 Michael A. Schmid
Michael A. Schmid Michael S. Diamond
Michael S. Diamond Eva Harris
Eva Harris- 1Division of Infectious Diseases and Vaccinology, School of Public Health, University of California Berkeley, Berkeley, CA, USA
- 2Department of Medicine, Washington University School of Medicine, St. Louis, MO, USA
- 3Department of Molecular Microbiology, Washington University School of Medicine, St. Louis, MO, USA
- 4Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, MO, USA
Dendritic cells (DCs) are sentinels of the immune system and detect pathogens at sites of entry, such as the skin. In addition to the ability of DCs to control infections directly via their innate immune functions, DCs help to prime adaptive B- and T-cell responses by processing and presenting antigen in lymphoid tissues. Infected Aedes aegypti or Aedes albopictus mosquitoes transmit the four dengue virus (DENV) serotypes to humans while probing for small blood vessels in the skin. DENV causes the most prevalent arthropod-borne viral disease in humans, yet no vaccine or specific therapeutic is currently licensed. Although primary DENV infection confers life-long protective immunity against re-infection with the same DENV serotype, secondary infection with a different DENV serotype can lead to increased disease severity via cross-reactive T-cells or enhancing antibodies. This review summarizes recent findings in humans and animal models about DENV infection of DCs, monocytes, and macrophages. We discuss the dual role of DCs as both targets of DENV replication and mediators of innate and adaptive immunity, and summarize immune evasion strategies whereby DENV impairs the function of infected DCs. We suggest that DCs play a key role in priming DENV-specific neutralizing or potentially harmful memory B- and T-cell responses, and that future DC-directed therapies may help induce protective memory responses and reduce dengue pathogenesis.
Introduction
Dendritic Cells, Monocytes, and Macrophages: Location and Function
Dendritic cells (DCs) reside and migrate into barrier tissues such as the skin and mucosal epithelium that are the sites of pathogen invasion. In the steady state, DCs display high levels of phagocytic activity, take up antigen, and probe for pathogens via pattern-recognition receptors. DCs express Toll-like receptors (TLRs) and C-type lectins as transmembrane proteins as well as intracellular sensors, such as retinoic acid-inducible gene I (RIG-I), melanoma differentiation factor 5 (MDA-5), and cyclic GMP-AMP synthase (cGAS) that recognize conserved microbial patterns (1, 2). Upon pathogen recognition, DCs become activated, produce inflammatory cytokines and chemokines, migrate to lymph nodes (LNs), and present antigen to prime naïve T-cells (3).
Subsets of DCs, monocytes, and macrophages (MΦs) reside in different tissues and fulfill distinct functions. Classical DCs (cDCs) display the characteristic DC dendrites, are present in LNs, spleen, and bone marrow as well as skin, lung, liver, and intestine, and have the greatest ability to stimulate naïve T-cells (4). All cDCs can present endogenous antigen from the cytosol via MHC I to CD8+ T-cells (5). In addition, most subsets of cDCs present exogenous antigen via MHC II to CD4+ T-cells, whereas only specialized subsets can cross-present exogenous antigen via MHC I to CD8+ T-cells (6, 7). In addition, plasmacytoid DCs (pDCs) are another DC subset that reside in the spleen, bone marrow, and liver and circulate in the blood. During viral infections, pDCs migrate to infected tissues and secrete up to 1,000-fold higher amounts of interferon (IFN)-α/β than other cell types (8), although their capacity for antigen presentation is still debated (9). Nevertheless, the role of DCs in priming protective immune responses against many human pathogens and their potential contribution to pathogenesis and development of disease need further investigation.
Monocytes circulate in steady-state blood, patrol lymphoid, and non-lymphoid organs, and are recruited to inflamed tissues, where they phagocytize pathogens as well as infected or damaged cells (10). During inflammation, monocytes can differentiate to monocyte-derived DCs (moDCs) (11, 12). In vitro-generated human moDCs are used widely to study DC biology (13). Monocytes are isolated from human peripheral blood, differentiated in the presence of GM-CSF and IL-4 first to immature moDCs and after further stimulation with inflammatory cytokines or pathogen-associated microbial patterns (PAMPs) to mature moDCs (13, 14). The ability of moDCs to prime naïve T-cell responses remains controversial, as this function initially was attributed solely to cDCs (15). Nevertheless, recent studies demonstrated that moDCs can migrate to LNs and prime naïve T-cells during Leishmania major, influenza virus, and bacterial infections (12, 16, 17). In contrast to DCs, MΦs have limited ability to migrate and prime naïve T-cells. MΦs reside within tissues, where they phagocytize, secrete cytokines, present antigen to effector and memory T-cells, and contribute to the healing of injured tissue (18).
Dengue Epidemiology and Pathogenesis
Female Aedes aegypti and Aedes albopictus mosquitoes transmit the four dengue virus serotypes (DENV1–4) while feeding on blood vessels in the skin (19). The positive-sense RNA genome of the flavivirus DENV encodes three structural (C, prM/M, E) and seven non-structural (NS) proteins (20). DENV causes the most prevalent arthropod-borne viral disease of humans, with an estimated 390 million infections and 96 million apparent cases per year (21).
The acute febrile illness dengue fever (DF) can progress to a potentially life-threatening vascular leakage syndrome, dengue hemorrhagic fever/dengue shock syndrome (DHF/DSS), the latter characterized by hypotension and circulatory failure (22). At present, no vaccine or therapeutic against dengue is approved for use in humans. A major challenge in the development of vaccines and therapies is that although infection with one DENV serotype leads to long-lasting immunity against the same serotype, subsequent infection with a different (heterotypic) serotype is the major risk factor for severe disease (19). To date, the mechanisms by which the host immune response to DENV provides either protection or enhancement in secondary infection remain poorly understood. Antibodies can neutralize infection or conversely trigger “antibody-dependent enhancement” (ADE) (23, 24), whereby cross-reactive anti-DENV antibodies facilitate entry of DENV into Fcγ receptor (FcγR)-bearing cells and thus increase viral load and ultimately disease severity. Some DHF/DSS cases occur during primary (1°) infections, especially in infants 6–9 months of age (25). In this case, it is thought that maternal DENV-specific antibodies transferred via the placenta wane to levels that can enhance a newly acquired DENV infection (26). Thus, the quantity and quality of the antibody response influences the severity of a secondary DENV infection. Similarly, T-cells can provide protection (27–29), but cross-reactive T-cells have been implicated in disease pathogenesis (30–32). Nevertheless, most secondary DENV infections are asymptomatic or mild, suggesting that the immune system can mount protective responses against dengue.
Aedes mosquitoes that take a blood meal from a human with acute dengue viremia become infected and, after DENV spreads to the salivary glands, transmit the virus when feeding on a new individual. Mosquito saliva contains components that counteract the host hemostatic response and modulate immunity (33, 34). The addition of saliva from Ae. aegypti mosquitoes was found to decrease DENV infection of moDCs in vitro (35). In contrast, mosquito saliva or transmission via infected mosquitoes prolonged DENV serum viremia and fever in “humanized” mice as compared to inoculation with DENV alone (36). Furthermore, saliva that was inoculated by non-infected mosquitoes prior to needle inoculation of DENV blocked the upregulation of genes involved in innate pathogen recognition and increased serum viremia in mice deficient in IRF3 and IRF7 (37). Although certain in vivo studies suggest that mosquito saliva can facilitate DENV infection by generating an environment that favors early virus replication, the impact of saliva on skin DCs requires further study.
Targets of DENV Replication
DENV Infection in the Absence of Enhancing Antibodies
Identifying the targets of DENV infection is crucial for understanding virus spread and disease pathogenesis. Human autopsies revealed staining for DENV structural proteins and negative-sense viral RNA, indicative of virus replication, in MΦs in LNs, spleen, lung, and liver and monocytes in clotted blood from patients with lethal dengue disease (38, 39). Staining of the non-structural protein NS3 confirmed DENV replication in phagocytes (including monocytes, MΦs, and DCs) in LNs and spleen, as well as in MΦs in the lung in other autopsy studies (40, 41). In earlier stages of the disease, most DENV-infected cells in the peripheral blood of acute dengue patients were identified as CD14+ CD11c+ activated monocytes, with higher proportions of monocytes and DENV-infected total cells in the blood in DHF compared to DF patients (42).
Although in humans, DENV efficiently suppresses the IFN response, replicates, and causes disease, DENV fails to antagonize mouse IFN responses, and thus wild-type (WT) mice generally do not sustain DENV replication or develop disease (43). In comparison, mice deficient in IFN-α/β receptor (Ifnar−/−) and also -γ receptor (AG129) are susceptible to DENV infection and display a tropism similar to humans (40, 44–46). DENV replicated in murine MΦs that were isolated from the peritoneum (47), as well as in MΦs in LNs and spleens of AG129 mice (48, 49). Treatment with clodronate liposomes that deplete monocytes and MΦs decreased viral load in AG129 mice on day 2 but increased viral load on day 4 post-inoculation with DENV2 (48). Monocytes and MΦs thus play an important role as targets for early DENV replication as well as in subsequent control of DENV infection.
Human moDCs generated in vitro support DENV infection (50), with immature moDCs being more susceptible to DENV infection than mature moDCs, monocytes (51), or MΦs (52). Analogously, CD11chigh cells in the spleen of AG129 mice (47) that likely comprised both moDCs and cDCs supported DENV replication in vivo. Recent studies in Ifnar−/− mice have shown that early after infection, monocytes are recruited to the dermis and differentiate to moDCs, where they become primary targets for DENV replication (53). Although DENV can infect monocytes and MΦs directly, these studies emphasize the greater permissiveness of DCs to DENV infection in the absence of enhancing antibodies, such as during 1° infection conditions.
Surface expression of viral attachment factors determines the susceptibility to DENV infection. DC-SIGN (dendritic-cell-specific ICAM3-grabbing non-integrin, CD209) is a C-type lectin expressed on the surface of DCs and MΦs that recognizes mannose-type sugars on the surface of bacterial, fungal, and viral pathogens. Signals via DC-SIGN induce the phagocytosis of pathogens and contribute to host defense (54). However, DC-SIGN also interacts with carbohydrates on DENV glycoproteins and mediates the attachment of DENV to moDCs (55–57). Human immature moDCs express high levels of DC-SIGN and are highly susceptible to DENV infection (50, 58, 59). Of note, DC-SIGN mediates virus attachment to the cell surface, but not endocytosis into moDCs (60). Activation of immature moDCs via inflammatory cytokines results in downregulation of DC-SIGN, explaining in part why mature moDCs are less susceptible to DENV infection (51, 58). Further, human cDCs freshly isolated from blood do not express DC-SIGN and, accordingly, become highly susceptible to DENV infection only after culture with GM-CSF and IL-4 that induces DC-SIGN expression (61). Similarly, treatment of monocytes with IL-4 or IL-13 increased DC-SIGN expression and DENV infection (62), but it remains unclear whether these monocytes had differentiated to moDCs or MΦs. Consistent with these findings, higher levels of DC-SIGN expression on cDCs of different blood donors correlated with higher DENV infection (61). Furthermore, a polymorphic variant of the DC-SIGN promoter with a decreased transcriptional activity correlated with protection against DF in humans (63). In summary, changes in DC-SIGN expression on different myeloid cell subsets through differentiation correlate with DENV infection (Figure 1).
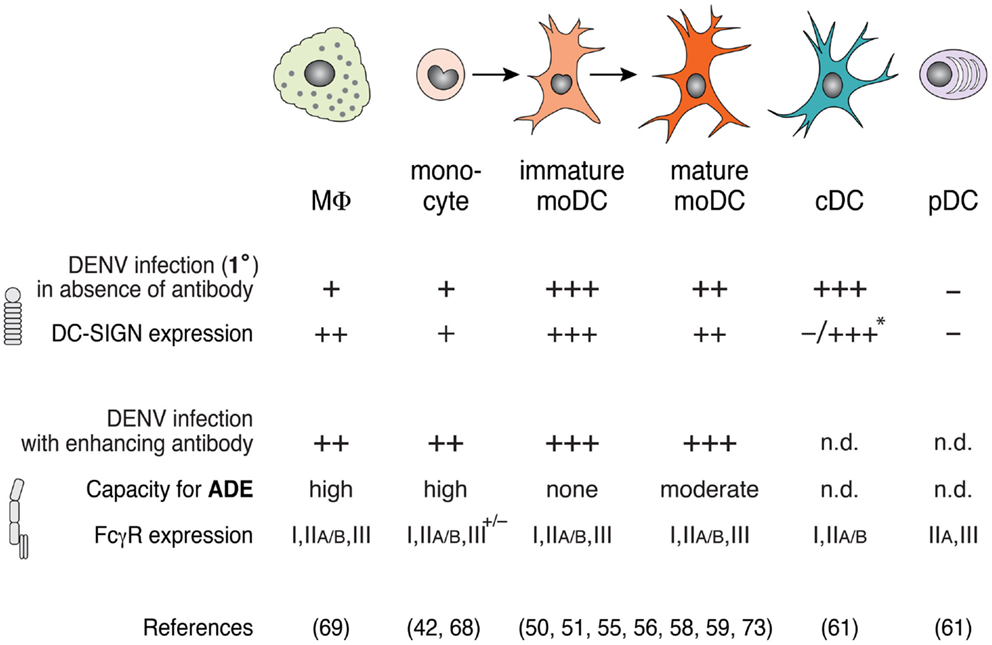
Figure 1. DENV infection varies among macrophages, monocyte, and dendritic cell (DC) subsets in the presence or absence of enhancing antibodies. DC-SIGN expression correlates with high infection in the absence of enhancing antibodies (i.e., 1° infection conditions), whereas FcγR expression modulates antibody-enhanced infection during ADE. Macrophages (MΦ) and monocytes express low levels of DC-SIGN, and show little DENV infection in the absence of enhancing antibody, but are highly infected in the presence of enhancing antibody. Under inflammatory conditions, monocytes differentiate to immature monocyte-derived DCs (moDCs) and, further, to mature moDCs after stimulation via PAMPs or inflammatory cytokines. While immature moDCs express high levels of DC-SIGN and can be infected with DENV in the absence of antibodies, mature moDCs express lower levels of DC-SIGN and show moderate permissiveness under these conditions. Accordingly, mature moDCs show a capacity for enhanced infection in the presence of subneutralizing anti-DENV antibodies. Classical DCs (cDCs) that are freshly isolated from human blood do not express DC-SIGN, but express high levels of DC-SIGN after stimulation with GM-CSF and IL-4 in vitro (*), which renders them highly susceptible to DENV infection without antibody, similar to immature moDCs. Plasmacytoid DCs (pDCs) do not express DC-SIGN or support DENV replication in the absence of antibody. cDCs and pDCs express FcγRs, but DENV infection of cDCs and pDCs during ADE has not been determined (n.d.).
Although Aedes mosquitoes transmit DENV when probing for blood vessels in the skin, most studies have focused on DENV infection in tissues after the virus has spread via the blood. Few studies have examined DENV infection and the immune response in the skin. DENV infects epidermal Langerhans cells (LCs) in healthy human skin explants in vitro (58, 64). Infection of LCs was confirmed in AG129 mice after intradermal inoculation of DENV2 (48). However, the dermis of intradermally inoculated Ifnar−/− mice contains 100-fold more DENV-infected cells than the epidermis (53). Recent studies indicate that dermal cDCs, and to a lesser extent MΦs, are the initial targets of DENV replication after intradermal inoculation of Ifnar−/− mice (53) or infection of skin explants from healthy human donors (65). Subsequently, de novo-recruited monocytes differentiate into moDCs, which become primary targets for DENV replication in the dermis (53).
Regarding the source of infectious virus, DENV produced in mosquito cells interacts with DC-SIGN and infected human immature moDCs in vitro (66). In contrast, DENV that was produced in human moDCs did not bind to DC-SIGN or infect moDCs but instead was infectious for cells expressing the homolog L-SIGN, such as monocytes and endothelial cells (66). The difference in binding to DC-SIGN or L-SIGN was likely due to different N-linked glycosylation patterns present on DENV particles produced in mosquito or mammalian cells. This may explain how DCs in the skin are the initial targets for DENV infection immediately after transmission. Characterization of the initial targets and immune response to DENV in the skin may foster new strategies to block DENV replication and abort pathogenesis.
Antibody-Enhanced DENV Infection
Dengue virus-specific adaptive immune responses, in particular subneutralizing concentrations of antibodies, can enhance DENV entry and infection. In cell culture, subneutralizing amounts of DENV-immune human serum or monoclonal antibodies enhance infection of monocytes (67, 68) and mature moDCs, but not immature moDCs (51). Human splenic MΦs showed low levels of DENV infection at baseline in vitro, but at least 10-fold greater infection in the presence of enhancing concentrations of diluted DENV-immune human (69). In vivo, CD14+ monocytes in the blood of acute dengue patients contained significantly higher levels of DENV genomic RNA in severe DHF compared to DF cases, and in secondary compared to 1° infections (70). Thus, ADE mediates efficient DENV infection of monocytes, mature moDCs, and MΦs.
FcγR expression determines the susceptibility of cells to ADE during DENV infection via uptake of virus-antibody complexes (Figure 1). Most myeloid cells express FcγRs, which bind the Fc region of antibodies and thus are an important link between cellular effector functions and antigen recognition via the antibody. Different types of FcγRs recognize distinct isotypes of IgG with varying affinity and can transmit activating or inhibitory signals to the cells (71). Attachment of DENV-antibody complexes to ectopically expressed FcγRI (CD64) (72) and FcγRIIA (CD32) (73) in fibroblast cell lines mediated ADE, independently of FcγR-signaling. DENV2 infection in primary human monocytes was increased 50-fold in the presence of enhancing DENV-immune human serum and depended on binding of antibody-virus complexes to FcγRI or FcγRIIA (68). Consistent with these data, blocking of FcγRIIA, but not of FcγRIIB, abrogated ADE of mature moDCs (51). Although mature and immature moDCs express similar levels of FcγRIIA, only mature moDCs sustain antibody-enhanced DENV infection because immature moDCs express high levels of DC-SIGN and thus do not require FcγR for DENV attachment or entry (51). Inflammation may lead to activation and differentiation of monocytes and moDCs, as well as altered expression of FcγRs and DC-SIGN, which modulates ADE of DENV infection in a cell-type specific manner.
Different classes of FcγRs transmit activating or inhibitory signals and play different roles during ADE. Although activating FcγRIIA and inhibitory FcγRIIB similarly bind DENV-antibody complexes, only FcγRIIA mediates enhanced DENV infection (74). In contrast to activating FcγRs, the inhibitory FcγRIIB may help prevent ADE. DENV-antibody aggregates cross-linked the inhibitory low-affinity FcγRIIB, which inhibited the phagocytosis and infection that would have occurred through activating FcγRs (75). This study showed evidence that the size of antibody-DENV aggregates may contribute to the neutralizing versus enhancing capacity of DENV-immune sera. More detailed studies on the expression of activating and inhibitory FcγRs are needed to understand the neutralization or enhancement of DENV infection of diverse cell subsets in different tissues. Along with greater infection in vitro, ADE may contribute to severe dengue disease in humans. Secondary DENV infection with a heterologous serotype is associated with an increased risk of DHF/DSS (76–79). Consistent with a possible role for ADE in vivo, polymorphisms in FcγR genes affect binding affinities for IgG subclasses and may influence the susceptibility to severe disease. Homozygotes for the arginine variant at position 131 (R/R131) of the FcRIIA gene, who have less capacity to opsonize IgG2 antibodies, showed reduced risk of developing DHF (80, 81). In contrast, the histidine variant H/H131 of FcRIIA was associated with an increased risk of developing DHF (81). Binding affinity of FcγRs to DENV-antibody complexes and the ratio of activating and inhibitory receptors likely determine DENV infection and disease outcome in the setting of pre-existing anti-DENV antibody.
Animal models also have been used to study ADE in vivo. AG129 mice develop mild disease after intravenous or intradermal inoculation with DENV2 in the absence of pre-existing antibody and lethal disease after passive transfer of subneutralizing levels of DENV-immune mouse or human serum prior to infection with otherwise sublethal doses of DENV (53, 82–84). The vascular leakage syndrome that develops in DENV-infected AG129 mice during ADE is similar to that observed after high-dose lethal DENV infection and recapitulates many features of severe dengue disease in humans (40, 44). Monoclonal antibodies directed against DENV protein E or prM can mediate ADE in vitro and in vivo (82, 83, 85). Addition of antibodies that block FcγR binding, F(ab)’2 fragments that lack the Fc domain, or recombinant monoclonal antibodies that lack the ability to bind FcγR all prevented ADE (82, 83). DENV-specific monoclonal antibodies with modified Fc domains that do not mediate ADE show therapeutic potential in vivo (85–87).
Mouse models have helped to define possible cellular targets for antibody-enhanced DENV infection. While the same cell types become DENV infected in the presence or absence of enhancing antibodies, DENV infection increases during ADE (82). In addition, MHC II+ cells in the intestinal lamina propria and sinusoidal endothelial cells in the liver of AG129 mice were infected by DENV mostly in the presence of enhancing concentrations of antibody (83). More detailed studies using human cells in vitro, clinical samples, and animal models are needed to clarify how cellular activation and differentiation modulate FcγR expression and impacts DENV infection and pathogenesis.
Innate Function of Dendritic Cells and Evasion by DENV
As first line of defense against virus infection, host cells recognize PAMPs (e.g., viral nucleic acids) and induce cell-intrinsic and cell-extrinsic innate immune responses. DENV infection stimulates responses via TLR7, TLR3, MDA5, and RIG-I (88–90) and induces the secretion of IFN-α/β that renders other host cells resistant to subsequent DENV infection (91, 92).
Plasmacytoid DCs recognize DENV via TLR7 in endocytic vesicles (61, 89), become activated, produce high amounts of IFN-α (93, 94), and may thus limit DENV replication. Further, pDCs sense DENV-infected cells by direct cell-to-cell contact, and immature DENV particles containing uncleaved prM were found to trigger higher IFN responses in pDCs compared to mature particles (95). Nevertheless, DENV does not infect human pDCs efficiently in vitro, and IFN-α production in pDCs appears independent of active viral replication within pDCs (61). This suggests that pDCs combat DENV without being susceptible to infection or to immune evasion mediated via cytosolic viral proteins. Because DENV cannot infect pDCs productively, its proteins cannot block the production of pDC-derived IFN-α that promotes transcription of IFN-stimulated genes (ISGs), which induce an antiviral state in neighboring cells. Patients with non-severe DF produced high levels of IFN-α in the serum (93, 96) and had an increased frequency of circulating pDCs compared to steady state (94). In contrast, numbers of pDCs declined early (day 3 or 4 of illness) in the blood of children who subsequently developed DHF (94) and, correspondingly, accumulated less in severe compared to non-severe adult cases (93). Furthermore, serum of patients with severe dengue contained less IFN-α than patients with non-severe manifestations (96). Consistent with this observation, peripheral blood mononuclear cells of patients that subsequently developed DSS expressed fewer transcripts of a set of ISGs than patients with mild disease (97, 98). Overall, pDCs protect against DENV pathogenesis by producing high amounts of IFN-α that prevent infection of additional target cells.
Dengue virus actively blocks the production and action of IFN-α/β in cell types that are susceptible to infection. The DENV non-structural protein NS2B/3 cleaves the human protein STING (also known as MITA) (99, 100), which is a key adaptor molecule in the cellular response to virus infection and in establishing the basal set-point of IRF3 signaling and IFN-α/β production (101). DENV NS5 protein induces targeted degradation of STAT2 via the proteasome (102–105) and NS4B blocks STAT1 activation (102, 106) and thus inhibits IFN-α/β and likely IFN-γ and IFN-λ receptor signaling in DENV-infected cells. However, non-infected cells remain capable of IFN production and IFNAR signaling, which may induce resistance to subsequent DENV infection. Indeed, pre-treatment of cells with IFN-α/β or IFN-γ prevents DENV infection (107–109).
Antibody-enhanced DENV infection also is believed to contribute to the evasion of the innate immune response through a mechanism termed “intrinsic ADE” (110). A monocytic cell line, THP-1, showed decreased production of inflammatory cytokines and mediators, such as IL-12, IFN-γ, TNFα, and nitric oxide radicals when they were infected with DENV in the presence but not in the absence of enhancing antibodies (111). Similarly, ADE infection suppressed TLR-mediated signals and the secretion of IFN-β, but increased the production of anti-inflammatory cytokines, such as IL-10, compared to DENV infection without antibodies (112). These effects depended on FcγR binding because antibodies blocking FcγRI or FcγRIIA restored IFN-β production (112). These findings were unexpected because activating FcγR signals should induce expression of ISGs and thus block DENV replication. Recent studies showed that the leukocyte immunoglobulin-like receptor-B1 binds DENV-antibody aggregates and blocks activating FcγR signals, which enabled DENV to evade the early antiviral response during ADE (113). These results suggest that antibody-mediated DENV entry also triggers intracellular signals that suppress innate responses in infected cells to increase viral production. This “intrinsic ADE” is complemented by “extrinsic ADE,” which refers to the enhanced FcγR-mediated binding and uptake of DENV described above.
Priming and Evasion of Adaptive Immune Responses
Dendritic cells link innate and adaptive immune responses by integrating innate signals from PAMPs with pathogen-derived antigens to induce antigen-specific T-cell and B-cell responses. DCs achieve this by (a) taking up and processing antigen and presenting antigen-derived peptides on MHC I to CD8+ T-cells or on MHC II to CD4+ T-cells; (b) expressing co-stimulatory molecules that activate T-cells; and (c) secreting chemokines and cytokines that attract T-cells and modulate the priming T-cell effector functions.
Because DC survival is required for optimal T-cell activation, DENV-induced apoptosis of DCs could antagonize the priming of immune responses. Bulk culture studies observed increased survival of moDCs (50, 114) and monocytes (115) after exposure to DENV. However, intracellular staining studies revealed a higher fraction of Annexin V+ apoptotic cells in those co-staining for DENV E protein (116). These results suggest that DENV induces apoptosis in infected cells, but increases survival in non-infected bystander cells. The impact of cell survival on the number of antigen-presenting cells and priming of DENV-specific adaptive immune responses warrants further study.
Pathogen recognition leads to DC maturation, which is characterized by increased expression of MHC II and co-stimulatory markers required for efficient priming of T-cell responses (117). After exposure to DENV, non-infected bystander moDCs upregulate MHC I and II molecules, as well as co-stimulatory molecules CD80 (B7-1), CD83, and CD86 (B7-2), although DENV blocks activation and maturation of infected moDCs within the same cultures (59, 116) (Figure 2A). Intracellular staining for DENV proteins revealed a block in activation of DENV-infected moDCs that was not observed in bulk culture (50, 59). Similarly, non-infected bystander monocytes, moDCs and cDCs, expressed higher levels of CD80 and CD86 than DENV-infected cells in the dermis of intradermally infected Ifnar−/− mice (53). In addition, DCs produce cytokines and chemokines to modulate T-cell responses. DENV-exposed moDCs (50, 59, 116) or cDCs (61), produce IL-6, IL-10, TNFα, and IFN-α. Furthermore, DENV-exposed moDCs secrete CXCL9, CXCL10, and CXCL11 (118) that bind the chemokine receptor CXCR3 and could attract effector and memory T-cells. However, it remains unclear whether it is the DENV-infected or non-infected DCs that produce these inflammatory mediators. Together, these data suggest that DENV blocks activation in infected DCs, which may decrease the priming of CD4+ or CD8+ T-cells, whereas non-infected bystander cells still can become activated.
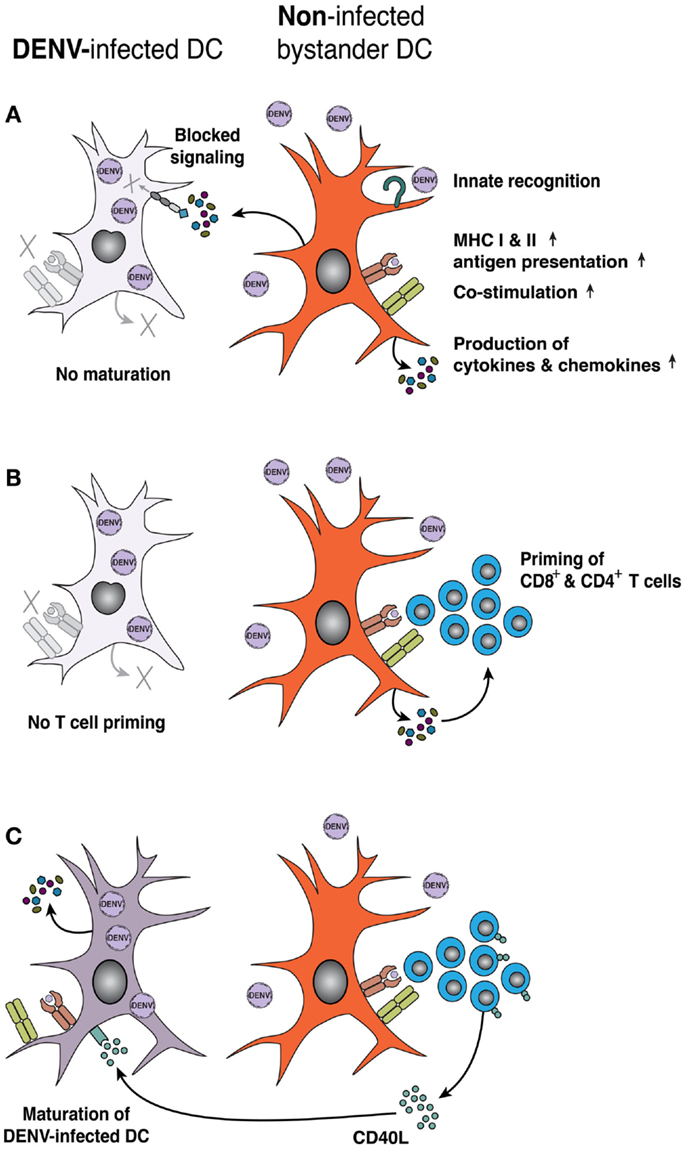
Figure 2. DENV infection impairs DC activation and priming of adaptive T-cell responses. (A) Maturation of human moDCs is inhibited by DENV infection, due to DENV non-structural proteins blocking induction and intracellular signaling of IFN-α/β. However, non-infected bystander DCs respond to PAMPs and/or cytokines associated with DENV infection and upregulate MHC class I and II molecules, co-stimulatory molecules, and the expression of inflammatory cytokines and chemokines. (B) Mature, bystander DCs efficiently prime adaptive T-cell responses, whereas DENV-infected DCs prime naïve T-cells less efficiently. (C) Activated T-cells display CD40L on their surface or secrete CD40L that acts on DENV-infected DCs and can restore DC maturation and function. Purple denotes DENV infection, while orange denotes inflammation/cell activation.
Mixed lymphocyte reactions (MLR) are a functional read-out for DC–T-cell interactions, in which DCs from MHC mismatched donors activate allogeneic T-cells. DENV infection decreased the capacity of moDCs (116, 119) as well as DCs isolated from human skin explants (65) to stimulate proliferation of DENV-naïve CD4+ T-cells in MLRs in vitro, suggesting that DENV-infected DCs are less capable of activating CD4+ T-cells (Figure 2B). Others have reported that DENV-infected cultures of moDCs can prime CD4+ T-cells, but with decreased T-cell effector functions, such as secretion of IFN-γ or TNFα (109). The impaired ability of DENV-infected moDCs to produce IFN-α and IFN-β may explain the decreased ability to prime T-cell responses (120). Although these surrogate MLR assays are interesting, little information exists as to how DCs prime DENV-specific T-cells. Non-infected moDCs that were pulsed with DENV E protein efficiently activated CD4+ or CD8+ CD45RO+ memory T-cells from DENV-immune but not from naïve individuals to produce IFN-γ (121). More study is needed to determine the capacity of DCs to prime DENV-specific naïve T-cells or to reactivate memory T-cells during re-exposure. Rapid progress should be possible given the publication of large numbers of HLA-restricted immunodominant DENV antigens (29, 31).
Also, activated T-cells can support the maturation of DCs. An initial study showed that co-culture of moDCs with a CD40L-transfected cell line restored the ability of DENV-infected moDCs to induce MLR responses (114) (Figure 2C). Subsequent work demonstrated that co-culture of activated T-cells with DENV-infected moDCs rescued the otherwise suppressed MLR response, and the activation of non-infected bystander moDCs depended on TNFα and IFN-α/β (118). Consequently, stimulation of the adaptive response to DENV infection requires signals from both cells in DC–T-cell interactions, likely via cell-to-cell contact. In clinical studies, gene expression analysis of acute dengue patients revealed that DHF cases expressed lower levels of genes linked to antigen processing, presentation, and T-cell activation compared to DF patients (122). Thus, impaired antigen presentation and functionality of DENV-infected DCs may reflect a viral immune escape strategy to dampen T-cell responses and impact disease severity.
To date, mostly in vitro-generated human moDCs have been used to study the role of DCs in inducing innate or adaptive immune responses during DENV infection. Nevertheless, diverse subsets of cDCs in lymphoid or non-lymphoid tissues execute specialized functions in presenting antigen and inducing CD4+ or CD8+ T-cell responses (123, 124). Expanding previous findings using human cDC subsets directly isolated from blood or tissues will be important to characterize the full spectrum of immune responses to DENV infection.
Concluding Remarks
Dengue virus infects the same cells (DCs, monocytes, and MΦs) that are essential for inducing and maintaining optimal innate and adaptive immune responses. This tropism of DENV appears to impair DC function, which may undermine the priming of DENV-specific memory responses. Is it possible to block DENV replication in DCs to reduce viral load and restore DC function, which could impact the generation of neutralizing or potentially harmful memory B- and T-cell responses? Can we as a field harness the knowledge gained about DC biology during DENV infection to prevent human disease? Can DC function be optimized in the context of live-attenuated DENV vaccines to stimulate protective immunity?
Significant progress has been made on characterizing DENV infection and activation of DCs, as well as protective or enhancing T-cell and B-cell responses. Nevertheless, early events after DENV transmission in the skin require further study, including a greater understanding of early virus replication, local immune responses at the site of transmission (i.e., the skin), and immunomodulatory effects of mosquito saliva. Further effort should focus on how responses of DCs impact disease outcome in an acute infection and prime immunological memory responses that will affect dengue pathogenesis and disease severity in secondary DENV infections. A collaborative effort using a multi-disciplinary approach among experts in dengue virology, medicine, vector biology, and immunology is called for to reach these goals.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health [grant R01-AI085607 (Eva Harris) and R01-AI077955 (Michael S. Diamond)], and the German Research Foundation, Deutsche Forschungsgemeinschaft [research fellowship SCHM 3011/1-1 (Michael A. Schmid)]. The authors declare no competing financial interests.
References
1. Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science (2010) 327(5963):291–5. doi:10.1126/science.1183021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Hornung V, Hartmann R, Ablasser A, Hopfner KP. OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat Rev Immunol (2014) 14(8):521–8. doi:10.1038/nri3719
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol (2013) 31:563–604. doi:10.1146/annurev-immunol-020711-074950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Satpathy AT, Wu X, Albring JC, Murphy KM. Re(de)fining the dendritic cell lineage. Nat Immunol (2012) 13(12):1145–54. doi:10.1038/ni.2467
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol (2007) 7(7):543–55. doi:10.1038/nri2103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Heath WR, Carbone FR. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat Immunol (2009) 10(12):1237–44. doi:10.1038/ni.1822
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Romani N, Clausen BE, Stoitzner P. Langerhans cells and more: langerin-expressing dendritic cell subsets in the skin. Immunol Rev (2010) 234(1):120–41. doi:10.1111/j.0105-2896.2009.00886.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol (2011) 29:163–83. doi:10.1146/annurev-immunol-031210-101345
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Villadangos JA, Young L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity (2008) 29(3):352–61. doi:10.1016/j.immuni.2008.09.002
10. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol (2011) 11(11):762–74. doi:10.1038/nri3070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol (2009) 27:669–92. doi:10.1146/annurev.immunol.021908.132557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell (2010) 143(3):416–29. doi:10.1016/j.cell.2010.09.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor [alpha]. J Exp Med (1994) 179:1109–18. doi:10.1084/jem.179.4.1109
14. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med (1992) 176(6):1693–702. doi:10.1084/jem.176.6.1693
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Sallusto F, Lanzavecchia A. Monocytes join the dendritic cell family. Cell (2010) 143(3):339–40. doi:10.1016/j.cell.2010.10.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity (2007) 26(4):519–31. doi:10.1016/j.immuni.2007.01.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Nakano H, Lin KL, Yanagita M, Charbonneau C, Cook DN, Kakiuchi T, et al. Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat Immunol (2009) 10(4):394–402. doi:10.1038/ni.1707
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science (2010) 327(5966):656–61. doi:10.1126/science.1178331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Halstead SB. Dengue. Lancet (2007) 370(9599):1644–52. doi:10.1016/S0140-6736(07)61687-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Pierson TC, Diamond MS. Flaviviruses. Sixth ed. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia, PA: Lippincott Williams and Wilkins (2013).
21. Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The global distribution and burden of dengue. Nature (2013) 496(7446):504–7. doi:10.1038/nature12060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. World Health Organization. Dengue Haemorrhagic Fever: Diagnosis, Treatment, Prevention, and Control. Second ed. Geneva: World Health Organization (1997).
23. Guzman MG, Kouri G, Martinez E, Bravo J, Riveron R, Soler M, et al. Clinical and serologic study of Cuban children with dengue hemorrhagic fever/dengue shock syndrome (DHF/DSS). Bull Pan Am Health Organ (1987) 21(3):270–9.
25. Kliks SC, Nimmanitya S, Nisalak A, Burke DS. Evidence that maternal dengue antibodies are important in the development of dengue hemorrhagic fever in infants. Am J Trop Med Hyg (1988) 38(2):411–9.
26. Halstead SB, Lan NT, Myint TT, Shwe TN, Nisalak A, Kalyanarooj S, et al. Dengue hemorrhagic fever in infants: research opportunities ignored. Emerg Infect Dis (2002) 8(12):1474–9. doi:10.3201/eid0812.020170
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Gagnon SJ, Ennis FA, Rothman AL. Bystander target cell lysis and cytokine production by dengue virus-specific human CD4(+) cytotoxic T-lymphocyte clones. J Virol (1999) 73(5):3623–9.
28. Hatch S, Endy TP, Thomas S, Mathew A, Potts J, Pazoles P, et al. Intracellular cytokine production by dengue virus-specific T cells correlates with subclinical secondary infection. J Infect Dis (2011) 203(9):1282–91. doi:10.1093/infdis/jir012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Weiskopf D, Angelo MA, de Azeredo EL, Sidney J, Greenbaum JA, Fernando AN, et al. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proc Natl Acad Sci U S A (2013) 110(22):E2046–53. doi:10.1073/pnas.1305227110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Mongkolsapaya J, Dejnirattisai W, Xu XN, Vasanawathana S, Tangthawornchaikul N, Chairunsri A, et al. Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat Med (2003) 9(7):921–7. doi:10.1038/nm887
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Duangchinda T, Dejnirattisai W, Vasanawathana S, Limpitikul W, Tangthawornchaikul N, Malasit P, et al. Immunodominant T-cell responses to dengue virus NS3 are associated with DHF. Proc Natl Acad Sci U S A (2010) 107(39):16922–7. doi:10.1073/pnas.1010867107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Rothman AL. Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nat Rev Immunol (2011) 11(8):532–43. doi:10.1038/nri3014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Ribeiro JM, Francischetti IM. Role of arthropod saliva in blood feeding: sialome and post-sialome perspectives. Annu Rev Entomol (2003) 48:73–88. doi:10.1146/annurev.ento.48.060402.102812
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Valenzuela JG. Exploring tick saliva: from biochemistry to ‘sialomes’ and functional genomics. Parasitology (2004) 129(Suppl):S83–94. doi:10.1017/S0031182004005189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Ader DB, Celluzzi C, Bisbing J, Gilmore L, Gunther V, Peachman KK, et al. Modulation of dengue virus infection of dendritic cells by Aedes aegypti saliva. Viral Immunol (2004) 17(2):252–65. doi:10.1089/0882824041310496
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Cox J, Mota J, Sukupolvi-Petty S, Diamond MS, Rico-Hesse R. Mosquito bite delivery of dengue virus enhances immunogenicity and pathogenesis in humanized mice. J Virol (2012) 86(14):7637–49. doi:10.1128/JVI.00534-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. McCracken MK, Christofferson RC, Chisenhall DM, Mores CN. Analysis of early dengue virus infection in mice as modulated by Aedes aegypti probing. J Virol (2014) 88(4):1881–9. doi:10.1128/JVI.01218-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Hall WC, Crowell TP, Watts DM, Barros VL, Kruger H, Pinheiro F, et al. Demonstration of yellow fever and dengue antigens in formalin-fixed paraffin-embedded human liver by immunohistochemical analysis. Am J Trop Med Hyg (1991) 45(4):408–17.
39. Jessie K, Fong MY, Devi S, Lam SK, Wong KT. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J Infect Dis (2004) 189:1411–8. doi:10.1086/383043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Balsitis SJ, Coloma J, Castro G, Alava A, Flores D, McKerrow JH, et al. Tropism of dengue virus in mice and humans defined by viral nonstructural protein 3-specific immunostaining. Am J Trop Med Hyg (2009) 80:416–24.
41. Aye KS, Charngkaew K, Win N, Wai KZ, Moe K, Punyadee N, et al. Pathologic highlights of dengue hemorrhagic fever in 13 autopsy cases from Myanmar. Hum Pathol (2014) 45(6):1221–33. doi:10.1016/j.humpath.2014.01.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Durbin A, Vargas MJ, Thumar B, Hammond SN, Gordon G, Rocha C, et al. Phenotyping of peripheral blood mononuclear cells during acute dengue illness demonstrates infection and increased activation of monocytes in severe cases compared to classic dengue fever. Virology (2008) 376:429–35. doi:10.1016/j.virol.2008.03.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Shresta S, Kyle JL, Snider HM, Basanavapatna M, Beatty R, Harris E. Interferon-dependent immunity is essential for resistance to primary dengue virus infection in mice, whereas T and B cell-dependent immunity is less critical. J Virol (2004) 78:2701–10. doi:10.1128/JVI.78.6.2701-2710.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Shresta S, Sharar KL, Prigozhin DM, Beatty PR, Harris E. A murine model for dengue lethal disease with increased vascular permeability. J Virol (2006) 80:10208–17. doi:10.1128/JVI.00062-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Kyle JL, Balsitis SB, Zhang L, Beatty PR, Harris E. Antibodies play a greater role than immune cells in heterologous protection against secondary dengue virus infection in a mouse model. Virology (2008) 380:296–303. doi:10.1016/j.virol.2008.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Orozco S, Schmid MA, Parameswaran P, Lachica R, Henn MR, Beatty R, et al. Characterization of a model of lethal dengue virus 2 infection in C57BL/6 mice deficient in the alpha/beta interferon receptor. J Gen Virol (2012) 93(Pt 10):2152–7. doi:10.1099/vir.0.045088-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Kyle JL, Beatty PR, Harris E. Dengue virus infects macrophages and dendritic cells in a mouse model of infection. J Infect Dis (2007) 195(12):1808–17. doi:10.1086/518007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Fink K, Ng C, Nkenfou C, Vasudevan SG, van Rooijen N, Schul W. Depletion of macrophages in mice results in higher dengue virus titers and highlights the role of macrophages for virus control. Eur J Immunol (2009) 39(10):2809–21. doi:10.1002/eji.200939389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Prestwood TR, May MM, Plummer EM, Morar MM, Yauch LE, Shresta S. Trafficking and replication patterns reveal splenic macrophages as major targets of dengue virus in mice. J Virol (2012) 86(22):12138–47. doi:10.1128/JVI.00375-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Ho LJ, Wang JJ, Shaio MF, Kao CL, Chang DM, Han SW, et al. Infection of human dendritic cells by dengue virus causes cell maturation and cytokine production. J Immunol (2001) 166:1499–506. doi:10.4049/jimmunol.166.3.1499
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Boonnak K, Slike BM, Burgess TH, Mason RM, Wu SJ, Sun P, et al. Role of dendritic cells in antibody-dependent enhancement of dengue virus infection. J Virol (2008) 82:3939–51. doi:10.1128/JVI.02484-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Kwan WH, Navarro-Sanchez E, Dumortier H, Decossas M, Barreto dos Santos F, Fridman HW, et al. Dermal-type macrophages show resistance to dengue virus infection despite CD209/DC-SIGN expression. PLoS Negl Trop Dis (2008) 2:e311. doi:10.1371/journal.pntd.0000311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Schmid MA, Harris E. Monocyte recruitment to the dermis and differentiation to dendritic cells increases the targets for dengue virus replication. PLoS Pathog (2014) 10(12):e1004541. doi:10.1371/journal.ppat.1004541
54. Figdor CG, van Kooyk Y, Adema GJ. C-type lectin receptors on dendritic cells and Langerhans cells. Nat Rev Immunol (2002) 2(2):77–84. doi:10.1038/nri827
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med (2003) 197:823–9. doi:10.1084/jem.20021840
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Navarro-Sanchez E, Altmeyer R, Amara A, Schwartz O, Fieschi F, Vierelizier JL, et al. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep (2003) 4(7):723–9. doi:10.1038/sj.embor.embor866
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Pokidysheva E, Zhang Y, Battisti AJ, Bator-Kelly CM, Chipman PR, Xiao C, et al. Cryo-EM reconstruction of dengue virus in complex with the carbohydrate recognition domain of DC-SIGN. Cell (2006) 124(3):485–93. doi:10.1016/j.cell.2005.11.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Wu SJ, Grouard-Vogel G, Sun W, Mascola JR, Brachtel E, Putvatana R, et al. Human skin Langerhans cells are targets of dengue virus infection. Nat Med (2000) 6(7):816–20. doi:10.1038/77553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Libraty DH, Pichyangkul S, Ajariyakhajorn C, Endy TP, Ennis FA. Human dendritic cells are activated by dengue virus infection: enhancement by gamma interferon and implications for disease pathogenesis. J Virol (2001) 75:3501–8. doi:10.1128/JVI.75.8.3501-3508.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Lozach PY, Burleigh L, Staropoli I, Navarro-Sanchez E, Harriague J, Virelizier JL, et al. Dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN)-mediated enhancement of dengue virus infection is independent of DC-SIGN internalization signals. J Biol Chem (2005) 280(25):23698–708. doi:10.1074/jbc.M504337200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Sun P, Fernandez S, Marovich MA, Palmer DR, Celluzzi CM, Boonnak K, et al. Functional characterization of ex vivo blood myeloid and plasmacytoid dendritic cells after infection with dengue virus. Virology (2009) 383(2):207–15. doi:10.1016/j.virol.2008.10.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Miller JL, deWet BJ, Martinez-Pomares L, Radcliffe CM, Dwek RA, Rudd PM, et al. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog (2008) 8:e17. doi:10.1371/journal.ppat.0040017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Sakuntabhai A, Turbpaiboon C, Casademont I, Chuansumrit A, Lowhnoo T, Kajaste-Rudnitski A, et al. A variant in the CD209 promoter is associated with severity of dengue disease. Nat Genet (2005) 37(5):507–13. doi:10.1038/ng1550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Limon-Flores AY, Perez-Tapia M, Estrada-Garcia I, Vaughan G, Escobar-Gutierrez A, Calderon-Amador J, et al. Dengue virus inoculation to human skin explants: an effective approach to assess in situ the early infection and the effects on cutaneous dendritic cells. Int J Exp Pathol (2005) 86(5):323–34. doi:10.1111/j.0959-9673.2005.00445.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Cerny D, Haniffa M, Shin A, Bigliardi P, Tan BK, Lee B, et al. Selective susceptibility of human skin antigen presenting cells to productive dengue virus infection. PLoS Pathog (2014) 10(12):e1004548. doi:10.1371/journal.ppat.1004548
66. Dejnirattisai W, Webb AI, Chan V, Jumnainsong A, Davidson A, Mongkolsapaya J, et al. Lectin switching during dengue virus infection. J Infect Dis (2011) 203(12):1775–83. doi:10.1093/infdis/jir173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Halstead SB, O’Rourke EJ. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J Exp Med (1977) 146(1):201–17. doi:10.1084/jem.146.1.201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Kou Z, Quinn M, Chen H, Rodrigo WW, Rose RC, Schlesinger JJ, et al. Monocytes, but not T or B cells, are the principal target cells for dengue virus (DV) infection among human peripheral blood mononuclear cells. J Med Virol (2008) 80(1):134–46. doi:10.1002/jmv.21051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Blackley S, Kou Z, Chen H, Quinn M, Rose RC, Schlesinger JJ, et al. Primary human splenic macrophages, but not T or B cells, are the principal target cells for dengue virus infection in vitro. J Virol (2007) 81(24):13325–34. doi:10.1128/JVI.01568-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Srikiatkhachorn A, Wichit S, Gibbons RV, Green S, Libraty DH, Endy TP, et al. Dengue viral RNA levels in peripheral blood mononuclear cells are associated with disease severity and preexisting dengue immune status. PLoS One (2012) 7(12):e51335. doi:10.1371/journal.pone.0051335
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity (2006) 24(1):19–28. doi:10.1016/j.immuni.2005.11.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Schlesinger JJ, Chapman SE. Influence of the human high-affinity IgG receptor FcgammaRI (CD64) on residual infectivity of neutralized dengue virus. Virology (1999) 260(1):84–8. doi:10.1006/viro.1999.9816
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Rodrigo WW, Jin X, Blackley SD, Rose RC, Schlesinger JJ. Differential enhancement of dengue virus immune complex infectivity mediated by signaling-competent and signaling-incompetent human Fcgamma RIA (CD64) or FcgammaRIIA (CD32). J Virol (2006) 80(20):10128–38. doi:10.1128/JVI.00792-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Boonnak K, Slike BM, Donofrio GC, Marovich MA. Human FcgammaRII cytoplasmic domains differentially influence antibody-mediated dengue virus infection. J Immunol (2013) 190(11):5659–65. doi:10.4049/jimmunol.1203052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Chan KR, Zhang SL, Tan HC, Chan YK, Chow A, Lim AP, et al. Ligation of Fc gamma receptor IIB inhibits antibody-dependent enhancement of dengue virus infection. Proc Natl Acad Sci U S A (2011) 108(30):12479–84. doi:10.1073/pnas.1106568108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Sangkawibha N, Rojanasuphot S, Ahandrik S, Viriyapongse S, Jatanasen S, Salitul V, et al. Risk factors in dengue shock syndrome: a prospective epidemiologic study in Rayong, Thailand. I. The 1980 outbreak. Am J Epidemiol (1984) 120(5):653–69.
77. Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, et al. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J Infect Dis (2000) 181:2–9. doi:10.1086/315215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Libraty DH, Endy TP, Houng HS, Green S, Kalayanarooj S, Suntayakorn S, et al. Differing influences of virus burden and immune activation on disease severity in secondary dengue-3 virus infections. J Infect Dis (2002) 185(9):1213–21. doi:10.1086/340365
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Guzman MG, Alvarez M, Halstead SB. Secondary infection as a risk factor for dengue hemorrhagic fever/dengue shock syndrome: an historical perspective and role of antibody-dependent enhancement of infection. Arch Virol (2013) 158(7):1445–59. doi:10.1007/s00705-013-1645-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Loke H, Bethell D, Phuong CX, Day N, White N, Farrar J, et al. Susceptibility to dengue hemorrhagic fever in Vietnam: evidence of an association with variation in the vitamin d receptor and Fc gamma receptor IIa genes. Am J Trop Med Hyg (2002) 67(1):102–6.
81. Garcia G, Sierra B, Perez AB, Aguirre E, Rosado I, Gonzalez N, et al. Asymptomatic dengue infection in a Cuban population confirms the protective role of the RR variant of the FcgammaRIIa polymorphism. Am J Trop Med Hyg (2010) 82(6):1153–6. doi:10.4269/ajtmh.2010.09-0353
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Balsitis SJ, Williams KL, Lachica R, Flores D, Kyle JL, Mehlhop E, et al. Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification. PLoS Pathog (2010) 6(2):e1000790. doi:10.1371/journal.ppat.1000790
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Zellweger RM, Prestwood TR, Shresta S. Enhanced infection of liver sinusoidal endothelial cells in a mouse model of antibody-induced severe dengue disease. Cell Host Microbe (2010) 7(2):128–39. doi:10.1016/j.chom.2010.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Williams KL, Wahala WM, Orozco S, de Silva AM, Harris E. Antibodies targeting dengue virus envelope domain III are not required for serotype-specific protection or prevention of enhancement in vivo. Virology (2012) 429(1):12–20. doi:10.1016/j.virol.2012.03.003
85. Williams KL, Sukupolvi-Petty S, Beltramello M, Johnson S, Sallusto F, Lanzavecchia A, et al. Therapeutic efficacy of antibodies lacking Fcgamma receptor binding against lethal dengue virus infection is due to neutralizing potency and blocking of enhancing antibodies [corrected]. PLoS Pathog (2013) 9(2):e1003157. doi:10.1371/journal.ppat.1003157
86. Beltramello M, Williams KL, Simmons CP, Macagno A, Simonelli L, Quyen NT, et al. The human immune response to dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe (2010) 8(3):271–83. doi:10.1016/j.chom.2010.08.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Brien JD, Sukupolvi-Petty S, Williams KL, Lam CY, Schmid MA, Johnson S, et al. Protection by immunoglobulin dual-affinity retargeting antibodies against dengue virus. J Virol (2013) 87(13):7747–53. doi:10.1128/JVI.00327-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi:10.1016/j.cell.2010.01.022
89. Wang JP, Liu P, Latz E, Golenbock DT, Finberg RW, Libraty DH. Flavivirus activation of plasmacytoid dendritic cells delineates key elements of TLR7 signaling beyond endosomal recognition. J Immunol (2006) 177(10):7114–21. doi:10.4049/jimmunol.177.10.7114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Nasirudeen AM, Wong HH, Thien P, Xu S, Lam KP, Liu DX. RIG-I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLoS Negl Trop Dis (2011) 5(1):e926. doi:10.1371/journal.pntd.0000926
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Kurane I, Ennis FA. Induction of interferon alpha from human lymphocytes by autologous, dengue virus-infected monocytes. J Exp Med (1987) 166(4):999–1010. doi:10.1084/jem.166.4.999
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Kurane I, Ennis FA. Production of interferon alpha by dengue virus-infected human monocytes. J Gen Virol (1988) 69(Pt 2):445–9. doi:10.1099/0022-1317-69-2-445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Gandini M, Gras C, Azeredo EL, Pinto LM, Smith N, Despres P, et al. Dengue virus activates membrane TRAIL relocalization and IFN-alpha production by human plasmacytoid dendritic cells in vitro and in vivo. PLoS Negl Trop Dis (2013) 7(6):e2257. doi:10.1371/journal.pntd.0002257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Pichyangkul S, Endy TP, Kalayanarooj S, Nisalak A, Yongvanitchit K, Green S, et al. A blunted blood plasmacytoid dendritic cell response to an acute systemic viral infection is associated with increased disease severity. J Immunol (2003) 171(10):5571–8. doi:10.4049/jimmunol.171.10.5571
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Decembre E, Assil S, Hillaire ML, Dejnirattisai W, Mongkolsapaya J, Screaton GR, et al. Sensing of immature particles produced by dengue virus infected cells induces an antiviral response by plasmacytoid dendritic cells. PLoS Pathog (2014) 10(10):e1004434. doi:10.1371/journal.ppat.1004434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Chen RF, Yang KD, Wang L, Liu JW, Chiu CC, Cheng JT. Different clinical and laboratory manifestations between dengue haemorrhagic fever and dengue fever with bleeding tendency. Trans R Soc Trop Med Hyg (2007) 101(11):1106–13. doi:10.1016/j.trstmh.2007.06.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Simmons CP, Popper S, Dolocek C, Chau TN, Griffiths M, Dung NT, et al. Patterns of host genome-wide gene transcript abundance in the peripheral blood of patients with acute dengue hemorrhagic fever. J Infect Dis (2007) 195(8):1097–107. doi:10.1086/512162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Popper SJ, Gordon A, Liu M, Balmaseda A, Harris E, Relman DA. Temporal dynamics of the transcriptional response to dengue virus infection in Nicaraguan children. PLoS Negl Trop Dis (2012) 6(12):e1966. doi:10.1371/journal.pntd.0001966
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Yu CY, Chang TH, Liang JJ, Chiang RL, Lee YL, Liao CL, et al. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog (2012) 8(6):e1002780. doi:10.1371/journal.ppat.1002780
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog (2012) 8(10):e1002934. doi:10.1371/journal.ppat.1002934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Schoggins JW. Interferon-stimulated genes: roles in viral pathogenesis. Curr Opin Virol (2014) 6:40–6. doi:10.1016/j.coviro.2014.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Ho LJ, Hung LF, Weng CY, Wu WL, Chou P, Lin YL, et al. Dengue virus type 2 antagonizes IFN-alpha but not IFN-gamma antiviral effect via down-regulating Tyk2-STAT signaling in the human dendritic cell. J Immunol (2005) 174(12):8163–72. doi:10.4049/jimmunol.174.12.8163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Ashour J, Laurent-Rolle M, Shi PY, Garcia-Sastre A. NS5 of dengue virus mediates STAT2 binding and degradation. J Virol (2009) 83(11):5408–18. doi:10.1128/JVI.02188-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Mazzon M, Jones M, Davidson A, Chain B, Jacobs M. Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J Infect Dis (2009) 200(8):1261–70. doi:10.1086/605847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Morrison J, Laurent-Rolle M, Maestre AM, Rajsbaum R, Pisanelli G, Simon V, et al. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog (2013) 9(3):e1003265. doi:10.1371/journal.ppat.1003265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Munoz-Jordan JL, Sanchez-Burgos GG, Laurent-Rolle M, Garcia-Sastre A. Inhibition of interferon signaling by dengue virus. Proc Natl Acad Sci U S A (2003) 100(24):14333–8. doi:10.1073/pnas.2335168100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Diamond MS, Roberts TG, Edgil D, Lu B, Ernst J, Harris E. Modulation of dengue virus infection in human cells by alpha, beta, and gamma interferons. J Virol (2000) 74:4957–66. doi:10.1128/JVI.74.17.7814-7823.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Diamond MS, Harris E. Interferon inhibits dengue virus infection by preventing translation of viral RNA through a PKR-independent mechanism. Virology (2001) 289:297–311. doi:10.1006/viro.2001.1114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Chase AJ, Medina FA, Munoz-Jordan JL. Impairment of CD4+ T cell polarization by dengue virus-infected dendritic cells. J Infect Dis (2011) 203(12):1763–74. doi:10.1093/infdis/jir197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Halstead SB, Mahalingam S, Marovich MA, Ubol S, Mosser DM. Intrinsic antibody-dependent enhancement of microbial infection in macrophages: disease regulation by immune complexes. Lancet Infect Dis (2010) 10(10):712–22. doi:10.1016/S1473-3099(10)70166-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Chareonsirisuthigul T, Kalayanarooj S, Ubol S. Dengue virus (DENV) antibody-dependent enhancement of infection upregulates the production of anti-inflammatory cytokines, but suppresses anti-DENV free radical and pro-inflammatory cytokine production, in THP-1 cells. J Gen Virol (2007) 88(Pt 2):365–75. doi:10.1099/vir.0.82537-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Modhiran N, Kalayanarooj S, Ubol S. Subversion of innate defenses by the interplay between DENV and pre-existing enhancing antibodies: TLRs signaling collapse. PLoS Negl Trop Dis (2010) 4(12):e924. doi:10.1371/journal.pntd.0000924
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Chan KR, Ong EZ, Tan HC, Zhang SL, Zhang Q, Tang KF, et al. Leukocyte immunoglobulin-like receptor B1 is critical for antibody-dependent dengue. Proc Natl Acad Sci U S A (2014) 111(7):2722–7. doi:10.1073/pnas.1317454111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Sun P, Celluzzi CM, Marovich M, Subramanian H, Eller M, Widjaja S, et al. CD40 ligand enhances dengue viral infection of dendritic cells: a possible mechanism for T cell-mediated immunopathology. J Immunol (2006) 177(9):6497–503. doi:10.4049/jimmunol.177.9.6497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Wong KL, Chen W, Balakrishnan T, Toh YX, Fink K, Wong SC. Susceptibility and response of human blood monocyte subsets to primary dengue virus infection. PLoS One (2012) 7(5):e36435. doi:10.1371/journal.pone.0036435
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Palmer DR, Sun P, Celluzzi C, Bisbing J, Pang S, Sun W, et al. Differential effects of dengue virus on infected and bystander dendritic cells. J Virol (2005) 79(4):2432–9. doi:10.1128/JVI.79.4.2432-2439.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol (2006) 311:17–58. doi:10.1007/3-540-32636-7-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Dejnirattisai W, Duangchinda T, Lin CL, Vasanawathana S, Jones M, Jacobs M, et al. A complex interplay among virus, dendritic cells, T cells, and cytokines in dengue virus infections. J Immunol (2008) 181(9):5865–74. doi:10.4049/jimmunol.181.9.5865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Nightingale ZD, Patkar C, Rothman AL. Viral replication and paracrine effects result in distinct, functional responses of dendritic cells following infection with dengue 2 virus. J Leukoc Biol (2008) 84(4):1028–38. doi:10.1189/jlb.0208105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Rodriguez-Madoz JR, Bernal-Rubio D, Kaminski D, Boyd K, Fernandez-Sesma A. Dengue virus inhibits the production of type I interferon in primary human dendritic cells. J Virol (2010) 84(9):4845–50. doi:10.1128/JVI.02514-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Sun P, Beckett C, Danko J, Burgess T, Liang Z, Kochel T, et al. A dendritic cell-based assay for measuring memory T cells specific to dengue envelope proteins in human peripheral blood. J Virol Methods (2011) 173(2):175–81. doi:10.1016/j.jviromet.2011.01.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Sun P, Garcia J, Comach G, Vahey MT, Wang Z, Forshey BM, et al. Sequential waves of gene expression in patients with clinically defined dengue illnesses reveal subtle disease phases and predict disease severity. PLoS Negl Trop Dis (2013) 7(7):e2298. doi:10.1371/journal.pntd.0002298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol (2012) 12(8):557–69. doi:10.1038/nri3254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Haniffa M, Collin M, Ginhoux F. Identification of human tissue cross-presenting dendritic cells: a new target for cancer vaccines. Oncoimmunology (2013) 2(3):e23140. doi:10.4161/onci.23140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: dengue virus, dendritic cells, monocytes, macrophages, innate immunity, antibody-dependent enhancement, immune evasion
Citation: Schmid MA, Diamond MS and Harris E (2014) Dendritic cells in dengue virus infection: targets of virus replication and mediators of immunity. Front. Immunol. 5:647. doi: 10.3389/fimmu.2014.00647
Received: 09 October 2014; Paper pending published: 05 November 2014;
Accepted: 04 December 2014; Published online: 17 December 2014.
Edited by:
Stipan Jonjic, University of Rijeka, CroatiaReviewed by:
Dina Weilhammer, Lawrence Livermore National Laboratory, USAKatja Fink, Singapore Immunology Network (SIgN), Singapore
Copyright: © 2014 Schmid, Diamond and Harris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael A. Schmid, Division of Infectious Diseases and Vaccinology, 510D Li Ka Shing Center, School of Public Health, University of California, 1951 Oxford Street, Berkeley, CA 94720-3370, USA e-mail:bWljaGFlbC5hLnNjaG1pZEBiZXJrZWxleS5lZHU=;
Eva Harris, Division of Infectious Diseases and Vaccinology, 185 Li Ka Shing Center, School of Public Health, University of California, 1951 Oxford Street, Berkeley, CA 94720-3370, USA e-mail:ZWhhcnJpc0BiZXJrZWxleS5lZHU=