 Michael Peled*
Michael Peled* Edward A. Fisher*
Edward A. Fisher*- The Marc and Ruti Bell Program in Vascular Biology, Department of Medicine, Leon H. Charney Division of Cardiology, New York University School of Medicine, New York, NY, USA
It is well recognized that macrophages in many contexts in vitro and in vivo display a spectrum of inflammatory features and functional properties. A convenient system to group together different subsets of macrophages has been the M1 (inflammatory)/M2 (anti-inflammatory) classification. In addition to other sites of inflammation, it is now established that atherosclerotic plaques contain both M1 and M2 macrophages. We review results made possible by a number of recent mouse models of atherosclerotic regression that, taken with other literature, have shown the M1/M2 balance in plaques to be dynamic, with M1 predominating in disease progression and M2 in regression. The regulation of the macrophage phenotype in plaques and the functional consequences of the M1 and M2 states in atherosclerosis will also be discussed.
Introduction
Atherosclerotic cardiovascular diseases, which include myocardial infraction and stroke, are the most common causes of morbidity and mortality in western society and will soon be the same world-wide. Atherosclerosis represents a failure to resolve the inflammatory response in the arterial wall initiated by the retention of apolipoprotein B (apoB)-containing lipoproteins (1). These lipoproteins are taken up by tissue macrophages, which ultimately become engorged with cholesterol (foam cells) and activated. The continuing stimulus of the entry and retention of apoB-lipoproteins fuels not only the accumulation of foam cells to form a plaque, but also the chronicity and amplification of the inflammatory response, which contribute to the vulnerability of some plaques to rupture and cause acute tissue ischemia. The central role of macrophages in atherosclerosis pathophysiology has, therefore, focused attention on their properties in plaque initiation and progression, and more recently, in regression (2–5).
The study of macrophages over the past decade is characterized by a remarkable expansion of knowledge concerning their origin, functional properties, and potential to both protect from and contribute to disease [e.g., see in Ref. (6, 7)]. Admitting the complexity of macrophage biology, for the purposes of this review, we have restricted ourselves to considering how aspects of macrophage polarization in the M1/M2 classification system (8) relate to atherosclerosis progression and regression. In this system, which is influenced by the Th1 and Th2 classification of lymphocytes, macrophages can be grossly divided into pro-inflammatory, M1 cells and anti-inflammatory, M2 cells based mainly on in vitro criteria (9). It is important to note, however, that while the classification of lymphocytes into Th1 and Th2 preceded the classification scheme of macrophages as M1/M2, the Th1 and Th2-like responses result from polarization of macrophages to M1 and M2 states, respectively. Furthermore, M1/M2 polarization is not dependent on T cells, as has been demonstrated in Rag KO and other immune deficient mice (8).
Polarization toward the M1 state is induced by several stimuli in vitro, including Toll-like receptor (TLR) ligands (such as lipopolysaccharide, LPS) and interferon γ (potential endogenous stimuli in atherosclerosis will be discussed below). M1 macrophages express several pro-inflammatory mediators, such as inducible nitric oxide synthase, tumor necrosis factor-a (TNF-a), interleukin-1b (IL-1b), IL-6, IL-12, and proteolytic enzymes. M1 macrophages have been found in both human and mouse atherosclerosis, and their secretion of the pro-inflammatory mediators is thought to maintain local inflammation and the degradation of extracellular matrix components, resulting in disease progression [e.g., Ref. (10)], and ultimately, unstable plaques in humans. As alluded to earlier, unstable plaques are at increased risk of rupture and causing thrombosis, resulting in myocardial infarction and stroke (11–14).
On the other side of the polarization spectrum, M2 macrophages are induced in vitro by Th2-type cytokines, such as IL-4 and IL-13. T regulatory cells (Tregs) have also been implicated in the induction of M2 polarization (15), possibly through IL-10. M2 polarized macrophages have been characterized by their expression of CD163, mannose receptor 1 (also known as CD206), FIZZ1, and high levels of arginase 1. In addition, they secrete anti-inflammatory cytokines, such as transforming growth factorβ, IL-1 receptor antagonist, IL-10, and have increased secretion of collagen. Based particularly on the role of M2 macrophages in wound healing, the combination of the factors they express are thought to be particularly suited for tissue repair, which is consistent with their increased presence in regressing plaques in mouse models of atherosclerosis (16, 17).
In addition to the M1 and M2 macrophages, oxidized phospholipids present in oxidized LDL induce a macrophage phenotype that is distinct from M1 or M2 phenotypes and that has been termed Mox; these macrophages are characterized by the increases in the expression of nuclear factor erythroid 2-related factor 2 (NRF2)-dependent genes and in reactive oxygen species, and are found in the progressing plaques (18). Their role in atherosclerosis regression, however, has not been established.
With this background, we will now turn to a more complete consideration of the inflammatory states of macrophages in atherosclerosis progression and regression.
Atherosclerosis Progression
Drivers of Macrophage Inflammation in Progressing Plaques
As mentioned above, M1 macrophages are thought to have significant roles in progressing and vulnerable plaques (19). As noted above, a potent inducer of the M1 state in vitro is LPS, which is part of the outer membrane of Gram-negative bacteria, and which binds and activates TLR4 (20). Dozens of studies have demonstrated associations among high-fat diet, the metabolic syndrome, and endotoxemia (i.e., increased levels of LPS in the plasma) (21–23). It appears that a high-fat diet, the consumption of which leads to metabolic syndrome in mouse models, also induces a change in the gut bacterial flora, which, in turn, causes an increase in the LPS plasma levels. Circumstantial considerations that support a link between the metabolic syndrome and LPS signaling include the increased cardiovascular risk in patients with the metabolic syndrome and the positive association of TLR4 activity with atherosclerosis progression in mice and human beings (24–28). Despite the evidence for association between LPS, atherosclerosis progression, and M1 polarization, a study in germ-free apoE-deficient mice on low-fat chow diet showed increased atherosclerosis progression (29), suggesting that some bacteria in the gut flora have an anti-inflammatory effect (30).
Cholesterol and macrophage activation
The most accepted and robust risk factor for atherosclerosis is low-density lipoprotein-cholesterol (LDL-C). Thus, several studies have tried to understand how cholesterol can induce inflammation in general, and specifically to an activated state. The different mechanisms by which cholesterol can drive macrophage activation could be divided into those direct – how cholesterol affects macrophages, and indirect – how cholesterol affects other cell types through which activation could be induced, for example by the secretion of pro-inflammatory cytokines from T cells (Figure 1).
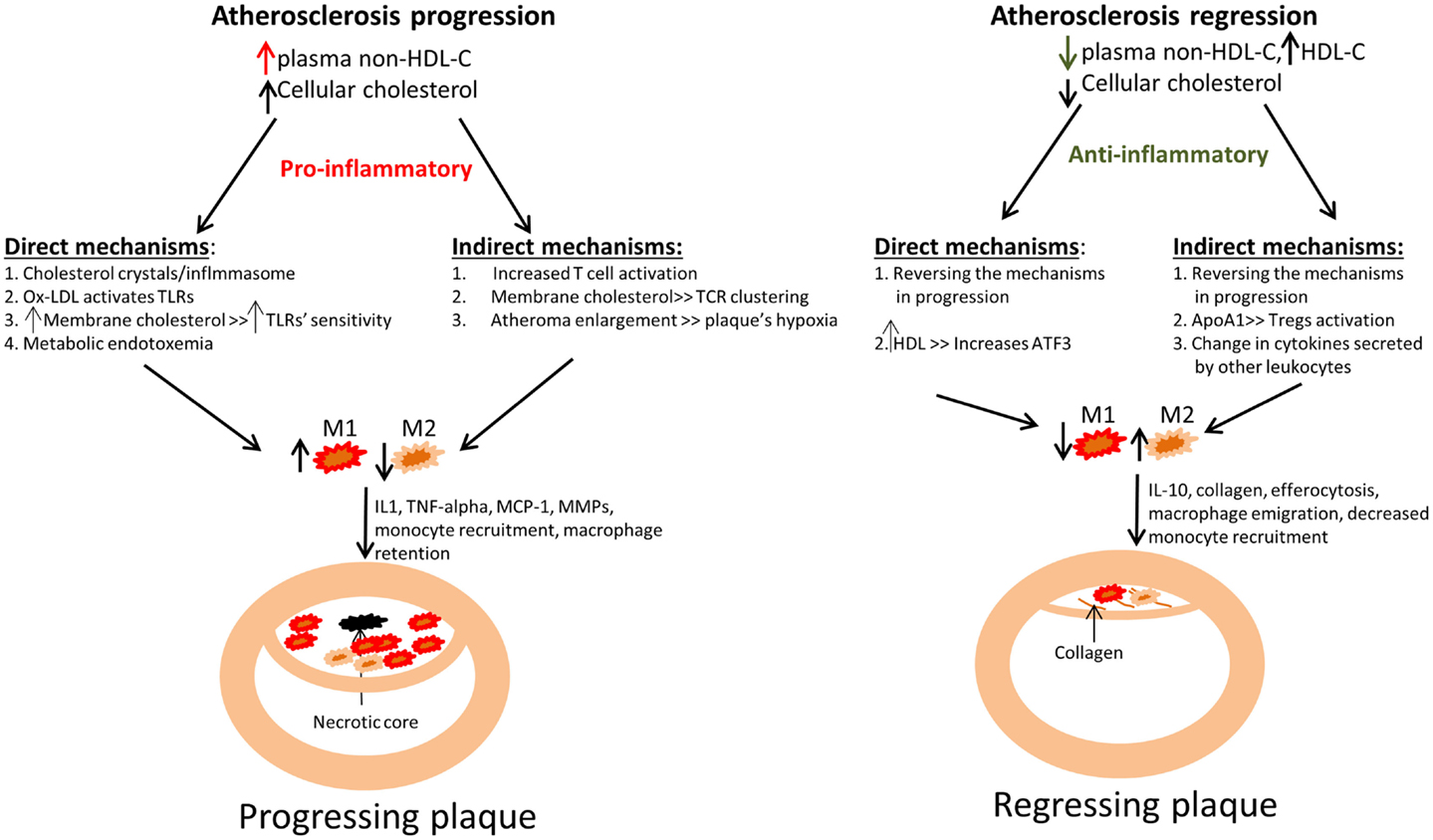
Figure 1. Summary of how changes in plasma cholesterol level and in cellular cholesterol content affect macrophage polarization and kinetics in atherosclerotic plaque progression and regression. Left panel: An increase in non-HDL cholesterol (mainly VLDL cholesterol and LDL cholesterol) in mouse models has been linked to an increase in monocyte recruitment into atherosclerotic plaques, with their subsequent polarization to M1 macrophages, which are retained. This ultimately leads to atherosclerotic plaque progression, as evident by plaque enlargement. The failure to clear dead macrophages by efferocytosis results in the appearance of the necrotic core. Right panel: An opposite effect has been demonstrated in atherosclerosis regression models, where a reduction in non-HDL-C or a selective increase in HDL-C (representing an increase in functional HDL particles) induces a decrease in plaque size and macrophage content (from decreased monocyte recruitment and macrophage retention), as well as enrichment in the expression of markers of the M2 state. Improved efferocytosis is also expected under these conditions, with shrinkage of the necrotic core. There is an increase in collagen content, likely from decreased MMP production by the macrophages. It is also likely that in a regression environment there are decreases in the secretion of inflammatory cytokines and chemokines by the macrophages as a result of the polarization of macrophages toward a M2-like state. The different mechanisms by which cholesterol can drive macrophage activation and polarization are divided into those direct – how cholesterol affects macrophages, and indirect – how cholesterol affects other cell types, for example by the secretion of pro-inflammatory cytokines from T cells.
Direct mechanisms that link cholesterol to macrophage inflammation. Accumulation of cholesterol leads to the formation of crystals that are both intra- and extracellular. The presence of cholesterol crystals in early lesions in Apoe-/- mice was recently demonstrated (31, 32); in addition, it was shown that both macrophage engulfment of cholesterol crystals and de novo formation of intracellular cholesterol crystals activate the NLRP3 (NOD-, LRR- and pyrin domain-containing 3) inflammasome (33). Activation of NLRP3, in turn, results in the secretion of the pro-inflammatory cytokine IL-1β.This pathway appears to be necessary for atherosclerosis progression, as LDL receptor (Ldlr)-/- mice, transplanted with bone marrow cells deficient in IL-1β or in components of the NLRP3 inflammasome, had reduced plaque progression (31, 34).
A second direct mechanism that can explain macrophage activation by cholesterol is mediated through oxidized LDL. Oxidized LDL is present in both human and mouse atheromas. LDL oxidation is thought to be mediated by enzymes (such as 12/15-lipoxygenase and myeloperoxidase) and by free radicals that are abundant in the atherosclerotic plaque (35). Several studies have demonstrated that oxidized LDL can act as a ligand for both the scavenger receptors and TLRs on macrophages. Depending on the extent of oxidation, species of oxidized LDL were found to be agonists of CD14–TLR4–MD2 or CD36–TLR4–TLR6 complexes in vitro; these complexes can induce a pro-inflammatory signaling cascade involving IL-1 receptor-associated kinase 4 (IRAK4) (36, 37), myeloid differentiation primary-response protein 88 (MYD88) (28, 38) and other signaling proteins, culminating in activation of Nf-kB targets. Whole-body knock out mice of TLR2, TLR4 and some of the signaling proteins mentioned here were demonstrated to have reduced atherosclerosis progression (28, 39).
A third direct mechanism points toward the increase in plasma membrane cholesterol. The change in the microenvironment of TLRs (40, 41) renders them more sensitive to ligands, thereby heightening the inflammatory responses.
Despite the predominance of data that show a direct link between cholesterol and macrophage inflammation, there are a number of studies that show the opposite – an anti-inflammatory phenotype induced by intracellular cholesterol. The basis for this is rooted in the LXRs and PPARs, which are important nuclear receptors. The ligands for these receptors include lipids, and in addition to regulating many steps of their metabolism, LXRs and PPARs can also suppress inflammatory signaling in macrophages (42). For example, a recent study demonstrated that cholesterol-loading induced LXR-dependent downregulation of inflammatory gene expression in macrophages as a consequence of the accumulation of the cholesterol precursor desmosterol (43).
Indirect mechanisms that link cholesterol to macrophage inflammation. It has been long known that T cells participate in plaque inflammation (4). For example, using flow cytometry of whole aortic digests, Galkina et al. could demonstrate the presence of diverse cell-types of the adaptive immune system in the atherosclerotic plaque and the surrounding adventitia (44). Moreover, the same group showed that antigen presentation to CD4+ T cells in the arterial wall causes local T cell activation and the secretion of pro-inflammatory cytokines, which promote atherosclerosis by maintaining chronic inflammation and inducing foam cell formation (45). Other studies have shown a direct pro-inflammatory role of intracellular cholesterol in T cells, mediated by cholesterol-induced nano-clustering of T cell receptors (46). In addition, mice lacking T cells can have a significant reduction in atherosclerosis progression (47). Some studies, however, have demonstrated only a minor role for T cells in atherosclerosis progression (48).
Another indirect effect on macrophage polarization/activation by cholesterol is related to its being a major structural component of progressing plaques, so its ongoing accumulation will contribute to enlargement of the atheroma. This, in turn, is likely to contribute to a hypoxic environment because as the atheroma grows, the distance between the intimal cells from an oxygen supply will increase, particularly in mice, which have little capacity to form vasa vasorum. Hypoxia triggers a heavy reliance on glycolysis for energy production and it has been recently recognized that M1 cells are more glycolytic and M2 cells are more fatty acid oxidizing, and that factors that promote one pathway of energy generation over the other will promote the polarization state corresponding to the favored pathway (49). In addition, hypoxia can contribute to the formation of the necrotic core, a characteristic feature of advanced plaques that in humans increases the possibility of rupture (50). In response to the hypoxic conditions in the arterial wall, the development of vasa vasorum is enhanced, and this has also been related to atherosclerosis progression, possibly by the recruitment of monocytes to the plaque through this vascular route (51).
Macrophage Polarization and Location in Progressing Plaques
Recent studies have found that although both M1 and M2 macrophage numbers are increased during human plaque progression, M1 macrophages were the predominant phenotype in rupture-prone shoulder regions, whereas M2 markers were predominant in the adventitia and in stable cell-rich areas the of plaque (19). In another study of human plaques, using the mannose receptor as a marker, M2 macrophages were found located far from the lipid core of the plaque (52).
In mouse plaques, M1 macrophages tend to be diffusely distributed in, and characteristic of, progressing plaques at the usual age that atherosclerotic mice are examined in detail (typically after 12–16 of the consumption of a high-fat, high-cholesterol diet, or ~16–20 weeks of age). Khallou-Laschet et al. have found in apoE-deficient mice of a similar age, but with less advanced atherosclerosis because they were maintained on a low-fat, chow diet that these early plaques were infiltrated by M2 macrophages, with M1 macrophages appearing later. Disease progression correlated with the dominance of M1 over the M2 phenotype (53). Based on serial histologic examination, they further propose that the M2 → M1 shift in balance was due to a phenotypic switch of the infiltrated cells, but the data to support this were indirect and did not exclude the possibility of replacement of macrophages by new ones with a different phenotype, or the local proliferation of M2-like tissue-resident cells, which has been reported in other contexts as well (54, 55). In regard to the location of the different macrophage phenotypes in the plaque, they show that M2 macrophages accumulated in the luminal side of the plaques in young mice, while at 55 weeks of age, both M1 and M2 labeling was evenly distributed across the plaque.
Functional Consequences of M1 Polarization in Progressing Plaques
The secretion of a wide range of cytokines and chemokines (e.g., IL-1, TNFalpha, MCP-1) by M1 macrophages serves to further activate macrophages, as well as other cell types in the atheroma, such as endothelial and smooth muscle cells. There are also effects on cellular lipid metabolism. In one study of human plaques, macrophages with a marker of M2 macrophages had small lipid droplets and in studies in vitro, this was associated with a decreased ability to efflux cholesterol (52). In a study of murine macrophages, when the cells were polarized to the M1 state, there was downregulation of ABCA1 and reduced cholesterol efflux (56). If this were to happen in vivo, a vicious cycle would be formed – cholesterol accumulation would contribute to macrophage activation and M1 polarization, which would further induces cholesterol accumulation. Before concluding that this happens, given the divergent results on the effects of the M1 and M2 phenotypes on cholesterol efflux, more research in this area is clearly needed.
M1 macrophages also secrete chemokines (such as MCP-1) and cytokines (such as IL-12) that induce chemotaxis of other white blood cells (57, 58). In addition, M1 macrophages secrete several matrix metallo-proteinases (MMPs), such as MMP2 and MMP9, that can degrade the extracellular matrix in the plaque, which is thought to lead to destabilization and rupture. Indeed, MMPs were shown to co-localize with M1 macrophages in atherosclerotic plaques (59).
Atherosclerosis Regression
Fatty streaks, the initial phase in the development of plaques, were found in children as young as 3 years of age (60), with well-established plaques developing by adolescence (61). Thus, while most studies are focused on the progression of atherosclerosis and finding means to delay it, the more frequent clinical scenario is that by the time the patient comes for treatment of cardiovascular risk factors, as a frequently middle-aged adult, he or she may already have a significant burden of atherosclerosis, making the optimal goal of therapy the induction of plaque regression.
Plaque regression can be defined in various ways, such as a reduction in plaque size, plaque cholesterol content, plaque macrophage number/percentage, or a decreased inflammatory state. Of course, multiple changes can occur simultaneously, but not in every case. For example, if the plaque macrophage content decreases, while collagen content increases, as we have observed experimentally in some models of regression [e.g., Ref. (16)], the size may not change, but there will be less inflammation and more stabilizing material. Nevertheless, though size changes may vary, one consistent finding in various mouse models of atherosclerosis regression in which the issue of macrophage polarization was examined, as will be summarized below, is that the plaque content of M1 markers decreased, while those of M2 markers increased [e.g., Ref. (16, 17, 62)].
Atherosclerosis Regression Models and Changes in Plaque Macrophages
Some of the currently available regression models include aortic arch transplantation model (17, 63), Reversa mouse model (16, 64), adenoviral gene transfer of the LDL receptor, apoA1 or apoE, a “hypomorphic apoE” model, administration of an inhibitor of MTP, and infusion of apoA-I (the major protein in HDL) or apoA-I mimetics (5, 65–69). By necessity, all models begin with a progression phase, in which the total plasma cholesterol, and in particular, LDL-C and VLDL-C, are very high. After a certain period of time, preferably at least 12–16 weeks of western diet (rich in saturated fat and cholesterol) in order to accelerate in apoE-deficient-based or enable in Ldl receptor-deficient-based models the development of a complex atherosclerotic plaque, regression is typically induced by a major change in the plasma lipid profile. This change is either a reduction of LDL/VLDL-C or an increase in apoA1/HDL, both of which would decrease the plaque content of cholesterol.
In the transplantation model, the plaque-containing aortic arch from a donor apoE-deficient (apoE−/−) or Ldl receptor-deficient (ldlr−/−) mouse fed a high-fat diet for 16 weeks is transferred into the abdominal aorta of a normo-lipidemic wild-type recipient mouse or an apoE−/− mouse made transgenic in human apoA1 (“hA1/EKO”) (17, 63, 70, 71). In either case, the regression of the plaque in the transplanted arch occurs within a few days. The advantage of the transplant model is that it can be used to test the effects of specific genes on regression by using knock out or transgenic strains, either for the donor or recipient mice, as well as to conveniently study leukocyte trafficking in and out of the plaques if the donors and recipients are mismatched in isoforms of the pan-leukocyte marker for CD45. The disadvantage lies in the technical difficulty in performing such a surgery in the mouse, thereby limiting the throughput. In addition, there is inherent inflammation induced by the surgery itself, which theoretically can affect the process of regression, though control transplants into apoE−/− mice are used as a standard control for these and other effects related to the surgical procedures.
As noted above, HDL and its major protein, apoA1, can be increased by injections of apoA1, apoA1 mimetics, or an adenoviral vector expressing apoA1 (69, 72). In advanced atherosclerotic plaques of apoE−/− mice, every other day injections of native human apoA-I over only 1 week led to atherosclerosis regression, as demonstrated by significant decreases in plaque lipid content, macrophage number, and an increase in collagen content; moreover, apoA1 injections led to a significant reduction in the plaques of inflammatory M1 and an increase in anti-inflammatory M2 macrophage markers, mannose receptor 1 and arginase 1 (73). Treating mice with a recombinant adenovirus encoding human apoA1 with relatively early atherosclerotic plaques resulted in a 70% reduction in aortic lesion area characterized by a significant decrease in the fraction of lesions occupied by macrophages and macrophage-derived foam cells. The inflammatory status of this population of cells was not reported (69).
Another example for plaque regression induced by an increase in HDL was shown with our collaborators using an inhibitor of microRNA-33 (miR-33). miR-33 suppresses HDL formation in the liver and its ability to efflux cholesterol from macrophages by suppressing the expression of cholesterol transporter ATP-binding cassette transporter 1 (ABCA1) (74). It was hypothesized that inhibiting it by an antagomir (anti-miR-33) would promote atherosclerosis regression. Ldlr−/− mice with established plaques were treated with anti-miR-33 over 4 weeks. As expected, anti-miR-33 treatment led to increased reverse cholesterol transport through an increase in HDL levels and expression of ABCA1 in the liver and macrophages. Consistent with that, and consistent with the apoA1 injection study, atherosclerotic lesions regressed by anti-miR-33 treatment, as shown by reduced plaque size, lipid and macrophage content, increased collagen content and a diminished inflammatory state of the macrophages in the plaque (75).
The Reversa (Ldlr−/−ApoB100/100Mttpfl/flMx1Cre+/+) mouse is a non-surgical regression model, based on the Ldlr−/− mouse, in which the hyperlipidemia can be reversed by inducing the conditional knock out of the microsomal triglyceride transfer protein (MTTP) gene (64). MTTP is required for the proper assembly of VLDL, the precursor of LDL (76). The reversal of hyperlipidemia by inactivation of MTTP leads to regression of atherosclerosis over a few weeks accompanied by favorable changes in the composition of the atherosclerotic plaque. Again, plaque lipid content decreases, collagen content increases, and M1 markers are decreased while M2 markers are increased in the plaque macrophages (16). The advantage of the Reversa model is that it does not require any surgery in order to get extreme reduction in LDL/VLDL-C. In addition, it is important to note that unlike the transplant models, there is no increase in HDL after inducing the conditional knock out of the MTTP gene. This might be part of the reason for the reduced regression rate that can be seen in the Reversa model compared with the transplant model. The disadvantage of the model lies in the complicated genetic manipulations that were performed to create it – there are four different gene insertions/deletions in the Reversa mouse, and thus breeding it with another transgenic/knock out mouse to test the importance of a specific gene for regression is extremely time-intensive, making bone marrow transfer for myeloid-specific factors a more convenient manipulation.
The hepatic overexpression of apoE in apoE−/− or ldlr in ldlr−/− mice is two gene transfer strategies to induce regression, again by normalization of the lipid profile (5). The drawback of this method is a potential immune response of the host after the adenoviral gene transfer (77), which might complicate the interpretation of the inflammatory state of cells in the plaque. Also, especially with early versions of viral vectors, there can be limitations related to the duration or amplitude of expression.
Inducing regression just by a diet change, from western diet to chow, in the ldlr−/− mouse model has also been tried. Many times, no significant changes have been observed, presumably because the plasma cholesterol levels remain elevated, and perhaps, the experiments were not continued long enough. In one recent report, we and our collaborators have observed a reduction in plaque macrophage content and inflammatory state over 4 weeks (78). Notably, these favorable changes were impaired by hyperglycemia, consistent with our previous report using Reversa mice (79).
Mechanisms for M2 macrophage enrichment in atherosclerosis regression
There are two major questions we will consider in this section – the origin of the M2 macrophages in regressing plaques and the mechanisms for their increase.
One possibility is that the re-balancing from enrichment in M1 to M2 markers in regressing plaques represents either a change in an individual cell, as can be accomplished in vitro by changing the cytokine environment, or as proposed to happen in the “early to advanced” plaque transition in apoE−/− mice. Indeed, it is generally accepted that the phenotype of a macrophage is quite “plastic” and responsive to microenvironmental changes (9). It is also possible that M1-like macrophages leave and are replaced by M2-like cells, as occurs in wound healing. Support for this scenario is our demonstration, particularly in the aortic transplantation model, of emigration of macrophages from, and the ongoing recruitment of monocytes to, plaques in the regression environment (80). A third possibility is the induction of the proliferation of a resident population of yolk-sac-derived M2 macrophages, as observed in other settings (54). Support for this possibility is the recent demonstration in progressing plaques for macrophage proliferation (55).
It is tempting to speculate that the pro-activating direct and indirect effects of cholesterol described above are reversed under conditions of regression, under which the plaque content of cellular and extracellular cholesterol is typically reduced. Little experimental evidence, however, is available in vivo to prove this. Even for factors with known cholesterol-removing function, such as HDL, there could be lipid-independent reasons for their effects. For example, we have recently reported that murine bone marrow derived-macrophages (BMDM) not loaded with cholesterol will increase their M2 marker expression when incubated with HDL (81) [though in human monocytes this was not found to be the case (82)], and Latz and colleagues have found that HDL will induce in macrophages the transcriptional regulator ATF3, a repressor of a number of inflammatory factors (83). There is also a study in which injection of apoA1 reduced skin inflammation in ldlr−/−, apoA1−/− DKO mice (which show signs of autoimmunity) by augmenting the effectiveness of the lymph node Treg cells (84); Tregs were shown to induce an M2 polarization in yet another study (15). In this example, there may have been a lipid-removal aspect in that apoA1−/− mice are deficient in cholesterol efflux because of reduced numbers of HDL particles. Although unrelated directly to macrophage polarization, it has been shown that defects in cholesterol efflux can enrich the plasma membranes of monocyte precursors in the bone marrow with cholesterol, which results in their greater proliferation, circulating monocytosis, and increased entry into plaques of monocytes, which subsequently promotes atherogenesis (85). Importantly, we have recently found that this mechanism might also be related to the impairment of atherosclerosis regression in diabetes (7).
Turning to the issue of what regulates the enrichment in M2 macrophages in regressing plaques, there are no results to discuss at this time, but there are a number of possibilities. As mentioned earlier, potent cytokines that polarize macrophages are IL-4 and IL-13. These can be derived from a variety of leukocytes, namely Th2 lymphocytes, eosinophils, and basophils. Even if one or more of these types of leukocytes were the source of polarizing signals, there is still the mystery of how the change in the lipoprotein/lipid environment causes either the recruitment of the cells to the plaque or the stimulation of secretion from either pre-existing or newly recruited cells.
Functional contribution of M2 polarized cells to atherosclerosis regression
Another major and incompletely understood area is the requirement for, and the function of, the enrichment in M2 macrophages in atherosclerosis regression. Because their properties include tissue remodeling and inflammation resolution, it is tempting to attribute such changes in regressing plaques to M2 macrophages. This would be consistent with studies in which treatment of ldlr−/− mice with IL-13 resulted in pre-existing plaques the following: an increase in collagen, a reduction in vascular cell adhesion molecule-1 (VCAM-1)-dependent monocyte recruitment, decreased macrophage content, and the induction of M2 macrophages, despite ongoing hyperlipidemia (86). Another reflection of the importance of the M2 macrophages for regression comes from our studies in diabetic Reversa mice, in which hyperglycemia impaired their enrichment in plaques despite lipid lowering and limited the favorable changes in macrophage content and inflammatory state (79).
The promotion of the resolution of inflammation by M2 macrophages in regressing plaques is likely a consequence of their secretion of IL-10. The plaque remodeling may represent at least two other properties of M2 macrophages, namely their secretion of collagen (87) and their enhanced capacity for efferocytosis (88). Efferocytosis is the clearance of apoptotic cells by macrophages. During atherosclerosis progression, the ability to store cholesterol in plaque macrophages in the form of cholesteryl ester lipid droplets wanes and free cholesterol accumulates, causing ER stress and apoptosis (89). If efferocytosis of the dying cells does not keep up with their formation, the cellular debris that are not cleared would be expected to accumulate and become a necrotic core. Indeed, in both mouse and human plaques, As plaques advance, the efferocytotic activity of their macrophages decreases (50). Thus, an enrichment in M2 macrophages with enhanced efferocytosis would be expected to clear apoptotic cells and thereby stop, and even reverse, necrotic core expansion.
Concluding Remarks
During both the progression and the regression of atherosclerosis, macrophages have central roles. While macrophage phenotypes are diverse and form a continuum (90), we chose a simple and dichotomous approach in order to emphasize the differences between macrophage properties in atherosclerosis progression and regression. M1 macrophages predominate in progression and contribute to the inflammatory state, whereas M2 macrophages are enriched in many models of regression and appear to participate in inflammation resolution and plaque remodeling. Interestingly, M2 macrophages seem to have a beneficial role even when plasma cholesterol levels remain high [e.g., Ref. (17, 86)], and the opposite is also true – M1 macrophages, though activated by direct and indirect effects of cholesterol, have also been linked to an increase in atherosclerosis progression despite similar levels of plasma cholesterol, for example, in diabetes (91). Thus, there are complex interactions between macrophage phenotypes and plasma cholesterol levels, a situation likely to also exist with other known (and yet to be discovered) risk factors.
As noted throughout this review, there are many areas in which our knowledge of macrophage biology in plaques is inadequate. Yet, it is already clear that the inflammatory state of these cells is dynamically influenced by multiple metabolic, genetic, and pharmacologic factors. Deeper understanding of how these factors effect changes in plaque macrophages will likely advance the development of new strategies to reduce the huge burden of cardiovascular morbidity and mortality that persists with existing therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all of our lab members, past and present, as well as our collaborators, who have contributed to our studies cited in this review. Support for these studies comes from the following NIH grants: HL098055, HL084312, DK95684, and HL117226.
References
1. Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol (1995) 15(5):551–61. doi: 10.1161/01.ATV.15.5.551
2. Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med (1999) 340(2):115–26. doi:10.1056/NEJM199901143400207
3. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med (2011) 17(11):1410–22. doi:10.1038/nm.2538
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol (2011) 12(3):204–12. doi:10.1038/ni.2001
5. Williams KJ, Feig JE, Fisher EA. Rapid regression of atherosclerosis: insights from the clinical and experimental literature. Nat Clin Pract Cardiovasc Med (2008) 5(2):91–102. doi:10.1038/ncpcardio1086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity (2014) 41(1):21–35. doi:10.1016/j.immuni.2014.06.013
7. Distel E, Barrett TJ, Chung K, Girgis NM, Parathath S, Essau CC, et al. miR33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice. Circ Res (2014) 115(9):759–69. doi:10.1161/CIRCRESAHA.115.304164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (2000) 164(12):6166–73. doi:10.4049/jimmunol.164.12.6166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity (2010) 32(5):593–604. doi:10.1016/j.immuni.2010.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, et al. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res (2012) 110(3):416–27. doi:10.1161/CIRCRESAHA.111.253377
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Davies MJ. Stability and instability: two faces of coronary atherosclerosis. The Paul Dudley White lecture 1995. Circulation (1996) 94(8):2013–20. doi:10.1161/01.CIR.94.8.2013
12. Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis, American heart association. Arterioscler Thromb Vasc Biol (1995) 15(9):1512–31. doi:10.1161/01.ATV.15.9.1512
13. Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol (2000) 20(5):1262–75. doi:10.1161/01.ATV.20.5.1262
14. Kolodgie FD, Narula J, Burke AP, Haider N, Farb A, Hui-Liang Y, et al. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am J Pathol (2000) 157(4):1259–68. doi:10.1016/S0002-9440(10)64641-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A (2007) 104(49):19446–51. doi:10.1073/pnas.0706832104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Feig JE, Parathath S, Rong JX, Mick SL, Vengrenyuk Y, Grauer L, et al. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation (2011) 123(9):989–98. doi:10.1161/CIRCULATIONAHA.110.984146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, et al. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci U S A (2011) 108(17):7166–71. doi:10.1073/pnas.1016086108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res (2010) 107(6):737–46. doi:10.1161/CIRCRESAHA.109.215715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Stöger JL, Gijbels MJ, van der Velden S, Manca M, der Loos CM, Biessen EA, et al. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis (2012) 225(2):461–8. doi:10.1016/j.atherosclerosis.2012.09.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine (2008) 42(2):145–51. doi:10.1016/j.cyto.2008.01.006
21. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes (2007) 56(7):1761–72. doi:10.2337/db06-1491
22. Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, et al. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr (2008) 87(5):1219–23.
23. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes (2008) 57(6):1470–81. doi:10.2337/db07-1403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet (2000) 25(2):187–91. doi:10.1038/76048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E, et al. Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med (2002) 347(3):185–92. doi:10.1056/NEJMoa012673
26. Kiechl S, Wiedermann CJ, Willeit J. Toll-like receptor 4 and atherogenesis. Ann Med (2003) 35(3):164–71. doi:10.1080/07853890310008215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Ameziane N, Beillat T, Verpillat P, Chollet-Martin S, Aumont MC, Seknadji P, et al. Association of the toll-like receptor 4 gene Asp299Gly polymorphism with acute coronary events. Arterioscler Thromb Vasc Biol (2003) 23(12):e61–4. doi:10.1161/01.ATV.0000101191.92392.1D
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, et al. Lack of toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A (2004) 101(29):10679–84. doi:10.1073/pnas.0403249101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Stepankova R, Tonar Z, Bartova J, Nedorost L, Rossman P, Poledne R, et al. Absence of microbiota (germ-free conditions) accelerates the atherosclerosis in ApoE-deficient mice fed standard low cholesterol diet. J Atheroscler Thromb (2010) 17(8):796–804. doi:10.5551/jat.3285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A (2008) 105(43):16731–6. doi:10.1073/pnas.0804812105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464(7293):1357–61. doi:10.1038/nature08938
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Lim RS, Suhalim JL, Miyazaki-Anzai S, Miyazaki M, Levi M, Potma EO, et al. Identification of cholesterol crystals in plaques of atherosclerotic mice using hyperspectral CARS imaging. J Lipid Res (2011) 52(12):2177–86. doi:10.1194/jlr.M018077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol (2013) 14(8):812–20. doi:10.1038/ni.2639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol (2003) 23(4):656–60. doi:10.1161/01.ATV.0000064374.15232.C3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res (2011) 108(2):235–48. doi:10.1161/CIRCRESAHA.110.223875
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Kim TW, Febbraio M, Robinet P, Dugar B, Greene D, Cerny A, et al. The critical role of IL-1 receptor-associated kinase 4-mediated NF-kappaB activation in modified low-density lipoprotein-induced inflammatory gene expression and atherosclerosis. J Immunol (2011) 186(5):2871–80. doi:10.4049/jimmunol.1002242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Rekhter M, Staschke K, Estridge T, Rutherford P, Jackson N, Gifford-Moore D, et al. Genetic ablation of IRAK4 kinase activity inhibits vascular lesion formation. Biochem Biophys Res Commun (2008) 367(3):642–8. doi:10.1016/j.bbrc.2007.12.186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Björkbacka H, Kunjathoor VV, Moore KJ, Koehn S, Ordija CM, Lee MA, et al. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med (2004) 10(4):416–21. doi:10.1038/nm1008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by toll-like receptor 2. J Clin Invest (2005) 115(11):3149–56. doi:10.1172/JCI25482
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, et al. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation (2008) 118(18):1837–47. doi:10.1161/CIRCULATIONAHA.108.793869
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem (2008) 283(34):22930–41. doi:10.1074/jbc.M801408200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Chawla A. Control of macrophage activation and function by PPARs. Circ Res (2010) 106(10):1559–69. doi:10.1161/CIRCRESAHA.110.216523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell (2012) 151(1):138–52. doi:10.1016/j.cell.2012.06.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med (2006) 203(5):1273–82. doi:10.1084/jem.20052205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Koltsova EK, Garcia Z, Chodaczek G, Landau M, McArdle S, Scott SR, et al. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. J Clin Invest (2012) 122(9):3114–26. doi:10.1172/JCI61758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Molnár E, Swamy M, Holzer M, Beck-García K, Worch R, Thiele C, et al. Cholesterol and sphingomyelin drive ligand-independent T-cell antigen receptor nanoclustering. J Biol Chem (2012) 287(51):42664–74. doi:10.1074/jbc.M112.386045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Emeson EE, Shen ML, Bell CG, Qureshi A. Inhibition of atherosclerosis in CD4 T-cell-ablated and nude (nu/nu) C57BL/6 hyperlipidemic mice. Am J Pathol (1996) 149(2):675–85.
48. Dansky HM, Charlton SA, Harper MM, Smith JD. T and B lymphocytes play a minor role in atherosclerotic plaque formation in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci U S A (1997) 94(9):4642–6. doi:10.1073/pnas.94.9.4642
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol (2011) 6:275–97. doi:10.1146/annurev-pathol-011110-130138
50. Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol (2009) 86(5):1089–95. doi:10.1189/jlb.0209115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Moulton KS, Vakili K, Zurakowski D, Soliman M, Butterfield C, Sylvin E, et al. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci U S A (2003) 100(8):4736–41. doi:10.1073/pnas.0730843100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARgamma and LXRalpha pathways. Circ Res (2011) 108(8):985–95. doi:10.1161/CIRCRESAHA.110.233775
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston AT, Clement M, et al. Macrophage plasticity in experimental atherosclerosis. PLoS One (2010) 5(1):e8852. doi:10.1371/journal.pone.0008852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol (2013) 14(10):986–95. doi:10.1038/ni.2705
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med (2013) 19(9):1166–72. doi:10.1038/nm.3258
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Maitra U, Parks JS, Li L. An innate immunity signaling process suppresses macrophage ABCA1 expression through IRAK-1-mediated downregulation of retinoic acid receptor alpha and NFATc2. Mol Cell Biol (2009) 29(22):5989–97. doi:10.1128/MCB.00541-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Devaraj S, Jialal I. C-reactive protein polarizes human macrophages to an M1 phenotype and inhibits transformation to the M2 phenotype. Arterioscler Thromb Vasc Biol (2011) 31(6):1397–402. doi:10.1161/ATVBAHA.111.225508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Iwasaki M, Mukai T, Gao P, Park WR, Nakajima C, Tomura M, et al. A critical role for IL-12 in CCR5 induction on T cell receptor-triggered mouse CD4(+) and CD8(+) T cells. Eur J Immunol (2001) 31(8):2411–20. doi:10.1002/1521-4141(200108)31:8<2411::AID-IMMU2411>3.0.CO;2-Y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Huang WC, Sala-Newby GB, Susana A, Johnson JL, Newby AC. Classical macrophage activation up-regulates several matrix metalloproteinases through mitogen activated protein kinases and nuclear factor-kappaB. PLoS One (2012) 7(8):e42507. doi:10.1371/journal.pone.0042507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Holman RL, McGILL HC Jr, STRONG JP, GEER JC. The natural history of atherosclerosis: the early aortic lesions as seen in New Orleans in the middle of the 20th century. Am J Pathol (1958) 34(2):209–35.
61. McMahan CA, Gidding SS, McGill HC Jr. Coronary heart disease risk factors and atherosclerosis in young people. J Clin Lipidol (2008) 2(3):118–26. doi:10.1016/j.jacl.2008.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, et al. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci U S A (2006) 103(10):3781–6. doi:10.1073/pnas.0511043103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Chereshnev I, Trogan E, Omerhodzic S, Itskovich V, Aguinaldo JG, Fayad ZA, et al. Mouse model of heterotopic aortic arch transplantation. J Surg Res (2003) 111(2):171–6. doi:10.1016/S0022-4804(03)00039-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Lieu HD, Withycombe SK, Walker Q, Rong JX, Walzem RL, Wong JS, et al. Eliminating atherogenesis in mice by switching off hepatic lipoprotein secretion. Circulation (2003) 107(9):1315–21. doi:10.1161/01.CIR.0000054781.50889.0C
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J Clin Invest (2011) 121(5):2025–36. doi:10.1172/JCI43802
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Van Craeyveld E, Gordts SC, Nefyodova E, Jacobs F, De Geest B. Regression and stabilization of advanced murine atherosclerotic lesions: a comparison of LDL lowering and HDL raising gene transfer strategies. J Mol Med (Berl) (2011) 89(6):555–67. doi:10.1007/s00109-011-0722-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Raffai RL, Loeb SM, Weisgraber KH. Apolipoprotein E promotes the regression of atherosclerosis independently of lowering plasma cholesterol levels. Arterioscler Thromb Vasc Biol (2005) 25(2):436–41. doi:10.1161/01.ATV.0000152613.83243.12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Hewing B, Parathath S, Mai CK, Fiel MI, Guo L, Fisher EA. Rapid regression of atherosclerosis with MTP inhibitor treatment. Atherosclerosis (2013) 227(1):125–9. doi:10.1016/j.atherosclerosis.2012.12.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Tangirala RK, Tsukamoto K, Chun SH, Usher D, Puré E, Rader DJ. Regression of atherosclerosis induced by liver-directed gene transfer of apolipoprotein A-I in mice. Circulation (1999) 100(17):1816–22. doi:10.1161/01.CIR.100.17.1816
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Reis ED, Li J, Fayad ZA, Rong JX, Hansoty D, Aguinaldo JG, et al. Dramatic remodeling of advanced atherosclerotic plaques of the apolipoprotein E-deficient mouse in a novel transplantation model. J Vasc Surg (2001) 34(3):541–7. doi:10.1067/mva.2001.115963
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Rong JX, Li J, Reis ED, Choudhury RP, Dansky HM, Elmalem VI, et al. Elevating high-density lipoprotein cholesterol in apolipoprotein E-deficient mice remodels advanced atherosclerotic lesions by decreasing macrophage and increasing smooth muscle cell content. Circulation (2001) 104(20):2447–52. doi:10.1161/hc4501.098952
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Zhao Y, Black AS, Bonnet DJ, Maryanoff BE, Curtiss LK, Leman LJ, et al. In vivo efficacy of HDL-like nanolipid particles containing multivalent peptide mimetics of apolipoprotein A-I. J Lipid Res (2014) 55(10):2053–63. doi:10.1194/jlr.M049262
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Hewing B, Parathath S, Barrett T, Chung WK, Astudillo YM, Hamada T, et al. Effects of native and myeloperoxidase-modified apolipoprotein a-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler Thromb Vasc Biol (2014) 34(4):779–89. doi:10.1161/ATVBAHA.113.303044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Rayner KJ, Suárez Y, Dávalos A, Parathath S, Fitzgerald ML, Tamehiro N, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science (2010) 328(5985):1570–3. doi:10.1126/science.1189862
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest (2011) 121(7):2921–31. doi:10.1172/JCI57275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Olofsson SO, Asp L, Boren J. The assembly and secretion of apolipoprotein B-containing lipoproteins. Curr Opin Lipidol (1999) 10(4):341–6. doi:10.1097/00041433-199908000-00008
77. Ritter T, Lehmann M, Volk HD. Improvements in gene therapy: averting the immune response to adenoviral vectors. BioDrugs (2002) 16(1):3–10. doi:10.2165/00063030-200216010-00001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab (2013) 17(5):695–708. doi:10.1016/j.cmet.2013.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Parathath S, Grauer L, Huang LS, Sanson M, Distel E, Goldberg IJ, et al. Diabetes adversely affects macrophages during atherosclerotic plaque regression in mice. Diabetes (2011) 60(6):1759–69. doi:10.2337/db10-0778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Llodrá J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci U S A (2004) 101(32):11779–84. doi:10.1073/pnas.0403259101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Sanson M, Distel E, Fisher EA. HDL induces the expression of the M2 macrophage markers arginase 1 and Fizz-1 in a STAT6-dependent process. PLoS One (2013) 8(8):e74676. doi:10.1371/journal.pone.0074676
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Colin S, Fanchon M, Belloy L, Bochem AE, Copin C, Derudas B, et al. HDL does not influence the polarization of human monocytes toward an alternative phenotype. Int J Cardiol (2014) 172(1):179–84. doi:10.1016/j.ijcard.2013.12.168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. De Nardo D, Labzin LI, Kono H, Seki R, Schmidt SV, Beyer M, et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat Immunol (2014) 15(2):152–60. doi:10.1038/ni.2784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Wilhelm AJ, Zabalawi M, Owen JS, Shah D, Grayson JM, Major AS, et al. Apolipoprotein A-I modulates regulatory T cells in autoimmune LDLr−/−, ApoA-I−/− mice. J Biol Chem (2010) 285(46):36158–69. doi:10.1074/jbc.M110.134130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Murphy AJ, Westerterp M, Yvan-Charvet L, Tall AR. Anti-atherogenic mechanisms of high density lipoprotein: effects on myeloid cells. Biochim Biophys Acta (2012) 1821(3):513–21. doi:10.1016/j.bbalip.2011.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Cardilo-Reis L, Gruber S, Schreier SM, Drechsler M, Papac-Milicevic N, Weber C, et al. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol Med (2012) 4(10):1072–86. doi:10.1002/emmm.201201374
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Schnoor M, Cullen P, Lorkowski J, Stolle K, Robenek H, Troyer D, et al. Production of type VI collagen by human macrophages: a new dimension in macrophage functional heterogeneity. J Immunol (2008) 180(8):5707–19. doi:10.4049/jimmunol.180.8.5707
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol (2012) 189(7):3508–20. doi:10.4049/jimmunol.1200662
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol (2003) 5(9):781–92. doi:10.1038/ncb1035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8(12):958–69. doi:10.1038/nri2448
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Vikramadithyan RK, Hu Y, Noh HL, Liang CP, Hallam K, Tall AR, et al. Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. J Clin Invest (2005) 115(9):2434–43. doi:10.1172/JCI24819
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: macrophages, atherosclerosis, regression, cholesterol, polarization
Citation: Peled M and Fisher EA (2014) Dynamic aspects of macrophage polarization during atherosclerosis progression and regression. Front. Immunol. 5:579. doi: 10.3389/fimmu.2014.00579
Received: 25 September 2014; Accepted: 29 October 2014;
Published online: 12 November 2014.
Edited by:
Charles Dudley Mills, BioMedical Consultants, USAReviewed by:
Klaus Ley, La Jolla Institute for Allergy & Immunology, USAJanos G. Filep, University of Montreal, Canada
Copyright: © 2014 Peled and Fisher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Peled and Edward A. Fisher, The Marc and Ruti Bell Program in Vascular Biology, Department of Medicine, Leon H. Charney Division of Cardiology, New York University School of Medicine, Smilow 7, 522 First Avenue, New York, NY 10016, USA e-mail:bWljaGFlbC5wZWxlZEBueXVtYy5vcmc=;ZWR3YXJkLmZpc2hlckBueXVtYy5vcmc=