 Eric Muraille
Eric Muraille Oberdan Leo
Oberdan Leo Muriel Moser
Muriel Moser- 1Laboratory of Parasitology, Faculty of Medicine, Université Libre de Bruxelles, Brussels, Belgium
- 2Laboratory of Immunobiology, Faculty of Sciences, Université Libre de Bruxelles, Gosselies, Belgium
The classical view of the Th1/Th2 paradigm posits that the pathogen nature, infectious cycle, and persistence represent key parameters controlling the choice of effector mechanisms operating during an immune response. Thus, efficient Th1 responses are triggered by replicating intracellular pathogens, while Th2 responses would control helminth infection and promote tissue repair during the resolution phase of an infectious event. However, this vision does not account for a growing body of data describing how pathogens exploit the polarization of the host immune response to their own benefit. Recently, the study of macrophages has illustrated a novel aspect of this arm race between pathogens and the immune system, and the central role of macrophages in homeostasis, repair and defense of all tissues is now fully appreciated. Like T lymphocytes, macrophages differentiate into distinct effectors including classically (M1) and alternatively (M2) activated macrophages. Interestingly, in addition to represent immune effectors, M1/M2 cells have been shown to represent potential reservoir cells to a wide range of intracellular pathogens. Subversion of macrophage cell metabolism by microbes appears as a recently uncovered immune escape strategy. Upon infection, several microbial agents have been shown to activate host metabolic pathways leading to the production of nutrients necessary to their long-term persistence in host. The purpose of this review is to summarize and discuss the strategies employed by pathogens to manipulate macrophage differentiation, and in particular their basic cell metabolism, to favor their own growth while avoiding immune control.
Th1/Th2 Paradigm and Infectious Immunology
Historically, immunology emerged as an independent scientific discipline whose aim was to understand and possibly ameliorate the development of vaccines, a medical practice that was mostly pioneered, in an empirical fashion, by microbiologists of the end of the 19th century. Notably, however, the establishment of fundamental immunological concepts and paradigms, such as B and T cell collaboration or tolerance, was based on a reductionist (or analytical) approach that departed from a classical “host-pathogen” view of immunology, by putting much emphasis on self/non-self discrimination concepts explored with simpler protein antigens such as ovalbumin or keyhole limpet hemocyanin (KLH).
The discovery that CD4+ T cells can be characterized by distinct “cytokine signatures” by Mosmann and Coffman in 1986 (1) marked the great return of infection in immunology, leading to emergence of the Th1/Th2 “division of labor” paradigm in 1989, and its application to the field of vaccinology since the early 1990s. This concept led to a critical appraisal of the multiple and diverse sets of immune effectors promoting the now well established idea that the best immune response is often not the most intense nor the most specific but rather the best adapted to counteract or control the pathogen infectious cycle in the host. In consequence, identification of the nature of pathogens, selection of the appropriate immune response, repression of damaging or inadequate immune response are now recognized as crucial steps for the successful control of an infectious event. Thus, the immune reaction must be adapted to eliminate or control the pathogen and to restore homeostatic conditions during an infection to prevent severe tissue damage (2); for review see Ref. (3). The high frequency of chronic infections observed in nature demonstrates that pathogens are, however, able to manipulate these responses to evade immune control or even subvert immune reactions to their own advantage.
This review highlights current understanding of the interplay between pathogens and macrophages with a special emphasis on the role of metabolism regulation in the control of infections in mouse models unless otherwise stated. A clear understanding of the importance of macrophage polarization may reveal novel strategies for controlling infectious diseases.
Classically and Alternatively Activated Macrophage
Myelomonocytic cells are an essential component of innate immunity and represent the first line of defense against pathogens. Myelomonocytic cells fulfill a variety of homeostatic functions, which go beyond host defense and include tissue remodeling during embryogenesis, wound healing (fibrin dissolution, removal of dead tissues, fibroblast recruitment and growth and connective-tissue remodeling), as well as orchestration and contribution to metabolic activity [reviewed in Ref. (4–6)]. Myelomonocytic cell function needs to be tailored to their tissue of residence, an adaptation that is driven by tissue-derived factors and by the physiological environment.
The myelomonocytic cell populations are particularly dynamic during inflammation or infection. Under such conditions, blood monocytes are recruited into the tissues, where they differentiate into macrophages or dendritic cells. Depending on the microenvironment, macrophages can acquire distinct functional phenotypes. The concept of macrophage polarization was first defined in 1992 with the discovery that IL-4 inhibits the respiratory burst of macrophages while enhancing expression of MHC-II and mannose receptors (CD206) on their cell surface (7). Since then, two opposite and competing phenotypes, often referred to as classically activated macrophages (M1 macrophages) and alternatively activated macrophages (M2 macrophages) [reviewed in Ref. (8–10)] have been defined and identified in several physiological settings. The M1/M2 nomenclature is derived from the Th1 and Th2 cytokines that are associated with these macrophage phenotypes.
M1 macrophages differentiate under the influence of IFN-γ and/or LPS and display functional subdivisions depending on stimulation (11). They are usually characterized by increased microbicidal activity, as witnessed by the secretion of high levels of pro-inflammatory cytokines such as TNFα and IL-6, production of reactive oxygen intermediates (ROI) and nitric oxide synthase-2 (NOS-2/iNOS)-dependent reactive nitrogen intermediates (RNI); high antigen-presenting activity and increased production of IL-12. These characteristics are promoted by IFN-γ-mediated Janus kinase–signal transducer and activator of transcription (JAK–STAT) signaling or directly by pathogen associated molecular patterns (PAMPs) such as LPS. M1 macrophages constitute the first line of defense against intracellular pathogens and promote or amplify Th1 polarization of CD4+ lymphocytes by IL-12 production.
M2 macrophages have been initially identified in the context of infection by helminths, differentiating from monocytic precursors under the influence of IL-4 and IL-13 produced during a strongly Th2-polarized response. M2-like cells have been described in different pathological conditions such as infections by intracellular bacteria or virus, allergy, diabetes, and cancer [reviewed in Ref. (8, 9, 12)]. They are characterized by the selective expression of markers such as arginase 1 (Arg1), chitinase-like protein (for example, Ym1), Fizz1 (Found in Inflammatory Zone 1), CD36 (fatty acid translocase), and CD206, as well as the production of low levels of IL-12 and iNOS. Depending on the context, differences in the expression of M2-associated phenotypic markers have been recognized, leading to a redefinition of this cell population to accommodate a diverse set of subtypes: M2a (induced by exposure to IL-4 or IL-13) and M2b [induced by stimulation with immune complexes, TLR, or the IL-1 receptor antagonist (IL-1ra)] macrophages drive Th2 responses, whereas M2c cells (generated by stimulation with IL-10) play a predominant role in the suppression of immune responses and tissue remodeling [reviewed in Ref. (13)].
In addition, M1 and M2 macrophages differentially express a panel of co-stimulatory receptors of the B7 family. M1 macrophages have been shown to express higher levels of CD86 and PD-L1, whereas M2 macrophages display elevated levels of PD-L2. The expression differences are less clear for CD80 (14, 15).
It should be noted that macrophages with intermediate or overlapping phenotypes have been observed in vivo. For example, adipose tissue macrophages from obese mice have a mixed profile, with upregulation of several M1 and M2 gene transcripts (16). Taken together, these observations suggest that the prototypical M1 and M2 phenotypes probably represent extremes of a continuum spectrum of functionally distinct cell types. In this review, we will focus mainly on the role of M1 and M2a macrophages, these latter identified on the basis of high Arg1 expression and STAT6 pathways dependence.
Macrophage Polarization is Associated to a Shift of Metabolic Program
M1 and M2 macrophages display a drastic shift in the amino acid, glucose, lipid, and iron metabolism [reviewed in Ref. (6, 17)]. We discuss several aspects of metabolic shift in relation to pathogen growth control.
Amino Acid Metabolic Shift
Amino acid catabolism represents a key mechanism whereby M1 and M2-types macrophages exert their anti-microbial and immunoregulatory roles. Tryptophan and arginine degradation by innate immune cells represents the most studied examples of functional links between amino acid metabolism and immune function. This relationship is quite complex, because selected degradation of amino acids can not only affect an immune response through the depletion of important precursors to protein synthesis, but also generates new catabolites endowed with immunoregulatory functions. In keeping with the general purpose of this review, we will mostly discuss herein the known links between expression and function of amino acid catabolizing enzymes and M1/M2 differentiation.
Among the three different enzymes, indoleamine 2,3-dioxygenase 1 (IDO1), 2 (IDO2), and tryptophan 2,3-dioxygenase (TDO), which catalyze oxidative Trp catabolism, IDO1 represents the best known and widely studied example illustrating how amino acid metabolism and immune regulation interface. IDO1 has been shown to confer potent anti-microbial activities in vitro. Through the depletion of the essential amino acid tryptophan, IDO1 can restrict in vitro the growth of a wide range of pathogens, including viruses, bacteria, and protozoa (18–22). Although IDO1 is constitutively expressed by numerous tissues including the epididymis, uterus, spleen, small intestines, and prostate (23), its expression is highly inducible by both type I and type II interferons, with IFN-γ considered as one of the strongest inducers of IDO expression and activity. As a consequence, and despite the fact that IDO1 expression is not restricted to hematopoietic cells, some authors have come to consider IDO1 as a prototypical M1 marker (11).
The role of IDO1 as potent anti-microbial agent in vivo remains, however, questionable, due in particular to the well-known immunosuppressive function of IDO1-expressing cells. Increased tryptophan catabolism has been indeed observed in the tumor microenvironment and in the placenta, contributing to tolerance to, respectively, tumor and fetal antigens. Although apparently at odds with the anti-microbial properties of IDO1, recent observations seem to concur with the notion that despite its association to an M1-like state, IDO1 may indeed primarily mediate anti-inflammatory/regulatory roles in vivo opposing pathogen clearance. Indeed, genetic IDO1 ablation or pharmacological inhibition of IDO reduced parasitic load in Leishmania major infected mice (24). Similarly, in vivo administration of a pharmacological inhibitor of IDO to Toxoplasma gondii infected mice or infection of an IDO1-deficient mouse strain led to a more efficient control of parasite growth by IDO1-impaired hosts (25). In keeping with this prevalent immunoregulatory role of IDO1 in vivo, inhibition of IDO1 activity enhanced both CD4 and CD8 T cell responses to influenza virus infection, globally improving the antigen-specific memory response to this virus (26, 27).
Recent observations, albeit performed exclusively in vitro, have shed a new light on the possible role of IDO1 in the M1 vs M2 differentiation pathways (28). In this study, the authors demonstrate that forced expression of IDO1 in the human acute monocyte leukemia cell line THP-1 is sufficient to induce an M2-like profile, characterized by high IL-10 and low IL-12 expression pattern. Similarly, siRNA-mediated inhibition of IDO1 expression led to an increased expression of M1 markers, suggesting an important and cell autonomous role for IDO1 in regulating macrophage polarization. Collectively, and although the molecular and cellular mechanisms need to be identified, the available evidence suggests the existence of negative feedback regulatory loop whereby expression of IDO1 under M1-polarizing conditions contributes to the attenuation of a T cell mediated inflammatory response while favoring a M1 to M2 shift in macrophages.
Elevated arginine catabolism has also been linked to immunoregulation and anti-microbial immunity. Noteworthy, two of the prototypic M1 and M2 markers (respectively, iNOS and Arg1) have the capacity to use l-arginine as a substrate, leading to the production of l-citrulline and nitric oxide (NO), or l-ornithine and urea, respectively. The finding that concentrations of l-arginine at site of inflammation often decline to undetectable levels suggests an important role for l-arginine catabolism during an immune response (29, 30).
Products of Arg1 vs iNOS appear to fulfill diametrically opposed functions (Figure 1). Arg1 enhances collagen synthesis and cell growth via l-ornithine production, while iNOS opposes cell viability and proliferation. Moreover, iNOS protein translation appears as particularly sensitive to l-arginine levels, providing a further mechanism, in addition to competition for the same substrate, of counter-regulation between these two enzymes (31). This substrate competition between iNOS and Arg1 has been extensively described in mouse models (32), and further studies may be required to validate the universality of this concept in other species. Arginase activity has been found in lesions of patients with cutaneous leishmaniasis and in human tuberculous granulomas, suggesting an evolutionary conserved response linking arginine metabolism to infection (33–35).
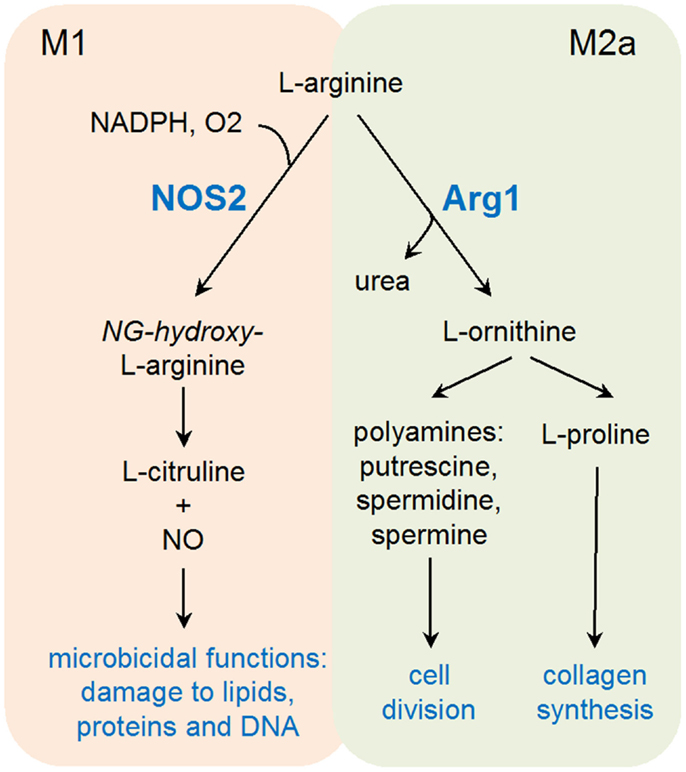
Figure 1. Schematic representation of l-arginine catabolism in M1 and M2a-type macrophages.
M1-derived NO represents a major effector molecule in macrophage-mediated cytotoxicity, playing an important role in controlling bacterial and parasitic infections (36–38). In keeping with the previously discussed antagonistic role of Arg1 and iNOS, induction of arginase activity represents an efficient immune escape strategy developed by several pathogens. As an example, active induction of Arg1 gene transcription in infected macrophages through manipulation of the STAT6 signaling pathway has been demonstrated for both T. gondii and Leishmania donovanii (39–41). The discovery that T cells are particularly sensitive to local arginine concentrations has further confirmed the important immunoregulatory role of Arg1, able therefore to inhibit both innate (NO production) and adaptive immune responses to parasites (30, 32). In addition to its immunoregulatory role, increased arginine metabolism has been shown to directly promote intracellular growth of several Leishmania species, notably through the accumulation of polyamines (42, 43). A compelling argument in favor of a role of arginine catabolism in promoting parasite growth is the expression of endogenous arginase by several parasites including Leishmania, Crithidia, and Leptomonas [see Ref. (44) for review].
Most of the available evidence links therefore Arg1 expression, and thus the M2 phenotype, to a disease-promoting activity. Noteworthy, however, expression of Arg1 in macrophages also plays an important role in protecting the host against the lethal effects of chronic Th2 pathology, as recently demonstrated in mice infected by Schistosoma mansoni (45). A similar role for Arg1 in suppressing pathologic inflammation has been derived from studies conducted in acutely S. mansoni-infected Arg1-deficient bone marrow chimeric mice (46). Finally, the importance of Arg1 in wound healing has been recently demonstrated using both pharmacological inhibition of arginase activity and genetic deletion of Arg1 in both haematopoietic and endothelial cells, further illustrating the important and positive role of this enzyme in tissue repair and homeostasis (47).
Although expression of Arg1 is often associated to an immunoregulatory/pathogen-growth promoting setting, recent studies have uncovered a previously unsuspected effector role for this enzyme against nematode infection. In models of secondary infections, arginase activity was found to play a key role in the acquired protective immunity against reinfection with an intestinal helminth. Upon primary exposure to Nippostrongylus brasiliensis, mice developed the capacity to retain larvae in the infected skin, thus reducing the worm burden in the lungs (48). In this model, Arg1 was shown to play a key role in larvae trapping, confirming previously suspected antiparasitic effects of arginase activity against Heligmosomoides polygyrus (49). The capacity of the arginine catabolite l-ornithine to inhibit H. polygyrus larvae mobility suggests a plausible mechanism whereby Arg1 may protect the host during parasitic infections (50).
In summary, catabolism of both tryptophan and l-arginine lays at the heart of the M1/M2 dichotomy. Arginine catabolism represents an important immune mechanism limiting pathogen growth and spread, exploited by both M1 (through iNOS) and M2 (via Arg1) – like macrophages. Through distinct and mutually exclusive enzymatic reactions, the same amino acid can therefore be exploited to generate effector molecules tailored to different pathogens. Moreover, both iNOS and Arg1 have also been shown to dampen excessive inflammatory and T-cell mediated reactions. Whether these counter-regulatory properties have been evolutionary selected to control unwanted immune activity or represent unintended side effects remains to be established. In marked contrast, it is difficult at present to evaluate the precise role of IDO1 as an anti-microbial effector. Despite a large body of in vitro data, a role for IDO1 in limiting pathogen growth in vivo is still lacking, suggesting that despite being associated to an M1-like subpopulation of cells, tryptophan catabolism may primarily serve a regulatory role in vivo. In conclusion, amino acid catabolism elegantly illustrates the dual function of M1 and M2 macrophages, both subsets being able to express anti-microbial and immunoregulatory functions.
Hypoxia
Decrease in oxygen pressure (hypoxia) in tissues can result from various causes such as mechanical or infection induced inflammation, intense metabolic activity but also obesity and tumor growth [reviewed in Ref. (51)]. The transcription factor hypoxia-inducible factor-1 (HIF-1) is a central mediator of hypoxic adaptation. In normoxia, HIF-1 is repressed primarily through the action of a family of hydroxylases, which targets HIF-1 subunits for degradation in an oxygen-dependent manner. In hypoxia, HIF-1 is rapidly stabilized in cells and induces the expression of hundreds of genes, which regulate angiogenesis, metabolism, growth and survival (50).
In 1938, Kempner was the first to associate cellular metabolism and inflammation: he examined the chemical composition of control or inflamed tissues and reported chemical changes that he interpreted as mainly due to the aerobic glycolysis of blood cells (52). There is indeed growing evidence that tissue foci of inflammation display a declining oxygen gradient, as compared to oxygen-rich blood stream, leading to increased cellular HIF-1α levels particularly in phagocytes, and activation of genes required to maintain viability and activity in these demanding conditions (low oxygen and glucose). It is noteworthy that HIF-1α may also exert regulatory effects in normoxia, in particular during bacterial infection, where HIF-1α expression is stimulated through TLR receptor engagement and cell signaling pathways (such as NF-κB and MAPK) [for review see Ref. (53)].
Exposure of primary human monocyte-derived macrophages to hypoxia for 16 h has been shown to result in increased mRNA levels for vascular endothelial growth factor (VEGF), glucose transporter (GLUT-1), and matrix metalloproteinase 7 (MMP-7) (54). Induction of VEGF in hypoxic conditions is involved in the pro-angiogenic activities of macrophages in tumors, whereas GLUT-1, a glucose transporter, may favor macrophage survival in ischemic tissues by increasing glucose uptake for glycolytic production of energy (Figure 2). The increase in mRNA coding for MMP-7 could have several consequences, as MMP-7, the smallest MMP, is able to digest many components of the basement membrane and extracellular matrix, and is involved in proteolytic processing, leading to activation of TNF-α, defensins, and other MMPs.
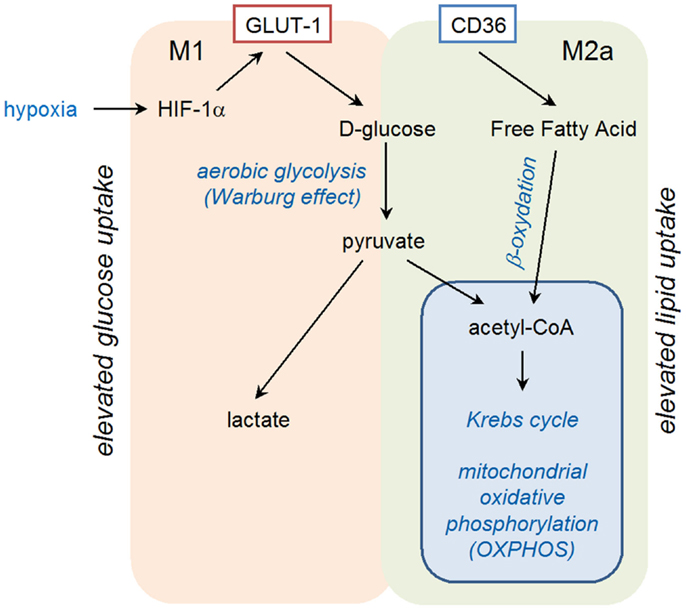
Figure 2. Schematic representation of polarized metabolism in M1 and M2a-type macrophages.
A number of reports have shown that hypoxia profoundly affects macrophage recruitment and development of M1 associated anti-microbial functions. Specific deletion of HIF-1α in the myeloid lineage resulted in lower ATP levels caused by decreased glycolysis. This metabolic defect results in profound impairment of myeloid cell aggregation, motility, invasiveness, and bacterial killing (55). Analysis of intracellular killing of Group B streptococci by bone marrow-derived macrophages revealed approximately sevenfold more viable bacteria within the HIF-1α deficient macrophages as compared with the wild-type cells. In vivo, loss of HIF-1α in myeloid cells impaired infiltration at the dermal-epidermal border of inflamed skin and prevented resolution of passively induced arthritis. HIF-1α agonist mimosine and the pharmacological agent AKB-4924 (which stabilizes HIF-1) were both shown to enhance the antibacterial activity of phagocytes and to kill the pathogen Staphylococcus aureus in vitro and in vivo (56, 57). Conversely, mice lacking HIF-1α in their myeloid lineage developed larger necrotic skin lesions upon subcutaneous infection with group A Streptococcus (58). Murine macrophages exposed to low oxygen tensions have been shown to display M1 phenotype as suggested by increased antigen-presenting and phagocytic functions that involved IFN-γ production (59).
Collectively, these observations indicate that HIF-1α critically regulates pathways essential for the maintenance of energy homeostasis and function of macrophages in sites deprived of oxygen. However, a recent report contradicts this conclusion, as the authors found that hypoxia in L. major skin lesions impaired the NO-dependent leishmanicidal activity of macrophages in a reversible manner. The mechanism was found to involve impaired translation of NOS protein (60). This latter study is in accordance with two recent reports showing (i) that tumor-infiltrating macrophages in hypoxic areas displayed an altered pro-tumoral phenotype, characterized by impaired M1-type function (61) and (ii) that tumor-derived lactic acid induced vegf and Arg1 and the M2-like polarization of tumor-associated macrophages in a HIF-1α-dependent manner (62).
Iron Metabolism
Iron is an ideal redox catalyst, accepting or donating electrons, implicated in numerous cellular processes such as respiration, DNA replication but also host defenses by the production of reactive oxygen and nitrogen intermediates. Almost all living organisms from archaea to eukaryotes display absolute requirement for iron [reviewed in Ref. (63–65)]. Erythropoiesis is the most avid consumer of iron in the mammal organism as erythrocytes contain approximately 1 billion of ferric atoms. Approximately 60–70% of the human adult body iron is bound within hemoglobin (2.5 g). Macrophages play a central role in regulating iron metabolism since they recycle heme iron from senescent erythrocytes and regulate its storage. On the other hand, microbes have evoked multiple strategies to utilize iron because a sufficient supply of this metal is linked to pathogen proliferation, virulence and persistence. The expression of iron uptake systems is linked to virulence in a broad range of pathogens including bacteria, protozoa and fungi. The control over iron homeostasis is thus of central importance in host–pathogen interaction, in which both opponents compete for iron. A complex network of host proteins renders this valuable nutrient largely inaccessible to pathogens, a concept usually known as “nutritional immunity.” Of course, control of intra- and extracellular pathogens requires distinct mechanisms of iron restriction in different compartments. M1 macrophages, by repressing ferroportin (a cellular iron exporter) and CD163 (a hemoglobin scavenger receptor) and inducing ferritin (which favors iron intracellular sequestration and storage), reduce the labile iron pool (LIP), the metabolically active fraction of cytosolic iron that is available for metabolic purposes. In contrast, through the upregulation of ferroportin and the downregulation of ferritin, M2 macrophages have reduced iron storage and enhanced release of iron (66). Sequestration of iron by M1 macrophages would have a bacteriostatic effect (since iron is essential to support bacterial growth) and thus represents a host protective response. Conversely, iron release from M2 macrophages would favor tissue repair. Interestingly, iron is also an important regulator of immune effector functions, immune cell proliferation and cytokine production. Iron antagonizes the IFN-γ-induced expression of MHC-II, iNOS, and TNF-α and shifts the Th1/Th2 differentiation toward a Th2 reaction.
Lipid Oxidative Metabolism
Peroxisome proliferator activated receptors (PPARs) are lipid-activated transcription factors of the nuclear hormone receptor superfamily. Endogenous ligands that regulate the PPAR family remain poorly characterized and include free native and modified fatty acids. PPARs regulate expression of target genes as obligate heterodimers with the retinoic X receptors (RXRs). In mammals, there are three PPAR isoforms (PPARα, -γ, -δ) expressed in a large variety of cells and playing pleiotropic functions in fatty acid metabolism including transport, synthesis storage, mobilization, and oxidation [reviewed in Ref. (67)].
Binding of IL-4 or IL-13 to their cognate receptors, IL-4Rα/IL-2Rγ and IL-4Rα/IL-13Rα1, respectively, initiates a cytoplasmic signaling cascade leading to tyrosine phosphorylation of transcription factor STAT6. Phosphorylated STAT6 dimerizes and translocates to the nucleus, where it induces the expression of numerous target genes. Among them, PPARγ, PPARδ, and PPARγ-coactivator-1β (PGC-1β) appear particularly important. PPARγ mediates the expression of the main M2 phenotype markers such as Arg1 and CD36 (favoring the uptake of fatty acid and apoptotic cells) and increases oxidation metabolism of fatty acid [reviewed in Ref. (68)] (Figure 2). By its transrepressive action, PPARγ blocks the expression of numerous pro-inflammatory mediators induced by LPS and IFN-γ such as IL-1β and iNOS. Although PPARγ is not essential for monocyte/macrophage differentiation, it functions as an important modulator of macrophage lipid metabolism and a fine-tuner of immune functions (Figure 3). Whereas the instructions for alternative macrophage activation are provided by IL-4 and IL-13, the acquisition and long-term maintenance of this phenotype implicate PPARγ and PPARδ. PPARγ KO mice are deficient in M2 macrophages, develop spontaneous chronic Th1 inflammation in lung (69) and are more susceptible to obesity and insulin resistance, suggesting that homeostatic functions performed by M2 macrophages might allow animals to more efficiently store and oxidize incoming lipids, thereby maintaining insulin sensitivity and glucose tolerance by attenuating inflammation (70).
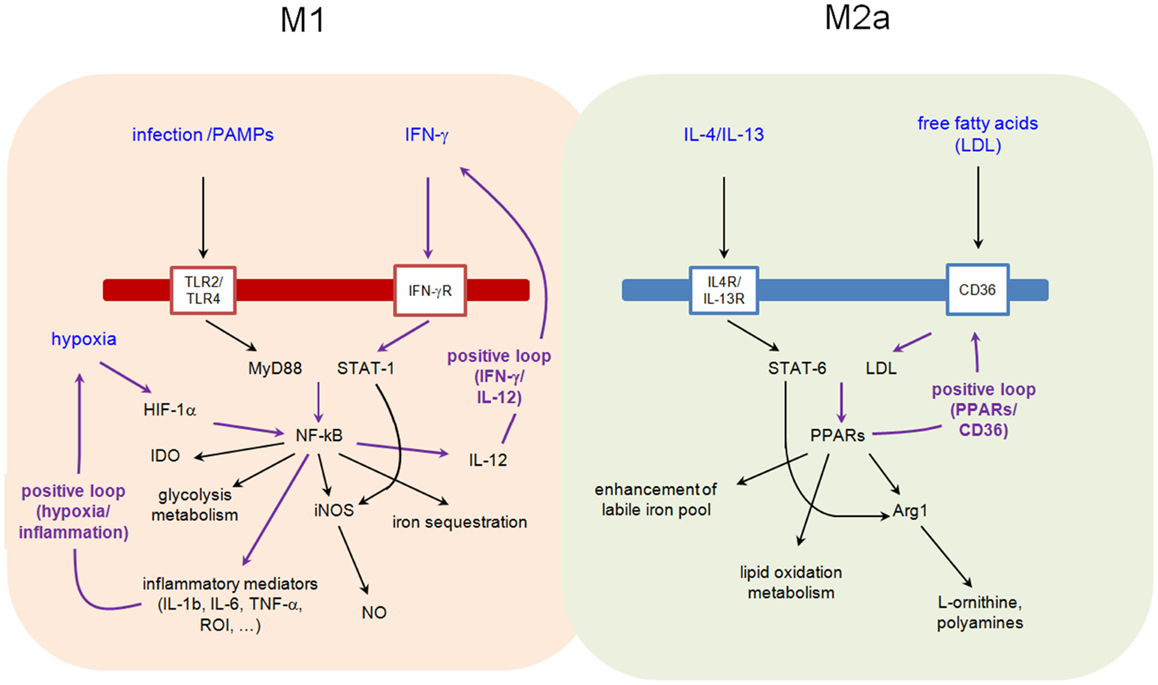
Figure 3. Interplay between metabolic pathways and M1 and M2a functions.
Reciprocally, fatty acids facilitate the acquisition and maintenance of M2 phenotype (Figure 3). In a PPARδ-dependent manner, monounsaturated fatty acids, such as oleic acid, were found to synergize with IL-4 to enhance the expression of alternative activation signature genes such as Arg1 in macrophages. Arg1 promoter is directly activated by PPAR/RXR heterodimers [reviewed in Ref. (67)] providing a molecular explanation for the observed synergy between nutrients and Il-4/IL-13 signaling pathways.
The Energy Sensors: AMPK and mTOR
The AMP-activated kinase (AMPK) represents a well described energy sensor able to adapt cell metabolism to available substrates and/or cellular energetic requirements. Accumulation of cellular AMP, indicative of a reduction in the energy status of the cell, activates the AMPK, which in turn will phosphorylate a series of substrates to favor metabolic reactions (such as stimulation of mitochondrial biogenesis, oxidative metabolism and autophagy), while decreasing ATP consuming, biosynthetic pathways. The mammalian target of rapamycin (mTOR) plays a similar role, by adapting metabolic reactions (such as protein synthesis) to nutrient availability in order to preserve cellular homeostasis. Pathogens have been shown to manipulate these regulatory pathways to highjack cell metabolism to their own benefit [reviewed in Ref. (71, 72)]. Collectively, these studies have highlighted how, by inhibiting AMPK and/or maintaining an activated mTOR pathway, pathogens generate essential substrates for their own replication. Notably, a link between macrophage metabolism and inflammation has also been recently established. A reduction in macrophage AMPK activity is associated with M1-like, pro-inflammatory status, while increased AMPK activity promotes an anti-inflammatory, M2-type, response [for review see Ref. (73)]. Inhibition of AMPK expression or activity promotes expression of pro-inflammatory mediators in macrophages activated by LPS (74). Similarly, AMPKα1 KO macrophages failed to acquire an M2 phenotype, retaining high levels of iNOS expression even when stimulated in the presence of M2-polarizing factors such as IL-4 (75).
In summary, polarization states and functional properties of macrophages largely depend on environmental conditions, such as hypoxia, cytokines, pathogen-derived TLR-ligands, and lipid mediators. Metabolism shift associated to macrophage polarization appears highly adapted to microbicidal and tissue regeneration function of M1 and M2 macrophages, respectively.
M2-Like Polarization as a Pathogen-Induced Immune Escape Strategy
The interaction between pathogenic microbes and their host is determined by survival strategies to compete for essential nutrients. During evolution, microbes have developed strategies to access selected nutrients from the host, while these, in return, have devised defensive means to restrict their availability to pathogens. Long-term pathogen persistence is frequently the consequence of a subtle equilibrium between the nutritive needs of host and pathogens. M2 macrophages appear as a favorable niche for long-term persistence of numerous intracellular pathogens. Some evidences suggest that intracellular pathogens can induce or benefit from PPAR expression and/or high iron availability in infected macrophages.
Mycobacterium tuberculosis, the agent of tuberculosis, is well adapted to survive within the hostile macrophage environment. Current evidence (76) indicates that M. tuberculosis and BCG infection causes a time-dependent upregulation of PPARγ expression in infected macrophages. Even uptake of dead bacteria triggered PPARγ expression in macrophages, suggesting a pattern recognition receptor-mediated triggering mechanism. Accordingly, recent studies have shown the implication of TLR2 (77) and mannose receptor (CD206) (78) in this process. PPARγ expression and activation led to increased lipid droplet formation, expression of M2 markers and downmodulation of bactericidal M1 response including respiratory burst and NO production. Involvement of CD36 in lipid droplet formation was further confirmed by decreased BCG-induced lipid droplet formation in CD36 deficient macrophages (77). These observations suggest that PPARγ expression may not only represent an escape mechanism to circumvent the protective host response, but may also provide the nutrient rich environment required for mycobacterial growth and survival by promoting host lipid metabolism.
A key anti-pathogen effector of M1 macrophages is NO, which is involved in direct killing of pathogens, such as M. tuberculosis. Interestingly, a report has shown that Arg1 was induced in macrophages in vitro via a STAT6 independent and TLR dependent pathway, and prevented NO production. The absence of Arg1 was associated with increased NO expression and enhanced control of mycobacteria. These observations suggest that chronic infection could be linked to pathogen-induced Arg1 expression (37). Conversely, abrogation of Arg1 expression in hypoxic macrophages where iNOS was rendered ineffective resulted in exacerbated lung granuloma pathology and bacterial burden, suggesting that Arg1 could also play a positive role in the control of M. tuberculosis (79).
Iron availability in macrophage is also crucial for Mycobacteria growth (64, 65). Iron overload enhanced susceptibility while iron-poor diets reduced Mycobacteria virulence in mice (80). The depletion of intracellular iron occurs in M1 polarized macrophages. However, Mycobacterium produces siderophores (mycobactins) with incredible high affinity to iron able to remove iron from transferrin and lactoferrin, leading to augmentation of iron concentrations in infected macrophages favoring Mycobacterium growth. As previously mentioned, intracellular iron accumulation could also downregulate M1 phenotype by negatively affecting iNOS expression (81).
Brucella spp. are facultative intracellular α-proteobacteria and the causative agent of the world’s leading zoonotic disease brucellosis (82). Its pathogenesis is mainly based on its ability to survive and multiply inside macrophages. Treating Brucella abortus-infected mice with a PPARγ inhibitor led to a significant decrease in splenic colonization during the chronic phase of infection (83). In contrast, treatment of B. abortus-infected mice with PPARγ agonist led to increased expression of M2 markers in splenic macrophages and enhanced bacterial count in the spleen during the chronic infection phase (83). These results strongly suggest that pathways downstream of PPARγ contribute to generate a niche for persistence of B. abortus. In the same study (83), further analysis of in vitro infected macrophages suggested that PPARγ favors intracellular glucose availability for Brucella by shifting the energetic cell metabolism from a typical M1-associated glycolytic pathway to a glucose-sparing lipid oxidation associated to M2 differentiation.
Although iron is needed by bacteria for growth, it also plays an important role in the anti-microbial M1 activities of macrophages such as generation of hydroxyl radicals. Brucella appears well equipped to deal with the iron-poor environment in M1 macrophages. Brucella expresses two siderophores [2,3-dihydroxybenzoic acid (2,3-DHBA) and 2,3-DHBA-based molecule brucebactin] and a heme oxygenase (BhuO) allowing the use of heme as an iron source [reviewed in Ref. (84)]. It has been observed that iron supplementation increased the ability of macrophages to control intracellular Brucella (85) suggesting that iron chelation may represent a valuable strategy whereby this bacterium impedes the generation of toxic hydroxyl radicals.
Salmonella are Gram-negative bacteria infecting hosts via the gastrointestinal tract. Host-adapted strains of Salmonella enterica cause systemic infections (typhoid fever) and have the ability to asymptomatically persist within host tissues for long periods of time in 1–6% of patients [reviewed in Ref. (86)]. Early protective Th1 responses are followed by pathogen-permissive Th2 responses. Salmonella infection caused accumulation and persistence of hemophagocyte macrophages (characterized by the ingestion of non-apoptotic cells) expressing M2 markers (CD36 and CD206) in spleen (87). During later stages of infection in vivo, Salmonella was found preferentially associated with these cells (88), possibly representing a specific niche for persistent infection. PPARδ was found upregulated in Salmonella infected macrophages in vitro. PPARδ deficiency dramatically inhibited Salmonella replication in vitro and in vivo and its pharmacological activation enhanced bacterial growth in mice. Absence of PPARδ was associated to a decrease of available glucose necessary to Salmonella persistence in macrophages (88). How macrophages acquire and/or maintain an M2 phenotype in vivo during Salmonella infection is presently unknown.
Leishmania represent obligate intracellular protozoan parasites that are transmitted in the dermis of the mammalian host by blood-feeding sand flies. After transmission, these parasites invade mainly macrophages, which are decisive effector cells that either kill or host the intracellular parasites depending on the balance of iNOS and Arg1 [reviewed in Ref. (89)]. Control of parasite growth was associated with the induction of strong Th1 responses and induction of iNOS-expressing M1 macrophage. Accordingly, there was a clear direct correlation between the parasite load and the arginase activity in the lesions. Inhibition of arginase activity during the course of infection reduced parasite growth (43). In vitro, parasitized dendritic cells showed coordinated transcriptional modulations that correlated in part to PPARχ upregulation and promoted the generation and storage of neutral lipids, such as triacyl-sn-glycerol and cholesteryl esters (90), that can be important for the synthesis of key parasite membrane components. Accumulation of neutral lipid has been also shown to reduce antigen processing and presentation to effector T cells (91). Leishmania parasites are coated with phosphatidylserine (92), a major surface characteristic of apoptotic cells (93) recognized by CD36 expressed by M2 macrophages (94). As engulfment of apoptotic cells by CD36 pathways leads to induction of PPAR, it is possible that Leishmania actively promotes M2 polarization of macrophages as a virulence strategy. Of note, a recent report shows that the macrophage response upon visceral infection with Leishmania infantum is characterized by an M2b-like phenotype, identified by expression of C-type lectin receptors signature (95).
Francisella tularensis, a gram-negative intracellular bacteria and the causative agent of tularemia, induces acute and frequently lethal pulmonary infection. F. tularensis replicates in myeloid cells and alveolar epithelial cells in lung of infected mice [reviewed in (96)]. It has been observed that virulent strains induce M2 activation markers on macrophages in wild type but not in IL-4Rα or STAT6 deficient mice (97). Lipid isolated from virulent strain but not from attenuated strain appeared able to induce a TLR2/PPARγ-dependent M2 polarization of macrophage (98), suggesting that redirection of macrophage polarization by F. tularensis could constitute an escape immune mechanism.
Listeria monocytogenes is a facultative intracellular Gram-positive bacteria able of serious acute infection. Listeria infection of macrophages rapidly led to increased expression of PPARγ (99). Selective loss of PPARγ in myeloid cells resulted in enhanced innate immune defense against Listeria both, in vitro and in vivo suggesting that PPARγ-dependent M2 phenotype could favor L. monocytogenes multiplication.
T. gondii represents one of clear examples of how an intracellular parasite can subvert macrophage polarization to enhance its infectivity. This parasite secretes into the cytoplasm of infected macrophage several virulence factors including a protein, ROP16, displaying protein kinase activity. Remarkably, this protein interferes with the host signaling machinery through the direct phosphorylation of STAT6 (and STAT3), bypassing therefore early cytokine receptor proximal events. Notably, ROP16 was shown to phosphorylate STAT6 on the critical activation residue Tyr641 (39), resulting in the productive transcription of M2-associated genes including Arg1 (41, 100).
In conclusion, numerous evidences suggest that some intracellular bacterial and protozoan pathogens responsible for chronic (Mycobacteria, Brucella, Leishmania, and Toxoplasma) but also acute (Francisella and Listeria) infection actively manipulate STAT6-PPARγ/δ pathways to avoid M1 polarization of macrophages and/or benefit from a nutrient rich environment associated to lipid oxidation metabolism. Although the possible impact of macrophage polarization on chronic viral infections remains largely unknown, recent studies suggest that HBV-induced M2 macrophage polarization can participate to immune impairment and pathology (101). Thus, PPARγ/δ pathways hold a pivotal role in the establishment of chronic infection and their ligands may be used as combination therapeutics to limit host pathology or pathogen persistence.
Beneficial Role of Th2 Response and Alternatively Activated Macrophage during Infection
It is well appreciated that M2 macrophages can act to limit disease severity and protect the host from detrimental effects of an excessive Th1 response, making symbiotic survival between host and parasites more likely. For example, infection of mice or humans with the trematode S. mansoni results in a Th2 dependent response. Neutralization of IL-4 or genetic invalidation of IL-4/IL-13 led to increased rates of mortality during acute schistosomiasis, illustrating the protective role of Th2 in this experimental model. Mice rendered selectively deficient for IL-4R in macrophages were extremely susceptible to infection with 100% mortality, suggesting that M2 macrophages are essential during schistosomiasis for protection against organ injury.
In addition, M2 macrophages can also display anti-microbial effector function and actively reduce the level of infection by various pathogens.
Infective larvae of the nematode N. brasiliensis enter host animals through skin penetration and further migrate in the lungs and intestine. Primary protective response against N. brasiliensis relied mostly on an IL-13-dependent goblet cell hyperplasia leading to increased mucus production in the intestine. Accordingly, IL-4Rα and STAT6 deficient mice failed to expulse the gastrointestinal nematode parasite (102). Interestingly, protective memory response against N. brasiliensis implicated neutralization of larva migration from skin to lung (48). Larval trapping was dependent on induction of M2 macrophage differentiation from recruited monocytes as demonstrated by the fact that neutralization of IL-4 or Arg1 abolished larval trapping.
The central pathophysiologic events in severe malaria caused by Plasmodium falciparum are the inability of the host defenses to control parasite replication resulting in the excessive release of pro-inflammatory cytokines. Mice treated with an agonist of PPARγ had reduced parasitemia of Plasmodium and decreased inflammatory response (103). This effect was mediated by CD36 that served as ligand for P. falciparum erythrocyte membrane protein 1 (PfEMP-1) expressed by the parasite (104). CD36-mediated internalization by M2 macrophages facilitated removal of neutrophils and dead tissues to resolve inflammation as well as clearing of the parasite. P. falciparum used host hemoglobin as iron source and sequestered heme into a pigment known as hemozoin. It has been showed that hemozoin reacted with membrane phospholipids to generate hydroxy-polyunsaturated fatty acids which are ligands of PPARγ (105), suggesting that the parasite can actively modulate its growth by a PPARγ pathway.
The cestode Mesocestoides corti induces a central nervous system infection in mice dominated by M2 macrophages. Absence of STAT6 signaling resulted in enhanced susceptibility to infection coinciding with increased parasite burden in the brain (106) suggesting that M2 macrophages actively control infection.
The ubiquitous fungus Aspergillus fumigatus causes invasive and allergenic disease. Host defense relied on the ability of the respiratory immune system to restrict spore germination into invasive hyphae and to limit fungus-induced or inflammation-induced damage in infected tissues. Infection induced a M2 polarization of alveolar macrophages. Control of infection appeared dependent on dectin-1 mediated phagocytosis of fungus by M2 macrophages (107) as dectin-1 deficiency or elimination of M2 macrophages was associated to increased fungal burden in mice.
Plasticity of Macrophage Polarization during Infection: Two Opposite Examples
Several infectious experimental models demonstrate that immune polarization may display high plasticity, suggesting that M1 and M2 phenotypes may not represent terminally differentiated cells.
Classically, M1 macrophages are implicated in initiating and sustaining inflammation, whereas M2 macrophages differentiate later and are involved in resolution of inflammation and tissue regeneration. In particular, the early and late phases of Trypanosoma congolense infection are characterized by M1 and M2 polarization of macrophages, respectively. The shift to M2 appears indispensable to control inflammation and limit tissue injury (105).
However, the opposite scenario has also been reported. Cryptococcus neoformans, the etiological agent of cryptococcosis constitutes a well-documented case of M2-to-M1 shift in vivo. After inhalation, C. neoformans resides primarily in the alveolar spaces, where it can survive and replicate in the extracellular lung environment. The immune response in C. neoformans-infected C57BL/6 wild type mice is predominantly Th2 biased and associated with development of M2 macrophages. These latter serve as intracellular reservoirs for the microbe and promote the development of lung pathology (108). In this model, neutralization of IFN-γ resulted in more severe pulmonary infections and lesions (109). In contrast, IL-4 (109) or IL-13 (110) deletion led to Th1-dominated immune responses associated with M1 macrophage granuloma formation displaying iNOS dependent fungicidal activity. Interestingly, the lung immune polarization environment changed overtime (108). Following a strong initial induction of Th2 cytokines in the lungs at 2 and 3 weeks post-infection, a considerable increase in Th1 cytokines occurred at 5 weeks post-infection. This increase in Th1 cytokines was accompanied by a decrease in Th2 cytokines, indicating that the immune system is spontaneously capable of a Th2–Th1 shift. However, if it is now appreciated that polarized T cells exhibit previously unsuspected flexibility and plasticity (111), it remains unclear whether macrophage polarization switches involve recruitment of new precursors or de-differentiation of macrophages in situ.
Collectively, these observations suggest that macrophages can undergo dynamic transitions between different functional states. This polarization switch may provide protection to uncontrolled inflammation or constitute an indispensable adaptation to rapid phenotypic change of pathogen during its cycle.
Conclusions
In summary, the available evidences are compatible with the original view of a “division of labor” between M1 and M2 macrophages. M1 macrophages do display an increased microbicidal activity against a wide range of intracellular parasites, while differentiation toward an M2-like state is often observed during the resolution phase of an inflammatory response, favoring in particular tissue repair. This purely dualistic view needs, however, to be considered with care, since M1 macrophages are also able to negatively control an inflammatory response (through notably NO production and IDO1-mediated tryptophan catabolism), while M2 macrophages display anti-microbial activities via expression of Arg1 (Figure 4). The M1/M2 cell paradigm represents, therefore, an additional example of pathogen-tailored immune effectors, endowed with regulatory properties.
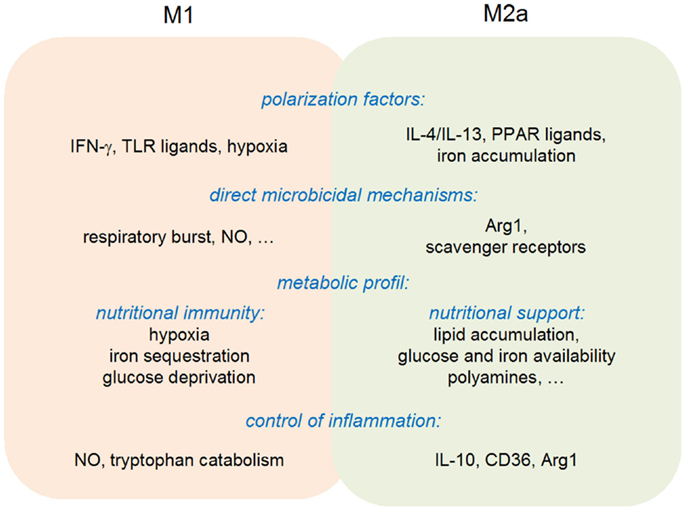
Figure 4. Multipolar view of M1 and M2a polarization.
A growing number of clinically relevant infectious diseases are characterized by pathogen persistence in the host. Chronic and recurrent infections implicate long-lasting and costly therapy and are the cause of an important morbidity in the world. Therefore, there is a strong interest in understanding the biology of pathogen persistence.
Numerous reports demonstrate that chronic bacterial persistence in vivo depends on the ability of bacteria to resist the anti-microbial activity but also to reconfigure local host environment to their profit (112). Pathogens exploit host regulatory pathways limiting damaging inflammatory responses for the benefit of tolerance. They also reprogram host cell metabolism to produce nutrients necessary for their long-term persistence (88, 113). Similar results have been obtained in parasitic infections (12, 89) suggesting that induction of a metabolic shift in infected cells constitutes a general strategy to favor persistence. In this paradigm, PPARs family seems key actor in the hijacking of macrophages by pathogens and an interesting target for therapeutic strategy.
Targeting microbial nutrient requirements represents a promising therapeutic strategy. Both anti-microbial compounds and vaccine have been created to specifically target pathogen iron acquisition systems [reviewed in Ref. (65)]. Unfortunately, microbes often possess redundant mechanisms for nutrient acquisition. A better understanding of these mechanisms may lead to the development of new strategies to control infection.
The importance of nutrient availability in control of infection also leads to focus our attention on the multifactorial and complex impact of modern fat rich diet on chronic infection. Commensal and mutualistic microorganisms are present in all mucosal compartments and form the microbiota. These symbiotic organisms compete with pathogenic microorganisms for nutrients, thereby preventing pathogenic colonization and invasion. As discussed above, obesity can affect macrophage polarization but also drastically change the composition of host microbial community and thus its ability to compete with pathogen for host nutriment availability. A better knowledge of the relationship between nutrition, microbiota, and immune defense could provide guidelines for prophylactic nutritional measures to protect against and/or treat infections.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol (1986) 136:2348–57.
2. Guilliams M, Movahedi K, Bosschaerts T, VandenDriessche T, Chuah MK, Herin M, et al. IL-10 dampens TNF/inducible nitric oxide synthase-producing dendritic cell-mediated pathogenicity during parasitic infection. J Immunol (2009) 182:1107–18. doi: 10.4049/jimmunol.182.2.1107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nat Rev Immunol (2007) 7:875–88. doi:10.1038/nri2189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol (2013) 229:176–85. doi:10.1002/path.4133
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Mahdavian Delavary B, van der Veer WM, van Egmond M, Niessen FB, Beelen RHJ. Macrophages in skin injury and repair. Immunobiology (2011) 216:753–62. doi:10.1016/j.imbio.2011.01.001
6. Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell Metab (2012) 15:432–7. doi:10.1016/j.cmet.2011.11.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med (1992) 176:287–92. doi:10.1084/jem.176.1.287
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol (2010) 11:889–96. doi:10.1038/ni.1937
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Gordon S. Alternative activation of macrophages. Nat Rev Immunol (2003) 3:23–35. doi:10.1038/nri978
10. Sica A, Mantovani A. Science in medicine macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122:787–95. doi:10.1172/JCI59643DS1
11. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi:10.1016/j.immuni.2014.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Raes G, Beschin A, Ghassabeh GH, De Baetselier P. Alternatively activated macrophages in protozoan infections. Curr Opin Immunol (2007) 19:454–9. doi:10.1016/j.coi.2007.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci (2008) 13:453–61. doi:10.2741/2692
14. Parsa R, Andresen P, Gillett A, Mia S, Zhang XM, Mayans S, et al. Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice. Diabetes (2012) 61:2881–92. doi:10.2337/db11-1635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Cavnar MJ, Zeng S, Kim TS, Sorenson EC, Ocuin LM, Balachandran VP, et al. KIT oncogene inhibition drives intratumoral macrophage M2 polarization. J Exp Med (2013) 210:2873–86. doi:10.1084/jem.20130875
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Shaul ME, Bennett G, Strissel KJ, Greenberg AS, Obin MS. Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet – induced obesity in mice. Diabetes (2010) 59:1171–81. doi:10.2337/db09-1402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Shapiro H, Lutaty A, Ariel A. Macrophages, meta-inflammation, and immuno-metabolism. ScientificWorldJournal (2011) 11:2509–29. doi:10.1100/2011/397971
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Prachason T, Konhan K, Pongnarin P, Chatsiricharoenkul S, Suputtamongkol Y, Limwongse C. Activation of indoleamine 2,3-dioxygenase in patients with scrub typhus and its role in growth restriction of Orientia tsutsugamushi. PLoS Negl Trop Dis (2012) 6(7):e1731. doi:10.1371/journal.pntd.0001731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Meisel R, Brockers S, Heseler K, Degistirici O, Bülle H, Woite C, et al. Human but not murine multipotent mesenchymal stromal cells exhibit broad-spectrum antimicrobial effector function mediated by indoleamine 2,3-dioxygenase. Leukemia (2011) 25:648–54. doi:10.1038/leu.2010.310
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Spekker K, Czesla M, Ince V, Heseler K, Schmidt SK, Schares G, et al. Indoleamine 2,3-dioxygenase is involved in defense against Neospora caninum in human and bovine cells. Infect Immun (2009) 77:4496–501. doi:10.1128/IAI.00310-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Adams O, Besken K, Oberdörfer C, MacKenzie CR, Takikawa O, Däubener W. Role of indoleamine-2,3-dioxygenase in alpha/beta and gamma interferon-mediated antiviral effects against herpes simplex virus infections. J Virol (2004) 78:2632–6. doi:10.1128/JVI.78.5.2632-2636.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Daubener W, Spors B, Hucke C, Adam R, Stins M, Kim KS, et al. Restriction of Toxoplasma gondii growth in human brain microvascular endothelial cells by activation of indoleamine 2,3-dioxygenase. Infect Immun (2001) 69:6527–31. doi:10.1128/IAI.69.10.6527-6531.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Dai X, Zhu BT. Indoleamine 2,3-dioxygenase tissue distribution and cellular localization in mice: implications for its biological functions. J Histochem Cytochem (2010) 58:17–28. doi:10.1369/jhc.2009.953604
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Makala LHC, Baban B, Lemos H, El-Awady AR, Chandler PR, Hou D-Y, et al. Leishmania major attenuates host immunity by stimulating local indoleamine 2,3-dioxygenase expression. J Infect Dis (2011) 203:715–25. doi:10.1093/infdis/jiq095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Murakami Y, Hoshi M, Hara A, Takemura M, Arioka Y, Yamamoto Y, et al. Inhibition of increased indoleamine 2,3-dioxygenase activity attenuates Toxoplasma gondii replication in the lung during acute infection. Cytokine (2012) 59:245–51. doi:10.1016/j.cyto.2012.04.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Fox JM, Sage LK, Huang L, Barber J, Klonowski KD, Mellor AL, et al. Inhibition of indoleamine 2,3-dioxygenase enhances the T-cell response to influenza virus infection. J Gen Virol (2013) 94:1451–61. doi:10.1099/vir.0.053124-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Sage LK, Fox JM, Mellor AL, Tompkins SM, Tripp RA. Indoleamine 2,3-dioxygenase (IDO) activity during the primary immune response to influenza infection modifies the memory T cell response to influenza challenge. Viral Immunol (2014) 27:112–23. doi:10.1089/vim.2013.0105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Wang XF, Wang HS, Wang H, Zhang F, Wang KF, Guo Q, et al. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: focus on macrophage polarization of THP-1 cells. Cell Immunol (2014) 289:42–8. doi:10.1016/j.cellimm.2014.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Albina JE, Mills CD, Henry WL, Caldwell MD. Temporal expression of different pathways of 1-arginine metabolism in healing wounds. J Immunol (1990) 144:3877–80.
30. Modolell M, Choi B-S, Ryan RO, Hancock M, Titus RG, Abebe T, et al. Local suppression of T cell responses by arginase-induced l-arginine depletion in nonhealing leishmaniasis. PLoS Negl Trop Dis (2009) 3:e480. doi:10.1371/journal.pntd.0000480
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. El-Gayar S, Thuring-Nahler H, Pfeilschifter J, Rollinghoff M, Bogdan C. Translational control of inducible nitric oxide synthase by IL-13 and arginine availability in inflammatory macrophages. J Immunol (2003) 171:4561–8. doi:10.4049/jimmunol.171.9.4561
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Munder M, Choi B-S, Rogers M, Kropf P. l-Arginine deprivation impairs Leishmania major-specific T-cell responses. Eur J Immunol (2009) 39:2161–72. doi:10.1002/eji.200839041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Pessanha AP, Martins RA, Mattos-Guaraldi AL, Vianna A, Moreira LO. Arginase-1 expression in granulomas of tuberculosis patients. FEMS Immunol Med Microbiol (2012) 66:265–8. doi:10.1111/j.1574-695X.2012.01012.x
34. Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol (2013) 191:773–84. doi:10.4049/jimmunol.1300113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Abebe T, Hailu A, Woldeyes M, Mekonen W, Bilcha K, Cloke T, et al. Local increase of arginase activity in lesions of patients with cutaneous leishmaniasis in Ethiopia. PLoS Negl Trop Dis (2012) 6:e1684. doi:10.1371/journal.pntd.0001684
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Serbina NV, Salazar-mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection University of Texas at Austin. Immunity (2003) 19:59–70. doi:10.1016/S1074-7613(03)00171-7
37. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol (2008) 9:1399–406. doi:10.1038/ni.1671
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Adams LB, Hibbs JB, Taintor RR, Krahenbuhl JL. Microbiostatic effect of murine-activated macrophages for Toxoplasma gondii. Role for synthesis of inorganic nitrogen oxides from l-arginine. J Immunol (1990) 144:2725–9.
39. Ong Y-C, Reese ML, Boothroyd JC. Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J Biol Chem (2010) 285:28731–40. doi:10.1074/jbc.M110.112359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Osorio EY, Zhao W, Espitia C, Saldarriaga O, Hawel L, Byus CV, et al. Progressive visceral leishmaniasis is driven by dominant parasite-induced STAT6 activation and STAT6-dependent host arginase 1 expression. PLoS Pathog (2012) 8:e1002417. doi:10.1371/journal.ppat.1002417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Butcher BA, Fox BA, Rommereim LM, Kim SG, Maurer KJ, Yarovinsky F, et al. Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog (2011) 7:e1002236. doi:10.1371/journal.ppat.1002236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Iniesta V, Carcelén J, Molano I, Pablo M, Peixoto V, Redondo E, et al. Arginase I induction during Leishmania major infection mediates the development of disease. Infect Immun (2005) 73(9):6085–90. doi:10.1128/IAI.73.9.6085
43. Kropf P, Fuentes JM, Fähnrich E, Arpa L, Herath S, Weber V, et al. Arginase and polyamine synthesis are key factors in the regulation of experimental leishmaniasis in vivo. FASEB J (2005) 19:1000–2. doi:10.1096/fj.04-3416fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Da Silva MFL, Floeter-Winter LM. Arginase in Leishmania. Subcell Biochem (2014) 74:103–17. doi:10.1007/978-94-007-7305-9_4
45. Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog (2009) 5:e1000371. doi:10.1371/journal.ppat.1000371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Herbert DR, Orekov T, Roloson A, Ilies M, Perkins C, O’Brien W, et al. Arginase I suppresses IL-12/IL-23p40-driven intestinal inflammation during acute schistosomiasis. J Immunol (2010) 184:6438–46. doi:10.4049/jimmunol.0902009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Campbell L, Saville CR, Murray PJ, Cruickshank SM, Hardman MJ. Local arginase 1 activity is required for cutaneous wound healing. J Invest Dermatol (2013) 133:2461–70. doi:10.1038/jid.2013.164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Obata-Ninomiya K, Ishiwata K, Tsutsui H, Nei Y, Yoshikawa S, Kawano Y, et al. The skin is an important bulwark of acquired immunity against intestinal helminths. J Exp Med (2013) 210:2583–95. doi:10.1084/jem.20130761
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Anthony RM, Urban JF, Alem F, Hamed HA, Rozo CT, Boucher J-L, et al. Memory T(H)2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat Med (2006) 12:955–60. doi:10.1038/nm1451
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Esser-von Bieren J, Mosconi I, Guiet R, Piersgilli A, Volpe B, Chen F, et al. Antibodies trap tissue migrating helminth larvae and prevent tissue damage by driving IL-4Rα-independent alternative differentiation of macrophages. PLoS Pathog (2013) 9:e1003771. doi:10.1371/journal.ppat.1003771
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Rahat MA, Bitterman H, Lahat N. Molecular mechanisms regulating macrophage response to hypoxia. Front Immunol (2011) 2:45. doi:10.3389/fimmu.2011.00045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Kempner BYW. The nature of leukemic blood cells as determined by metabolism. J Clin Invest (1938) 18(3):291–300. doi:10.1172/JCI101045
53. Shehade H, Oldenhove G, Moser M. Hypoxia in the intestine or solid tumors: a beneficial or deleterious alarm signal? Eur J Immunol (2014) 44(9):2550–7. doi:10.1002/eji.201444719
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Burke B, Giannoudis A, Corke KP, Gill D, Wells M, Ziegler-Heitbrock L, et al. Hypoxia-induced gene expression in human macrophages: implications for ischemic tissues and hypoxia-regulated gene therapy. Am J Pathol (2003) 163:1233–43. doi:10.1016/S0002-9440(10)63483-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell (2003) 112:645–57. doi:10.1016/S0092-8674(03)00154-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Zinkernagel AS, Peyssonnaux C, Johnson RS, Nizet V. Pharmacologic augmentation of hypoxia-inducible factor-1alpha with mimosine boosts the bactericidal capacity of phagocytes. J Infect Dis (2008) 197:214–7. doi:10.1086/524843
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Okumura CYM, Hollands A, Tran DN, Olson J, Dahesh S, von Köckritz-Blickwede M, et al. A new pharmacological agent (AKB-4924) stabilizes hypoxia inducible factor-1 (HIF-1) and increases skin innate defenses against bacterial infection. J Mol Med (Berl) (2012) 90:1079–89. doi:10.1007/s00109-012-0882-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1 α expression regulates the bactericidal capacity of phagocytes. J Clin Invest (2005) 115:1806–15. doi:10.1172/JCI23865.1806
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Acosta-Iborra B, Elorza A, Olazabal IM, Martín-Cofreces NB, Martin-Puig S, Miró M, et al. Macrophage oxygen sensing modulates antigen presentation and phagocytic functions involving IFN-gamma production through the HIF-1 alpha transcription factor. J Immunol (2009) 182:3155–64. doi:10.4049/jimmunol.0801710
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Mahnke A, Meier RJ, Schatz V, Hofmann J, Castiglione K, Schleicher U, et al. Hypoxia in Leishmania major skin lesions impairs the NO-dependent leishmanicidal activity of macrophages. J Invest Dermatol (2014) 134(9):2339–46. doi:10.1038/jid.2014.121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell (2013) 24:695–709. doi:10.1016/j.ccr.2013.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi:10.1038/nature13490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Nairz M, Schroll A, Sonnweber T, Weiss G. The struggle for iron – a metal at the host-pathogen interface. Cell Microbiol (2010) 12:1691–702. doi:10.1111/j.1462-5822.2010.01529.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Silva-Gomes S, Vale-Costa S, Appelberg R, Gomes MS. Iron in intracellular infection: to provide or to deprive? Front Cell Infect Microbiol (2013) 3:96. doi:10.3389/fcimb.2013.00096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Cassat JE, Skaar EP. Iron in infection and immunity. Cell Host Microbe (2013) 13:509–19. doi:10.1016/j.chom.2013.04.010
66. Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol (2010) 40:824–35. doi:10.1002/eji.200939889
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Chawla A. Control of macrophage activation and function by PPARs. Circ Res (2010) 106:1559–69. doi:10.1161/CIRCRESAHA.110.216523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Kiss M, Czimmerer Z, Nagy L. The role of lipid-activated nuclear receptors in shaping macrophage and dendritic cell function: from physiology to pathology. J Allergy Clin Immunol (2013) 132:264–86. doi:10.1016/j.jaci.2013.05.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Malur A, McCoy AJ, Arce S, Barna BP, Kavuru MS, Malur AG, et al. Deletion of PPAR gamma in alveolar macrophages is associated with a Th-1 pulmonary inflammatory response. J Immunol (2009) 182:5816–22. doi:10.4049/jimmunol.0803504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature (2007) 447:1116–20. doi:10.1038/nature05894
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Tsalikis J, Croitoru DO, Philpott DJ, Girardin SE. Nutrient sensing and metabolic stress pathways in innate immunity. Cell Microbiol (2013) 15:1632–41. doi:10.1111/cmi.12165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Brunton J, Steele S, Ziehr B, Moorman N, Kawula T. Feeding uninvited guests: mTOR and AMPK set the table for intracellular pathogens. PLoS Pathog (2013) 9:e1003552. doi:10.1371/journal.ppat.1003552
73. Steinberg GR, Schertzer JD. AMPK promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: implications for diabetes and cardiovascular disease. Immunol Cell Biol (2014) 92:340–5. doi:10.1038/icb.2014.11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Sag D, Carling D, Stout RD, Suttles J. Adenosine 5’-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol (2008) 181:8633–41. doi:10.4049/jimmunol.181.12.8633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Mounier R, Théret M, Arnold L, Cuvellier S, Bultot L, Göransson O, et al. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab (2013) 18:251–64. doi:10.1016/j.cmet.2013.06.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Almeida PE, Carneiro AB, Silva AR, Bozza PT. PPARγ expression and function in mycobacterial infection: roles in lipid metabolism, immunity, and bacterial killing. PPAR Res (2012) 2012:383829. doi:10.1155/2012/383829
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Almeida PE, Roque NR, Magalhães KG, Mattos KA, Teixeira L, Maya-Monteiro C, et al. Differential TLR2 downstream signaling regulates lipid metabolism and cytokine production triggered by Mycobacterium bovis BCG infection. Biochim Biophys Acta (2014) 1841:97–107. doi:10.1016/j.bbalip.2013.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Rajaram MVS, Brooks MN, Morris JD, Torrelles JB, Azad AK, Schlesinger LS. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J Immunol (2010) 185:929–42. doi:10.4049/jimmunol.1000866
79. Duque-Correa MA, Kühl AA, Rodriguez PC, Zedler U, Schommer-Leitner S, Rao M, et al. Macrophage arginase-1 controls bacterial growth and pathology in hypoxic tuberculosis granulomas. Proc Natl Acad Sci U S A (2014) 111(38):E4024–32. doi:10.1073/pnas.1408839111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Gomes MS, Boelaert JR, Appelberg R. Role of iron in experimental Mycobacterium avium infection. J Clin Virol (2001) 20(3):117–22. doi:10.1016/S1386-6532(00)00135-9
81. Weiss G, Werner-Felmayer G, Werner ER, Grünewald K, Wachter H, Hentze MW. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J Exp Med (1994) 180:969–76. doi:10.1084/jem.180.3.969
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV. The new global map of human brucellosis. Lancet Infect Dis (2006) 6:91–9. doi:10.1016/S1473-3099(06)70382-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Xavier MN, Winter MG, Spees AM, Den Hartigh AB, Nguyen K, Roux CM, et al. PPARγ-mediated increase in glucose availability sustains chronic Brucella abortus infection in alternatively activated macrophages. Cell Host Microbe (2013) 14:159–70. doi:10.1016/j.chom.2013.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Roop RM. Metal acquisition and virulence in Brucella. Anim Health Res Rev (2012) 13:10–20. doi:10.1017/S1466252312000047
85. Jiang X, Baldwin CL. Iron augments macrophage-mediated killing of Brucella abortus alone and in conjunction with interferon-gamma. Cell Immunol (1993) 148:397–407. doi:10.1006/cimm.1993.1121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Monack DM, Mueller A, Falkow S. Persistent bacterial infections: the interface of the pathogen and the host immune system. Nat Rev Microbiol (2004) 2:747–65. doi:10.1038/nrmicro955
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. McCoy MW, Moreland SM, Detweiler CS. Hemophagocytic macrophages in murine typhoid fever have an anti-inflammatory phenotype. Infect Immun (2012) 80:3642–9. doi:10.1128/IAI.00656-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Eisele NA, Ruby T, Jacobson A, Manzanillo PS, Cox JS, Lam L, et al. Salmonella require the fatty acid regulator PPARδ for the establishment of a metabolic environment essential for long-term persistence. Cell Host Microbe (2013) 14:171–82. doi:10.1016/j.chom.2013.07.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Chan MM, Adapala N, Chen C. Peroxisome proliferator-activated receptor-γ-mediated polarization of macrophages in Leishmania infection. PPAR Res (2012) 2012:796235. doi:10.1155/2012/796235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Lecoeur H, Giraud E, Prévost MC, Milon G, Lang T. Reprogramming neutral lipid metabolism in mouse dendritic leucocytes hosting live Leishmania amazonensis amastigotes. PLoS Negl Trop Dis (2013) 7(6):e2276. doi:10.1371/journal.pntd.0002276
91. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med (2010) 16:880–6. doi:10.1038/nm.2172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Wanderley JLM, Moreira MEC, Benjamin A, Bonomo AC, Barcinski MA. Mimicry of apoptotic cells by exposing phosphatidylserine participates in the establishment of amastigotes of Leishmania (L) amazonensis in mammalian hosts. J Immunol (2006) 176:1834–9. doi:10.4049/jimmunol.176.3.1834
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol (1992) 148:2207–16.
94. Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med (2006) 203:2613–25. doi:10.1084/jem.20060370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Lefèvre L, Lugo-Villarino G, Meunier E, Valentin A, Olagnier D, Authier H, et al. The C-type lectin receptors dectin-1, MR, and SIGNR3 contribute both positively and negatively to the macrophage response to Leishmania infantum. Immunity (2013) 38:1038–49. doi:10.1016/j.immuni.2013.04.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Cowley SC, Elkins KL. Immunity to Francisella. Front Microbiol (2011) 2:26. doi:10.3389/fmicb.2011.00026
97. Shirey KA, Cole LE, Keegan AD, Vogel SN. Francisella tularensis live vaccine strain induces macrophage alternative activation as a survival mechanism. J Immunol (2008) 181:4159–67. doi:10.4049/jimmunol.181.6.4159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Crane DD, Ireland R, Alinger JB, Small P, Bosio CM. Lipids derived from virulent Francisella tularensis broadly inhibit pulmonary inflammation via toll-like receptor 2 and peroxisome proliferator-activated receptor α. Clin Vaccine Immunol (2013) 20:1531–40. doi:10.1128/CVI.00319-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Abdullah Z, Geiger S, Nino-Castro A, Böttcher JP, Muraliv E, Gaidt M, et al. Lack of PPARγ in myeloid cells confers resistance to Listeria monocytogenes infection. PLoS One (2012) 7(5):e37349. doi:10.1371/journal.pone.0037349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Jensen KDC, Wang Y, Wojno EDT, Shastri AJ, Hu K, Cornel L, et al. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe (2011) 9:472–83. doi:10.1016/j.chom.2011.04.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Washburn ML, Bility MT, Zhang L, Kovalev GI, Buntzman A, Frelinger JA, et al. A humanized mouse model to study hepatitis C virus infection, immune response, and liver disease. Gastroenterology (2011) 140:1334–44. doi:10.1053/j.gastro.2011.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Urban JF, Noben-Trauth N, Donaldson DD, Madden KB, Morris SC, Collins M, et al. IL-13, IL-4Ra, and Stat6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis. Immunity (1998) 8:255–64. doi:10.1016/S1074-7613(00)80477-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Serghides L, Patel SN, Ayi K, Lu Z, Gowda DC, Liles WC, et al. Rosiglitazone modulates the innate immune response to Plasmodium falciparum infection and improves outcome in experimental cerebral malaria. J Infect Dis (2009) 199:1536–45. doi:10.1086/598222
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Serghides L, Kain KC. Peroxisome proliferator-activated receptor gamma-retinoid X receptor agonists increase CD36-dependent phagocytosis of Plasmodium falciparum-parasitized erythrocytes and decrease malaria-induced TNF-alpha secretion by monocytes/macrophages. J Immunol (2001) 166:6742–8. doi:10.4049/jimmunol.166.11.6742
105. Skorokhod OA, Alessio M, Mordmüller B, Arese P, Schwarzer E. Hemozoin (malarial pigment) inhibits differentiation and maturation of human monocyte-derived dendritic cells: a peroxisome proliferator-activated receptor-gamma-mediated effect. J Immunol (2004) 173:4066–74. doi:10.4049/jimmunol.173.6.4066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Mishra BB, Gundra UM, Teale JM. STAT6−/− mice exhibit decreased cells with alternatively activated macrophage phenotypes and enhanced disease severity in murine neurocysticercosis. J Neuroimmunol (2011) 232:26–34. doi:10.1016/j.jneuroim.2010.09.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Bhatia S, Fei M, Yarlagadda M, Qi Z, Akira S, Saijo S, et al. Rapid host defense against Aspergillus fumigatus involves alveolar macrophages with a predominance of alternatively activated phenotype. PLoS One (2011) 6(1):e15943. doi:10.1371/journal.pone.0015943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio (2013) 4:e264–213. doi:10.1128/mBio.00264-13.Editor
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Arora S, Olszewski MA, Tsang TM, McDonald RA, Toews GB, Huffnagle GB. Effect of cytokine interplay on macrophage polarization during chronic pulmonary infection with Cryptococcus neoformans. Infect Immun (2011) 79:1915–26. doi:10.1128/IAI.01270-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Müller U, Stenzel W, Köhler G, Werner C, Polte T, Hansen G, et al. IL-13 induces disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with Cryptococcus neoformans. J Immunol (2007) 179:5367–77. doi:10.4049/jimmunol.179.8.5367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science (2010) 327:1098–102. doi:10.1126/science.1178334
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Corsetti PP, de Almeida LA, Carvalho NB, Azevedo V, Silva TMA, Teixeira HC, et al. Lack of endogenous IL-10 enhances production of proinflammatory cytokines and leads to Brucella abortus clearance in mice. PLoS One (2013) 8(9):e74729. doi:10.1371/journal.pone.0074729
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog (2008) 4(11):e1000204. doi:10.1371/journal.ppat.1000204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: macrophage polarization, metabolic switch, amino acid metabolism, hypoxia, iron, PPARs, infection, immune escape strategy
Citation: Muraille E, Leo O and Moser M (2014) Th1/Th2 paradigm extended: macrophage polarization as an unappreciated pathogen-driven escape mechanism? Front. Immunol. 5:603. doi: 10.3389/fimmu.2014.00603
Received: 23 July 2014; Accepted: 10 November 2014;
Published online: 26 November 2014.
Edited by:
Dragana Jankovic, National Institutes of Health, USAReviewed by:
Christopher E. Rudd, University of Cambridge, UKAntónio Gil Castro, University of Minho, Portugal
Hans Acha-Orbea, Center of Immunity and Infection Lausanne (CIIL), Switzerland
Thomas E. Morrison, University of Colorado School of Medicine, USA
Copyright: © 2014 Muraille, Leo and Moser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muriel Moser, Université Libre de Bruxelles, Laboratory of Immunobiology, Rue des Pr Jeener et Brachet, 12, Gosselies 6041, Belgium e-mail:bW1vc2VyQHVsYi5hYy5iZQ==