 Brittany D. Conroy1†
Brittany D. Conroy1† Tyler A. Herek1†
Tyler A. Herek1† Timothy D. Shew1†
Timothy D. Shew1† Matthew Latner1Joshua J. Larson1
Matthew Latner1Joshua J. Larson1 Laura Allen1
Laura Allen1 Paul H. Davis1
Paul H. Davis1 Tomáš Helikar2
Tomáš Helikar2 Christine E. Cutucache1*
Christine E. Cutucache1*- 1Department of Biology, University of Nebraska at Omaha, Omaha, NE, USA
- 2Department of Biochemistry, University of Nebraska at Lincoln, Lincoln, NE, USA
Caveolin-1 (CAV1) is a vital scaffold protein heterogeneously expressed in both healthy and malignant tissue. We focus on the role of CAV1 when overexpressed in T-cell leukemia. Previously, we have shown that CAV1 is involved in cell-to-cell communication, cellular proliferation, and immune synapse formation; however, the molecular mechanisms have not been elucidated. We hypothesize that the role of CAV1 in immune synapse formation contributes to immune regulation during leukemic progression, thereby warranting studies of the role of CAV1 in CD4+ T-cells in relation to antigen-presenting cells. To address this need, we developed a computational model of a CD4+ immune effector T-cell to mimic cellular dynamics and molecular signaling under healthy and immunocompromised conditions (i.e., leukemic conditions). Using the Cell Collective computational modeling software, the CD4+ T-cell model was constructed and simulated under CAV1+/+, CAV1+/−, and CAV1−/− conditions to produce a hypothetical immune response. This model allowed us to predict and examine the heterogeneous effects and mechanisms of CAV1 in silico. Experimental results indicate a signature of molecules involved in cellular proliferation, cell survival, and cytoskeletal rearrangement that were highly affected by CAV1 knock out. With this comprehensive model of a CD4+ T-cell, we then validated in vivo protein expression levels. Based on this study, we modeled a CD4+ T-cell, manipulated gene expression in immunocompromised versus competent settings, validated these manipulations in an in vivo murine model, and corroborated acute T-cell leukemia gene expression profiles in human beings. Moreover, we can model an immunocompetent versus an immunocompromised microenvironment to better understand how signaling is regulated in patients with leukemia.
Introduction
Caveolae are cave-like invaginations comprised mostly of the protein caveolin-1 (CAV1). In addition to the traditional roles of CAV1 in endocytosis, CAV1 has been implicated in processes ranging from signal transduction (1, 2), to both oncogenesis (3–5), and tumor suppression (6–8). Recently, three new roles for CAV1 emerged, including regulating immune synapse formation, T-cell receptor (TCR) activation, and mediating actin polymerization (9–11). Caveolin-1 knockout studies show an attenuated immune synapse formation as observed by decreased F-actin staining and dysregulation of RAC1 and ARP2/3 pathways (9). When T-cells engage with antigen-presenting cells (APCs), decreased TCR-dependent T-cell proliferation is observed when CAV1 is prohibited from interacting with CD26 (12). Downstream signaling pathways affected by CAV1 knockdown include the organization of the KSR1 mediated Raf/MEK/ERK signal cascade (13) and ZAP70, p56lck, and TCRζ phosphorylation (14). This mechanism has been shown to be distinct from CD3/CD28 stimulation (15), where no proliferation defects were observed in Cav1−/− T-cells.
CAV1 acts as a scaffolding molecule thereby likely contributing to diverse events in the cell through CAV1-mediated recruitment of signaling complexes to the plasma membrane. Moreover, T-cell activation through the TCR and competent immune synapse formation are necessary for a healthy immune response. Misregulation of these processes can lead to deleterious effects, including cancer progression and a phenotype known as tumor-induced immunosuppression (16, 17). As CD4+ T-cells are vital for proper adaptive immune function, and CAV1 plays a role in immune synapse formation, we chose to further investigate the CAV1-mediated pathways in a CD4+ T-cell.
To better understand the intricate biology of CAV1 signaling in CD4+ T-cells, the development of a comprehensive in silico model is warranted. Through such a model, further identification of molecules associated with CAV1 signaling can occur.
Importantly, such a model allows for real-time simulations using computer software in an effort to identify specific mechanisms in the cell. In order to generate such a comprehensive and dynamic model, a systems biology approach is required (18). This approach provides a potentially greater understanding of the complex cellular functions that occur in living systems, allowing the use of computer models to conduct thousands of virtual experiments as well as make methodical predictions regarding proteins of interest (18–21).
Herein, we describe the construction and validation of a fully functional in silico CD4+ T-cell model using the Cell Collective, a web-based, open-source dynamic modeling platform that allows scientists to construct computational models in a non-mathematical fashion (20, 22). From this in silico starting position, comprehensive simulations were performed, allowing for predictions and hypotheses to be drawn for further in vitro/in vivo experimentation. To our knowledge, this is the first time a dynamic model of a CD4+ T-cell has been created to observe the downstream effects of CAV1+/+ (wild type), CAV1+/− (heterozygous), and CAV1−/− (knock down) upon cell signaling and intracellular networks as validated by in silico simulations and in vivo investigations.
Materials and Methods
Computational Model Construction with Cell Collective
The presented model was constructed using Cell Collective – a collaborative and interactive platform for modeling biological/biochemical systems (20, 22). The mathematical framework behind Cell Collective is based on a common qualitative (discrete) modeling technique where the regulatory mechanism of each node is described with a logical function [for more comprehensive information on logical modeling, see Ref. (23, 24)]. Cell Collective allows users to construct and simulate large-scale computational models of various biological processes based on qualitative interaction information extracted from previously published literature. The initial version of the model was structured after the previously published models (25, 26). The individual components and local interactions in the presented final model were retrieved manually from published literature. The model was subsequently validated against well-known experimentally demonstrated T-cell dynamics (see Model Validation), as well as new experiments presented in this paper. The Cell Collective’s Knowledge Base was used to catalog and annotate every interaction and regulatory mechanism (e.g., tyrosine phosphorylation on Y316) as mined from the primary literature. The model is freely available for simulations and further contributions by others directly in the platform. The model can be also downloaded in the SBML format (24) to be used within other software tools.
Model Validation
The model was constructed using local (e.g., protein–protein interaction) information from the primary literature. In other words, during the construction phase of the model, there was no attempt to determine the local interactions based on any other larger phenotypes or phenomena. However, after the model was completed, verification of the accuracy of the model involved testing it for the ability to reproduce complex input–output phenomena that have been observed in the laboratory. To do this, the T-cell model was simulated under a multitude of cellular conditions and analyzed in terms of input–output dose–response curves to determine whether the model behaves as expected [Figure 2; Ref. (27–33)], including various downstream effects as a result of activation of the TCR, G-protein-coupled receptor, cytokine, and integrin pathways. A total of 20 phenomena were used for the validation phase (data not shown).
In silico Simulations
The Cell Collective platform was utilized to perform all simulations for the CD4+ T-cell model. Virtual extracellular environments, composed of 20 CD4+ T-cell stimuli, were optimized for each in silico experiment based on immunocompetent versus immunocompromised (diseased) settings [Table 1; Ref. (9, 13, 30, 31, 34, 35)]. For each experiment, these values were analyzed and used to compare proteins most affected by CAV1+/+, CAV1+/−, and CAV1−/− in an immunocompetent (i.e., WT) versus varying degrees of immunosuppression (i.e., Diseases A versus B) condition. The model was simulated under hypothetical disease-causing environments in order to observe the changes, if any, in CAV1 regulatory activity. Disease A mimics an immunosuppressive disease condition, and disease B simulates varying degrees of immunosuppression, as controlled by varying IL-10 levels. Each experiment consisted of 1,000 simulations, with different activity levels randomly selected between 0 and 100 for the external stimuli representing the extracellular environment. Each simulation consisted of 800 iterations and activity level of the model species was calculated over the last 300 iterations using methods previously described (36). The aforementioned simulations were run under six separate conditions including wild-type tissue, diseased tissue (diseases A and B), wild-type blood, and diseased blood (diseases A and B). Wild-type and diseased blood simulations are not included due to inconclusive data. Each experimental environment was simulated under (1) healthy cellular conditions, (2) CAV1 knocked out, (3) CAV1 activated 50% of the time (CAV1+/−) and (4) CAV1 activated at random levels between 0 and 100. (To be able to artificially control the activity levels of CAV1 under environments 3 and 4, an external species, “CAV1 Activator,” was built into the model to activate CAV1 independently of the activity levels of its direct upstream regulators).
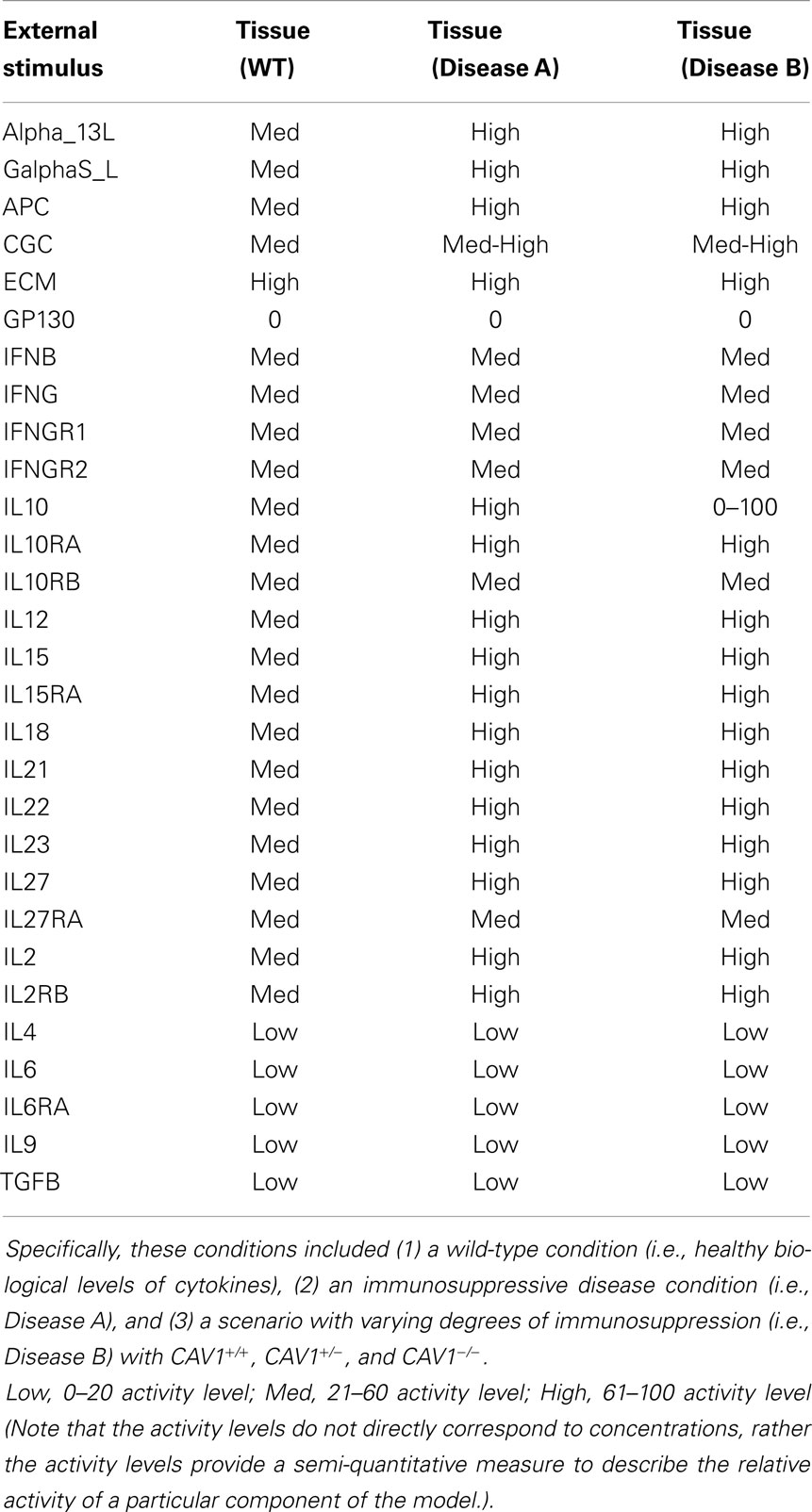
Table 1. A summary of the experimental conditions simulated.
Mouse Maintenance
Animals were housed in pathogen-free animal facilities, and all experimental protocols were reviewed and approved per the Institutional Animal Care and Use Committee at the University of Nebraska Medical Center/University of Nebraska at Omaha (IACUC# 13-056-08-EP). C57Bl/6J and B6.Cg-Cav1tm1Mls/J mice were purchased from the Jack-son Laboratory (Bar Harbor, ME, USA). Post-natal day 54 (±5 days) mice were used for all experiments.
Histological Staining
Spleen and lymph node tissues were sectioned and stained at the University of Nebraska Medical Center’s Tissue Science Facility. Spleen and lymph node tissues were sectioned, preserved in 10% formalin, and embedded onto slides in paraffin. Specifically, all tissues were sectioned after at least 48 h in fixative and stained with hematoxylin and eosin using standard protocol.
For immunohistochemical staining (IHC), tissues were deparrafinized in xylene for 3 min and rehydrated in decreasing concentrations of ethanol (100–50%). Antigen retrieval was performed by boiling sections in a solution of Sodium Citrate with 0.05% Tween-20. Blocking against non-specific binding and endogenous peroxidases was performed by incubation with 5% bovine serum albumin (BSA; Invitrogen) and 0.3% hydrogen peroxide, respectively. Primary antibody incubation was conducted for 90 min at room temperature in phosphate buffered saline (PBS; DIBCO). The following antibodies were used: Rac1 (Merck KGaA, Darmstadt, Germany); CD28 (BD Pharmingen); GATA3 (BD Pharmingen); CD26 (Abcam); BCL10 (Cell Applications, Inc.). Horse radish peroxidase-conjugated secondary antibodies (Cell Signaling Technology; BD Pharmingen; Abcam) were incubated for 1 h at room temperature. Slides were developed with a working solution 3,3-diaminobenzidine for 10 min at room temperature, followed by rinsing with distilled water and mounting the coverslip with Permount (Thermo Fisher Scientific).
Gene Expression Profiling
Microarray data were downloaded from the Gene Expression Omnibus, accession number GSE55851 (37). Data contained whole genome expression profiling of CD4+ T-cells, sorted based on a CADM1/CD7 phenotype. Samples were collected from patients diagnosed with Adult T-cell leukemia-lymphoma (ATL) subtypes: asymptomatic (n = 2), smoldering (n = 2), chronic (n = 1), acute (n = 2), and healthy controls (n = 3). A minimum of two samples were taken from each patient for microarray analyses. Molecules of interest, as established utilizing the Cell Collective, were selected within the microarray data and analyzed by fold-change from normal controls between ATL subtypes. Fold-change values were subjected to uncentered, average-linkage correlation using Cluster 3.0, and Java TreeView as described previously (9). Further, Pearson regression analyses were conducted to discern correlation among molecules of interest in relation to CAV1 expression across ATL subtypes.
Results
In silico Modeling of a CD4+ T-Cell
The completed CD4+ T-cell model consists of 188 nodes representing components of various signaling pathways and corresponding protein-to-protein, protein-phosphorylation, and kinase interactions (Figure 1A). These interactions correlate with preliminary data generated using Osprey software used to model CAV1 protein-to-protein interaction types (Figure 1B). Each method of analysis (both the Cell Collective and Osprey) implicates a role for CAV1 in phosphorylation, signal transduction, transport, and cytoskeletal arrangement. The regulation of these events by CAV1 is highly complex and dynamic, as illustrated in Figure 1C.
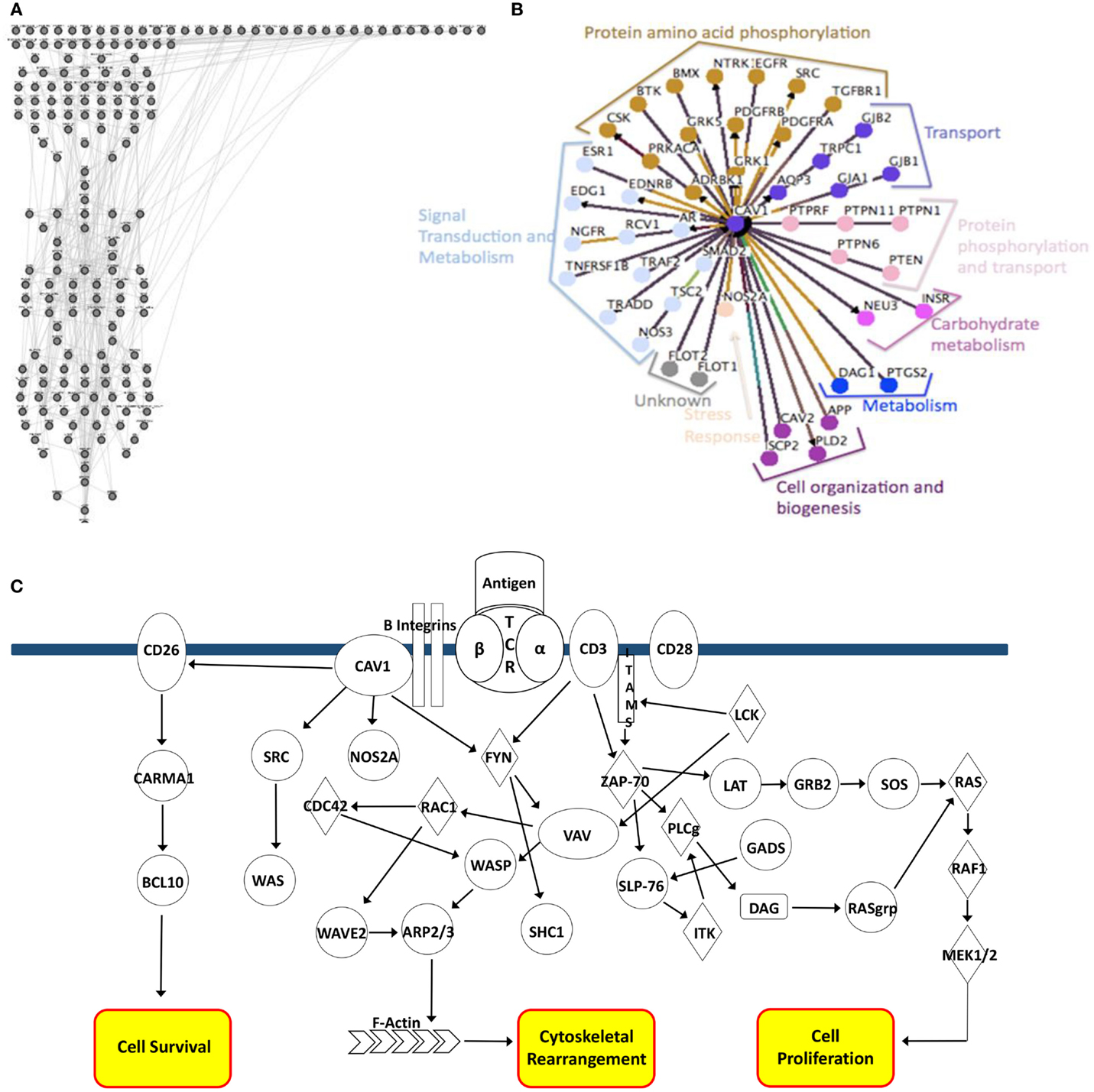
Figure 1. In silico modeling of a CD4+ T-cell. (A) Nodal representation of CD4+ T-cell signaling pathways constructed using the Bio-Logic builder inclusive within the Cell Collective. Linkages represent protein–protein, protein–phosphorylation, and kinase interactions. (B) Osprey modeling of predicted CAV1 protein–protein interactions and functions. Linkages are categorized by function and centrality to CAV1. (C) Graphical depiction of CAV1-associated interactions. Major pathway end-points include cell survival, cytoskeletal rearrangement, and cellular proliferation.
To verify that the local interactions built into the model were able to accurately mimic complex phenomena that have been produced in the laboratory, 20 validations were conducted via simulations in Cell Collective (six representative validations are shown in Figures 2A–F). As an example, simulated TCR activation by an APC leads to Erk activation, and the subsequent downstream effect of cellular proliferation as represented by the literature (Figure 2A, Figure S1 in Supplemental Material).
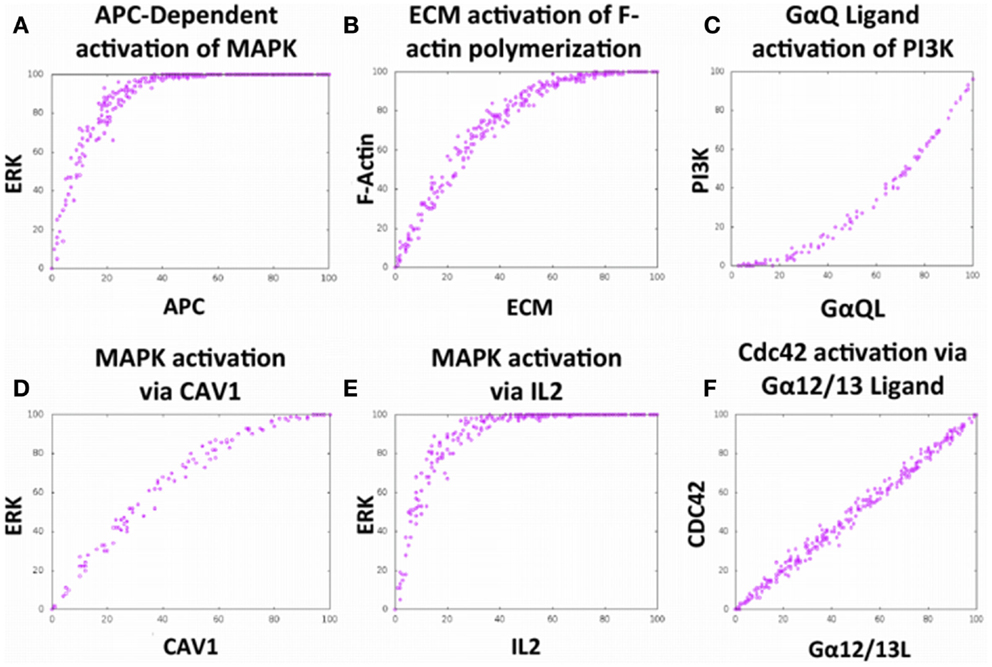
Figure 2. The Cell Collective accurately models complex cellular phenomena. (A–F) Certification of Bio-Logic built local interactions executing in accordance with primary literature findings. (A) Activation of the mitogen-activated protein kinase (MAPK) pathway via APC stimulation (27). (B) Positive relationship between filamentous actin polymerization in response to stimulation with extracellular matrix (ECM) components (28). (C) PI3-Kinase activation via binding of ligand to G protein-coupled receptor, GαQ (29). (D) Activation of the MAPK pathway via integrin-dependent ECM stimulation (9, 30). (E) Activation of the MAPK pathway via stimulation with interleukin-2 (IL2) (31, 32). (F) Activation of the small GTPase Cdc42 via binding of ligand to the G protein-coupled receptor, Gα12/13 (33); these results not shown in the graphic. Each dose–response curves appears to demonstrate a positive correlation with the stimulus.
Identification of Most Affected Proteins in silico
Experiments were simulated using the in silico model (as described in Table 1) to make rational predictions about how the system would function in the laboratory. Following 1,000 iterations, we observed the protein products most affected by CAV1+/+, CAV1+/−, and CAV1−/− in immounocompetent versus immunocompromised conditions (Figures 3A–C). In order to test the validity of the model, we chose to investigate expression levels of the proteins most affected in the knock down genotype as compared to a wild-type system. Specifically proteins disregulated by CAV1−/− across all three conditions (WT, Disease A, and Disease B) included CD26, CARMA1, FYN, SHC1, SOS, SHP2, NOS2A, BCL10, and GRB2 (Figure 3C). Based on findings from the model as well as preliminary in vitro data, we hypothesized that CAV1 expression regulates Ras-related C3 botulinum toxin substrate 1 (RAC1), B-cell lymphoma/leukemia 10 (BCL10), GATA-binding protein 3 (GATA3), CD26, and CD28 (Figures 1C and 3C).
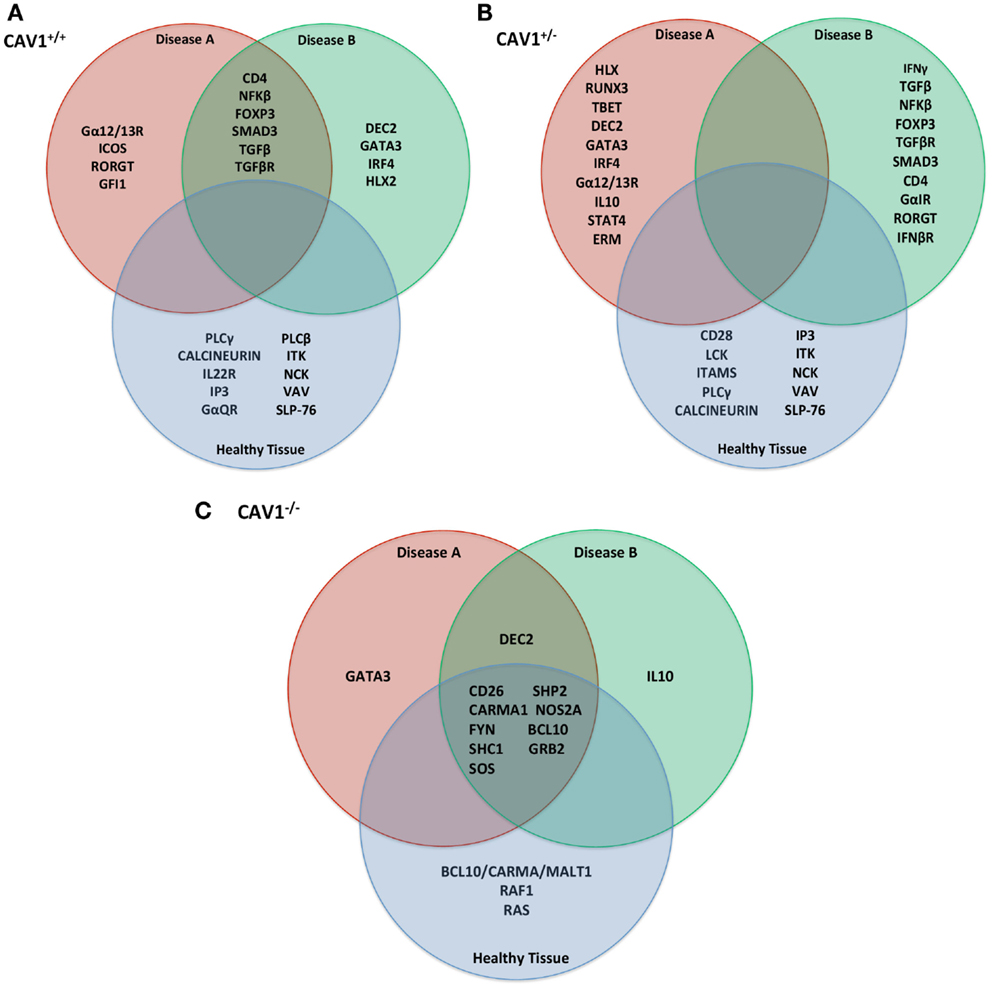
Figure 3. In silico predictions for translation into in vitro/in vivo experimentation. Following 1,000 iterations of simulation as described in Table 1, the most affected proteins (either up or downregulated) were compiled by the Cell Collective and ranked based on activity% ON. Specifically, those described were the top 15 most differentially expressed molecules in the (A) CAV1+/+, (B) Cav1+/−, (C) CAV1−/− genotype. These proteins were selected for further investigation with in vitro/in vivo verification.
The abovementioned results are indicative of CAV1-mediate regulation of a variety of cellular functions, notably those that are downstream of TCR and integrin pathways of which CAV1 serves as the scaffold. Given these results, we were able to make predictions about protein expression in relation to CAV1 and test them in the laboratory using in vitro and in vivo experiments. For example, we observed that CAV1 is involved in the integrin signaling pathway that ultimately activates the mitogen-activated protein kinase (MAPK) cascade (Figure 2); therefore, we can predict that cellular proliferation will be decreased if CAV1 is knocked down.
Verification of in Silico Predictions in vivo
To determine differences in morphology between CAV1−/− and wild-type mice, we examined tissues (including lymph nodes and spleen) stained with hematoxylin and eosin and observed no differences were observed in tissue architecture. Lymphoid organs (lymph nodes and spleen) were selected for their robustness of CD4+ T-cells, and liver was used as a control for tissue histology (i.e., to ensure mice were disease free). Furthermore, immunohistochemistry was utilized to biologically validate in silico predictions from our CD4+ T-cell model.
Top hit proteins: GATA3, RAC1, CD26, and BCL10 were selected for IHC to validate the model (Figure 4). RAC1 and GATA3 showed upregulation in the lymph node tissue of the CAV1−/− mice. We observed CD26 and BCL10 to be upregulated in both the lymph node and spleen tissues of the CAV1−/− mice. We also stained for CD28, as it is a well-characterized co-stimulatory protein involved in T-cell activation. CD28 showed no differential expression between the wild-type and CAV1−/− mice.
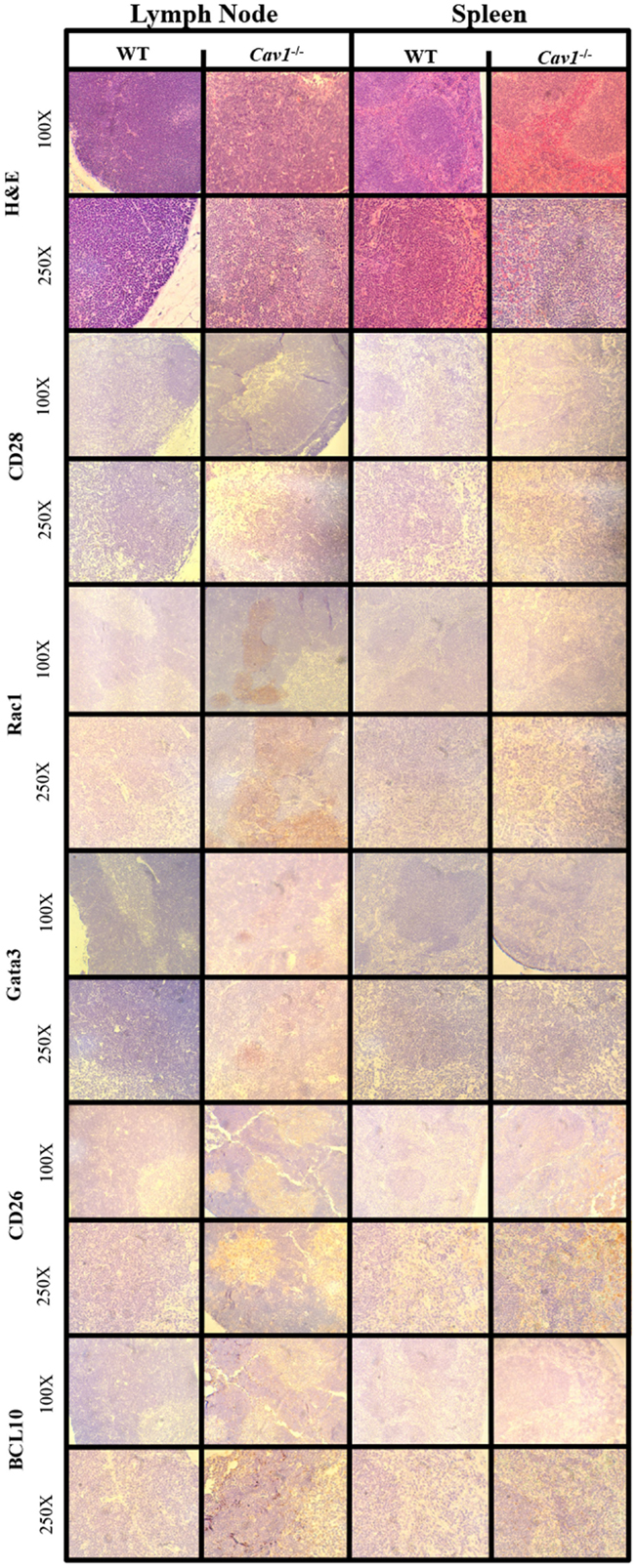
Figure 4. Immunohistochemistry from murine model validation of in silico-predicted differentially expressed molecules with CAV1 knockout. Molecules downstream of CAV1 that were predicted to be affected with CAV1 knockout using the Cell Collective were validated using lymphoid tissue histology from wild-type (WT) C57Bl/6 mice and CAV1−/− mice. The corresponding hematoxylin and eosin preparations are included in the top panel for morphological orientation; all tissues were sectioned subsequently.
Based on predictions from the model (Figure 3C), we investigated the differential expression, hierarchal clustering (Figure 5A), and regression analyses (Figure 5B) of top hit proteins using microarray data from ATL cases (GSE55851). Comparison of ATL subtypes for identification of potential molecular signatures in relation to CAV1 expression reveal seven molecules, including CAV1, clustered together based upon gene expression profiles following hierarchal clustering (Figure 5A). We observed a positive correlation (R = 0.78), with distinct signatures displayed between healthy, asymptomatic, smoldering, chronic, and acute patients (Figure 5B). Pearson regression analyses were conducted between each molecule of interest in relation to CAV1 expression across ATL subtypes (Figure 5B). Specifically, we observed strong correlations between CAV1 and the following molecules: BCL10 (R = 0.947), DEC2/BHLHB3 (R = 0.782), SHP2/PTPN11 (R = 0.742), and GATA3 (R = 0.694). Conversely, SOS1 (R = −0.981), FYN (R = −0.949), SOS2 (R = -0.825), and CD26 (R = −0.740) were most negatively correlated to CAV1 expression. These data corroborate those described and simulated in the model (Figure 3); therefore, it is translated to in vivo, leukemic conditions.
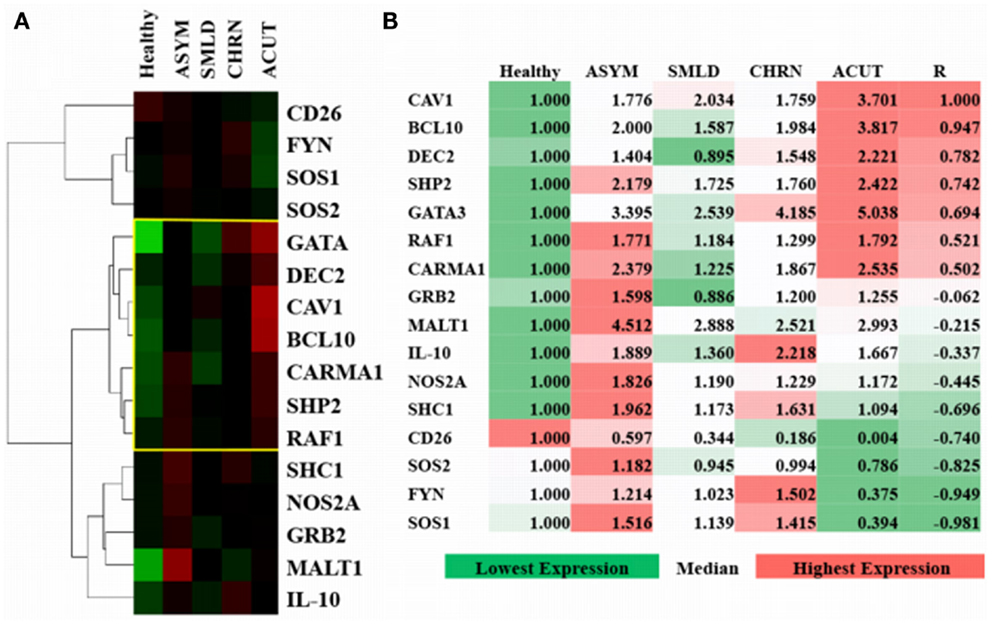
Figure 5. Verification and comparison of in silico results with gene expression profiling from adult T-cell leukemia-lymphoma (ATL). (A) Uncentered average-linkage correlation of fold-change values from top affected proteins in ATL patients. The yellow-boxed region represents a CAV1-associated molecular signature (R = 0.78). Healthy (n = 3), asymptomatic (ASYM) (n = 2), smoldering (SMLD) (n = 2), chronic (CHRN) (n = 1), and acute (ACUT) (n = 2) cases are shown. A minimum of two samples were taken from each patient for microarray analyses. (B) Pearson regression analyses of top affected proteins in relation to CAV1 expression across ATL subtypes.
Discussion
Herein, we present a comprehensive, computational model of a CD4+ T-cell, including CAV1 regulatory pathways. This model incorporates experimentally validated interactions to posit the role of CAV1 in healthy CD4+ cells and CD4+ cells in the context of T-cell leukemia/lymphoma (i.e., when the immune response is skewed). CD4+ T-cells are a vital component of the immune system, as they protect against cancer, infection, and play a role in autoimmunity. CAV1 has been shown to be upregulated in numerous types of malignancies. Consequently, we built a CD4+ T-cell model to better understand basic T-cell biology and to address the role(s) of CAV1 within immunocompetent versus immunocompromised conditions. Inclusively, we investigated the role of CAV1 in the regulation of cellular processes, including cell cycle progression, cell proliferation, actin polymerization, and immune synapse formation. This model was successfully constructed using the Cell Collective platform that allows users to build cellular models capable of mimicking actual cellular systems in the laboratory (19–22, 38). The accuracy of the model was then successfully validated through the comparison of simulations with well-established, global input–output relationships as previously observed experimentally. Most importantly, new in silico predictions were validated in vitro/in vivo using both murine models and gene expression profiles from patients with a T-cell leukemia due to the previously observed role of Cav1 in lymphocytes (39–46).
Using Cell Collective, we were able to perform virtual experiments in order to make predictions as to how a CD4+ T-cell would behave when the expression levels of CAV1 were altered and when CAV1 was knocked down. These experiments provided insight as to which proteins, and ultimately which cellular functions, might be regulated by CAV1. Based on in silico results, we observed that BCL10, CD26, FYN, CARMA1, SHC1, SOS, SHP2, NOS2A, GRB2, and GATA3 are strongly influenced by CAV1 expression (Figure 3C). Consequently, the expression of these molecules in vivo was investigated using cluster analyses of microarray data to measure gene expression (Figure 5). Finally, four key proteins were selected for further verification of the predictions of the model using immunohistochemistry on mouse tissue with and without Cav1 (Figure 4).
In addition to the four key proteins, CD28 was chosen because it is known to regulate T-cell activation independent of CAV1 expression (15). RAC1 and GATA3 expression were upregulated in the lymph node tissue of the CAV1−/− mice. These results were expected based on studies showing the role of CAV1 in lymphoid tissue (9). We observed both CD26 and BCL10 upregulation, especially in the germinal centers of the lymph node and spleen tissue of the CAV1−/− mice. These validations of the in silico predictions show that the CD4+ T-cell model is biologically relevant. These validations of the in silico predictions show that the CD4+ T-cell model is biologically relevant. Therefore, we suggest that based on the validations in vivo the T-cell in silico model is predictive of biological processes.
We then translated the in silico predictions to gene expression profiles of patients with a T-cell malignancy (i.e., ATL). Of the top 15 most differentially expressed molecules in CAV1-mediated pathways, the top 4 most highly correlated molecules to CAV1 expression were BCL10, DEC2, SHP2, and GATA3 (R value > 0.74; Figure 5B). Obstinately, SOS1, FYN, SOS2, and CD26 were negatively correlated with CAV1 (R < 0.74; Figure 5B). Additionally, we observed unique clustering of these 15 conserved molecules across subtypes of ATL patients. Interestingly, there was a distinguishable differential expression of MALT1 in asymptomatic ATL cases as compared with healthy individuals (Figure 5A). Therefore, further investigation into the plausibility of these correlated molecules being used for diagnostic purposes is warranted. We hope that our other ongoing studies will shed light as to their role in ATL. In short, our data regarding the role of CAV1 in cell signaling (as demonstrated using an in silico software and subsequently validated experimentally) corroborate that of the existing literature.
Specifically, CAV1 participates in processes including actin polymerization, cell proliferation, and cell survival (9, 12, 15, 47–49).
The usefulness of the in silico approach combined with that of in vivo/in vitro approaches provide rapid information as to the cell signaling networks in healthy and leukemic cells. There are currently many therapies being used to treat leukemia that target specific proteins in order to inhibit cellular pathways. These treatment modalities are advanced when comprehensive, molecular models allow the researcher to observe the direct mechanism of action of gene targeting as well as downstream consequences of gene/protein knockdown. The CD4+ T-cell model will hopefully be able to provide insight to for both T-cell biology (as demonstrated herein) as well as possible targets for lymphocytic leukemia treatments. The importance of in silico approaches combined with immunoinformatics as well as in vivo validation cannot be understated (18). With the often-prohibitive cost of drug design, it is imperative to use computational approaches to derive and test hypotheses. Current therapy regimens for T-cell malignancies can be modeled in silico initially in an effort to understand mechanisms and potential outcomes.
A comprehensive model of the CD4+ cells has the potential to provide substantial insight into cancer treatment, immunotherapy, and cellular biology. This twofold approach incorporating in silico and in vivo investigations has the potential to translate diagnostics and therapeutic targets from bench to bedside.
Author Contributions
Brittany D. Conroy, Tyler A. Herek, and Timothy D. Shew are equally contributing first authors. Brittany D. Conroy wrote the paper, built the model, and did the simulations and verifications. Timothy D. Shew and Tyler A. Herek did bioinformatics studies, immunohistochemistry, verifications, and wrote the paper. Joshua J. Larson assisted in bioinformatics studies and mouse work. Laura Allen and Matthew Latner built the model, ran validations, and performed data analyses. Paul H. Davis provided suggestions for experimental design and helped implement experiments. Christine E. Cutucache and Tomáš Helikar conceptualized and designed the experiment, assisted in implementation, oversaw evaluation, and wrote the paper.
Conflict of Interest Statement
Tomáš Helikar is a founder and scientific advisor to Discovery Collective, Inc. Discovery Collective holds a license to use the Cell Collective software. All other authors declare no conflict of interest.
Acknowledgments
We thank Andrew Pulfer for his work in helping to construct the initial CD4+ T-cell model. Thanks to the Tissue Core Facility at the University of Nebraska Medical Center for assistance with histology. Additionally, we thank Nebraska NASA for Fellowships (Brittany D. Conroy and Christine E. Cutucache), the University of Nebraska at Omaha’s Sponsored Programs Office for internal funding through an UCRCA grant (Tomáš Helikar and Christine E. Cutucache), and thanks to Dr. George Haddix and the Nebraska University Foundation.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fimmu.2014.00599/abstract
References
1. Mercier I, Jasmin JF, Pavlides S, Minetti C, Flomenberg N, Pestell RG, et al. Clinical and translational implications of the caveolin gene family: lessons from mouse models and human genetic disorders. Lab Invest (2009) 89(6):614–23. doi: 10.1038/labinvest.2009.23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Lajoie P, Nabi IR. Lipid rafts, caveolae, and their endocytosis. Int Rev Cell Mol Biol (2010) 282:135–63. doi:10.1016/S1937-6448(10)82003-9
3. Kato K, Hida Y, Miyamoto M, Hashida H, Shinohara T, Itoh T, et al. Overexpression of caveolin-1 in esophageal squamous cell carcinoma correlates with lymph node metastasis and pathologic stage. Cancer (2002) 94(4):929–33. doi:10.1002/cncr.10329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Williams T, Lisanti M. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am J Physiol (2005) 288(3):C494–506. doi:10.1152/ajpcell.00458.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Yeh D, Chen C, Sun MZ, Shao S, Hao L, Song Y, et al. Caveolin-1 is an important factor for the metastasis and proliferation of human small cell lung cancer NCI-H446 cell. Anat Rec (2009) 292(10):1584–92. doi:10.1002/ar.20974
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Wiechen K, Sers C, Agoulnik A, Arlt K, Dietel M, Schlag PM, et al. Down-regulation of caveolin-1, a candidate tumor suppressor gene, in sarcomas. Am J Pathol (2001) 158(3):833–9. doi:10.1016/S0002-9440(10)64031-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Capozza F, Williams TM, Schubert W, McClain S, Bouzahzah B, Sotgia F, et al. Absence of caveolin-1 sensitizes mouse skin to carcinogen-induced epidermal hyperplasia and tumor formation. Am J Pathol (2003) 162(6):2029–39. doi:10.1016/S0002-9440(10)64335-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Kim EJ, Sohn JH, Kim M, Chae SW, Lee HS, Coo EY, et al. The loss of expression of caveolin-1 in gastrointestinal stromal tumors. Korean J Pathol (2005) 39(5):338–44.
9. Gilling CE, Mittal AK, Chaturvedi N, Iqbal J, Aoun P, Bierman P, et al. Lymph node-induced immune tolerance in chronic lymphocytic leukaemia: a role for caveolin-1. Br J Haematol (2012) 158(2):216–31. doi:10.1111/j.1365-2141.2012.09148.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Gilling CE, Mittal AK, Joshi SS. Immune synapse malformation: role for CAV1 and PAK1. FASEB J (2012) 26:522.7. doi:10.1096/fj.1530-6860
11. Proust R, Bertoglio J, Gesbert F. The adaptor protein SAP directly associates with CD3ζ chain and regulates T cell receptor signaling. PLoS One (2012) 7:8. doi:10.1371/journal.pone.0043200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Ohnuma K, Uchiyama M, Yamochi T, Nishibashi K, Hosono O, Takahashi N, et al. Caveolin-1 triggers T-cell activation via CD26 in association with CARMA1. J Biol Chem (2007) 282(13):10117–31. doi:10.1074/jbc.M609157200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Kortum RL, Fernandez MR, Costanzo-Garvey DL, Johnson HJ, Fisher KW, Volle DJ, et al. Caveolin-1 is required for kinase suppressor of Ras 1 (KSR1)-mediated ERK1/2 activation, H-rasv12-induced senescence and transformation. Mol Cell Biol (2014) 34(18):3461–72. doi:10.1128/MCB.01633-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Ishii T, Ohnuma K, Murakami A, Takasawa N, Kobayashi S, Dang NH, et al. CD26-mediated signaling for T cell activation occurs in lipid rafts through its association with CD45RO. Proc Natl Acad Sci U S A (2001) 98(21):12138–43. doi:10.1073/pnas.211439098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Tomassian T, Humphries LA, Liu SD, Silva O, Brooks DG, Miceli MC. Caveolin-1 orchestrates TCR synaptic polarity, signal specificity, and function in CD8 T cells. J Immunol (2011) 187(6):2993–3002. doi:10.4049/jimmunol.1101447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Burger JA, Kipps TJ. Chemokine receptors and stromal cells in the homing and homeostasis of chronic lymphocytic leukemia B cells. Leuk Lymphoma (2002) 43(3):461–6. doi:10.1080/10428190290011921
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Cutucache CE. Tumor-induced host immunosuppression: special focus on CLL. Int Immunopharmacol (2013) 17(1):35–41. doi:10.1016/j.intimp.2013.05.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Tomar N, De RK. Immunoinformatics: a brief review. Methods Mol Biol (2014) 1184:23–55. doi:10.1007/978-1-4939-1115-8_3
19. Helikar T, Konvalina J, Heidel J, Rogers JA. Emergent decision-making in biological signal transduction networks. Proc Natl Acad Sci U S A (2008) 105(6):1913–8. doi:10.1073/pnas.0705088105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Helikar T, Kowal B, McClenathan S, Bruckner M, Rowley T, Madrahimov A, et al. The cell collective: toward an open and collaborative approach to systems biology. BMC Syst Biol (2012) 6(1):96. doi:10.1186/1752-0509-6-96
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Madrahimov A, Helikar T, Kowal B, Lu G, Rogers J. Dynamics of influenza virus and human host interactions during infection and replication cycle. Bull Math Biol (2013) 75(6):988–1011. doi:10.1007/s11538-012-9777-2
22. Helikar T, Kochi N, Kowal B, Dimri M, Naramura M, Raja SM, et al. A comprehensive, multi-scale dynamical model of ErbB receptor signal transduction in human mammary epithelial cells. PLoS One (2013) 8:4. doi:10.1371/journal.pone.0061757
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Morris MK, Saez-Rodriguez J, Sorger PK, Lauffenburger DA. Logic-based models for the analysis of cell signaling networks. Biochemistry (2010) 49(15):3216–24. doi:10.1021/bi902202q
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Helikar T, Kowal B, Madrahimov A, Shrestha M, Pedersen J, Limbu K, et al. Bio-Logic Builder: a nontechnical tool for building dynamical, qualitative models. PLoS One (2012) 7(10):e46417. doi:10.1371/journal.pone.0046417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Naldi A, Carneiro J, Chaouiya C, Thieffry D. Diversity and plasticity of Th cell types predicted from regulatory network modelling. PLoS Comput Biol (2010) 6(9):e1000912. doi:10.1371/journal.pcbi.1000912
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Chaouiya C, Bérenguier D, Keating SM, Naldi A, van Iersel MP, Rodriguez N, et al. SBML qualitative models: a model representation format and infrastructure to foster interactions between qualitative modelling formalisms and tools. BMC Syst Biol (2013) 7:135. doi:10.1186/1752-0509-7-135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Alonso MA, Millán J. The role of lipid rafts in signaling and membrane trafficking in T lymphocytes. J Cell Sci (2001) 114:3957–65.
28. Kumar CC. Signaling by integrin receptors. Oncogene (1998) 17:1365–73. doi:10.1038/sj.onc.1202172
29. Shi CS, Kehrl JR. PYK2 links G(q)alpha and G(13)alpha signaling to NF-kappa B activation. J Biol Chem (2001) 276(34):31845–50. doi:10.1074/jbc.M101043200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Change F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta (2007) 1773(8):1263–84. doi:10.1016/j.bbamcr.2006.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Lundin Brockdorff J, Woetmann A, Mustelin T, Kaltoft K, Zhang Q, Wasik MA, et al. SHP2 regulates IL-2 induced MAPK activation, but not STAT3 or STAT5 tyrosine phosphorylation, incutaneous T cell lymphoma cells. Cytokine (2002) 4:141–7. doi:10.1006/cyto.2002.1986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol (2007) 178:280–90. doi:10.4049/jimmunol.178.1.280
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Hofmann C, Shepelev M, Chernoff J. The genetics of Pak. J Cell Sci (2004) 117(19):4343–54. doi:10.1242/jcs.01392
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Iqbal J, Kucuk C, Deleeuw RJ, Srivastava G, Tam W, Geng H, et al. Genomic analyses reveal global functional alterations that promote tumor growth and novel tumor suppressor genes in natural killer-cell malignancies. Leukemia (2009) 23(6):1139–51. doi:10.1038/leu.2009.3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Mittal AK, Gilling CE, Iqbal J, Bociek RG, Aoun P, Bierman P, et al. Clinical heterogeneity of CLL: role for immune dysregulation mediated by the lymph node microenvironment. Blood (2009) 546:112.
36. Helikar T, Rogers JA. ChemChains: a platform for simulation and analysis of biochemical networks aimed to laboratory scientists. BMC Syst Biol (2009) 3:58. doi:10.1186/1752-0509-3-58
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Kobayashi S, Nakano K, Watanabe E, Ishigaki T, Ohno N, Yuji K, et al. CADM1 expression and stepwise downregulation of CD7 are closely associated with clonal expansion of HTLV-1-infected cells in adult T-cell leukemia/lymphoma. Clin Cancer Res (2014). doi:10.1158/1078-0432.CCR-13-3169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Todd R, Helikar T. Ergodic sets as cell phenotype of budding yeast cell cycle. PLoS One (2012) 7(10):e45780. doi:10.1371/journal.pone.0045780
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Fischer KD, Kong YY, Nishina H, Tedford K, Marengere LEM, Kozieradzki I, et al. Vav is a regulator of cytoskeletal reorganization mediated by the T-cell receptor. Curr Biol (1998) 8(10):554–62. doi:10.1016/S0960-9822(98)70224-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Wary KK, Mariotti A, Zurzolo C, Giancotti FG. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell (1998) 94(5):625–34. doi:10.1016/S0092-8674(00)81604-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signaling to actin cytoskeletal dynamics. Nat Cell Biol (1999) 1(5):253–9. doi:10.1038/12963
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Egawa T, Albrecht B, Favier B, Sunshine MJ, Mirchandani K, O’Brien W, et al. Requirement for CARMA1 in antigen receptor-induced NF-κB activation and lymphocyte proliferation. Curr Biol (2003) 13(14):1252–8. doi:10.1016/S0960-9822(03)00491-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-κB functions in synaptic signaling and behavior. Nat Neurosci (2003) 6(10):1072–8. doi:10.1038/nn1110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Dutt P, Nguyen N, Toksoz D. Role of Lbc RhoGEF in Ga12/13-induced signals to Rho GTPase. Cell Signal (2004) 16(2):201–9. doi:10.1016/S0898-6568(03)00132-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Görgün G, Holderried TA, Zahrieh D, Neuberg D, Gribben JG. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J Clin Invest (2005) 115(7):1797–805. doi:10.1172/JCI24176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Garcia-Closas M, Troester MA, Qi Y, Langerød A, Yeager M, Lissowska J, et al. Common genetic variation in GATA-binding protein 3 and differential susceptibility to breast cancer by estrogen receptor alpha tumor status. Cancer Epidemiol Biomarkers Prev (2007) 16(11):2269–75. doi:10.1158/1055-9965.EPI-07-0449
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Lee H, Volonte D, Galbiati F, Iyengar P, Lublin DM, Bregman DB, et al. Constitutive and growth factor-regulated phosphorylation of caveolin-1 occurs at the same site (Tyr-14) in vivo: identification of a c-Src/Cav-1/Grb7 signaling cassette. Mol Endocrinol (2000) 14(11):1750–75. doi:10.1210/mend.14.11.0553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Sasahara Y, Rachid R, Byrne MJ, de la Fuente MA, Abraham RT, Ramesh N, et al. Mechanism of recruitment of WASP to the immunological synapse and of its activation following TCR ligation. Mol Cell (2002) 10(6):1269–81. doi:10.1016/S1097-2765(02)00728-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: caveolin-1, CD4+ T-lymphocyte, the cell collective, adult T-cell leukemia, immunosuppression, immunotherapy, computational biology, logical models
Citation: Conroy BD, Herek TA, Shew TD, Latner M, Larson JJ, Allen L, Davis PH, Helikar T and Cutucache CE (2014) Design, assessment, and in vivo evaluation of a computational model illustrating the role of CAV1 in CD4+ T-lymphocytes. Front. Immunol. 5:599. doi: 10.3389/fimmu.2014.00599
Received: 25 August 2014; Accepted: 07 November 2014;
Published online: 05 December 2014.
Edited by:
Sergio Quezada, University College London Cancer Institute, UKCopyright: © 2014 Conroy, Herek, Shew, Latner, Larson, Allen, Davis, Helikar and Cutucache. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christine E. Cutucache, Department of Biology, University of Nebraska at Omaha, Allwine Hall 413, 6001 Dodge Street, Omaha, NE 68182, USA e-mail:Y2N1dHVjYWNoZUB1bm9tYWhhLmVkdQ==
†Co-first authors of this manuscript