119136
138. Johnson RM. Murine oviduct epithelial cell cytokine responses to Chlamydia muridarum infection include interleukin-12-p70 secretion. Infect Immun (2004) 72:3951–60. doi:10.1128/IAI.72.7.3951-3960.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Buchholz KR, Stephens RS. Activation of the host cell proinflammatory interleukin-8 response by Chlamydia trachomatis. Cell Microbiol (2006) 8:1768–79. doi:10.1111/j.1462-5822.2006.00747.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Witko-Sarsat V, Rieu P, Descamps-Latscha B, Lesavre P, Halbwachs-Mecarelli L. Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest (2000) 80:617–53. doi:10.1038/labinvest.3780067
141. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science (2011) 331:44–9. doi:10.1126/science.1198687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Yong EC, Klebanoff SJ, Kuo CC. Toxic effect of human polymorphonuclear leukocytes on Chlamydia trachomatis. Infect Immun (1982) 37:422–6.
143. Register KB, Morgan PA, Wyrick PB. Interaction between Chlamydia spp. and human polymorphonuclear leukocytes in vitro. Infect Immun (1986) 52:664–70.
144. Barteneva N, Theodor I, Peterson EM, De La Maza LM. Role of neutrophils in controlling early stages of a Chlamydia trachomatis infection. Infect Immun (1996) 64:4830–3.
145. Salamone G, Giordano M, Trevani AS, Gamberale R, Vermeulen M, Schettinni J, et al. Promotion of neutrophil apoptosis by TNF-alpha. J Immunol (2001) 166:3476–83. doi:10.4049/jimmunol.166.5.3476
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13:159–75. doi:10.1038/nri3399
147. Bradley LM, Douglass MF, Chatterjee D, Akira S, Baaten BJ. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog (2012) 8:e1002641. doi:10.1371/journal.ppat.1002641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. van Zandbergen G, Gieffers J, Kothe H, Rupp J, Bollinger A, Aga E, et al. Chlamydia pneumoniae multiply in neutrophil granulocytes and delay their spontaneous apoptosis. J Immunol (2004) 172:1768–76. doi:10.4049/jimmunol.172.3.1768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Sukhotnik I, Voskoboinik K, Lurie M, Coran AG, Greenblatt R, Shiloni E, et al. Effect of testicular ischemia-reperfusion on recruitment of neutrophils, E-selectin expression and germ cell apoptosis in the contralateral testis in a rat. Pediatr Surg Int (2007) 23:479–85. doi:10.1007/s00383-006-1854-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Nagaosa K, Nakashima C, Kishimoto A, Nakanishi Y. Immune response to bacteria in seminiferous epithelium. Reproduction (2009) 137:879–88. doi:10.1530/REP-08-0460
151. Shegarfi H, Sydnes K, Lovik M, Inngjerdingen M, Rolstad B, Naper C. The role of natural killer cells in resistance to the intracellular bacterium Listeria monocytogenes in rats. Scand J Immunol (2009) 70:238–44. doi:10.1111/j.1365-3083.2009.02292.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol (2013) 10:230–52. doi:10.1038/cmi.2013.10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Tompkins AB, Hutchinson P, De Kretser DM, Hedger MP. Characterization of lymphocytes in the adult rat testis by flow cytometry: effects of activin and transforming growth factor beta on lymphocyte subsets in vitro. Biol Reprod (1998) 58:943–51. doi:10.1095/biolreprod58.4.943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. doi:10.1038/32588
155. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, et al. Immunobiology of dendritic cells. Annu Rev Immunol (2000) 18:767–811. doi:10.1146/annurev.immunol.18.1.767
156. Hackstein H, Thomson AW. Dendritic cells: emerging pharmacological targets of immunosuppressive drugs. Nat Rev Immunol (2004) 4:24–34. doi:10.1038/nri1256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Flores-Romo L. In vivo maturation and migration of dendritic cells. Immunology (2001) 102:255–62. doi:10.1046/j.1365-2567.2001.01204.x
158. Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, et al. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med (2003) 9:1484–90. doi:10.1038/nm960
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med (2003) 198:1527–37. doi:10.1084/jem.20030966
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Fijak M, Iosub R, Schneider E, Linder M, Respondek K, Klug J, et al. Identification of immunodominant autoantigens in rat autoimmune orchitis. J Pathol (2005) 207:127–38. doi:10.1002/path.1828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Fijak M, Meinhardt A. The testis in immune privilege. Immunol Rev (2006) 213:66–81. doi:10.1111/j.1600-065X.2006.00438.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Hedger MP, Eddy EM. The heterogeneity of isolated adult rat Leydig cells separated on Percoll density gradients: an immunological, cytochemical, and functional analysis. Endocrinology (1987) 121:1824–38. doi:10.1210/endo-121-5-1824
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Pollanen P, Niemi M. Immunohistochemical identification of macrophages, lymphoid cells and HLA antigens in the human testis. Int J Androl (1987) 10:37–42. doi:10.1111/j.1365-2605.1987.tb00163.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Tung KS, Yule TD, Mahi-Brown CA, Listrom MB. Distribution of histopathology and Ia positive cells in actively induced and passively transferred experimental autoimmune orchitis. J Immunol (1987) 138:752–9.
165. Haas GG Jr, D’Cruz OJ, De Bault LE. Distribution of human leukocyte antigen-ABC and -D/DR antigens in the unfixed human testis. Am J Reprod Immunol Microbiol (1988) 18:47–51.
166. Pollanen P, Maddocks S. Macrophages, lymphocytes and MHC II antigen in the ram and the rat testis. J Reprod Fertil (1988) 82:437–45. doi:10.1530/jrf.0.0820437
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Pollanen P, Jahnukainen K, Punnonen J, Sainio-Pollanen S. Ontogeny of immunosuppressive activity, MHC antigens and leukocytes in the rat testis. J Reprod Immunol (1992) 21:257–74. doi:10.1016/0165-0378(92)90030-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Lustig L, Lourtau L, Perez R, Doncel GF. Phenotypic characterization of lymphocytic cell infiltrates into the testes of rats undergoing autoimmune orchitis. Int J Androl (1993) 16:279–84. doi:10.1111/j.1365-2605.1993.tb01192.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Itoh M, De Rooij DG, Jansen A, Drexhage HA. Phenotypical heterogeneity of testicular macrophages/dendritic cells in normal adult mice: an immunohistochemical study. J Reprod Immunol (1995) 28:217–32. doi:10.1016/0165-0378(95)00923-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Hoek A, Allaerts W, Leenen PJ, Schoemaker J, Drexhage HA. Dendritic cells and macrophages in the pituitary and the gonads. Evidence for their role in the fine regulation of the reproductive endocrine response. Eur J Endocrinol (1997) 136:8–24. doi:10.1530/eje.0.1360008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Derrick EK, Barker JN, Khan A, Price ML, Macdonald DM. The tissue distribution of factor XIIIa positive cells. Histopathology (1993) 22:157–62. doi:10.1111/j.1365-2559.1993.tb00095.x
172. Sainio-Pollanen S, Saari T, Simell O, Pollanen P. CD28-CD80/CD86 interactions in testicular immunoregulation. J Reprod Immunol (1996) 31:145–63. doi:10.1016/0165-0378(96)00983-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Li Y, Xue WJ, Tian XH, Feng XS, Ding XM, Song HJ, et al. Study on systemic immune tolerance induction in rat islet transplantation by intravenous infusion of Sertoli cells. Transplantation (2010) 89:1430–7. doi:10.1097/TP.0b013e3181da607e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Selawry HP, Cameron DF. Sertoli cell-enriched fractions in successful islet cell transplantation. Cell Transplant (1993) 2:123–9.
175. Sanberg PR, Borlongan CV, Saporta S, Cameron DF. Testis-derived Sertoli cells survive and provide localized immunoprotection for xenografts in rat brain. Nat Biotechnol (1996) 14:1692–5. doi:10.1038/nbt1296-1692
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Korbutt GS, Elliott JF, Rajotte RV. Cotransplantation of allogeneic islets with allogeneic testicular cell aggregates allows long-term graft survival without systemic immunosuppression. Diabetes (1997) 46:317–22. doi:10.2337/diab.46.2.317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Nakanishi Y, Shiratsuchi A. Phagocytic removal of apoptotic spermatogenic cells by Sertoli cells: mechanisms and consequences. Biol Pharm Bull (2004) 27:13–6. doi:10.1248/bpb.27.13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Grandjean V, Vincent S, Martin L, Rassoulzadegan M, Cuzin F. Antimicrobial protection of the mouse testis: synthesis of defensins of the cryptdin family. Biol Reprod (1997) 57:1115–22. doi:10.1095/biolreprod57.5.1115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Lennartsson A, Pieters K, Vidovic K, Gullberg U. A murine antibacterial ortholog to human bactericidal/permeability-increasing protein (BPI) is expressed in testis, epididymis, and bone marrow. J Leukoc Biol (2005) 77:369–77. doi:10.1189/jlb.0304159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Sang Y, Ortega MT, Blecha F, Prakash O, Melgarejo T. Molecular cloning and characterization of three beta-defensins from canine testes. Infect Immun (2005) 73:2611–20. doi:10.1128/IAI.73.5.2611-2620.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Riccioli A, Starace D, Galli R, Fuso A, Scarpa S, Palombi F, et al. Sertoli cells initiate testicular innate immune responses through TLR activation. J Immunol (2006) 177:7122–30. doi:10.4049/jimmunol.177.10.7122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Girling JE, Hedger MP. Toll-like receptors in the gonads and reproductive tract: emerging roles in reproductive physiology and pathology. Immunol Cell Biol (2007) 85:481–9. doi:10.1038/sj.icb.7100086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Palladino MA, Johnson TA, Gupta R, Chapman JL, Ojha P. Members of the toll-like receptor family of innate immunity pattern-recognition receptors are abundant in the male rat reproductive tract. Biol Reprod (2007) 76:958–64. doi:10.1095/biolreprod.106.059410
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Wu H, Wang H, Xiong W, Chen S, Tang H, Han D. Expression patterns and functions of toll-like receptors in mouse sertoli cells. Endocrinology (2008) 149:4402–12. doi:10.1210/en.2007-1776
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. doi:10.1146/annurev.immunol.20.083001.084359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol (2005) 6:551–7. doi:10.1038/ni1206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol (2005) 17:1–14. doi:10.1093/intimm/dxh186
188. Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity (1999) 11:443–51. doi:10.1016/S1074-7613(00)80119-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol (2000) 165:5392–6. doi:10.4049/jimmunol.165.10.5392
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature (2001) 413:732–8. doi:10.1038/35099560
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Akashi S, Nagai Y, Ogata H, Oikawa M, Fukase K, Kusumoto S, et al. Human MD-2 confers on mouse toll-like receptor 4 species-specific lipopolysaccharide recognition. Int Immunol (2001) 13:1595–9. doi:10.1093/intimm/13.12.1595
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol (2002) 3:667–72. doi:10.1038/ni809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature (2001) 410:1099–103. doi:10.1038/35074106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Beatty WL, Belanger TA, Desai AA, Morrison RP, Byrne GI. Tryptophan depletion as a mechanism of gamma interferon-mediated chlamydial persistence. Infect Immun (1994) 62:3705–11.
195. Yang J, Murphy TL, Ouyang W, Murphy KM. Induction of interferon-gamma production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. Eur J Immunol (1999) 29:548–55. doi:10.1002/(SICI)1521-4141(199902)29:02<548::AID-IMMU548>3.0.CO;2-Z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Cotter TW, Ramsey KH, Miranpuri GS, Poulsen CE, Byrne GI. Dissemination of Chlamydia trachomatis chronic genital tract infection in gamma interferon gene knockout mice. Infect Immun (1997) 65:2145–52.
197. Johansson M, Schon K, Ward M, Lycke N. Genital tract infection with Chlamydia trachomatis fails to induce protective immunity in gamma interferon receptor-deficient mice despite a strong local immunoglobulin A response. Infect Immun (1997) 65:1032–44.
198. Perry LL, Feilzer K, Caldwell HD. Immunity to Chlamydia trachomatis is mediated by T helper 1 cells through IFN-gamma-dependent and -independent pathways. J Immunol (1997) 158:3344–52.
199. Ito JI, Lyons JM. Role of gamma interferon in controlling murine chlamydial genital tract infection. Infect Immun (1999) 67:5518–21.
200. Rottenberg ME, Gigliotti Rothfuchs AC, Gigliotti D, Svanholm C, Bandholtz L, Wigzell H. Role of innate and adaptive immunity in the outcome of primary infection with Chlamydia pneumoniae, as analyzed in genetically modified mice. J Immunol (1999) 162:2829–36.
201. Rottenberg ME, Gigliotti Rothfuchs A, Gigliotti D, Ceausu M, Une C, Levitsky V, et al. Regulation and role of IFN-gamma in the innate resistance to infection with Chlamydia pneumoniae. J Immunol (2000) 164:4812–8. doi:10.4049/jimmunol.164.9.4812
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
202. Byrne GI, Lehmann LK, Landry GJ. Induction of tryptophan catabolism is the mechanism for gamma-interferon-mediated inhibition of intracellular Chlamydia psittaci replication in T24 cells. Infect Immun (1986) 53:347–51.
203. Carlin JM, Borden EC, Byrne GI. Interferon-induced indoleamine 2,3-dioxygenase activity inhibits Chlamydia psittaci replication in human macrophages. J Interferon Res (1989) 9:329–37. doi:10.1089/jir.1989.9.329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. Rapoza PA, Tahija SG, Carlin JP, Miller SL, Padilla ML, Byrne GI. Effect of interferon on a primary conjunctival epithelial cell model of trachoma. Invest Ophthalmol Vis Sci (1991) 32:2919–23.
205. Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J (1991) 5:2516–22.
206. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol (2004) 4:762–74. doi:10.1038/nri1457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. Jalili RB, Forouzandeh F, Bahar MA, Ghahary A. The immunoregulatory function of indoleamine 2, 3 dioxygenase and its application in allotransplantation. Iran J Allergy Asthma Immunol (2007) 6:167–79.
208. King NJ, Thomas SR. Molecules in focus: indoleamine 2,3-dioxygenase. Int J Biochem Cell Biol (2007) 39:2167–72. doi:10.1016/j.biocel.2007.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest (2007) 117:1147–54. doi:10.1172/JCI31178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol (2008) 8:74–80. doi:10.1038/nri2233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
211. Xu H, Zhang GX, Ciric B, Rostami A. IDO: a double-edged sword for T(H)1/T(H)2 regulation. Immunol Lett (2008) 121:1–6. doi:10.1016/j.imlet.2008.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
212. Carlin JM, Borden EC, Sondel PM, Byrne GI. Biologic-response-modifier-induced indoleamine 2,3-dioxygenase activity in human peripheral blood mononuclear cell cultures. J Immunol (1987) 139:2414–8.
213. Leonhardt RM, Lee SJ, Kavathas PB, Cresswell P. Severe tryptophan starvation blocks onset of conventional persistence and reduces reactivation of Chlamydia trachomatis. Infect Immun (2007) 75:5105–17. doi:10.1128/IAI.00668-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
214. Woods ML II, Mayer J, Evans TG, Hibbs JB Jr. Antiparasitic effects of nitric oxide in an in vitro murine model of Chlamydia trachomatis infection and an in vivo murine model of Leishmania major infection. Immunol Ser (1994) 60:179–95.
215. Chen B, Stout R, Campbell WF. Nitric oxide production: a mechanism of Chlamydia trachomatis inhibition in interferon-gamma-treated RAW264.7 cells. FEMS Immunol Med Microbiol (1996) 14:109–20. doi:10.1111/j.1574-695X.1996.tb00277.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
216. Igietseme JU. Molecular mechanism of T-cell control of Chlamydia in mice: role of nitric oxide in vivo. Immunology (1996) 88:1–5. doi:10.1046/j.1365-2567.1996.d01-655.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
217. Igietseme JU. The molecular mechanism of T-cell control of Chlamydia in mice: role of nitric oxide. Immunology (1996) 87:1–8. doi:10.1046/j.1365-2567.1996.d01-655.x
218. Igietseme JU, Uriri IM, Hawkins R, Rank RG. Integrin-mediated epithelial-T cell interaction enhances nitric oxide production and increased intracellular inhibition of Chlamydia. J Leukoc Biol (1996) 59:656–62.
219. Igietseme JU, Uriri IM, Chow M, Abe E, Rank RG. Inhibition of intracellular multiplication of human strains of Chlamydia trachomatis by nitric oxide. Biochem Biophys Res Commun (1997) 232:595–601. doi:10.1006/bbrc.1997.6335
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
220. Al-Younes HM, Rudel T, Brinkmann V, Szczepek AJ, Meyer TF. Low iron availability modulates the course of Chlamydia pneumoniae infection. Cell Microbiol (2001) 3:427–37. doi:10.1046/j.1462-5822.2001.00125.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
221. Larue RW, Dill BD, Giles DK, Whittimore JD, Raulston JE. Chlamydial Hsp60-2 is iron responsive in Chlamydia trachomatis serovar E-infected human endometrial epithelial cells in vitro. Infect Immun (2007) 75:2374–80. doi:10.1128/IAI.01465-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
222. Dill BD, Dessus-Babus S, Raulston JE. Identification of iron-responsive proteins expressed by Chlamydia trachomatis reticulate bodies during intracellular growth. Microbiology (2009) 155:210–9. doi:10.1099/mic.0.022731-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
223. Bourgeade MF, Silbermann F, Thang MN, Besancon F. Reduction of transferrin receptor expression by interferon gamma in a human cell line sensitive to its antiproliferative effect. Biochem Biophys Res Commun (1988) 153:897–903. doi:10.1016/S0006-291X(88)81312-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
224. Taetle R, Honeysett JM. Gamma-interferon modulates human monocyte/macrophage transferrin receptor expression. Blood (1988) 71:1590–5.
225. Byrd TF, Horwitz MA. Interferon gamma-activated human monocytes downregulate transferrin receptors and inhibit the intracellular multiplication of Legionella pneumophila by limiting the availability of iron. J Clin Invest (1989) 83:1457–65. doi:10.1172/JCI114038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
226. Ryu SY, Jeong KS, Kang BN, Park SJ, Yoon WK, Kim SH, et al. Modulation of transferrin synthesis, transferrin receptor expression, iNOS expression and NO production in mouse macrophages by cytokines, either alone or in combination. Anticancer Res (2000) 20:3331–8.
227. Nairz M, Fritsche G, Brunner P, Talasz H, Hantke K, Weiss G. Interferon-gamma limits the availability of iron for intramacrophage Salmonella typhimurium. Eur J Immunol (2008) 38:1923–36. doi:10.1002/eji.200738056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
228. Zhong GM, de la Maza LM. Activation of mouse peritoneal macrophages in vitro or in vivo by recombinant murine gamma interferon inhibits the growth of Chlamydia trachomatis serovar L1. Infect Immun (1988) 56:3322–5.
229. Zhong GM, Peterson EM, Czarniecki CW, De La Maza LM. Recombinant murine gamma interferon inhibits Chlamydia trachomatis serovar L1 in vivo. Infect Immun (1988) 56:283–6.
230. Dejucq N, Dugast I, Ruffault A, Van Der Meide PH, Jegou B. Interferon-alpha and -gamma expression in the rat testis. Endocrinology (1995) 136:4925–31. doi:10.1210/endo.136.11.7588226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
231. Kimura T, Nakayama K, Penninger J, Kitagawa M, Harada H, Matsuyama T, et al. Involvement of the IRF-1 transcription factor in antiviral responses to interferons. Science (1994) 264:1921–4. doi:10.1126/science.8009222
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
232. Yu-Lee LY. Molecular actions of prolactin in the immune system. Proc Soc Exp Biol Med (1997) 215:35–52. doi:10.3181/00379727-215-44111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
233. Kanzaki M, Morris PL. Identification and regulation of testicular interferon-gamma (IFNgamma) receptor subunits: IFNgamma enhances interferon regulatory factor-1 and interleukin-1beta converting enzyme expression. Endocrinology (1998) 139:2636–44. doi:10.1210/endo.139.5.5975
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
234. Guazzone VA, Jacobo P, Theas MS, Lustig L. Cytokines and chemokines in testicular inflammation: a brief review. Microsc Res Tech (2009) 72:620–8. doi:10.1002/jemt.20704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
235. Gao Y, Lui WY. Synergistic effect of interferon-gamma and tumor necrosis factor-alpha on coxsackievirus and adenovirus receptor expression: an explanation of cell sloughing during testicular inflammation in mice. Biol Reprod (2014) 90:59. doi:10.1095/biolreprod.113.113407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
236. Jrad-Lamine A, Henry-Berger J, Gourbeyre P, Damon-Soubeyrand C, Lenoir A, Combaret L, et al. Deficient tryptophan catabolism along the kynurenine pathway reveals that the epididymis is in a unique tolerogenic state. J Biol Chem (2011) 286:8030–42. doi:10.1074/jbc.M110.172114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
237. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol (2010) 11:373–84. doi:10.1038/ni.1863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
238. Morrison SG, Morrison RP. In situ analysis of the evolution of the primary immune response in murine Chlamydia trachomatis genital tract infection. Infect Immun (2000) 68:2870–9. doi:10.1128/IAI.68.5.2870-2879.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
239. Beagley KW, Huston WM, Hansbro PM, Timms P. Chlamydial infection of immune cells: altered function and implications for disease. Crit Rev Immunol (2009) 29:275–305. doi:10.1615/CritRevImmunol.v29.i4.10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
240. Bas S, Neff L, Vuillet M, Spenato U, Seya T, Matsumoto M, et al. The proinflammatory cytokine response to Chlamydia trachomatis elementary bodies in human macrophages is partly mediated by a lipoprotein, the macrophage infectivity potentiator, through TLR2/TLR1/TLR6 and CD14. J Immunol (2008) 180:1158–68. doi:10.4049/jimmunol.180.2.1158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
241. Yilma AN, Singh SR, Fairley SJ, Taha MA, Dennis VA. The anti-inflammatory cytokine, interleukin-10, inhibits inflammatory mediators in human epithelial cells and mouse macrophages exposed to live and UV-inactivated Chlamydia trachomatis. Mediators Inflamm (2012) 2012:520174. doi:10.1155/2012/520174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
242. Yasir M, Pachikara ND, Bao X, Pan Z, Fan H. Regulation of chlamydial infection by host autophagy and vacuolar ATPase-bearing organelles. Infect Immun (2011) 79:4019–28. doi:10.1128/IAI.05308-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
243. Sun HS, Eng EW, Jeganathan S, Sin AT, Patel PC, Gracey E, et al. Chlamydia trachomatis vacuole maturation in infected macrophages. J Leukoc Biol (2012) 92:815–27. doi:10.1189/jlb.0711336
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
244. Al-Zeer MA, Al-Younes HM, Lauster D, Abu Lubad M, Meyer TF. Autophagy restricts Chlamydia trachomatis growth in human macrophages via IFNG-inducible guanylate binding proteins. Autophagy (2013) 9:50–62. doi:10.4161/auto.22482
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
245. Crotzer VL, Blum JS. Autophagy and its role in MHC-mediated antigen presentation. J Immunol (2009) 182:3335–41. doi:10.4049/jimmunol.0803458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
246. Hedger MP. Macrophages and the immune responsiveness of the testis. J Reprod Immunol (2002) 57:19–34. doi:10.1016/S0165-0378(02)00016-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
247. Hutson JC. Development of cytoplasmic digitations between Leydig cells and testicular macrophages of the rat. Cell Tissue Res (1992) 267:385–9. doi:10.1007/BF00302977
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
248. Hutson JC. Physiologic interactions between macrophages and Leydig cells. Exp Biol Med (Maywood) (2006) 231:1–7.
249. Bergh A, Damber JE, Van Rooijen N. Liposome-mediated macrophage depletion: an experimental approach to study the role of testicular macrophages in the rat. J Endocrinol (1993) 136:407–13. doi:10.1677/joe.0.1360407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
250. Gaytan F, Bellido C, Aguilar E, Van Rooijen N. Requirement for testicular macrophages in Leydig cell proliferation and differentiation during prepubertal development in rats. J Reprod Fertil (1994) 102:393–9. doi:10.1530/jrf.0.1020393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
251. Gaytan F, Bellido C, Morales C, Reymundo C, Aguilar E, Van Rooijen N. Selective depletion of testicular macrophages and prevention of Leydig cell repopulation after treatment with ethylene dimethane sulfonate in rats. J Reprod Fertil (1994) 101:175–82. doi:10.1530/jrf.0.1010175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
252. Gaytan F, Bellido C, Morales C, Reymundo C, Aguilar E, Van Rooijen N. Effects of macrophage depletion at different times after treatment with ethylene dimethane sulfonate (EDS) on the regeneration of Leydig cells in the adult rat. J Androl (1994) 15:558–64.
253. Gaytan F, Bellido C, Romero JL, Morales C, Reymundo C, Aguilar E. Decreased number and size and the defective function of testicular macrophages in long-term hypophysectomized rats are reversed by treatment with human gonadotrophins. J Endocrinol (1994) 140:399–407. doi:10.1677/joe.0.1400399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
254. Meinhardt A, Bacher M, Metz C, Bucala R, Wreford N, Lan H, et al. Local regulation of macrophage subsets in the adult rat testis: examination of the roles of the seminiferous tubules, testosterone, and macrophage-migration inhibitory factor. Biol Reprod (1998) 59:371–8. doi:10.1095/biolreprod59.2.371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
255. Wang J, Wreford NG, Lan HY, Atkins R, Hedger MP. Leukocyte populations of the adult rat testis following removal of the Leydig cells by treatment with ethane dimethane sulfonate and subcutaneous testosterone implants. Biol Reprod (1994) 51:551–61. doi:10.1095/biolreprod51.3.551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
256. Cohen PE, Nishimura K, Zhu L, Pollard JW. Macrophages: important accessory cells for reproductive function. J Leukoc Biol (1999) 66:765–72.
257. Kern S, Maddocks S. Indomethacin blocks the immunosuppressive activity of rat testicular macrophages cultured in vitro. J Reprod Immunol (1995) 28:189–201. doi:10.1016/0165-0378(95)91391-Q
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
258. Bergh A, Damber JE, Van Rooijen N. The human chorionic gonadotrophin-induced inflammation-like response is enhanced in macrophage-depleted rat testes. J Endocrinol (1993) 136:415–20. doi:10.1677/joe.0.1360415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
259. Moore C, Hutson JC. Physiological relevance of tumor necrosis factor in mediating macrophage-Leydig cell interactions. Endocrinology (1994) 134:63–9. doi:10.1210/endo.134.1.8275970
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
260. Kern S, Robertson SA, Mau VJ, Maddocks S. Cytokine secretion by macrophages in the rat testis. Biol Reprod (1995) 53:1407–16. doi:10.1095/biolreprod53.6.1407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
261. Hayes R, Chalmers SA, Nikolic-Paterson DJ, Atkins RC, Hedger MP. Secretion of bioactive interleukin 1 by rat testicular macrophages in vitro. J Androl (1996) 17:41–9.
262. Soder O, Syed V, Callard GV, Toppari J, Pollanen P, Parvinen M, et al. Production and secretion of an interleukin-1-like factor is stage-dependent and correlates with spermatogonial DNA synthesis in the rat seminiferous epithelium. Int J Androl (1991) 14:223–31. doi:10.1111/j.1365-2605.1991.tb01084.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
263. Meinhardt A, Bacher M, Mcfarlane JR, Metz CN, Seitz J, Hedger MP, et al. Macrophage migration inhibitory factor production by Leydig cells: evidence for a role in the regulation of testicular function. Endocrinology (1996) 137:5090–5. doi:10.1210/endo.137.11.8895383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
264. Meinhardt A, Bacher M, O’Bryan MK, Mcfarlane JR, Mallidis C, Lehmann C, et al. A switch in the cellular localization of macrophage migration inhibitory factor in the rat testis after ethane dimethane sulfonate treatment. J Cell Sci (1999) 112(Pt 9):1337–44.
265. Meinhardt A, Bacher M, Wennemuth G, Eickhoff R, Hedger M. Macrophage migration inhibitory factor (MIF) as a paracrine mediator in the interaction of testicular somatic cells. Andrologia (2000) 32:46–8.
266. O’Bryan MK, Schlatt S, Phillips DJ, De Kretser DM, Hedger MP. Bacterial lipopolysaccharide-induced inflammation compromises testicular function at multiple levels in vivo. Endocrinology (2000) 141:238–46. doi:10.1210/endo.141.1.7240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
267. Soder O, Sultana T, Jonsson C, Wahlgren A, Petersen C, Holst M. The interleukin-1 system in the testis. Andrologia (2000) 32:52–5.
268. Filippini A, Riccioli A, Padula F, Lauretti P, D’Alessio A, De Cesaris P, et al. Control and impairment of immune privilege in the testis and in semen. Hum Reprod Update (2001) 7:444–9. doi:10.1093/humupd/7.5.444
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
269. Hutson JC. Testicular macrophages. Int Rev Cytol (1994) 149:99–143. doi:10.1016/S0074-7696(08)62087-2
270. Hedger MP, Meinhardt A. Cytokines and the immune-testicular axis. J Reprod Immunol (2003) 58:1–26. doi:10.1016/S0165-0378(02)00060-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
271. Gerdprasert O, O’Bryan MK, Muir JA, Caldwell AM, Schlatt S, De Kretser DM, et al. The response of testicular leukocytes to lipopolysaccharide-induced inflammation: further evidence for heterogeneity of the testicular macrophage population. Cell Tissue Res (2002) 308:277–85. doi:10.1007/s00441-002-0547-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
272. Gerdprasert O, O’Bryan MK, Nikolic-Paterson DJ, Sebire K, De Kretser DM, Hedger MP. Expression of monocyte chemoattractant protein-1 and macrophage colony-stimulating factor in normal and inflamed rat testis. Mol Hum Reprod (2002) 8:518–24. doi:10.1093/molehr/8.6.518
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
273. O’Bryan MK, Gerdprasert O, Nikolic-Paterson DJ, Meinhardt A, Muir JA, Foulds LM, et al. Cytokine profiles in the testes of rats treated with lipopolysaccharide reveal localized suppression of inflammatory responses. Am J Physiol Regul Integr Comp Physiol (2005) 288:R1744–55. doi:10.1152/ajpregu.00651.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
274. Suescun MO, Rival C, Theas MS, Calandra RS, Lustig L. Involvement of tumor necrosis factor-alpha in the pathogenesis of autoimmune orchitis in rats. Biol Reprod (2003) 68:2114–21. doi:10.1095/biolreprod.102.011189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
275. Bryniarski K, Szczepanik M, Maresz K, Ptak M, Ptak W. Subpopulations of mouse testicular macrophages and their immunoregulatory function. Am J Reprod Immunol (2004) 52:27–35. doi:10.1111/j.1600-0897.2004.00178.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
276. Rank RG, Soderberg LS, Barron AL. Chronic chlamydial genital infection in congenitally athymic nude mice. Infect Immun (1985) 48:847–9.
277. Van Voorhis WC, Barrett LK, Sweeney YT, Kuo CC, Patton DL. Analysis of lymphocyte phenotype and cytokine activity in the inflammatory infiltrates of the upper genital tract of female macaques infected with Chlamydia trachomatis. J Infect Dis (1996) 174:647–50. doi:10.1093/infdis/174.3.647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
278. Penttila JM, Anttila M, Puolakkainen M, Laurila A, Varkila K, Sarvas M, et al. Local immune responses to Chlamydia pneumoniae in the lungs of BALB/c mice during primary infection and reinfection. Infect Immun (1998) 66:5113–8.
279. Rank RG, Bowlin AK, Kelly KA. Characterization of lymphocyte response in the female genital tract during ascending Chlamydial genital infection in the guinea pig model. Infect Immun (2000) 68:5293–8. doi:10.1128/IAI.68.9.5293-5298.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
280. Goodall JC, Beacock-Sharp H, Deane KH, Gaston JS. Recognition of the 60 kilodalton cysteine-rich outer membrane protein OMP2 by CD4(+) T cells from humans infected with Chlamydia trachomatis. Clin Exp Immunol (2001) 126:488–93. doi:10.1046/j.1365-2249.2001.01709.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
281. Goodall JC, Yeo G, Huang M, Raggiaschi R, Gaston JS. Identification of Chlamydia trachomatis antigens recognized by human CD4+ T lymphocytes by screening an expression library. Eur J Immunol (2001) 31:1513–22. doi:10.1002/1521-4141(200105)31:5<1513::AID-IMMU1513>3.0.CO;2-U
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
282. Holland MJ, Conway DJ, Blanchard TJ, Mahdi OM, Bailey RL, Whittle HC, et al. Synthetic peptides based on Chlamydia trachomatis antigens identify cytotoxic T lymphocyte responses in subjects from a trachoma-endemic population. Clin Exp Immunol (1997) 107:44–9. doi:10.1046/j.1365-2249.1997.2511129.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
283. Kim SK, Devine L, Angevine M, Demars R, Kavathas PB. Direct detection and magnetic isolation of Chlamydia trachomatis major outer membrane protein-specific CD8+ CTLs with HLA class I tetramers. J Immunol (2000) 165:7285–92. doi:10.4049/jimmunol.165.12.7285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
284. Ortiz L, Angevine M, Kim SK, Watkins D, Demars R. T-cell epitopes in variable segments of Chlamydia trachomatis major outer membrane protein elicit serovar-specific immune responses in infected humans. Infect Immun (2000) 68:1719–23. doi:10.1128/IAI.68.3.1719-1723.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
285. Li W, Murthy AK, Chaganty BK, Guentzel MN, Seshu J, Chambers JP, et al. Immunization with dendritic cells pulsed ex vivo with recombinant chlamydial protease-like activity factor induces protective immunity against genital chlamydiamuridarum challenge. Front Immunol (2011) 2:73. doi:10.3389/fimmu.2011.00073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
286. Olive AJ, Gondek DC, Starnbach MN. CXCR3 and CCR5 are both required for T cell-mediated protection against C. trachomatis infection in the murine genital mucosa. Mucosal Immunol (2011) 4:208–16. doi:10.1038/mi.2010.58
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
287. Johnson RM, Yu H, Kerr MS, Slaven JE, Karunakaran KP, Brunham RC. PmpG303-311, a protective vaccine epitope that elicits persistent cellular immune responses in Chlamydia muridarum-immune mice. Infect Immun (2012) 80:2204–11. doi:10.1128/IAI.06339-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
288. Starnbach MN, Bevan MJ, Lampe MF. Protective cytotoxic T lymphocytes are induced during murine infection with Chlamydia trachomatis. J Immunol (1994) 153:5183–9.
289. Starnbach MN, Bevan MJ, Lampe MF. Murine cytotoxic T lymphocytes induced following Chlamydia trachomatis intraperitoneal or genital tract infection respond to cells infected with multiple serovars. Infect Immun (1995) 63:3527–30.
290. Johansson M, Schon K, Ward M, Lycke N. Studies in knockout mice reveal that anti-chlamydial protection requires TH1 cells producing IFN-gamma: is this true for humans? Scand J Immunol (1997) 46:546–52. doi:10.1046/j.1365-3083.1997.d01-167.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
291. Roan NR, Gierahn TM, Higgins DE, Starnbach MN. Monitoring the T cell response to genital tract infection. Proc Natl Acad Sci U S A (2006) 103:12069–74. doi:10.1073/pnas.0603866103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
292. Marks E, Verolin M, Stensson A, Lycke N. Differential CD28 and inducible costimulatory molecule signaling requirements for protective CD4+ T-cell-mediated immunity against genital tract Chlamydia trachomatis infection. Infect Immun (2007) 75:4638–47. doi:10.1128/IAI.00465-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
293. Gondek DC, Roan NR, Starnbach MN. T cell responses in the absence of IFN-gamma exacerbate uterine infection with Chlamydia trachomatis. J Immunol (2009) 183:1313–9. doi:10.4049/jimmunol.0900295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
294. Morrison SG, Farris CM, Sturdevant GL, Whitmire WM, Morrison RP. Murine Chlamydia trachomatis genital infection is unaltered by depletion of CD4+ T cells and diminished adaptive immunity. J Infect Dis (2011) 203:1120–8. doi:10.1093/infdis/jiq176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
295. Su H, Caldwell HD. CD4+ T cells play a significant role in adoptive immunity to Chlamydia trachomatis infection of the mouse genital tract. Infect Immun (1995) 63:3302–8.
296. Cunningham KA, Carey AJ, Timms P, Beagley KW. CD4+ T cells reduce the tissue burden of Chlamydia muridarum in male BALB/c mice. Vaccine (2010) 28:4861–3. doi:10.1016/j.vaccine.2010.05.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
297. Johansson M, Ward M, Lycke N. B-cell-deficient mice develop complete immune protection against genital tract infection with Chlamydia trachomatis. Immunology (1997) 92:422–8. doi:10.1046/j.1365-2567.1997.00378.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
298. Gondek DC, Olive AJ, Stary G, Starnbach MN. CD4+ T cells are necessary and sufficient to confer protection against Chlamydia trachomatis infection in the murine upper genital tract. J Immunol (2012) 189:2441–9. doi:10.4049/jimmunol.1103032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
299. Beatty PR, Stephens RS. CD8+ T lymphocyte-mediated lysis of Chlamydia-infected L cells using an endogenous antigen pathway. J Immunol (1994) 153:4588–95.
300. Igietseme JU, Magee DM, Williams DM, Rank RG. Role for CD8+ T cells in antichlamydial immunity defined by Chlamydia-specific T-lymphocyte clones. Infect Immun (1994) 62:5195–7.
301. Wizel B, Nystrom-Asklin J, Cortes C, Tvinnereim A. Role of CD8(+)T cells in the host response to Chlamydia. Microbes Infect (2008) 10:1420–30. doi:10.1016/j.micinf.2008.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
302. Buzoni-Gatel D, Guilloteau L, Bernard F, Bernard S, Chardes T, Rocca A. Protection against Chlamydia psittaci in mice conferred by Lyt-2+ T cells. Immunology (1992) 77:284–8.
303. Magee DM, Williams DM, Smith JG, Bleicker CA, Grubbs BG, Schachter J, et al. Role of CD8 T cells in primary Chlamydia infection. Infect Immun (1995) 63:516–21.
304. Morrison RP, Feilzer K, Tumas DB. Gene knockout mice establish a primary protective role for major histocompatibility complex class II-restricted responses in Chlamydia trachomatis genital tract infection. Infect Immun (1995) 63:4661–8.
305. Hedger MP, Meinhardt A. Local regulation of T cell numbers and lymphocyte-inhibiting activity in the interstitial tissue of the adult rat testis. J Reprod Immunol (2000) 48:69–80. doi:10.1016/S0165-0378(00)00071-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
306. Hedger MP, Wang J, Lan HY, Atkins RC, Wreford NG. Immunoregulatory activity in adult rat testicular interstitial fluid: relationship with intratesticular CD8+ lymphocytes following treatment with ethane dimethane sulfonate and testosterone implants. Biol Reprod (1998) 58:935–42. doi:10.1095/biolreprod58.4.935
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
307. Dai Z, Nasr IW, Reel M, Deng S, Diggs L, Larsen CP, et al. Impaired recall of CD8 memory T cells in immunologically privileged tissue. J Immunol (2005) 174:1165–70. doi:10.4049/jimmunol.174.3.1165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
308. Nasr IW, Wang Y, Gao G, Deng S, Diggs L, Rothstein DM, et al. Testicular immune privilege promotes transplantation tolerance by altering the balance between memory and regulatory T cells. J Immunol (2005) 174:6161–8. doi:10.4049/jimmunol.174.10.6161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
309. Jawetz E, Rose L, Hanna L, Thygeson P. Experimental inclusion conjunctivitis in man: measurements of infectivity and resistance. JAMA (1965) 194:620–32. doi:10.1001/jama.1965.03090190042012
310. Barenfanger J, MacDonald AB. The role of immunoglobulin in the neutralization of trachoma infectivity. J Immunol (1974) 113:1607–17.
311. Peeling R, Maclean IW, Brunham RC. In vitro neutralization of Chlamydia trachomatis with monoclonal antibody to an epitope on the major outer membrane protein. Infect Immun (1984) 46:484–8.
312. Peterson EM, Cheng X, Markoff BA, Fielder TJ, De La Maza LM. Functional and structural mapping of Chlamydia trachomatis species-specific major outer membrane protein epitopes by use of neutralizing monoclonal antibodies. Infect Immun (1991) 59:4147–53.
313. Cotter TW, Meng Q, Shen ZL, Zhang YX, Su H, Caldwell HD. Protective efficacy of major outer membrane protein-specific immunoglobulin A (IgA) and IgG monoclonal antibodies in a murine model of Chlamydia trachomatis genital tract infection. Infect Immun (1995) 63:4704–14.
314. Finco O, Frigimelica E, Buricchi F, Petracca R, Galli G, Faenzi E, et al. Approach to discover T- and B-cell antigens of intracellular pathogens applied to the design of Chlamydia trachomatis vaccines. Proc Natl Acad Sci U S A (2011) 108:9969–74. doi:10.1073/pnas.1101756108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
315. Bartolini E, Ianni E, Frigimelica E, Petracca R, Galli G, Berlanda Scorza F, et al. Recombinant outer membrane vesicles carrying Chlamydia muridarum HtrA induce antibodies that neutralize chlamydial infection in vitro. J Extracell Vesicles (2013) 2. doi:10.3402/jev.v2i0.20181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
316. Moore T, Ananaba GA, Bolier J, Bowers S, Belay T, Eko FO, et al. Fc receptor regulation of protective immunity against Chlamydia trachomatis. Immunology (2002) 105:213–21. doi:10.1046/j.0019-2805.2001.01354.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
317. Igietseme JU, Eko FO, He Q, Black CM. Antibody regulation of T cell immunity: implications for vaccine strategies against intracellular pathogens. Expert Rev Vaccines (2004) 3:23–34. doi:10.1586/14760584.3.1.23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
318. Su H, Feilzer K, Caldwell HD, Morrison RP. Chlamydia trachomatis genital tract infection of antibody-deficient gene knockout mice. Infect Immun (1997) 65:1993–9.
319. Williams DM, Grubbs BG, Pack E, Kelly K, Rank RG. Humoral and cellular immunity in secondary infection due to murine Chlamydia trachomatis. Infect Immun (1997) 65:2876–82.
320. Morrison SG, Su H, Caldwell HD, Morrison RP. Immunity to murine Chlamydia trachomatis genital tract reinfection involves B cells and CD4(+) T cells but not CD8(+) T cells. Infect Immun (2000) 68:6979–87. doi:10.1128/IAI.68.12.6979-6987.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
321. Morrison SG, Morrison RP. A predominant role for antibody in acquired immunity to chlamydial genital tract reinfection. J Immunol (2005) 175:7536–42. doi:10.4049/jimmunol.175.11.7536
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
322. Lin-Xi L, McSorley S. Revisiting the role of B cells in Chlamydia muridarum genital tract infection. J Immunol Suppl (2014) 192.
323. Hogan RJ, Mathews SA, Mukhopadhyay S, Summersgill JT, Timms P. Chlamydial persistence: beyond the biphasic paradigm. Infect Immun (2004) 72:1843–55. doi:10.1128/IAI.72.4.1843-1855.2004
324. Clark RB, Schatzki PF, Dalton HP. Ultrastructural analysis of the effects of erythromycin on the morphology and developmental cycle of Chlamydia trachomatis HAR-13. Arch Microbiol (1982) 133:278–82. doi:10.1007/BF00521290
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
325. Wolf K, Fischer E, Hackstadt T. Ultrastructural analysis of developmental events in Chlamydia pneumoniae-infected cells. Infect Immun (2000) 68:2379–85. doi:10.1128/IAI.68.4.2379-2385.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
326. Mehta SJ, Miller RD, Ramirez JA, Summersgill JT. Inhibition of Chlamydia pneumoniae replication in HEp-2 cells by interferon-gamma: role of tryptophan catabolism. J Infect Dis (1998) 177:1326–31. doi:10.1086/515287
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
328. Coles AM, Reynolds DJ, Harper A, Devitt A, Pearce JH. Low-nutrient induction of abnormal chlamydial development: a novel component of chlamydial pathogenesis? FEMS Microbiol Lett (1993) 106:193–200. doi:10.1111/j.1574-6968.1993.tb05958.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
329. Akers JC, Tan M. Molecular mechanism of tryptophan-dependent transcriptional regulation in Chlamydia trachomatis. J Bacteriol (2006) 188:4236–43. doi:10.1128/JB.01660-05
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
330. Harper A, Pogson CI, Jones ML, Pearce JH. Chlamydial development is adversely affected by minor changes in amino acid supply, blood plasma amino acid levels, and glucose deprivation. Infect Immun (2000) 68:1457–64. doi:10.1128/IAI.68.3.1457-1464.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
331. Raulston JE. Response of Chlamydia trachomatis serovar E to iron restriction in vitro and evidence for iron-regulated chlamydial proteins. Infect Immun (1997) 65:4539–47.
332. Matsumoto A, Manire GP. Electron microscopic observations on the effects of penicillin on the morphology of Chlamydia psittaci. J Bacteriol (1970) 101:278–85.
333. Jones ML, Gaston JS, Pearce JH. Induction of abnormal Chlamydia trachomatis by exposure to interferon-gamma or amino acid deprivation and comparative antigenic analysis. Microb Pathog (2001) 30:299–309. doi:10.1006/mpat.2001.0433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
334. Beatty WL, Byrne GI, Morrison RP. Morphologic and antigenic characterization of interferon gamma-mediated persistent Chlamydia trachomatis infection in vitro. Proc Natl Acad Sci U S A (1993) 90:3998–4002. doi:10.1073/pnas.90.9.3998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
335. Caldwell HD, Wood H, Crane D, Bailey R, Jones RB, Mabey D, et al. Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J Clin Invest (2003) 111:1757–69. doi:10.1172/JCI17993
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
336. Nanagara R, Li F, Beutler A, Hudson A, Schumacher HR Jr. Alteration of Chlamydia trachomatis biologic behavior in synovial membranes. Suppression of surface antigen production in reactive arthritis and Reiter’s syndrome. Arthritis Rheum (1995) 38:1410–7. doi:10.1002/art.1780381008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
337. Mazzoli S, Bani D, Salvi A, Ramacciotti I, Romeo C, Bani T. In vivo evidence of Chlamydia trachomatis miniature reticulary bodies (MRB) as persistence markers in patients with chronic chlamydial prostatits. Proc Eur Soc Chlamydia Res (2000) 4:40.
338. Phillips DM, Swenson CE, Schachter J. Ultrastructure of Chlamydia trachomatis infection of the mouse oviduct. J Ultrastruct Res (1984) 88:244–56. doi:10.1016/S0022-5320(84)90122-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
339. Dean D, Suchland RJ, Stamm WE. Evidence for long-term cervical persistence of Chlamydia trachomatis by omp1 genotyping. J Infect Dis (2000) 182:909–16. doi:10.1086/315778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
340. Katz B, Fortenberry D, Orr D. Factors affecting chlamydial persistence or recurrence one and three months after treatment. In: Stephens RS, Byrne GI, Christiansen G, Clarke I, Grayston JT, Rank RG, et al., editors. Proceedings of the Ninth International Symposium on Human Chlamydial Infections. San Franciso, CA: Berkeley University Press (1998). p. 35–8.
341. Fan T, Lu H, Hu H, Shi L, Mcclarty GA, Nance DM, et al. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med (1998) 187:487–96. doi:10.1084/jem.187.4.487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
342. Airenne S, Surcel HM, Tuukkanen J, Leinonen M, Saikku P. Chlamydia pneumoniae inhibits apoptosis in human epithelial and monocyte cell lines. Scand J Immunol (2002) 55:390–8. doi:10.1046/j.1365-3083.2002.01075.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
343. Perfettini JL, Darville T, Gachelin G, Souque P, Huerre M, Dautry-Varsat A, et al. Effect of Chlamydia trachomatis infection and subsequent tumor necrosis factor alpha secretion on apoptosis in the murine genital tract. Infect Immun (2000) 68:2237–44. doi:10.1128/IAI.68.4.2237-2244.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
344. Byrne GI, Ojcius DM. Chlamydia and apoptosis: life and death decisions of an intracellular pathogen. Nat Rev Microbiol (2004) 2:802–8. doi:10.1038/nrmicro1007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
345. Miyairi I, Byrne GI. Chlamydia and programmed cell death. Curr Opin Microbiol (2006) 9:102–8. doi:10.1016/j.mib.2005.12.004
346. Roan NR, Starnbach MN. Immune-mediated control of Chlamydia infection. Cell Microbiol (2008) 10:9–19. doi:10.1111/j.01069.x
347. Zhong G, Fan T, Liu L. Chlamydia inhibits interferon gamma-inducible major histocompatibility complex class II expression by degradation of upstream stimulatory factor 1. J Exp Med (1999) 189:1931–8. doi:10.1084/jem.189.12.1931
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
348. Zhong G, Liu L, Fan T, Fan P, Ji H. Degradation of transcription factor RFX5 during the inhibition of both constitutive and interferon gamma-inducible major histocompatibility complex class I expression in chlamydia-infected cells. J Exp Med (2000) 191:1525–34. doi:10.1084/jem.191.9.1525
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
349. Zhong G, Fan P, Ji H, Dong F, Huang Y. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J Exp Med (2001) 193:935–42. doi:10.1084/jem.193.8.935
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
350. Belland R, Ojcius DM, Byrne GI. Chlamydia. Nat Rev Microbiol (2004) 2:530–1. doi:10.1038/nrmicro931
351. Roos D. The involvement of oxygen radicals in microbicidal mechanisms of leukocytes and macrophages. Klin Wochenschr (1991) 69:975–80. doi:10.1007/BF01645142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
352. Ogilvie KM, Held Hales K, Roberts ME, Hales DB, Rivier C. The inhibitory effect of intracerebroventricularly injected interleukin 1beta on testosterone secretion in the rat: role of steroidogenic acute regulatory protein. Biol Reprod (1999) 60:527–33. doi:10.1095/biolreprod60.2.527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
353. Riedel W, Lang U, Oetjen U, Schlapp U, Shibata M. Inhibition of oxygen radical formation by methylene blue, aspirin, or alpha-lipoic acid, prevents bacterial-lipopolysaccharide-induced fever. Mol Cell Biochem (2003) 247:83–94. doi:10.1023/A:1024142400835
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
354. Victor VM, De La Fuente M. Several functions of immune cells in mice changed by oxidative stress caused by endotoxin. Physiol Res (2003) 52:789–96.
355. Victor VM, De La Fuente M. Changes in the superoxide production and other macrophage functions could be related to the mortality of mice with endotoxin-induced oxidative stress. Physiol Res (2003) 52:101–10.
356. Hales DB, Diemer T, Hales KH. Role of cytokines in testicular function. Endocrine (1999) 10:201–17. doi:10.1007/BF02738619
357. Allen JA, Diemer T, Janus P, Hales KH, Hales DB. Bacterial endotoxin lipopolysaccharide and reactive oxygen species inhibit Leydig cell steroidogenesis via perturbation of mitochondria. Endocrine (2004) 25:265–75. doi:10.1385/ENDO:25:3:265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
358. Abdul-Sater AA, Said-Sadier N, Lam VM, Singh B, Pettengill MA, Soares F, et al. Enhancement of reactive oxygen species production and chlamydial infection by the mitochondrial Nod-like family member NLRX1. J Biol Chem (2010) 285:41637–45. doi:10.1074/jbc.M110.137885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
359. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal (2014) 20:1126–67. doi:10.1089/ars.2012.5149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
360. MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol (1997) 15:323–50. doi:10.1146/annurev.immunol.15.1.323
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
361. Bogdan C. Nitric oxide and the immune response. Nat Immunol (2001) 2:907–16. doi:10.1038/ni1001-907
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
362. Li P, Chan HC, He B, So SC, Chung YW, Shang Q, et al. An antimicrobial peptide gene found in the male reproductive system of rats. Science (2001) 291:1783–5. doi:10.1126/science.1056545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
363. Yamaguchi Y, Nagase T, Makita R, Fukuhara S, Tomita T, Tominaga T, et al. Identification of multiple novel epididymis-specific beta-defensin isoforms in humans and mice. J Immunol (2002) 169:2516–23. doi:10.4049/jimmunol.169.5.2516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
364. Yenugu S, Hamil KG, Radhakrishnan Y, French FS, Hall SH. The androgen-regulated epididymal sperm-binding protein, human beta-defensin 118 (DEFB118) (formerly ESC42), is an antimicrobial beta-defensin. Endocrinology (2004) 145:3165–73. doi:10.1210/en.2003-1698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
365. Zhou CX, Zhang YL, Xiao L, Zheng M, Leung KM, Chan MY, et al. An epididymis-specific beta-defensin is important for the initiation of sperm maturation. Nat Cell Biol (2004) 6:458–64. doi:10.1038/ncb1127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
366. Jalkanen J, Huhtaniemi I, Poutanen M. Discovery and characterization of new epididymis-specific beta-defensins in mice. Biochim Biophys Acta (2005) 1730:22–30. doi:10.1016/j.bbaexp.2005.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
367. Patil AA, Cai Y, Sang Y, Blecha F, Zhang G. Cross-species analysis of the mammalian beta-defensin gene family: presence of syntenic gene clusters and preferential expression in the male reproductive tract. Physiol Genomics (2005) 23:5–17. doi:10.1152/physiolgenomics.00104.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
368. Biswas B, Yenugu S. Lipopolysaccharide induces epididymal and testicular antimicrobial gene expression in vitro: insights into the epigenetic regulation of sperm-associated antigen 11e gene. Immunogenetics (2013) 65:239–53. doi:10.1007/s00251-012-0674-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
369. Barabas N, Rohrl J, Holler E, Hehlgans T. Beta-defensins activate macrophages and synergize in pro-inflammatory cytokine expression induced by TLR ligands. Immunobiology (2013) 218:1005–11. doi:10.1016/j.imbio.2012.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Chlamydia, infection, male fertility, inflammation, persistence
Citation: Redgrove KA and McLaughlin EA (2014) The role of the immune response in Chlamydia trachomatis infection of the male genital tract: a double-edged sword. Front. Immunol. 5:534. doi: 10.3389/fimmu.2014.00534
Received: 30 June 2014; Paper pending published: 23 August 2014;
Accepted: 09 October 2014; Published online: 27 October 2014.
Edited by:
Ken Beagley, Queensland University of Technology, AustraliaReviewed by:
Adrianus Wilhelmus Maria Van Der Velden, Stony Brook University, USAHeinrich Korner, Menzies Research Institute Tasmania, Australia
Copyright: © 2014 Redgrove and McLaughlin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eileen A. McLaughlin, Priority Research Centre in Reproductive Biology and Chemical Biology, University of Newcastle, University Drive, Callaghan, NSW 2308, Australia e-mail:RWlsZWVuLk1jTGF1Z2hsaW5AbmV3Y2FzdGxlLmVkdS5hdQ==