 Stanley Gititu Kimani1
Stanley Gititu Kimani1 Ke Geng1Canan Kasikara1
Ke Geng1Canan Kasikara1 Sushil Kumar1
Sushil Kumar1 Ganapathy Sriram1
Ganapathy Sriram1 Yi Wu2,3Raymond B. Birge1*
Yi Wu2,3Raymond B. Birge1*- 1Department of Biochemistry and Molecular Biology, Rutgers School of Biomedical and Health Sciences – Cancer Center, Newark, NJ, USA
- 2Cyrus Tang Hematology Center, Jiangsu Institute of Hematology, First Affiliated Hospital, Soochow University, Suzhou, China
- 3Sol Sherry Thrombosis Research Center, Temple University School of Medicine, Philadelphia, PA, USA
The rapid and efficient clearance of apoptotic cells results in the elimination of auto-antigens and provides a strong anti-inflammatory and immunosuppressive signal to prevent autoimmunity. While professional and non-professional phagocytes utilize a wide array of surface receptors to recognize apoptotic cells, the recognition of phosphatidylserine (PS) on apoptotic cells by PS receptors on phagocytes is the emblematic signal for efferocytosis in metazoans. PS-dependent efferocytosis is associated with the production of anti-inflammatory factors such as IL-10 and TGF-β that function, in part, to maintain tolerance to auto-antigens. In contrast, when apoptotic cells fail to be recognized and processed for degradation, auto-antigens persist, such as self-nucleic acids, which can trigger immune activation leading to autoantibody production and autoimmunity. Despite the fact that genetic mouse models clearly demonstrate that loss of PS receptors can lead to age-dependent auto-immune diseases reminiscent of systemic lupus erythematosus (SLE), the link between PS and defective clearance in chronic inflammation and human autoimmunity is not well delineated. In this perspective, we review emerging questions developing in the field that may be of relevance to SLE and human autoimmunity.
Introduction
The clearance of apoptotic cells by phagocytic cells (a process now called efferocytosis to distinguish the processing of apoptotic cells from other phagocytic processes) is critically important to maintain homeostasis in multicellular organisms. Efficient efferocytosis not only allows for the removal and degradation of effete and damaged cells, but has an equally important function in the resolution of inflammation by protecting tissue from harmful exposure to the inflammatory and immunogenic contents of dying cells (1–4). There is now considerable genetic evidence supported by mouse knockout studies that failed or delayed efferocytosis results in the release of auto-antigens that can contribute to the etiology of auto-immune diseases such as systemic lupus erythematosus (SLE) (5). In addition, macrophages derived from SLE patients also exhibit defects in efferocytosis (6, 7). Elucidating the genetic basis for defective clearance in relation to human autoimmunity is clearly a topical and important area of research.
Concomitant with caspase activation and cell death, apoptotic cells display a wide array of nascent and modified molecular determinants on their plasma membranes that act as “eat-me” signals for phagocytes. While these determinants result from a combination of re-localized proteins, modified carbohydrates, and from collapse of phospholipid asymmetry at the plasma membrane, the externalization of phosphatidylserine (PS) is arguably the most emblematic event associated with the early phase of apoptotic program (8–10). If apoptotic cells escape immediate clearance, a second wave of late apoptotic cells clearance is mediated by opsonins that includes nuclear materials (11), C1q (12), ficolins (13), and pentraxins (14–16). The late apoptotic cells bound by these opsonins are then recognized and cleared via phagocytic receptors including FcγRIIA, C1q receptor, CR1, CD91, and calreticulin (CRT), helping to avoid inflammation (17, 18). Although our discussion here focuses on cross-interactions between different PS receptors and opsonins, the crosstalk between different recognition systems (such as PS and modified carbohydrates and PS and protein neoepitopes) is likely equally important.
The fact that blockage of PS on the apoptotic cell prevents many of the anti-inflammatory consequences of efferocytosis, combined with observations that knockout of several PS receptors and PS opsonins (soluble factors that link PS on apoptotic cells to receptors) lead to failed efferocytosis, chronic inflammation, and age-dependent autoimmunity (4) has led many investigators to a conceptual framework that externalized PS functions as a dampening platform for negative immune regulation. In this capacity, externalized PS functions both as an “eat-me” signal for efferocytosis, but also as an “inflammo-suppression” signal that promotes tolerance for both immune cells and non-immune bystander cells that come in direct contact with PS externalized membranes (2, 19, 20). Despite convincing evidence as gleaned from knockout studies in mouse, identifying links between defective PS recognition and/or signaling and human autoimmunity has been surprisingly enigmatic (Table 1).
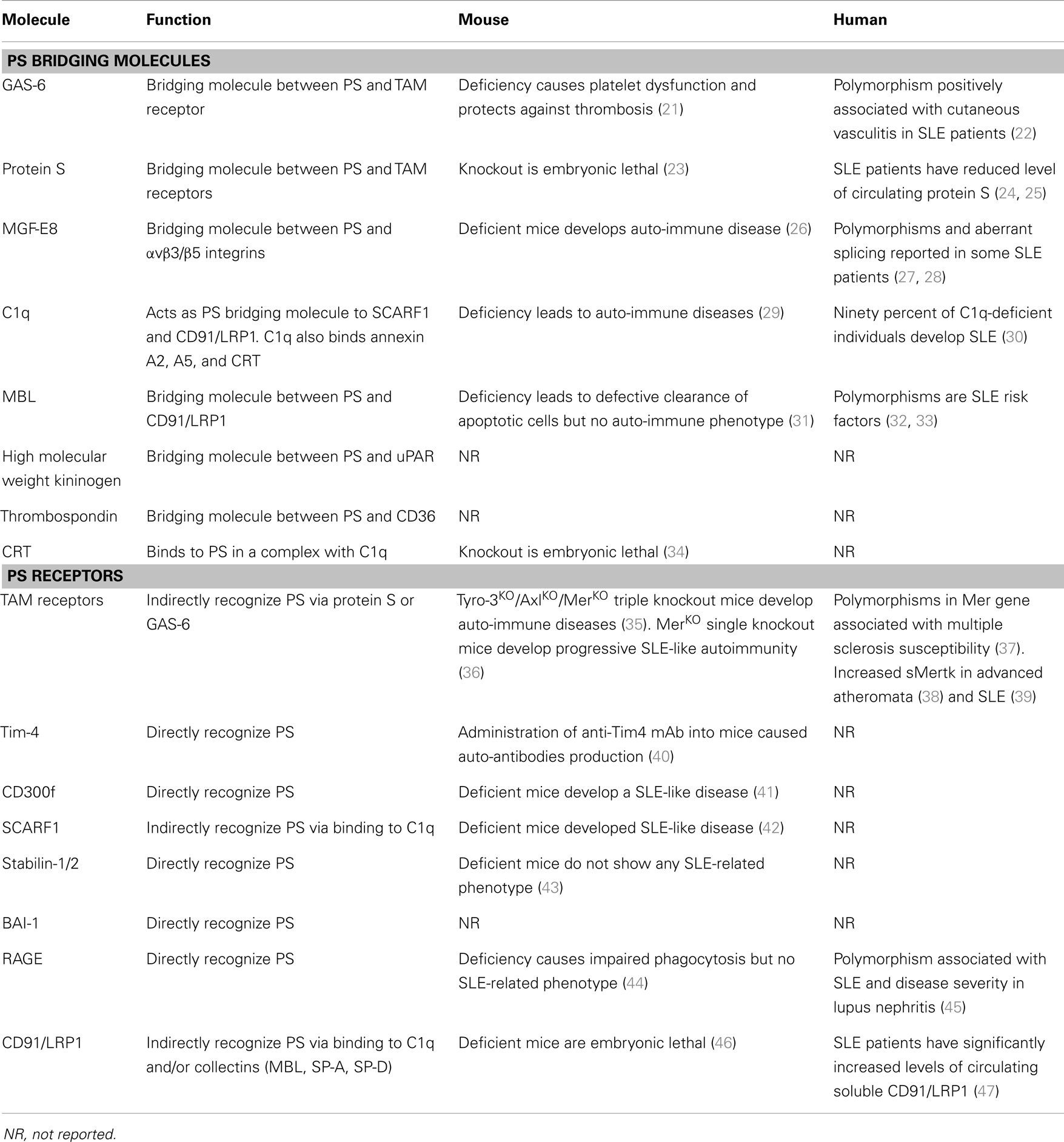
Table 1. Summary of PS receptors and soluble PS binding proteins and their relationship to autoimmunity in mouse and human systems.
Mice Lacking PS Receptors are Prone to Lupus-Like Auto-Immune Conditions
Over the past decade, a diverse array of PS receptors and soluble PS bridging proteins that link apoptotic cells to phagocytes have been identified (48–50) (Table 1). Although this suggests significant redundancy at the biochemical level, PS receptors do not appear to act in a compensatory capacity by loss-of-function. For example, on certain genetic backgrounds, single knockouts of Mer (36), Tim-1 (51), Tim-4 (40), SCARF1 (42), and CD300f (41) all have a common phenotype that include defective apoptotic cell clearance, the subsequent production of auto-antibodies, and SLE-like autoimmunity. Similarly, a knockout of MFG-E8 (26), a PS opsonin that bridges apoptotic cells to αvβ5 and αvβ3 integrin, also produces a strong SLE-like phenotype. While in some cases dual targeting of PS receptors can compound phenotypic outcomes [for example Tim-4 and MFG-E8 (52) develop autoimmunity at an earlier age, or triple knockout of TAM (Tyro3, Axl, and Mer) (35) have a more potent onset of disease than Mer alone), collectively these data suggest, at least in the mouse, that PS receptors are not functionally redundant. One possible interpretation is that PS receptors, analogously to the immunological synapse for T cell signaling, comprise a multi-protein signaling receptor complex, perhaps akin to a PS phagocytic synapse, where loss-of-function of any single component disrupts the higher order functional unit (53, 54). Several of the known PS receptors, such as αvβ5 integrin and Mertk, are known to synergize in order to activate intracellular signaling pathways such as Rac1 (55, 56) also supporting the idea of receptor crosstalk. However, while attractive to speculate, such a multi-protein structure (aka, the “engulfosome”) has not been identified at a biochemical level.
Clearly then, an obvious question is whether the aforementioned PS circuitry fails, or is a genetic risk factor for human auto-immune disease such as SLE. Presently, the answer is still not clear, although of the major PS recognition receptors that give rise to autoimmunity in mice (Mer, Tim-1, Tim-4, SCARF1, and CD300f), their involvement in human autoimmunity is not yet obvious from genetic linkage analysis. Although MFG-E8 mutations have been identified in a small subset of lupus patients (28), and a case-control study of MFG-E8 genetic polymorphisms showed some genetic linkage (27), these events appear to be rare. Likewise in the case of TAMs (Mer) and their ligands, it was shown that in SLE patients, TAM levels do not appear to be compromised (57, 58), and in some patients, serum levels of Mer and TAM ligands actually appear to be elevated (59–61).
The recent studies by Ramirez-Ortiz and colleagues, identifying the scavenger receptor SCARF1 (SREC1, CED-1) as a PS receptor that recognizes a PS in the context of complement component C1q (42) might have relevance to human SLE. In vivo, SCARF1 (−/−) mice in develop systemic SLE-like disease, including the generation of auto-antibodies and glomerulonephritis that closely mimics human SLE (42). Interestingly, while SCARF1 was shown to bind via PS, apoptotic cells deficient in C1q were notably impaired in their ability to bind to and activate SCARF1, suggesting the C1q acts as a requisite bridging molecule for PS. In addition to SCARF1, C1q also binds to PS-opsonized CRT (62) on the surface of apoptotic cells (a ligand for CD91/LRP1 on the phagocyte), as well as other PS-binding proteins that include Annexin A5 and Annexin A2 (63). Although genetic deficiency of C1q is quite rare (<100 known cases have been reported), over 90% of these individuals develop SLE (30), and monocytes (64, 65) derived from these patients have impaired ability to clear apoptotic cells suggesting a defect in the apoptotic cell clearance machinery. In addition, apoptotic cells derived from SLE patients also show greatly diminished capacity to bind C1q (66) suggesting one or more of the determinants on the apoptotic cell that bind C1q is also deficient in SLE. Although monocytes isolated from SLE patients showed only a modest decrease in CD91/LRP1 levels, patients with rheumatoid arthritis or SLE showed significantly elevated levels of soluble CD91/LRP1 cleaved by ADAM17 in response to inflammation (47). Possibly related, excessive protease cleavage of Mertk from macrophages has also been linked to inefficient clearance in the development of advanced atheromata (38) and SLE (39). Clearly, it will be of interest to ascertain at the genetic level whether loss-of-function mutations occur at CD91/LRP1 or SCARF1 receptor loci that result in risk associations for human auto-immune diseases.
Taken together, while loss-of-function genetic ablation studies in mouse models clearly show a link between systemic autoimmunity and loss-of-function of PS receptors, translating this biology into human SLE pathology still remains somewhat of a mystery. Future studies should address whether PS receptor biology is arranged differently in humans in comparison to mice PS receptors, allowing for more redundancy, or whether defective PS signaling in human is part of a multi-genic signature that acts as a cohort with other risk factors. Another caveat on relying on expression analysis is that many SLE and auto-immune patients are chronically treated with glucocorticoids and steroids, which may affect the levels of PS receptors or PS-opsonins. For example, Lauber and colleagues showed that MFG-E8 is transcriptionally regulated by dexamethasone, a steroid used to treat the chronic inflammation associated with lupus (67). In addition to MFG-E8, the TAM receptors are also subject to acute regulation by glucocorticoids but in a reciprocal fashion; Mer is up-regulated while Axl is down-regulated following dexamethasone treatment (68). This could also induce a feed-forward mechanism, where dexamethasone-induced increase in Mer levels could increase efferocytosis, which itself further increases Mer by the increased uptake of apoptotic cargo. Internalized apoptotic cells increase ingested cholesterol, which can activate LXR and activate the Mer promoter (69, 70). This idea that corticosteroids mediate their effects by manipulating PS biology might be interrogated via the development of more specific therapeutics for SLE.
Another possible reason for the discrepancy between the studies in mice and the observations in human autoimmunity is that defects in PSR signaling (generated in mouse models) may not be manifested as defects in PSRs or PS-opsonins in human autoimmunity but by mutations in genes involved in the mechanisms upstream such as PS externalization or modification. We explore facets of this hypothesis in the following three sections.
Scramblases, Flippases, and Upstream Mechanisms of PS Exposure
While the past decade has shown great strides in elucidating the repertoire of PS receptors that bind to and rely signals from PS on the apoptotic cell to phagocytic receptors, in recent years, there has also been a much greater appreciation for the genes and regulatory circuits that control PS externalization, including the realization that mutations in these genes can lead to pathologies related to dysfunctional PS biology. Novel scramblases and flippases responsible for PS externalization have been enumerated, opening up the possibility that genes that control externalization, and defects therein, may also contribute to chronic inflammation and autoimmunity.
Similar to other lipids, PS is synthesized in the endoplasmic reticulum and golgi apparatus and then transported to the plasma membrane by carrier proteins. Once PS reaches the plasma membrane, it is actively excluded from the extracellular milieu by several complementary enzymes. These enzymes, in part, maintain membrane asymmetry, with the choline-containing phospholipids; PC and SM predominantly maintained in the outer leaflet, and the amino-phospholipids; PS, PE, and PI predominately on the inner leaflet (71). To maintain PS asymmetry under homeostatic conditions, three main types of enzymes operate at equilibrium, but each can be perturbed during apoptosis and during cell stress. Flippases and Floppases translocate phospholipids from the outer surface to the inner surface and from the inner surface to the outer surface, respectively, and both require ATP for this activity (72). A third, and least understood class of lipid transporters that regulate PS topology are called scramblases, and as their name implies, when activated, collapse membrane asymmetry, and in the context of PS biology, promote the accumulation of PS to the external side of the membrane.
Although phospholipid scramblases do not show selectivity for the phospholipid species or for the direction of movement, the scramblase-mediated exposure of PS has important consequences for several biological events that include coagulation, neurotransmitter release, sperm capacitation, and apoptosis (73). While PS is externalized during both platelet activation and during apoptosis, the recent characterization of two scramblases, Transmembrane protein 16F (TMEM16F) (74, 75) and Xkr8 (76), provide some conceptual relief to this field, highlighting that cells externalize PS through different activation and regulatory mechanisms, but of equal significance, that not all externalized PS has the same biological function.
Transmembrane protein 16F is an eight-transmembrane spanning aminophospholipid scramblase that is critical for the calcium-dependent externalization of PS in activated platelets. In the studies from Nagata and colleagues, these investigators developed a clever FACS sorting approach to characterize a Ba/F3 pro-B cell sub-line that can be trained to respond to sub-threshold concentrations of calcium. After repetitive sorting of PS-positive cells, a Ba/F3 sub-clone that contained a mutated TMEM16F and constitutively scrambled PS was identified (74). Further studies showed that loss of TMEM16F function, either via knockout or through mutation, impairs calcium-dependent PS scramblase activity, and when occurring in platelets, results in their inability to recruit and activate hemostasis factors that include factor V, factor X, and prothrombin to the platelet membrane (75). DNA sequence analysis further showed that Scott syndrome patients, which are characterized by a rare bleeding disorder that have defects on calcium-dependent phospholipid scrambling, carry loss-of-function mutations in both Tmem16 alleles. Functionally, other members of TMEM16 family, including 16C, 16D, 16F, 16G, and 16J are also capable of scrambling PS, but further studies will be required to ascertain whether different family members are specific for different cell types (77).
Notably, the above-mentioned Ca2+-stimulated PS externalization induced by TMEM16F is readily reversible upon restoration of Ca2+ homeostasis, while the PS externalized during caspase-mediated apoptosis is distinct and separable from TMEM16F, as PS externalization is maintained in apoptotic TMEM16F (−/−) cells treated with Fas-L to induce apoptosis (77). Remarkably, when a mutant TMEM16F was introduced into a mouse lymphoma cell (W3-Ildm) to achieve constitutive PS exposure, these PS-positive tumor cells were not targets of efferocytosis, even by professional DCs, and only became phagocytic competent after treatment with Fas-L to activate caspase 3 (78). These data offer a molecular explanation as to why activated cells, such as during platelet aggregation, T cell activation, and during mast cell degranulation, externalize PS but fail to be engulfed. Conceptually, these data suggest that PS externalization, per se, while necessary, is not sufficient to promote clearance (Figure 1).
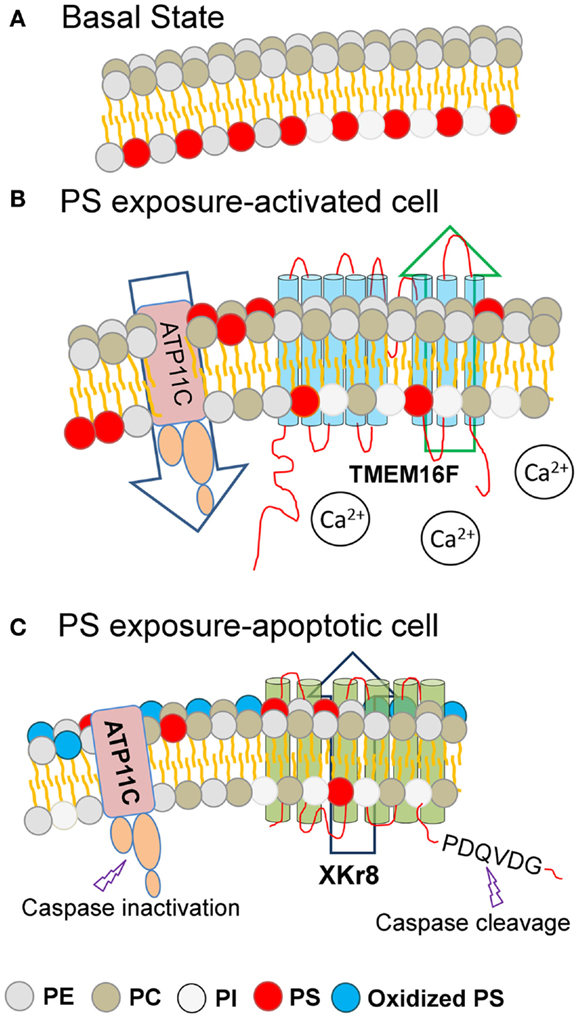
Figure 1. Different modes of PS externalization by lipid scramblases. Under basal resting conditions, the amino-phospholipids (PE and PS) are restricted to the inner surface of the plasma membrane (A). During conditions of cell stress or platelet activation (B) or during apoptosis (C), membrane asymmetry is lost and PS is externalized to the extrafacial surface (depicted in red). Under basal conditions (A), plasma membrane asymmetry is maintained by the combined activity of cellular flippases and floppases. During cell stress or during platelet activation, intracellular calcium levels rise, resulting in the activation of TMEM16F, and PS exposure to the extracellular leaflet (B). During apoptosis and the activation of caspases, executioner caspases are able to cleave and activate Xkr8, as well as cleave and inactivate ATP11C, resulting in PS exposure to the extracellular leaflet. Since PS externalized PS via TMEM16F and Xkr8/ATP11C are differentially recognized as eat-me signals, it is likely that the density of PS, or the oxidation state of the PS, provide assurance signals for efferocytosis. Key: PE, phosphatidylethanolamine; PC, phosphatidylcholine; PI, phosphatidylinositol; PS, phosphatidylserine; Oxidized PS, oxidized phosphatidylserine.
To identify scramblases associated with apoptosis, Nagata and colleagues used expression cloning to identify scramblases strictly dependent on caspase activity (i.e., inhibited by zFAD-fmk but not dependent on calcium). Based on these screens, a novel scramblase called Xkr8 was identified. Analogous to TMEM16F, over-expression of Xkr8 significantly increased PS exposure, but in stark contrast to TMEM16F, Xkr8 cells that express PS were recognized as an eat-me signal and engulfed. At the molecular level, Xkr8 is cleaved at a DEVD site near its C-termini by caspase 3 and caspase 7 during apoptosis, to activate a PS scramblase activity (76). Xkr8 is a mammalian homolog of the CED8 in Caenorhabditis elegans (79) and has an evolutionarily conserved function and is cleaved by CED-3, the homolog of caspase 3, during developmental apoptosis.
Adding complexity to the issue of PS externalization during apoptosis, new studies indicate that a net accumulation of externalized PS is also achieved by a dynamic and systematic interplay between PS scramblases (such as Xkr8) and specific flippases, such as ATP11C (a member of the P4-type ATPase family that redirects PS from the outer membrane back to the inner membrane) (80). Analogous to Xkr8, ATP11C also contains a caspase cleavage site, but when ATP11C is cleaved by active caspases, the Flippase activity is inactivated preventing the return of PS to the inner membrane. Interestingly, when cells express ATP11C with a mutated caspase recognition site, cellular flippase activity remains high, and cells expressing mutant ATP11C do not sustain PS externalization or retain their ability to be engulfed. This presents a highly intricate scenario, whereby caspases can activate Xkr8 and inactivate ATP11C, to increase the steady-state density of externalized PS (Figure 1). In contrast, in the non-apoptotic context, high concentration of calcium activates TMEM16, but does not inactivate ATP11C, possibly explaining the reversibility of TMEM16-mediated PS externalization.
Using an LC MS/MS labeling approach to derivatize primary amines on externalized amino-phospholipids (PE and PS), recent studies by Clark et al. found that different molecular species of amino-phospholipids (according to their fatty acyl composition, saturation, length, and oxidative status) were simultaneously externalized during platelet activation versus apoptosis, and revealed an optimal PE fatty acyl chain length that supported coagulation (81). Similar types of MS-based characterization have been reported to define the molecular species of oxidized PS (oxPS) driven by cytochrome c/H202 (82). These kinds of analyses might be revealing to accesses changes in the PS lipidome in SLE patients, or which species of PS are targets of anti-PS or anti-phospholipid antibodies in SLE. Moreover, the recent development of PS reporter lines, such as the generation of chimeric reporter cells to study the PS-dependent dimerization and activation of TAM receptors (Tyro3-γR1, Axl-γR1, and Mer-γR1 cells) (83), or the use of SCARF1 chimeric receptors to access the contribution of PS to C1q signaling (42), would be very useful to explore the functional analysis for PS receptors and to screen apoptotic cells from different cells undergoing apoptosis (normal versus SLE patients). By expanding this kind of analysis, it might be possible to identify if (and how) PS signaling fails during different externalization itineraries. Together, these studies indicate that not all PS externalization is phenotypically equivalent, and relevant to the thesis developed in this perspective, whether the Xkr8/TMEM16F/ATP11c circuit is compromised or genetically linked to SLE or other human auto-immune disorders is an important and timely question in the field.
Oxidatively Modified PS may Provide an Assurance Signal for Efferocytosis
The aforementioned discussion between the PS externalization mechanisms of TMEM16F and Xkr8 is instructive, and highlights the fact that PS externalization, per se, is not sufficient for efferocytosis. Efferocytosis therefore must require an additional assurance signal, affirming that the cell has passed a caspase-dependent checkpoint and is ready to be engulfed and processed for degradation (84, 85). Although it is likely that other plasma membrane markers act in concert with externalized PS on apoptotic cell, one idea that has gained traction in recent years is that oxPS, generated in a caspase-dependent manner, provides a death-specific marker for PS receptors, marking cells for engulfment (86). oxPS might be expected to change the distribution of PS in the plasma membrane rendering the cell more palatable, or conversely, PS oxidation could serve as a better substrate for PS receptors (i.e., the “altered self” idea) (2).
Although both ideas appear plausible, in support of the latter, it has long been realized that antibodies specific to oxidized phospholipids can block macrophage efferocytosis (87). Moreover, in macrophages, the recognition of apoptotic cells via the scavenger receptor CD36 occurs almost exclusively through interactions with oxPS, and to a lesser extent oxidized PC (oxPC), but not non-oxPS. Interestingly, the specificity of CD36 to oxPS within the apoptotic membranes appears to be mediated by a structurally conserved recognition motif for CD36 that comprises a “sn-2 acyl group with a terminal γ-hydroxy (or oxo)-α, β-unsaturated carbonyl” whereas, the reduction of this acyl chain prevents the oxPS/CD36 receptor activation (88). Other scavenger receptors implicated in apoptotic cell clearance that includes; SRB1, SRA, LOX-1, CD68, and CD14 (2, 89) also appear to selectively recognize the oxidized sn-2 acyl group, suggesting this may be a conserved and universal epitope in the apoptotic program.
In addition to scavenger receptors, recent studies also show that some of the conventional PS-binding proteins and receptors, such as GAS-6 and BAI-1, preferentially interact with oxPS, although in the same study, it was also shown that non-oxPS preferentially bound CXCL16 and Tim-4 (90), suggesting variations on this theme. Although previous studies showed that the peroxidase function of caspase 3 could directly oxidize PS, PS can be oxidized during inflammation as a result of enhanced lipid peroxidation (88). The fact that various oxPS species may alter the repertoire and/or change the affinities of PS toward scavenger receptors and PS receptors provides an impetus to better understand the molecular basis of PS oxidation.
It is also noteworthy that oxysterols and oxPS can also indirectly impinge on efferocytosis. For example, the engulfment of apoptotic cells brings in large amounts of cellular lipids, including the oxidized lipids alluded to above, into the intracellular compartments of the phagocyte. Elegant studies have shown that these internalized lipids can activate PPAR-δ receptors (91) and the nuclear receptor LXR in macrophages (69), to induce engulfment receptors such as Mer and C1q. In mice, genetic ablation of PPAR-δ results in impaired apoptotic cell clearance and SLE-like disease (92), although the significance to human lupus still remains to be determined.
Lyso-PS, a Unique Form of PS, Binds Distinct Receptors and is Involved in the Clearance of Non-Apoptotic Neutrophils
Finally, in addition to (i) the modes of externalization, (ii) whether PS is covalently oxidized, and (iii) whether a PS receptor is available to bind exposed PS on the surface of the apoptotic cell, under certain circumstances PS can also be hydrolyzed under oxidative conditions by a PS-specific phospholipase (PS-PLA1) (93–95) to generate lyso-PS, a deacylated form of PS that serves as an endogenous anti-inflammatory mediator. Although lyso-PS can stimulate efferocytosis under certain conditions (96), this form of PS remarkably also stimulates the uptake of live cells, and has been implicated in the clearance of activated and aged live neutrophils in anticipation for the resolution of inflammation. Despite that PS and lyso-PS have the same anionic head group, lyso-PS does not bind conventional PS receptors such as TAMs and TIMs, but instead interacts with two G-protein coupled receptors, GPR34 and G2A (97), which are linked to novel anti-inflammatory molecules such as PGE2.
Lessons from Blocking PS in Cancer Models
In recent years, the idea that PS serves as a tolerogenic and global immunosuppressive checkpoint has been therapeutically exploited by the generation of anti-PS antibodies for cancer immunotherapy. These studies show that systemic treatment of Bavituximab (which recognizes a complex of β2-glycoprotein and PS), can activate immune checkpoints, and drive the polarization of macrophages from M2 to M1 and the activation of immature DCs to antigen presenting cells, while decreasing MDSCs and Tregs in tumor-bearing mice (98). As such, this pre-clinical finding has an unanticipated consequence to ask whether blocking PS is sufficient to induce autoimmunity. While the answers are not completely clear, the available pre-clinical and clinical biosafety studies using acute rather than chronic dosing regiments of Bavituximab (anti-PS antibodies), suggest that anti-PS antibodies are well tolerated and do not produce systemic autoimmunity or pulmonary thrombosis (99). Furthermore, vaccinating mice with apoptotic RMA lymphoma cells pre-treated with Annexin-V attenuated the ability of mice to reject a challenge with live RMA lymphoma cells (100). Whether systemic anti-PS treatment exacerbates auto-immune responses in lupus-prone individuals, or in individuals with anti-phospholipid antibody (syndrome), has not been investigated. It will be of interest to identify if patients that develop anti-PS antibodies in SLE might have naturally occurring decreased metastatic burden. Together, these data suggest that blockage of PS, per se, may not be causal for the development of lupus, but nonetheless re-activates specific arms of the immune response, which may be fortuitously exploited where immunosuppressive mechanisms operate within the tumor microenvironment. Future studies, in mice, should be aimed to test whether anti-PS antibodies augment lupus-like autoimmunity in genetic strains with a propensity toward disease progression, and conversely whether PS liposomes might also have unexpected therapeutic value. Finally, several enveloped viruses such as Dengue, HIV, and Ebola virus employ apoptotic (PS) mimicry to gain entry to host cells, and blocking PS may also offer therapeutic prospects to block viral entry and immune suppression (101–104).
Concluding Remarks
While the link between defective efferocytosis and auto-immune disease and advanced atherosclerosis has been made, and validated in experimental animal models, where and when this circuitry fails in human disease has not been firmly established by genetic causation studies. In recent years, new developments have emerged concerning the mechanisms of PS externalization, and the once seemingly simple paradigm that externalized PS provides a signal for efferocytosis and actively drives a resolution in acute inflammation has been refined by the fact that externalized PS can exist in different functional states. A challenging problem in the field will be to decode the different biological fates of externalized PS, and whether its ability to actively transmit signals is compromised in human autoimmunity. Once the specific conditions can be identified, how exactly PS negatively impinges on chronic inflammation can be elucidated further. These data would be helpful to understand what components of the PS pathways fail during chronic inflammation and autoimmunity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Maderna P, Godson C. Phagocytosis of apoptotic cells and the resolution of inflammation. Biochim Biophys Acta (2003) 1639:141–51. doi: 10.1016/j.bbadis.2003.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol (2002) 2:965–75. doi:10.1038/nri957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature (2000) 407:784–8. doi:10.1038/35037722
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature (1997) 390:350–1. doi:10.1038/37022
5. Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol (2010) 6:280–9. doi:10.1038/nrrheum.2010.46
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum (2002) 46:191–201. doi:10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Shao WH, Cohen PL. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther (2011) 13:202. doi:10.1186/ar3206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol (1992) 148:2207–16.
9. Martin SJ, Reutelingsperger CPM, Mcgahon AJ, Rader JA, Vanschie RCAA, Laface DM, et al. Early redistribution of plasma-membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus – inhibition by overexpression of Bcl-2 and Abl. J Exp Med (1995) 182:1545–56. doi:10.1084/jem.182.5.1545
10. Wu Y, Tibrewal N, Birge RB. Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol (2006) 16:189–97. doi:10.1016/j.tcb.2006.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Zirngibl M, Furnrohr BG, Janko C, Munoz LE, Voll RE, Gregory CD, et al. Loading of nuclear autoantigens prototypically recognized by SLE sera into late apoptotic vesicles requires intact microtubules and MLCK activity. Clin Exp Immunol (2014). doi:10.1111/cei.12342
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Liang YY, Arnold T, Michlmayr A, Rainprecht D, Perticevic B, Spittler A, et al. Serum-dependent processing of late apoptotic cells for enhanced efferocytosis. Cell Death Dis (2014) 5:e1264. doi:10.1038/cddis.2014.210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Schmid M, Hunold K, Weber-Steffens D, Mannel DN. Ficolin-B marks apoptotic and necrotic cells. Immunobiology (2012) 217:610–5. doi:10.1016/j.imbio.2011.10.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Bijl M, Horst G, Bijzet J, Bootsma H, Limburg PC, Kallenberg CG. Serum amyloid P component binds to late apoptotic cells and mediates their uptake by monocyte-derived macrophages. Arthritis Rheum (2003) 48:248–54. doi:10.1002/art.10737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Janko C, Franz S, Munoz LE, Siebig S, Winkler S, Schett G, et al. CRP/anti-CRP antibodies assembly on the surfaces of cell remnants switches their phagocytic clearance toward inflammation. Front Immunol (2011) 2:70. doi:10.3389/fimmu.2011.00070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. van Rossum AP, Fazzini F, Limburg PC, Manfredi AA, Rovere-Querini P, Mantovani A, et al. The prototypic tissue pentraxin PTX3, in contrast to the short pentraxin serum amyloid P, inhibits phagocytosis of late apoptotic neutrophils by macrophages. Arthritis Rheum (2004) 50:2667–74. doi:10.1002/art.20370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Franz S, Herrmann K, Furnrohr BG, Sheriff A, Frey B, Gaipl US, et al. After shrinkage apoptotic cells expose internal membrane-derived epitopes on their plasma membranes. Cell Death Differ (2007) 14:733–42. doi:10.1038/sj.cdd.4402066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Hart SP, Smith JR, Dransfield I. Phagocytosis of opsonized apoptotic cells: roles for ‘old-fashioned’ receptors for antibody and complement. Clin Exp Immunol (2004) 135:181–5. doi:10.1111/j.1365-2249.2003.02330.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RAB, Henson PM. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature (2000) 405:85–90. doi:10.1038/35011084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest (2002) 109:41–50. doi:10.1172/JCI200211638
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Angelillo-Scherrer A, de Frutos P, Aparicio C, Melis E, Savi P, Lupu F, et al. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nat Med (2001) 7:215–21. doi:10.1038/84667
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Wu CS, Hu CY, Chan CJ, Chang SK, Hsu PN. Genetic polymorphism of the growth arrest-specific 6 gene is associated with cutaneous vasculitis in Taiwanese patients with systemic lupus erythematosus. Clin Rheumatol (2012) 31:1443–8. doi:10.1007/s10067-012-2027-z
23. Burstyn-Cohen T, Heeb MJ, Lemke G. Lack of protein S in mice causes embryonic lethal coagulopathy and vascular dysgenesis. J Clin Invest (2009) 119:2942–53. doi:10.1172/JCI39325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Bertolaccini ML, Sanna G, Ralhan S, Gennari LC, Merrill JT, Khamashta MA, et al. Antibodies directed to protein S in patients with systemic lupus erythematosus: prevalence and clinical significance. Thromb Haemost (2003) 90:636–41. doi:10.1160/TH03-03-0151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Song KS, Park YS, Kim HK. Prevalence of anti-protein S antibodies in patients with systemic lupus erythematosus. Arthritis Rheum (2000) 43:557–60. doi:10.1002/1529-0131(200003)43:3<557::AID-ANR11>3.0.CO;2-O
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (2004) 304:1147–50. doi:10.1126/science.1094359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Hu CY, Wu CS, Tsai HF, Chang SK, Tsai WI, Hsu PN. Genetic polymorphism in milk fat globule-EGF factor 8 (MFG-E8) is associated with systemic lupus erythematosus in human. Lupus (2009) 18:676–81. doi:10.1177/0961203309103027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Yamaguchi H, Fujimoto T, Nakamura S, Ohmura K, Mimori T, Matsuda F, et al. Aberrant splicing of the milk fat globule-EGF factor 8 (MFG-E8) gene in human systemic lupus erythematosus. Eur J Immunol (2010) 40:1778–85. doi:10.1002/eji.200940096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet (1998) 19:56–9. doi:10.1038/ng0598-56
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Walport MJ, Davies KA, Botto M. C1q and systemic lupus erythematosus. Immunobiology (1998) 199:265–85. doi:10.1016/S0171-2985(98)80032-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Stuart LM, Takahashi K, Shi L, Savill J, Ezekowitz RAB. Mannose-binding lectin-deficient mice display defective apoptotic cell clearance but no autoimmune phenotype. J Immunol (2005) 174:3220–6. doi:10.4049/jimmunol.174.6.3220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Davies EJ, Snowden N, Hillarby MC, Carthy D, Grennan DM, Thomson W, et al. Mannose-binding protein gene polymorphism in systemic lupus erythematosus. Arthritis Rheum (1995) 38:110–4. doi:10.1002/art.1780380117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Ip WK, Chan SY, Lau CS, Lau YL. Association of systemic lupus erythematosus with promoter polymorphisms of the mannose-binding lectin gene. Arthritis Rheum (1998) 41:1663–8.
34. Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause KH, et al. Calreticulin is essential for cardiac development. J Cell Biol (1999) 144:857–68. doi:10.1083/jcb.144.5.857
35. Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science (2001) 293:306–11. doi:10.1126/science.1061663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, et al. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med (2002) 196:135–40. doi:10.1084/jem.20012094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Ma GZ, Stankovich J, Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene), Kilpatrick TJ, Binder MD, Field J. Polymorphisms in the receptor tyrosine kinase MERTK gene are associated with multiple sclerosis susceptibility. PLoS One (2011) 6:e16964. doi:10.1371/journal.pone.0016964
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol (2009) 86:1089–95. doi:10.1189/jlb.0209115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Zizzo G, Guerrieri J, Dittman LM, Merri JT, Cohen PL. Circulating levels of soluble MER in lupus reflect M2c activation of monocytes/macrophages, autoantibody specificities and disease activity. Arthritis Res Ther (2013) 15:R212. doi:10.1186/ar4407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature (2007) 450:435–9. doi:10.1038/nature06307
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Tian L, Choi SC, Murakami Y, Allen J, Morse HC III, Qi CF, et al. p85alpha recruitment by the CD300f phosphatidylserine receptor mediates apoptotic cell clearance required for autoimmunity suppression. Nat Commun (2014) 5:3146. doi:10.1038/ncomms4146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Ramirez-Ortiz ZG, Pendergraft WF III, Prasad A, Byrne MH, Iram T, Blanchette CJ, et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol (2013) 14:917–26. doi:10.1038/ni.2670
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Schledzewski K, Geraud C, Arnold B, Wang S, Grone HJ, Kempf T, et al. Deficiency of liver sinusoidal scavenger receptors stabilin-1 and -2 in mice causes glomerulofibrotic nephropathy via impaired hepatic clearance of noxious blood factors. J Clin Invest (2011) 121:703–14. doi:10.1172/JCI44740
44. He M, Kubo H, Morimoto K, Fujino N, Suzuki T, Takahasi T, et al. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep (2011) 12:358–64. doi:10.1038/embor.2011.28
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Martens HA, Nienhuis HL, Gross S, van der Steege G, Brouwer E, Berden JH, et al. Receptor for advanced glycation end products (RAGE) polymorphisms are associated with systemic lupus erythematosus and disease severity in lupus nephritis. Lupus (2012) 21:959–68. doi:10.1177/0961203312444495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Herz J, Clouthier DE, Hammer RE. Ldl receptor-related protein internalizes and degrades Upa-Pai-1 complexes and is essential for embryo implantation. Cell (1992) 71:411–21. doi:10.1016/0092-8674(92)90511-A
47. Gorovoy M, Gaultier A, Campana WM, Firestein GS, Gonias SL. Inflammatory mediators promote production of shed LRP1/CD91, which regulates cell signaling and cytokine expression by macrophages. J Leukoc Biol (2010) 88:769–78. doi:10.1189/jlb.0410220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Bratton DL, Henson PM. Apoptotic cell recognition: will the real phosphatidylserine receptor(s) please stand up? Curr Biol (2008) 18:R76–9. doi:10.1016/j.cub.2007.11.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol (2013) 5:a008748. doi:10.1101/cshperspect.a008748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Poon IKH, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol (2014) 14:166–80. doi:10.1038/nri3607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Xiao S, Brooks CR, Zhu C, Wu C, Sweere JM, Petecka S, et al. Defect in regulatory B-cell function and development of systemic autoimmunity in T-cell Ig mucin 1 (Tim-1) mucin domain-mutant mice. Proc Natl Acad Sci U S A (2012) 109:12105–10. doi:10.1073/pnas.1120914109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Miyanishi M, Segawa K, Nagata S. Synergistic effect of Tim4 and MFG-E8 null mutations on the development of autoimmunity. Int Immunol (2012) 24:551–9. doi:10.1093/intimm/dxs064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Kinchen JM, Ravichandran KS. Phagocytic signaling: you can touch, but you can’t eat. Curr Biol (2008) 18:R521–4. doi:10.1016/j.cub.2008.04.058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Mol Cell (2004) 14:277–87. doi:10.1016/S1097-2765(04)00237-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Finnemann SC, Nandrot EF. MerTK activation during RPE phagocytosis in vivo requires alphaVbeta5 integrin. Adv Exp Med Biol (2006) 572:499–503. doi:10.1007/0-387-32442-9_69
56. Wu Y, Singh S, Georgescu MM, Birge RB. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci (2005) 118:539–53. doi:10.1242/jcs.01632
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Wu J, Ekman C, Jonsen A, Sturfelt G, Bengtsson AA, Gottsater A, et al. Increased plasma levels of the soluble Mer tyrosine kinase receptor in systemic lupus erythematosus relate to disease activity and nephritis. Arthritis Res Ther (2011) 13:R62. doi:10.1186/ar3316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Zhu H, Sun X, Zhu L, Hu F, Shi L, Li Z, et al. The expression and clinical significance of different forms of mer receptor tyrosine kinase in systemic lupus erythematosus. J Immunol Res (2014). doi:10.1155/2014/431896
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Hilliard BA, Zizzo G, Ulas M, Linan MK, Schreiter J, Cohen PL. Increased expression of Mer tyrosine kinase in circulating dendritic cells and monocytes of lupus patients: correlations with plasma interferon activity and steroid therapy. Arthritis Res Ther (2014) 16:R76. doi:10.1186/ar4517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Majai G, Kiss E, Tarr T, Zahuczky G, Hartman Z, Szegedi G, et al. Decreased apopto-phagocytic gene expression in the macrophages of systemic lupus erythematosus patients. Lupus (2014) 23:133–45. doi:10.1177/0961203313511557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Suh CH, Hilliard B, Li S, Merrill JT, Cohen PL. TAM receptor ligands in lupus: protein S but not gas6 levels reflect disease activity in systemic lupus erythematosus. Arthritis Res Ther (2010) 12:R146. doi:10.1186/ar3088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Paidassi H, Tacnet-Delorme P, Verneret M, Gaboriaud C, Houen G, Duus K, et al. Investigations on the C1q-calreticulin-phosphatidylserine interactions yield new insights into apoptotic cell recognition. J Mol Biol (2011) 408:277–90. doi:10.1016/j.jmb.2011.02.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Martin M, Leffler J, Blom AM. Annexin A2 and A5 serve as new ligands for C1q on apoptotic cells. J Biol Chem (2012) 287:33733–44. doi:10.1074/jbc.M112.341339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Mikołajczyk T, Skiba D, Batko B, Krezelok M, Wilk G, Osmenda G, et al. Characterization of the impairment of the uptake of apoptotic polymorphonuclear cells by monocyte subpopulations in systemic lupus erythematosus. Lupus (2014):1–12. doi:10.1177/0961203314541316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Tas SW, Quartier P, Botto M, Fossati-Jimack L. Macrophages from patients with SLE and rheumatoid arthritis have defective adhesion in vitro, while only SLE macrophages have impaired uptake of apoptotic cells. Ann Rheum Dis (2006) 65:216–21. doi:10.1136/ard.2005.037143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Donnelly S, Roake W, Brown S, Young P, Naik H, Wordsworth P, et al. Impaired recognition of apoptotic neutrophils by the C1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum (2006) 54:1543–56. doi:10.1002/art.21783
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Lauber K, Keppeler H, Munoz LE, Koppe U, Schroder K, Yamaguchi H, et al. Milk fat globule-EGF factor 8 mediates the enhancement of apoptotic cell clearance by glucocorticoids. Cell Death Differ (2013) 20:1230–40. doi:10.1038/cdd.2013.82
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Zagorska A, Traves PG, Lew ED, Dransfield I, Lemke G. Diversification of TAM receptor tyrosine kinase function. Nat Immunol (2014) 15:920–8. doi:10.1038/ni.2986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity (2009) 31:245–58. doi:10.1016/j.immuni.2009.06.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Zahuczky G, Kristof E, Majai G, Fesus L. Differentiation and glucocorticoid regulated apopto-phagocytic gene expression patterns in human macrophages. Role of Mertk in enhanced phagocytosis. PLoS One (2011) 6:e21349. doi:10.1371/journal.pone.0021349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys (2010) 39:407–27. doi:10.1146/annurev.biophys.093008.131234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Daleke DL. Regulation of transbilayer plasma membrane phospholipid asymmetry. J Lipid Res (2003) 44:233–42. doi:10.1194/jlr.R200019-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Marino G, Kroemer G. Mechanisms of apoptotic phosphatidylserine exposure. Cell Res (2013) 23:1247–8. doi:10.1038/cr.2013.115
74. Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature (2010) 468:834–8. doi:10.1038/nature09583
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Yang H, Kim A, David T, Palmer D, Jin T, Tien J, et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell (2012) 151:111–22. doi:10.1016/j.cell.2012.07.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science (2013) 341:403–6. doi:10.1126/science.1236758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, Nagata S. Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J Biol Chem (2013) 288:13305–16. doi:10.1074/jbc.M113.457937
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Segawa K, Suzuki J, Nagata S. Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci U S A (2011) 108:19246–51. doi:10.1073/pnas.1114799108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Stanfield GM, Horvitz HR. The ced-8 gene controls the timing of programmed cell deaths in C. elegans. Mol Cell (2000) 5:423–33. doi:10.1016/S1097-2765(00)80437-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science (2014) 344:1164–8. doi:10.1126/science.1252809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Clark SR, Thomas CP, Hammond VJ, Aldrovandi M, Wilkinson GW, Hart KW, et al. Characterization of platelet aminophospholipid externalization reveals fatty acids as molecular determinants that regulate coagulation. Proc Natl Acad Sci U S A (2013) 110:5875–80. doi:10.1073/pnas.1222419110
82. Tyurin VA, Yanamala N, Tyurina YY, Klein-Seetharaman J, Macphee CH, Kagan VE. Specificity of lipoprotein-associated phospholipase A(2) toward oxidized phosphatidylserines: liquid chromatography-electrospray ionization mass spectrometry characterization of products and computer modeling of interactions. Biochemistry (2012) 51:9736–50. doi:10.1021/bi301024e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Tsou WI, Nguyen KQ, Calarese DA, Garforth SJ, Antes AL, Smirnov SV, et al. Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J Biol Chem (2014) 289:25750–63. doi:10.1074/jbc.M114.569020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Fadok VA, Bratton DL, Henson PM. Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J Clin Invest (2001) 108:957–62. doi:10.1172/JCI200114122
85. Kagan VE, Borisenko GG, Tyurina YY, Tyurin VA, Jiang J, Potapovich AI, et al. Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic Biol Med (2004) 37:1963–85. doi:10.1016/j.freeradbiomed.2004.08.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Kagan VE, Gleiss B, Tyurina YY, Tyurin VA, Elenström-Magnusson C, Liu S-X, et al. A role for oxidative stress in apoptosis: oxidation and externalization of phosphatidylserine is required for macrophage clearance of cells undergoing Fas-mediated apoptosis. J Immunol (2002) 169:487–99. doi:10.4049/jimmunol.169.1.487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Chang MK, Bergmark C, Laurila A, Horkko S, Han KH, Friedman P, et al. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: evidence that oxidation-specific epitopes mediate macrophage recognition. Proc Natl Acad Sci U S A (1999) 96:6353–8. doi:10.1073/pnas.96.11.6353
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine–CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med (2006) 203:2613–25. doi:10.1084/jem.20060370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Erwig LP, Henson PM. Clearance of apoptotic cells by phagocytes. Cell Death Differ (2008) 15:243–50. doi:10.1038/sj.cdd.4402184
90. Tyurin VA, Balasubramanian K, Winnica D, Tyurina YY, Vikulina AS, He RR, et al. Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic ‘eat-me’ signals: cleavage and inhibition of phagocytosis by Lp-PLA2. Cell Death Differ (2014) 21:825–35. doi:10.1038/cdd.2014.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med (2009) 15:1266–72. doi:10.1038/nm.2048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Roszer T, Menendez-Gutierrez MP, Lefterova MI, Alameda D, Nunez V, Lazar MA, et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol (2011) 186:621–31. doi:10.4049/jimmunol.1002230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Frasch SC, Bratton DL. Emerging roles for lysophosphatidylserine in resolution of inflammation. Prog Lipid Res (2012) 51:199–207. doi:10.1016/j.plipres.2012.03.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Frasch SC, Fernandez-Boyanapalli RF, Berry KAZ, Murphy RC, Leslie CC, Nick JA, et al. Neutrophils regulate tissue neutrophilia in inflammation via the oxidant-modified lipid lysophosphatidylserine. J Biol Chem (2013) 288:4583–93. doi:10.1074/jbc.M112.438507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Kitamura H, Makide K, Shuto A, Ikubo M, Inoue A, Suzuki K, et al. GPR34 is a receptor for lysophosphatidylserine with a fatty acid at the sn-2 position. J Biochem (2012) 151:511–8. doi:10.1093/jb/mvs011
96. Makide K, Aoki J. GPR34 as a lysophosphatidylserine receptor. J Biochem (2013) 153:327–9. doi:10.1093/jb/mvt010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Frasch SC, Berry KZ, Fernandez-Boyanapalli R, Jin H-S, Leslie C, Henson PM, et al. NADPH Oxidase-dependent generation of lysophosphatidylserine enhances clearance of activated and dying neutrophils via G2A. J Biol Chem (2008) 283:33736–49. doi:10.1074/jbc.M807047200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Yin Y, Huang X, Lynn KD, Thorpe PE. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res (2013) 1:256–68. doi:10.1158/2326-6066.CIR-13-0073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Gerber DE, Stopeck AT, Wong L, Rosen LS, Thorpe PE, Shan JS, et al. Phase I safety and pharmacokinetic study of bavituximab, a chimeric phosphatidylserine-targeting monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res (2011) 17:6888–96. doi:10.1158/1078-0432.CCR-11-1074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Bondanza A, Zimmermann VS, Rovere-Querini P, Turnay J, Dumitriu IE, Stach CM, et al. Inhibition of phosphatidylserine recognition heightens the immunogenicity of irradiated lymphoma cells in vivo. J Exp Med (2004) 200:1157–65. doi:10.1084/jem.20040327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Callahan MK, Popernack PM, Tsutsui S, Truong L, Schlegel RA, Henderson AJ. Phosphatidylserine on HIV envelope is a cofactor for infection of monocytic cells. J Immunol (2003) 170:4840–5. doi:10.4049/jimmunol.170.9.4840
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Meertens L, Carnec X, Lecoin MP, Ramdasi R, Guivel-Benhassine F, Lew E, et al. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe (2012) 12:544–57. doi:10.1016/j.chom.2012.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Moller-Tank S, Kondratowicz AS, Davey RA, Rennert PD, Maury W. Role of the phosphatidylserine receptor TIM-1 in enveloped-virus entry. J Virol (2013) 87:8327–41. doi:10.1128/JVI.01025-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Morizono K, Chen IS. Role of phosphatidylserine receptors in enveloped virus infection. J Virol (2014) 88:4275–90. doi:10.1128/JVI.03287-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: phosphatidylserine, apoptotic cells, scramblases, apoptotic versus non-apoptotic PS externalization, autoimmunity
Citation: Kimani SG, Geng K, Kasikara C, Kumar S, Sriram G, Wu Y and Birge RB (2014) Contribution of defective PS recognition and efferocytosis to chronic inflammation and autoimmunity. Front. Immunol. 5:566. doi: 10.3389/fimmu.2014.00566
Received: 30 August 2014; Paper pending published: 16 September 2014;
Accepted: 23 October 2014; Published online: 10 November 2014.
Edited by:
Martin Herrmann, Universitätsklinikum Erlangen, GermanyReviewed by:
Ian Dransfield, University of Edinburgh, UKRostyslav Bilyy, Institute of Cell Biology, Ukraine
Copyright: © 2014 Kimani, Geng, Kasikara, Kumar, Sriram, Wu and Birge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raymond B. Birge, Department of Biochemistry and Molecular Biology, Rutgers School of Biomedical and Health Sciences – Cancer Center, 205 South Orange Avenue, Newark, NJ 07103, USA e-mail:YmlyZ2VyYUBuam1zLnJ1dGdlcnMuZWR1