 Bimal N. Desai
Bimal N. Desai Norbert Leitinger
Norbert Leitinger- Department of Pharmacology, University of Virginia, Charlottesville, VA, USA
In addition to a fundamental role in cellular bioenergetics, the purine nucleotide adenosine triphosphate (ATP) plays a crucial role in the extracellular space as a signaling molecule. ATP and its metabolites serve as ligands for a family of receptors that are collectively referred to as purinergic receptors. These receptors were first described and characterized in the nervous system but it soon became evident that they are expressed ubiquitously. In the immune system, purinergic signals regulate the migration and activation of immune cells and they may also orchestrate the resolution of inflammation (1, 2). The intracellular signal transduction initiated by purinergic receptors is strongly coupled to Ca2+-signaling, and co-ordination of these pathways plays a critical role in innate immunity. In this review, we provide an overview of purinergic and Ca2+-signaling in the context of macrophage phenotypic polarization and discuss the implications on macrophage function in physiological and pathological conditions.
Purinergic Receptors in Macrophages
Purinergic receptors are divided into P1 and P2 receptors. The adenosine receptors are referred to as P1 receptors. P2 receptors are the receptors for adenosine triphosphate (ATP) and can be further subdivided into metabotropic P2Y receptors, which are G-protein-coupled receptors and ionotropic P2X receptors, which are cation-selective ion channels. Macrophages express a wide variety of P2X and P2Y receptors; analysis of mouse macrophages using a variety of techniques indicates the presence of P2X4, P2X7, P2Y1, P2Y2, P2Y4, and P2Y6 receptors (3). Additionally, the expression of P2Y12 receptor has been reported in microglia (4) and macrophages (5). Historically, macrophages were also thought to express an additional ATP-sensitive large conductance channel termed P2Z receptor (6). However, this receptor was later shown to be P2X7 by Surprenant et al. (7).
Release of ATP and Other Nucleotides into the Extracellular Space
Cytosolic ATP can be secreted through the release of ATP-loaded vesicles or through the activation of large conductance channels (8). A variety of inflammation-related biological processes result in ATP release from cells, and macrophages respond to this extracellular ATP rapidly. Elegant studies by Ravichandran and colleagues have shown that cells undergoing apoptosis release ATP as a find-me signal that attracts phagocytes (9). In the case of dying cells, the release of ATP and other nucleotides is accomplished through Pannexin 1, a hemi-channel that is activated through caspase-dependent cleavage (10). Interestingly, monocytes stimulated with pathogen-associated ligands or danger molecules, such as uric acid can secrete ATP, which may execute an autocrine signal that results in the activation of inflammasomes and secretion of IL-1β and IL-18 (11). Moreover, the activation of the complement cascade has also been shown to elicit ATP efflux from macrophages and subsequent autocrine activation of the NLRP3 inflammasome (12).
The Function of P2Y Receptors in Macrophages
In 1989, Dubyak and colleagues showed that treatment of macrophages with extracellular ATP elicits elevations in intracellular Ca2+ in a wide variety of myeloid cells but not in lymphocytes. These Ca2+-elevations correlated with the hydrolysis of inositol phospholipids suggesting that the ATP receptors were Gq-coupled (13). Over the course of the last two decades, it has become clear that the P2Y receptors on macrophages are Gq and Gi/o-coupled and that they perform a critical function in ATP-responsive chemotaxis. For instance, the chemotaxis of cultured microglia in response to extracellular ATP was shown to be dependent on Gi/o-coupled P2Y receptors by Honda et al. (14). This receptor was later identified as P2Y12. Microglia deficient in P2Y12 fail to polarize and migrate toward an ATP source in vitro and is unable to extend their processes toward sites of brain damage in mice (4). On a related note, microglial phagocytosis is triggered by UDP that is released by damaged neurons and is dependent on P2Y6 receptors (15). In monocytes and macrophages, P2Y2 plays a crucial chemotactic role in locating apoptotic cells releasing ATP (9). This study used a murine air-pouch model to demonstrate that cell supernatants from apoptotic cells were able to recruit monocytes and macrophages in vivo and that this recruitment was diminished in mice lacking the P2Y2 receptor. In models of lung inflammation, P2Y2 plays a prominent role in the chemotaxis of dendritic cells and eosinophils. Moreover, mice deficient in the P2Y2 receptor show reduced airway inflammation in lung inflammation models where ATP has been shown to accumulate in the airways (16). Macrophages navigating in a gradient of C5a secrete ATP and use a purinergic feedback loop that involves P2Y2, P2Y12, and P1 receptors to migrate (5). However, ATP-triggered chemotactic differences in M1 and M2 macrophages have not been explored and the functional contribution of various P2Y receptors in macrophage phenotypes remains uncharacterized (Table 1).
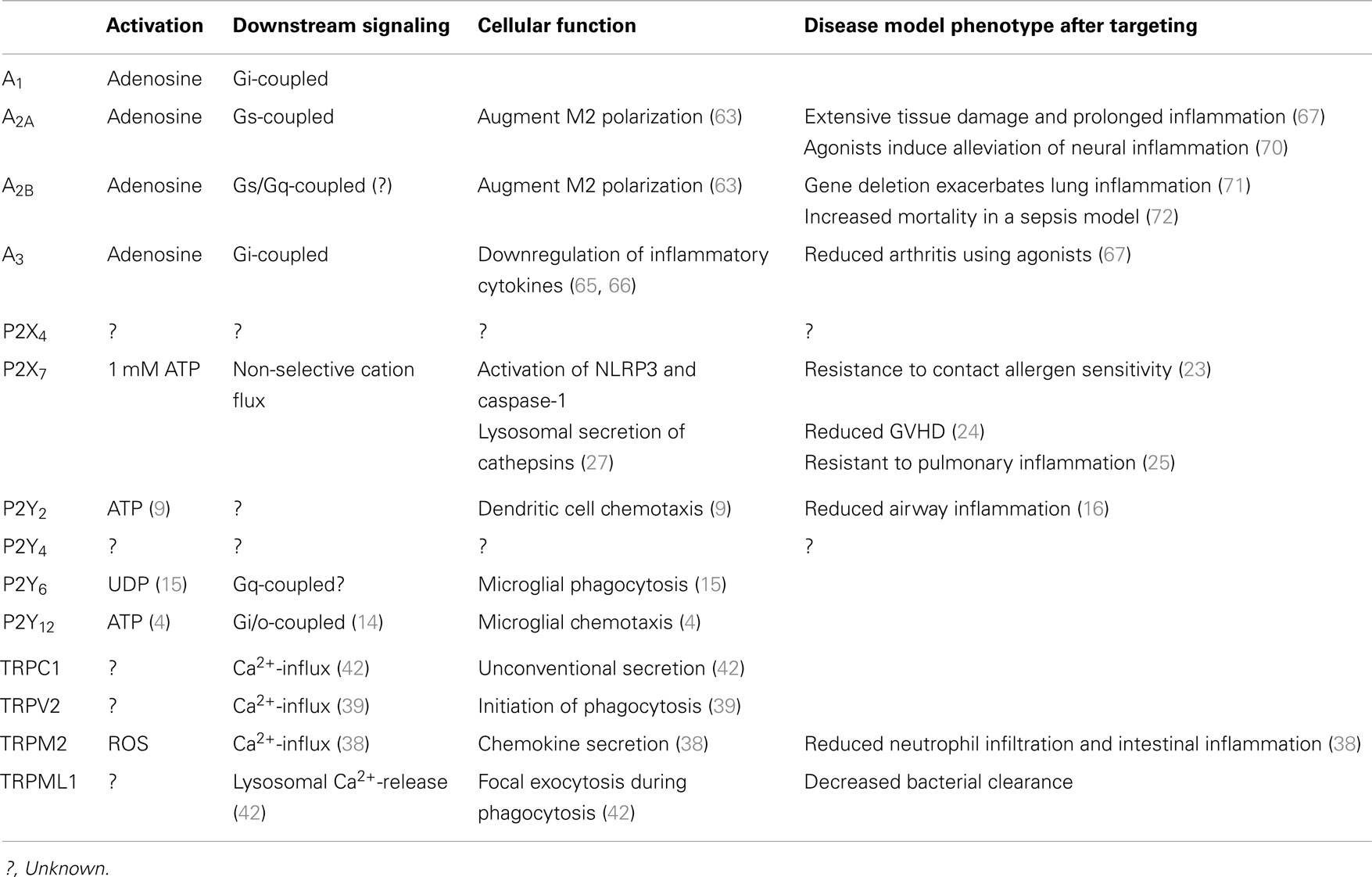
Table 1. Purinergic receptors and TRP channels in macrophages.
The Function of P2X Receptors in Macrophages
In comparison to P2Y receptors, the P2X receptors have a significantly lower affinity for ATP but their ability to respond to ATP is influenced by the ionic conditions. In the case of P2X7, replacement of Na+ with K+ greatly increases the responsiveness to ATP suggesting a physiological role in damaged tissues with altered ionic conditions (17). Activation of P2X7 by high concentrations of ATP mediates caspase-1-dependent cell death accompanied by the release of proinflammatory cytokines, such as IL-1β and IL-18. This process is greatly potentiated in macrophages activated by LPS (18). The processing of IL-1β and IL-18 by caspase-1 is followed by their unconventional secretion with or without accompanying pyroptosis, a caspase-1 mediated pathway of inflammatory cell death. These mechanisms appear to be greatly potentiated by the influx of extracellular Ca2+ through the P2X7 channels (19). Precisely how Ca2+ modulates the machinery mediating the secretion of IL-1β is not clear. Activation of P2X7 also induces membrane blebbing and activation of Rho-effector kinases but whether the influx of Ca2+ is essential for these processes is also not clear. Concomitantly, P2X7 is thought to regulate inflammasome-dependent activation of caspase-1 by mediating K+ efflux (20) and through the potentiation of an NFκB-driven transcriptional program (21). In a related process, P2X7 has been shown to control the secretion of MHC class II-containing exosomes in NLRP3-dependent but caspase-1-independent manner (22).
Due to the crucial role played by P2X7 in the regulation of the NLRP3 inflammasome, it has been implicated as a molecular target in a large variety of inflammatory diseases (Table 1). Mice deficient in P2X7 are not sensitized to contact allergens and fail to release IL-1β in response to LPS and ATP (23). This study suggests that the ligation of P2X7 by ATP is crucial for initiating skin inflammation. Similarly, P2X7 expression on antigen presenting cells appears to have a major impact on graft-versus-host disease (GVHD) (24). How P2X7 affects antigen processing and presentation is not clear yet. The P2X7-deficient mice have also been shown to be highly resistant to pulmonary inflammation induced by exposure to cigarette smoke (25). In the case of intestinal inflammation, mast cells expressing P2X7 have been shown to play a central role in initiating the inflammatory cascade (26). In this case, it seems likely that the influx of Ca2+ through P2X7 potentiates the degranulation of mast cells. A similar role for P2X7-mediated potentiation of lysosomal secretion of cathepsins has been reported in a mouse model of arthritis (27). Recently, it was discovered that monocytes from patients afflicted with Behcets disease, a severe auto-inflammatory disorder, have increased expression of P2X7 (28). Recent studies have also implicated P2X7 in the modulation of adaptive immunity through the control of antigen presentation on MHC class I molecules (29). In addition to P2X7, macrophages also express P2X1 channels but the functional significance is not yet clear (30). Although M1 macrophages are more efficient at ATP-induced secretion of IL-1β (31), no significant differences in the expression levels of P2X7 have been noted. It has been suggested that P2X7 activation is decoupled from IL-1β regulation in M2 macrophages (31).
Calcium Channels in Macrophages
As non-excitable cells, macrophages rely on Ca2+-permeable channels that are not gated by voltage. In addition to P2X channels, macrophages express the store-operated Orai channels and some members of the transient receptor potential (TRP) channel superfamily. The regulation and function of these ion channels remains a mystery and is an emerging topic of significance to inflammation (Table 1).
Orai Channels in Macrophages
Historically, the elevations of intracellular Ca2+ in macrophages were first observed in response to platelet-activating factor (PAF) (32). In accord with the classic store-operated Ca2+-entry, PAF first elicited the mobilization of intracellular Ca2+ stores through a Gq-coupled pathway. The emptying of the ER stores was then followed by the opening of the Ca2+-permeable channels in the plasma membrane, the so-called CRAC channels (33). For almost two decades, the identity of CRAC channels remained a mystery but we now know their molecular identities as Orai channels (34). Recent discoveries have unraveled the regulatory mechanisms of Orai channels but their functional role in macrophage biology remains undefined. Some observations have linked store-operated Ca2+ response to the production of reactive oxygen species (ROS) in macrophages but definitive work and mechanistic insights have not been forthcoming (35). The Ca2+-influx necessary for the engulfment of apoptotic cells by macrophages is thought to be mediated by Orai channels and genetic studies in Caenorhabditis elegans support this notion (36) but how these channels are activated when macrophages encounter apoptotic cells is not clear and the precise role of Ca2+ in the engulfment process has not been clarified. Since P2Y receptors can be Gq-linked, the subsequent depletion of Ca2+ stores through IP3 receptors should result in activation of Orai channels. Whether this actually occurs and whether Ca2+-influx through Orai channels is critical for the cellular outputs of P2Y receptor stimulation is not yet clear.
TRP Channels in Macrophages
The 28 members of TRP channel superfamily are subdivided into TRPC (seven members), TRPV (six members), TRPM (eight members), TRPML (three members), TRPP (three members), and TRPA (one member) families (37). TRP channels are cation-selective channels that are weakly voltage-sensitive and diversely gated by temperature, mechanical force, electrophiles, ligands, and internal cues, such as membrane composition and pH. Recent reports have highlighted the potent functional impact of these channels in macrophages. In monocytes lacking TRPM2, the Ca2+-influx in response to ROS is diminished and the cells are unable to produce chemokines necessary for the recruitment of other cells (38). In a mouse model of intestinal inflammation, ulceration and neutrophil infiltration were significantly attenuated in mice lacking TRPM2 (38).
Transient receptor potential channels have also been shown to play a major role in phagocytosis. Macrophages lacking TRPV2 are deficient in the triggering of phagocytosis when they encounter zymosan and IgG opsonized particles (39). Whether this function of TRPV2 is coupled to the influx of Ca2+ or other cations is not entirely clear but abnormalities in cytoskeletal rearrangements during phagocytosis were observed and the cells were also found to be deficient in chemotaxis. Mice lacking TRPV2 respond poorly when challenged with Listeria monocytogenes. They show increased mortality and greater bacterial load in their organs (39). During phagocytosis, macrophages replenish their membranes through a process termed focal exocytosis. This process was thought to be independent of Ca2+ (40), but for the phagocytosis of large particles, the process requires the activity of TRPML1 (41). Xu and colleagues have shown that TRPML1 is a lysosomal channel that is essential for the phagocytosis of large particles. Through the combined use of electrophysiology and live-cell imaging, authors show convincingly that TRPML1 mediates the release of lysosomal Ca2+ at the site of membrane uptake during large particle phagocytosis.
Recently, TRPC1 has been shown to play a role in restraining the unconventional secretion of IL-1β (42). Secretion of IL-1β is greatly potentiated after the degradation of TRPC1 by caspase-11 and macrophages lacking TRPC1 show increased secretion of IL-1β in response to inflammatory stimuli. The precise mechanism through which TRPC1 regulates this unconventional secretion machinery is not yet clear.
Coupling of Purinergic and Calcium Signaling in Macrophages
Extracellular ATP induces Ca2+ elevations in myeloid cells through the activation of Gq-coupled P2Y receptors and Ca2+-permeable P2X channels. P2Y receptors have a higher affinity for ATP and can elicit Ca2+ mobilization from the intracellular stores at low micromolar concentrations of extracellular ATP (13). In contrast, the P2X channels open at millimolar concentrations of ATP and mediate the influx of extracellular Ca2+ and other cations (43). Notably, the mobilization of Ca2+ stored in the endoplasmic reticulum is not sufficient for the activation of caspase-1 and secretion of IL-1β. When cells are stimulated with ATP in extracellular medium that is depleted of Ca2+, IL-1β secretion is nearly abolished (19). These observations indicate that activation of caspase-1 requires a sustained and more intense rise in intracellular Ca2+, which can be mediated by the activation of P2X7 channels but not P2Y receptors.
An alternative explanation for differential requirements of Ca2+ stores and Ca2+-entry in the activation of caspase-1 involves the efflux of K+ through the activated P2X7 channels. In this model, a concomitant efflux of K+ is necessary for the activation of caspase-1 and the rise in intracellular Ca2+ without K+ efflux is insufficient (44). In any case, although the activation of P2Y receptors by low concentrations of ATP is insufficient to activate caspase-1, the resulting Ca2+ oscillations have been shown to promote the transcription of proinflammatory cytokines such as IL-6 (45). The relative contributions of P2X and P2Y receptors in nucleotide-induced Ca2+-signaling have not been adequately defined but the use of knockout mice has provided useful insights into this complex aspect of inflammation (46). In myeloid cells, Ca2+-dependent activation of PKC plays a pivotal role in the NFκB pathway (47) and the cellular outputs at the site of inflammation are thus likely to be shaped by the purinergic microenvironment. Even in the absence of purinergic signals, Ca2+ stores can be mobilized by Toll-like receptors through the activation of tyrosine kinases and phospholipase C (48), but the presence of ATP in the microenvironment likely functions as a potent amplifying mechanism for inflammatory processes.
In addition to the regulation of proinflammatory gene expression and cytokine secretion, Ca2+-signaling plays a major role in phagosome maturation. This link is exploited by the internalized mycobacterium for immunoevasive block of phagosome maturation (49). Although the role of Ca2+ in phagosome maturation is incompletely defined, it is clear that the lysosomal synaptotagmin VII, a Ca2+-sensitive protein, is essential for the fusion of lysosomes with phagosomes (50, 51). Ca2+-influx is also essential for the engulfment of apoptotic cells and a subsequent anti-inflammatory response (36). Macrophage phenotypic polarization results in significant differences in the execution of phagocytosis and phagosome maturation but the associated differences in the role of purinergic and Ca2+-signals between differentially polarized macrophages remain undefined (52). Ca2+-influx has also been shown to be essential to maintain the leading-edge structure in migrating macrophages. In this context, Ca2+-influx may be necessary for the activity of PKCα, which is preferentially localized at the leading edge (53).
Chemotactic and Phagocytic Responses of Macrophages to Extracellular ATP
Macrophages and other cells of myeloid lineage respond at three basic levels to extracellular ATP gradients. First, they migrate toward increasing ATP concentrations; second, they use the ATP gradients emanating from dying cells as a “find-me” signal to locate and phagocytose the cell corpse; and third, at high concentrations of ATP, macrophages respond by robust secretion of proinflammatory cytokines.
Chemotactic responses to ATP were first convincingly demonstrated using cultured microglial cells. Extracellular ATP at micromolar concentrations induced pronounced membrane ruffling, chemokinesis, and chemotaxis (14). This aspect of purinergic response was not confined to ATP gradients emanating from a distant site, indicating ATP acted on cells in an autocrine manner. Indeed, migrating human neutrophils release ATP from their leading edges to amplify and steer their migration using an autocrine feedback loop that involves multiple types of purinergic receptors (54). In macrophages, the chemotactic response to C5a also utilizes an “autocrine purinergic loop” that involves the release of ATP at the leading edge and activation of multiple purinergic receptors (5). In asthmatic airway inflammation, ATP-induced chemotaxis appears to play a critical role in eosinophil and dendritic cell infiltration (55).
The analysis of dendritic cell responses to ATP clearly demonstrates that the chemotactic response to ATP is mechanistically dissociated from other cellular effects of ATP such as secretion of proinflammatory cytokines (16). However, the ATP-induced chemotactic response is intricately connected to the role of purinergic signaling in the location and phagocytosis of apoptotic cells. In the central nervous system, damaged neurons release UDP, which triggers the phagocytic response in neighboring microglial cells (15) and similar mechanisms are likely at work in other tissue-resident macrophages. A definitive role for extracellular ATP as a find-me signal for phagocytes was demonstrated by Elliott et al. (9). The release of ATP and UTP is dependent on the activation of caspases and Pannexin 1 during the early stages of apoptosis and the concentration gradient generated by such release is highly efficient at recruiting monocytes and macrophages. Whether different macrophage phenotypes migrate differently in response to ATP gradients has not been explored. Phagocytosis of dying cells that release ATP serves an anti-inflammatory role, and phagocytotic capacity is drastically inhibited in Mox macrophages that accumulate at the sites of oxidative tissue damage (56). In this context, it would be interesting to know whether alternatively activated M2 or M2-like macrophages show any significant specialization in locating dying cells using the ATP gradients.
Extracellular ATP as a Trigger for Proinflammatory Cytokine Secretion and Pyroptosis
Sustained exposure to relatively high concentrations of ATP has been shown to be a critical signal for the secretion of proinflammatory cytokines, such as IL-1β and IL-18. The unconventional secretion of these cytokines is accomplished through proteolytic processing by caspase-1. The activation of caspase-1 is regulated by large multimeric complexes called inflammasomes and the activation of one such inflammasome, the NLRP3 inflammasome, is highly sensitive to the presence of extracellular ATP. Early evidence for ATP-induced maturation of IL-1β came from studies involving apoptosis of peritoneal exudate cells when exposed to high concentrations of extracellular ATP. This form of apoptosis, which we now refer to as pyroptosis, was accompanied by proteolytic processing and release of IL-1β (57), and was especially pronounced in LPS-stimulated mouse peritoneal macrophages where exposure to millimolar concentrations of ATP resulted in rapid processing and release of IL-1β. It was further demonstrated that exposure to high concentrations of extracellular K+ prevented the processing and release of IL-1β (58), suggesting that depletion of intracellular K+ was essential for ATP-induced IL-1β processing. Subsequent studies of this phenomenon were greatly facilitated by the isolation of the human monocytic cell line THP-1, which was shown to be highly sensitive to purinergic stimulation of IL-1β processing. At least in human monocytes, the purinergic activation of IL-1β processing and secretion is also accompanied by release of proteolytically activated caspase-1 and a commitment to cell death (59).
A study conducted by Dixit and colleagues tested the role of channel-mediated ATP release by characterizing Pannexin 1-deficient mice in the context of inflammasome activation (60). Authors show that the activation of caspase-1 and secretion of IL-1β in response to a wide variety of stimuli including ATP is normal in Pannexin-1-deficient macrophages. In contrast, Pannexin 1-deficient thymocytes failed to recruit macrophages after undergoing apoptosis. Overall, these studies indicate that ATP released through Pannexin 1 is sufficient to reach extracellular concentrations that are of functional relevance to chemotaxis. However, the activation of caspase-1 typically requires high concentrations of ATP that are unlikely to be reached when cytosolic ATP is released through Pannexin 1. The role of vesicular release of ATP in the autocrine activation of inflammasome has not yet been ruled out and that may hold the key to reconcile these studies. A key component of vesicular ATP release is the vesicular nucleotide transporter VNUT (also known as SLC17A9), which is responsible for the accumulation of ATP into secretory vesicles (61). The human monocytic cell line THP-1 has been shown to express VNUT, which mediates the rapid secretion of ATP in response to LPS treatment (62). The function of vesicular ATP secretion in mouse macrophages and its physiological significance have not been reported yet. It is also not clear whether M1 and M2 macrophages exhibit mechanistic and functional differences in ATP release mechanisms.
The mechanisms through which activated caspase-1 and IL-1β are secreted have remained unclear but there is evidence for the involvement of Ca2+-influx elicited by ATP (19). Interestingly, although high concentrations of ATP are required for caspase-1 activation, low concentrations of ATP (10 μM) or UTP (10 μM) are sufficient to induce oscillations in intracellular Ca2+ and increased transcription of the proinflammatory cytokine IL-6 (45). An autocrine role for ATP-induced activation of inflammasome has also been suggested in the case of primary human monocytes that are stimulated by danger-associated uric acid. According to this model, the activation of inflammasome is dependent upon the initial release of ATP, which then acts on purinergic receptors in an autocrine manner. Whether such secretion results in local ATP concentrations that are high enough to activate the inflammasomes is not clear (11). In addition to the regulation of IL-1β and IL-18 secretion, ATP has also been shown to regulate the secretion of lysosomal cathepsins (27). These proteases are involved in the degradation of extracellular matrix and can result in auto-inflammatory tissue damage. Recently, ATP was shown to potentiate the release of IFNβ in LPS-stimulated macrophages (30).
With respect to macrophage phenotypes, a recent study provided evidence that inflammatory M1 macrophages are more sensitive to ATP and more efficient at ATP-induced IL-1β release when compared to M2 macrophages (31). While the physiological significance of these findings remains to be elucidated, this study provides the initial exploratory foray into these outstanding questions pertaining to how macrophage polarization fine-tunes the sensitivity to the purinergic microenvironment.
The ATP Metabolite Adenosine Regulates the Resolution of Inflammation
Many cells express membrane-bound ectonucleotidases that convert the extracellular ATP and ADP to adenosine. A common pathway involves the conversion of ATP and ADP to AMP by CD39 and subsequent conversion of AMP to adenosine by CD73. These enzymatic biochemical conversions have potent implications for the termination of inflammatory response due to the reduction in ATP levels. More significantly, adenosine serves as a ligand for G-protein coupled adenosine receptors or P1 receptors on myeloid cells (Table 1). The four P1 adenosine receptors (A1, A2A, A2B, and A3) transmit a “calm down” signal that may orchestrate the resolution of inflammation, a process conceptually different from anti-inflammatory signals that restrain the initiation of inflammatory process by preventing the recruitment and activation of immunocytes. Pertinently, extracellular adenosine has been shown to augment the polarization of macrophages toward the M2 phenotype (63).
Early studies showed that adenosine inhibited the secretion of TNFα, IL-6, and IL-8 by LPS-activated human monocytes (64). Subsequently, it was shown that the adenosine receptor A3 plays a major role in downregulating the synthesis of proinflammatory cytokines in monocytes and macrophages in response to extracellular adenosine (65, 66). Synthetic agonists of A3 adenosine receptor have been shown to have potent therapeutic effects in various mouse models of rheumatoid arthritis (67). A similar role for the A2A adenosine receptor has also been reported. Mice deficient in the A2A receptor show extensive tissue damage and prolonged inflammation to sub-threshold doses of inflammatory stimuli in three different disease models (68). In the context of infectious diseases and polymicrobial sepsis, A2A receptors are required for the control of IL-10 production by alternatively activated macrophages (69). The A2A receptors also play an anti-inflammatory role in neuroinflammation. The ligation of A2A receptors on the activated microglia has been shown to retract their processes and initiate the resolution of inflammation in the brain (70).
In the case of endotoxin-induced lung injury, the pharmacological inhibition or genetic deletion of A2B receptors greatly exacerbates lung injury. In contrast, the A2B receptor agonist attenuates endotoxin-induced lung inflammation (71). The role of A2B receptors in dampening endotoxin-induced inflammation is evident in mouse models of polymicrobial sepsis. Deletion of A2B receptors greatly increases the mortality of mice from cecal ligation and puncture-induced sepsis (72). The A2B-deficient mice showed increased levels of proinflammatory cytokines and chemokines in the serum, coincident with augmented activation of NFκB and p38 in the spleen.
In summary, the adenosine receptors provide crucial information to monocytes and macrophages and calibrate their response to the complex mix of purinergic stimuli in the inflammatory microenvironment (73). In this context, the ratio of ATP and adenosine may provide the crucial cues necessary for the polarization of macrophages toward the M2 phenotype induced by IL-4. The role of A2A and A2B receptors to augment this process of M2 polarization has been illuminated (63) but surprisingly, a recent study shows that adenosine may control this process independent of IL-4 (74). Similarly, a recent study provides evidence suggesting a role for adenosine in IL-10-induced STAT3 activation in alternatively activated phenotype termed M2c (75). Manipulation of ATP and adenosine levels in the tissue microenvironment is thus likely to emerge as a potent mechanism to guide the plasticity of macrophages and holds clinical potential for therapeutic intervention. This concept may find traction in a large variety of diseases where inflammation plays a major pathological role. A recent preclinical study exemplifies the application of this strategy in the treatment of osteolysis; the authors show that activation of A2A receptors prevents wear-induced osteolysis (76).
Extracellular ATP Plays a Prominent Role in Inflammatory Diseases
Some of the earliest evidence indicating a role for extracellular ATP in monocyte and macrophage function came from the findings that the cells of the myeloid lineage exhibit rapid elevations in intracellular Ca2+ when treated with micromolar amounts of ATP (13). The in vivo significance of this finding was further highlighted by the studies of Bertics and colleagues who showed that LPS-induced activation of macrophages was greatly enhanced by extracellular ATP (77) and that mice treated with the adenine nucleotide analog 2-methylthio-ATP were protected from endotoxic shock (78). Subsequent studies indicated a potent role for purinergic signals in controlling the inflammatory gene expression in response to LPS stimulation (79). The extensive contribution of purinergic signaling in inflammatory processes has now been established in a wide variety of pathologies (2), some of which are outlined below.
The tissue-resident macrophages of the central nervous system, the microglia, have been shown to be especially sensitive to extracellular ATP. In vitro studies with cultured microglia revealed that extracellular ATP and ADP stimulate chemotaxis and morphological changes (14). Local trauma in the brain, which results in cell death, is thought to increase the extracellular levels of ATP significantly and elegant studies using multiphoton imaging have demonstrated that microglia respond rapidly to local injury through dynamic changes in their morphology. The convergence of microglial processes at the injury site could also be stimulated by local injection of ATP and this response was demonstrated to be highly sensitive to the presence of ATP-hydrolyzing enzymes and blockers of purinergic receptors (80).
In the lungs of asthmatic patients, allergic challenges cause rapid accumulation of ATP and this has been modeled successfully in mice using experimentally induced asthma (55). Interestingly, hydrolysis of ATP in the airways through the application of apyrase greatly reduces eosinophil infiltration, production of Th2 cytokines, and bronchial hyper-reactivity. Corollary experiments show that exogenous ATP potentiates airway inflammation (55) and similar findings have demonstrated a role for purinergic signals in cigarette smoke-induced inflammation and emphysema (25). The significance of purinergic signals in allergic reactions is also evident in the skin. The accumulation of ATP in response to contact allergans is critical for the production of inflammatory cytokines by the myeloid cells and the subsequent sensitization process (23). Accumulation of ATP has also been observed in the ascites of patients and mice undergoing GVHD. In mice, the severity of GVHD is greatly reduced by neutralizing the ATP or by blocking purinergic signaling (24). Furthermore, recent studies have also implicated extracellular ATP in the development of intestinal inflammation in patients with Crohn’s disease. In corresponding mouse models, blocking purinergic signaling greatly reduces the activation of intestinal mast cells and thereby blocks the subsequent rise in proinflammatory cytokines and leukotrienes (26).
In summary, studies in human beings and mice provide conclusive evidence that purinergic signals play a major role in inflammation and tissue injury. Purinergic receptors are expressed ubiquitously and a comprehensive understanding of how purinergic signals influence the physiology and pathology is still in rudimentary stages. Purinergic control of macrophage function promises to play a central role in these processes and understanding the effects of purinergic signals on macrophage function provides an immediate window toward therapeutic intervention.
Concluding Remarks
Our understanding of the fundamental role played by purinergic and Ca2+ signaling in macrophage activity is increasing rapidly but the signaling mechanisms that drive specific cellular outputs still remain largely enigmatic. The close coupling of purinergic stimulation and Ca2+ influx suggests that the purinergic receptors, Orai channels, and TRP channels function in a co-ordinated network that responds rapidly to the changes in the inflammatory microenvironment. By virtue of being excellent drug targets, purinergic G-protein coupled receptors and ion channels offer an enticing pharmacological path to shape the plasticity of macrophage function in various diseases. To make this a reality, we will need to develop experimental models where the influence of the purinergic microenvironment and the resulting Ca2+-dynamics in macrophages can be interrogated in situ. All indications are that we have only just scratched the surface in this exciting area of innate immunity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH-P01HL120840.
References
1. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol (2011) 11(3):201–12. doi:10.1038/nri2938
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature (2014) 509(7500):310–7. doi:10.1038/nature13085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Coutinho-Silva R, Ojcius DM, Gorecki DC, Persechini PM, Bisaggio RC, Mendes AN, et al. Multiple P2X and P2Y receptor subtypes in mouse J774, spleen and peritoneal macrophages. Biochem Pharmacol (2005) 69(4):641–55. doi:10.1016/j.bcp.2004.11.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci (2006) 9(12):1512–9. doi:10.1038/nn1805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Kronlage M, Song J, Sorokin L, Isfort K, Schwerdtle T, Leipziger J, et al. Autocrine purinergic receptor signaling is essential for macrophage chemotaxis. Sci Signal (2010) 3(132):ra55. doi:10.1126/scisignal.2000588
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Falzoni S, Munerati M, Ferrari D, Spisani S, Moretti S, Di Virgilio F. The purinergic P2Z receptor of human macrophage cells. Characterization and possible physiological role. J Clin Invest (1995) 95(3):1207–16. doi:10.1172/JCI117770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science (1996) 272(5262):735–8. doi:10.1126/science.272.5262.735
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Lazarowski ER. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal (2012) 8(3):359–73. doi:10.1007/s11302-012-9304-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature (2009) 461(7261):282–6. doi:10.1038/nature08296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature (2010) 467(7317):863–7. doi:10.1038/nature09413
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A (2008) 105(23):8067–72. doi:10.1073/pnas.0709684105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J, et al. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood (2013) 122(20):3473–81. doi:10.1182/blood-2013-05-502229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Cowen DS, Lazarus HM, Shurin SB, Stoll SE, Dubyak GR. Extracellular adenosine triphosphate activates calcium mobilization in human phagocytic leukocytes and neutrophil/monocyte progenitor cells. J Clin Invest (1989) 83(5):1651–60. doi:10.1172/JCI114064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, et al. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci (2001) 21(6):1975–82.
15. Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature (2007) 446(7139):1091–5. doi:10.1038/nature05704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Muller T, Robaye B, Vieira RP, Ferrari D, Grimm M, Jakob T, et al. The purinergic receptor P2Y2 receptor mediates chemotaxis of dendritic cells and eosinophils in allergic lung inflammation. Allergy (2010) 65(12):1545–53. doi:10.1111/j.1398-9995.2010.02426.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Gudipaty L, Humphreys BD, Buell G, Dubyak GR. Regulation of P2X(7) nucleotide receptor function in human monocytes by extracellular ions and receptor density. Am J Physiol Cell Physiol (2001) 280(4):C943–53.
18. Le Feuvre RA, Brough D, Iwakura Y, Takeda K, Rothwell NJ. Priming of macrophages with lipopolysaccharide potentiates P2X7-mediated cell death via a caspase-1-dependent mechanism, independently of cytokine production. J Biol Chem (2002) 277(5):3210–8. doi:10.1074/jbc.M104388200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Gudipaty L, Munetz J, Verhoef PA, Dubyak GR. Essential role for Ca2+ in regulation of IL-1beta secretion by P2X7 nucleotide receptor in monocytes, macrophages, and HEK-293 cells. Am J Physiol Cell Physiol (2003) 285(2):C286–99. doi:10.1152/ajpcell.00070.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol (2004) 286(5):C1100–8. doi:10.1152/ajpcell.00494.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Kahlenberg JM, Lundberg KC, Kertesy SB, Qu Y, Dubyak GR. Potentiation of caspase-1 activation by the P2X7 receptor is dependent on TLR signals and requires NF-kappaB-driven protein synthesis. J Immunol (2005) 175(11):7611–22. doi:10.4049/jimmunol.175.11.7611
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Qu Y, Ramachandra L, Mohr S, Franchi L, Harding CV, Nunez G, et al. P2X7 receptor-stimulated secretion of MHC class II-containing exosomes requires the ASC/NLRP3 inflammasome but is independent of caspase-1. J Immunol (2009) 182(8):5052–62. doi:10.4049/jimmunol.0802968
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Weber FC, Esser PR, Muller T, Ganesan J, Pellegatti P, Simon MM, et al. Lack of the purinergic receptor P2X(7) results in resistance to contact hypersensitivity. J Exp Med (2010) 207(12):2609–19. doi:10.1084/jem.20092489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Wilhelm K, Ganesan J, Muller T, Durr C, Grimm M, Beilhack A, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med (2010) 16(12):1434–8. doi:10.1038/nm.2242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Lucattelli M, Cicko S, Muller T, Lommatzsch M, De Cunto G, Cardini S, et al. P2X7 receptor signaling in the pathogenesis of smoke-induced lung inflammation and emphysema. Am J Respir Cell Mol Biol (2011) 44(3):423–9. doi:10.1165/rcmb.2010-0038OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Kurashima Y, Amiya T, Nochi T, Fujisawa K, Haraguchi T, Iba H, et al. Extracellular ATP mediates mast cell-dependent intestinal inflammation through P2X7 purinoceptors. Nat Commun (2012) 3:1034. doi:10.1038/ncomms2023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Lopez-Castejon G, Theaker J, Pelegrin P, Clifton AD, Braddock M, Surprenant A. P2X(7) receptor-mediated release of cathepsins from macrophages is a cytokine-independent mechanism potentially involved in joint diseases. J Immunol (2010) 185(4):2611–9. doi:10.4049/jimmunol.1000436
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Castrichini M, Lazzerini PE, Gamberucci A, Capecchi PL, Franceschini R, Natale M, et al. The purinergic P2x7 receptor is expressed on monocytes in Behcet’s disease and is modulated by TNF-alpha. Eur J Immunol (2014) 44(1):227–38. doi:10.1002/eji.201343353
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Baroja-Mazo A, Barbera-Cremades M, Pelegrin P. P2X7 receptor activation impairs exogenous MHC class I oligopeptides presentation in antigen presenting cells. PLoS One (2013) 8(8):e70577. doi:10.1371/journal.pone.0070577
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Gavala ML, Liu YP, Lenertz LY, Zeng L, Blanchette JB, Guadarrama AG, et al. Nucleotide receptor P2RX7 stimulation enhances LPS-induced interferon-beta production in murine macrophages. J Leukoc Biol (2013) 94(4):759–68. doi:10.1189/jlb.0712351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Pelegrin P, Surprenant A. Dynamics of macrophage polarization reveal new mechanism to inhibit IL-1beta release through pyrophosphates. EMBO J (2009) 28(14):2114–27. doi:10.1038/emboj.2009.163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Randriamampita C, Trautmann A. Biphasic increase in intracellular calcium induced by platelet-activating factor in macrophages. FEBS Lett (1989) 249(2):199–206. doi:10.1016/0014-5793(89)80624-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Randriamampita C, Bismuth G, Trautmann A. Ca(2+)-induced Ca2+ release amplifies the Ca2+ response elicited by inositol trisphosphate in macrophages. Cell Regul (1991) 2(7):513–22.
34. Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol (2010) 28:491–533. doi:10.1146/annurev.immunol.021908.132550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Jin SW, Zhang L, Lian QQ, Yao SL, Wu P, Zhou XY, et al. Close functional coupling between Ca2+ release-activated Ca2+ channels and reactive oxygen species production in murine macrophages. Mediators Inflamm (2006) 2006(6):36192. doi:10.1155/MI/2006/36192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Gronski MA, Kinchen JM, Juncadella IJ, Franc NC, Ravichandran KS. An essential role for calcium flux in phagocytes for apoptotic cell engulfment and the anti-inflammatory response. Cell Death Differ (2009) 16(10):1323–31. doi:10.1038/cdd.2009.55
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Nilius B, Szallasi A. Transient receptor potential channels as drug targets: from the science of basic research to the art of medicine. Pharmacol Rev (2014) 66(3):676–814. doi:10.1124/pr.113.008268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med (2008) 14(7):738–47. doi:10.1038/nm1758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Link TM, Park U, Vonakis BM, Raben DM, Soloski MJ, Caterina MJ. TRPV2 has a pivotal role in macrophage particle binding and phagocytosis. Nat Immunol (2010) 11(3):232–9. doi:10.1038/ni.1842
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Di A, Nelson DJ, Bindokas V, Brown ME, Libunao F, Palfrey HC. Dynamin regulates focal exocytosis in phagocytosing macrophages. Mol Biol Cell (2003) 14(5):2016–28. doi:10.1091/mbc.E02-09-0626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Samie M, Wang X, Zhang X, Goschka A, Li X, Cheng X, et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell (2013) 26(5):511–24. doi:10.1016/j.devcel.2013.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Py BF, Jin M, Desai BN, Penumaka A, Zhu H, Kober M, et al. Caspase-11 controls interleukin-1beta release through degradation of TRPC1. Cell Rep (2014) 6(6):1122–8. doi:10.1016/j.celrep.2014.02.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Alonso-Torre SR, Trautmann A. Calcium responses elicited by nucleotides in macrophages. Interaction between two receptor subtypes. J Biol Chem (1993) 268(25):18640–7.
44. Brough D, Le Feuvre RA, Wheeler RD, Solovyova N, Hilfiker S, Rothwell NJ, et al. Ca2+ stores and Ca2+ entry differentially contribute to the release of IL-1 beta and IL-1 alpha from murine macrophages. J Immunol (2003) 170(6):3029–36. doi:10.4049/jimmunol.170.6.3029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Hanley PJ, Musset B, Renigunta V, Limberg SH, Dalpke AH, Sus R, et al. Extracellular ATP induces oscillations of intracellular Ca2+ and membrane potential and promotes transcription of IL-6 in macrophages. Proc Natl Acad Sci U S A (2004) 101(25):9479–84. doi:10.1073/pnas.0400733101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. del Rey A, Renigunta V, Dalpke AH, Leipziger J, Matos JE, Robaye B, et al. Knock-out mice reveal the contributions of P2Y and P2X receptors to nucleotide-induced Ca2+ signaling in macrophages. J Biol Chem (2006) 281(46):35147–55. doi:10.1074/jbc.M607713200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Zhou X, Yang W, Li J. Ca2+- and protein kinase C-dependent signaling pathway for nuclear factor-kappaB activation, inducible nitric-oxide synthase expression, and tumor necrosis factor-alpha production in lipopolysaccharide-stimulated rat peritoneal macrophages. J Biol Chem (2006) 281(42):31337–47. doi:10.1074/jbc.M602739200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Chun J, Prince A. Activation of Ca2+-dependent signaling by TLR2. J Immunol (2006) 177(2):1330–7. doi:10.4049/jimmunol.177.2.1330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Malik ZA, Thompson CR, Hashimi S, Porter B, Iyer SS, Kusner DJ. Cutting edge: Mycobacterium tuberculosis blocks Ca2+ signaling and phagosome maturation in human macrophages via specific inhibition of sphingosine kinase. J Immunol (2003) 170(6):2811–5. doi:10.4049/jimmunol.170.6.2811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Roy D, Liston DR, Idone VJ, Di A, Nelson DJ, Pujol C, et al. A process for controlling intracellular bacterial infections induced by membrane injury. Science (2004) 304(5676):1515–8. doi:10.1126/science.1098371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Czibener C, Sherer NM, Becker SM, Pypaert M, Hui E, Chapman ER, et al. Ca2+ and synaptotagmin VII-dependent delivery of lysosomal membrane to nascent phagosomes. J Cell Biol (2006) 174(7):997–1007. doi:10.1083/jcb.200605004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Canton J. Phagosome maturation in polarized macrophages. J Leukoc Biol (2014) 96(5):729–38. doi:10.1189/jlb.1MR0114-021R
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Evans JH, Falke JJ. Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proc Natl Acad Sci U S A (2007) 104(41):16176–81. doi:10.1073/pnas.0707719104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science (2006) 314(5806):1792–5. doi:10.1126/science.1132559
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MA, Muskens F, et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med (2007) 13(8):913–9. doi:10.1038/nm1617
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res (2010) 107(6):737–46. doi:10.1161/CIRCRESAHA.109.215715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Hogquist KA, Nett MA, Unanue ER, Chaplin DD. Interleukin 1 is processed and released during apoptosis. Proc Natl Acad Sci U S A (1991) 88(19):8485–9. doi:10.1073/pnas.88.19.8485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem (1994) 269(21):15195–203.
59. Laliberte RE, Eggler J, Gabel CA. ATP treatment of human monocytes promotes caspase-1 maturation and externalization. J Biol Chem (1999) 274(52):36944–51. doi:10.1074/jbc.274.52.36944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Qu Y, Misaghi S, Newton K, Gilmour LL, Louie S, Cupp JE, et al. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J Immunol (2011) 186(11):6553–61. doi:10.4049/jimmunol.1100478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Sawada K, Echigo N, Juge N, Miyaji T, Otsuka M, Omote H, et al. Identification of a vesicular nucleotide transporter. Proc Natl Acad Sci U S A (2008) 105(15):5683–6. doi:10.1073/pnas.0800141105
62. Sakaki H, Tsukimoto M, Harada H, Moriyama Y, Kojima S. Autocrine regulation of macrophage activation via exocytosis of ATP and activation of P2Y11 receptor. PLoS One (2013) 8(4):e59778. doi:10.1371/journal.pone.0059778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Csoka B, Selmeczy Z, Koscso B, Nemeth ZH, Pacher P, Murray PJ, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J (2012) 26(1):376–86. doi:10.1096/fj.11-190934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Bouma MG, Stad RK, van den Wildenberg FA, Buurman WA. Differential regulatory effects of adenosine on cytokine release by activated human monocytes. J Immunol (1994) 153(9):4159–68.
65. Sajjadi FG, Takabayashi K, Foster AC, Domingo RC, Firestein GS. Inhibition of TNF-alpha expression by adenosine: role of A3 adenosine receptors. J Immunol (1996) 156(9):3435–42.
66. McWhinney CD, Dudley MW, Bowlin TL, Peet NP, Schook L, Bradshaw M, et al. Activation of adenosine A3 receptors on macrophages inhibits tumor necrosis factor-alpha. Eur J Pharmacol (1996) 310(2–3):209–16. doi:10.1016/0014-2999(96)00272-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Baharav E, Bar-Yehuda S, Madi L, Silberman D, Rath-Wolfson L, Halpren M, et al. Antiinflammatory effect of A3 adenosine receptor agonists in murine autoimmune arthritis models. J Rheumatol (2005) 32(3):469–76.
68. Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature (2001) 414(6866):916–20. doi:10.1038/414916a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Csoka B, Nemeth ZH, Virag L, Gergely P, Leibovich SJ, Pacher P, et al. A2A adenosine receptors and C/EBPbeta are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood (2007) 110(7):2685–95. doi:10.1182/blood-2007-01-065870
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Orr AG, Orr AL, Li XJ, Gross RE, Traynelis SF. Adenosine A(2A) receptor mediates microglial process retraction. Nat Neurosci (2009) 12(7):872–8. doi:10.1038/nn.2341
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Schingnitz U, Hartmann K, Macmanus CF, Eckle T, Zug S, Colgan SP, et al. Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol (2010) 184(9):5271–9. doi:10.4049/jimmunol.0903035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Csoka B, Nemeth ZH, Rosenberger P, Eltzschig HK, Spolarics Z, Pacher P, et al. A2B adenosine receptors protect against sepsis-induced mortality by dampening excessive inflammation. J Immunol (2010) 185(1):542–50. doi:10.4049/jimmunol.0901295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Hasko G, Pacher P, Deitch EA, Vizi ES. Shaping of monocyte and macrophage function by adenosine receptors. Pharmacol Ther (2007) 113(2):264–75. doi:10.1016/j.pharmthera.2006.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, Outram S, et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Ralpha) signaling. Inflammation (2013) 36(4):921–31. doi:10.1007/s10753-013-9621-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Koscso B, Csoka B, Kokai E, Nemeth ZH, Pacher P, Virag L, et al. Adenosine augments IL-10-induced STAT3 signaling in M2c macrophages. J Leukoc Biol (2013) 94(6):1309–15. doi:10.1189/jlb.0113043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Mediero A, Frenkel SR, Wilder T, He W, Mazumder A, Cronstein BN. Adenosine A2A receptor activation prevents wear particle-induced osteolysis. Sci Transl Med (2012) 4(135):135ra65. doi:10.1126/scitranslmed.3003393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Tanke T, van de Loo JW, Rhim H, Leventhal PS, Proctor RA, Bertics PJ. Bacterial lipopolysaccharide-stimulated GTPase activity in RAW 264.7 macrophage membranes. Biochem J (1991) 277(Pt 2):379–85.
78. Proctor RA, Denlinger LC, Leventhal PS, Daugherty SK, van de Loo JW, Tanke T, et al. Protection of mice from endotoxic death by 2-methylthio- ATP. Proc Natl Acad Sci U S A (1994) 91(13):6017–20. doi:10.1073/pnas.91.13.6017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Hu Y, Fisette PL, Denlinger LC, Guadarrama AG, Sommer JA, Proctor RA, et al. Purinergic receptor modulation of lipopolysaccharide signaling and inducible nitric-oxide synthase expression in RAW 264.7 macrophages. J Biol Chem (1998) 273(42):27170–5. doi:10.1074/jbc.273.42.27170
80. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci (2005) 8(6):752–8. doi:10.1038/nn1472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: macrophages, calcium, purinergic receptors, inflammation, inflammasome activation
Citation: Desai BN and Leitinger N (2014) Purinergic and calcium signaling in macrophage function and plasticity. Front. Immunol. 5:580. doi: 10.3389/fimmu.2014.00580
Received: 15 August 2014; Accepted: 29 October 2014;
Published online: 27 November 2014.
Edited by:
Klaus Ley, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Geraldine Olivia Canny, Ecole Polytechnique Fédérale de Lausanne, SwitzerlandCaglar Cekic, Bilkent University, Turkey
Copyright: © 2014 Desai and Leitinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bimal N. Desai and Norbert Leitinger, Department of Pharmacology, University of Virginia, Charlottesville, VA, USA e-mail:YmRlc2FpQHZpcmdpbmlhLmVkdQ==;bmwycUB2aXJnaW5pYS5lZHU=